

Abstract
Background
Rigorous preclinical studies of chimeric antigen receptor (CAR) immunotherapy will require large quantities of consistent and high-quality CAR-transduced T (CART) cells that can be used in syngeneic mouse glioblastoma (GBM) models. To this end, we developed a novel transgenic (Tg) mouse strain with a fully murinized CAR targeting epidermal growth factor receptor variant III (EGFRvIII).
Methods
We first established the murinized version of EGFRvIII-CAR and validated its function using a retroviral vector (RV) in C57BL/6J mice bearing syngeneic SB28 GBM expressing EGFRvIII. Next, we created C57BL/6J-background Tg mice carrying the anti-EGFRvIII-CAR downstream of a Lox-Stop-Lox cassette in the Rosa26 locus. We bred these mice with CD4-Cre Tg mice to allow CAR expression on T cells and evaluated the function of the CART cells both in vitro and in vivo. To inhibit immunosuppressive myeloid cells within SB28 GBM, we also evaluated a combination approach of CART and an anti-EP4 compound (ONO-AE3-208).
Results
Both RV- and Tg-CART cells demonstrated specific cytotoxic activities against SB28-EGFRvIII cells. A single intravenous infusion of EGFRvIII-CART cells prolonged the survival of glioma-bearing mice when preceded by a lymphodepletion regimen with recurrent tumors displaying profound EGFRvIII loss. The addition of ONO-AE3-208 resulted in long-term survival in a fraction of CART-treated mice and those survivors demonstrated delayed growth of subcutaneously re-challenged both EGFRvIII+ and parental EGFRvIII− SB28.
Conclusion
Our new syngeneic CAR Tg mouse model can serve as a useful tool to address clinically relevant questions and develop future immunotherapeutic strategies.
Keywords: CART cells, EGFRvIII, glioblastoma, immunotherapy, transgenic
Key Points.
The transgenic CAR mice serve as a reliable and predictable source of CART cells.
The syngeneic and clinically relevant GBM model allows mechanistic research.
The tumor model provides the opportunity to evaluate combination therapies targeting immunosuppressive myeloid cells together with CART-cell therapy.
Importance of the Study.
The majority of preclinical studies evaluating CART therapy for GBM have utilized xenografts implanted into immunocompromised mice. Because the successful development of these strategies will depend on the understanding of critical interactions between therapeutic cells and the endogenous immune environment, it is essential to develop a novel immunocompetent system that allows us to study these interactions in a robust and reproducible manner. We created a Tg mouse strain in which all T cells express a murinized EGFRvIII-CAR. T cells derived from these mice consistently express EGFRvIII-specific CAR while traditional transduction with a CAR vector showed batch-to-batch variability. The Tg-CART mice represent a novel system for robust, and reproducible preclinical investigations.
CART (chimeric antigen receptor T cells) therapy represents a promising immunotherapeutic modality based on remarkable outcomes in treating hematologic malignancies.1 However, the development of effective CART therapies for glioblastoma (GBM) must overcome multiple barriers, including antigenic heterogeneity, CART cell homing to GBM, and suppressive tumor microenvironment (TME).2–4
Indeed, the vast majority of preclinical CART studies have utilized CAR-transduced human T cells and xenografts in immunocompromised mice. To understand the resistance mechanisms and develop effective CART therapy strategies, a clinically relevant and immunocompetent preclinical model is needed. To adequately evaluate the study questions in multiple arms, experiments often require a large number and consistent quality of syngeneic murine CART cells. However, transduction of murine T cells with viral CAR vectors may result in inconsistent batch-to-batch quality due to variable transduction efficiencies and the random genomic integration sites for the CAR transgene.
To overcome these challenges, we generated a novel Tg mouse strain with Rosa26-targeted genetic insertion of EGFRvIII-specific5 CAR cDNA. When bred with CD4-Cre Tg mice, all T cells derived from these Tg mice constitutively express a murinized EGFRvIII-CAR, thereby serving as a continuous, reliable, and predictable source of CART cells. We chose EGFRvIII as the CAR target because the first-in-human trial of i.v. administered EGFRvIII-CART cells in patients with recurrent GBM6 demonstrated both an encouraging safety profile and also important issues to overcome. i.v.-infused CART cells infiltrated GBM tumors and reduced the number of EGFRvIII-positive GBM cells, but failed to demonstrate clinical benefits associated with induction of immunoregulatory mechanisms in the TME.6
As a tumor model, we developed a C57BL/6J-background SB28 GBM cell line7,8 engineered to express EGFRvIII. We have shown that SB28 cells recapitulate key characteristics of human GBM, such as aggressive growth, low mutation burden, and resistance to immune checkpoint blockade therapy,7,8 allowing us to investigate many clinically relevant questions.9
The GBM microenvironment is largely composed of immunosuppressive myeloid cells, including myeloid-derived suppressor cells (MDSCs).10,11 As a mechanism promoting MDSC accumulation in gliomas, we and others have shown the critical role of prostaglandin-E2 (PGE2).7,12,13 Therefore, we evaluated a combination of i.v. EGFRvIII-CART therapy and inhibition of MDSCs using ONO-AE3-208, a selective inhibitor of the EP4 receptor,14 which is known to mediate PGE2-induced promotion of immunosuppressive myeloid cells.15
Materials and Methods
Rosa26-Targeting mCAR Vector
The 3C10 antibody-derived single-chain variable fragment (scFv, Supplementary Figure 1A) was linked with murine CD8α hinge and transmembrane domain. The intracellular transducing domains of the murine co-stimulating factors CD28 and 4-1BB were added followed by the murine CD3ζ-activating domain (Figure 1A). The entire murinized construct (mCAR) sequence was codon-optimized for expression efficiency and synthesized by GenScript. Binding sites for the restriction enzyme AscI were added to the 5′ and 3′ ends of the construct by PCR and used to insert the mCAR sequence into the Rosa26-targeting vector CTV [(Addgene #15912), Figure 2A].
Fig. 1.

A single intravenous infusion of EGFRvIII-specific CART cells prolongs survival of tumor-bearing mice but results in recurrence demonstrating target antigen loss. (A) mCAR domains. (B) Treatment protocol schematic and Kaplan-Meier survival curves. No treatment (n = 7; MS 19 days), LD (n = 7; MS 24 days), RV-CART (n = 8; MS 19 days), LD + RV-CART (n = 8; MS 28 days). (C) Tumor samples from mice at the endpoint were collected and analyzed by FC for EGFRvIII expression. (D) Summary of FC staining for myeloid populations in samples from (C). (E) Prospective study of tumor tissues 3-day post-LD + CART i.v. infusion. Representative FC diagrams for tumor single-cell suspensions are shown. Abbreviations: CART, chimeric antigen receptor-transduced T cells; FC, flow cytometry; LD, lymphodepletion; MS, median survival.
Fig. 2.

Generation of transgenic mice with the conditional EGFRvIII-CAR allele at the Rosa26 locus (A) The Rosa26 chromosome 6 locus and the location of the targeted insertion. Restriction sites for EcoRI and BgII enzymes and locations for Probes A and B are indicated. (B) Correct genomic targeting of the transgene at the 3′ end was confirmed by Southern blotting of BgII-digest genomic DNA with Probe B. (C) Overview of targeting the conditional allele to the Rosa26 locus of C57BL/6J embryonic stem (ES) cells and generation of founder mice.
Transgenic Mice
The CTV-mCAR construct was linearized using the AsiSI enzyme (New England Biolabs) and electroporated into PRXB6T (C57BL/6J) embryonic stem cells (ESC). Genomic DNA from multiple clones, resulting from G418 selection of the ESC, was screened by long-range PCR (Supplementary Figure 2A and B) and Southern blot (Figure 2B, Supplementary Figure 2C–E) to confirm the intended insertion of the CTV-mCAR construct at the Rosa26 locus. Selected ESC clones were injected into 3.5-day embryos from B6(Cg)-Tyrc2J/J (B6-albino; JAX 000058) by the Transgenic Gene Targeting Core at UCSF. A total of 62 embryos were injected and implanted into recipient B6-albino female mice. Eight chimeric male founder mice were born. The selected founder (26-4) was then bred with C57BL/6J females (JAX 000664) to establish the CAR knock-in (Ki) colony (Figure 2C).
The CAR Ki mice were bred with the CD4-Cre mouse strain (JAX 022071), and the resulting offspring were genotyped by PCR for expression of both transgenes. All mice used for breeding and experiments maintained Cre hemizygosity to minimize any unintended consequences of the Cre random insertion. The primer list is available in Supplementary Table 1.
Retroviral Vectors
Standard cloning methods were used to generate the pMSCV-m3C10-IRES-GFP, pMSCV-m3C10, and pBABE-hEGFRvIII retroviral vectors (RV). Method details are described in the Supplementary Materials and Methods.
Retroviral Supernatants
Standard packaging plasmid pCL-Eco and a gene-of-interest expressing retroviral plasmid were used to obtain retrovirus-containing medium used in all transduction assays. A detailed protocol is available in the Supplementary Materials and Methods.
Retroviral Transduction of Mouse T Cells
Procedure details are described in the Supplementary Materials and Methods section.
SB28-EGFRvIII Cell Line
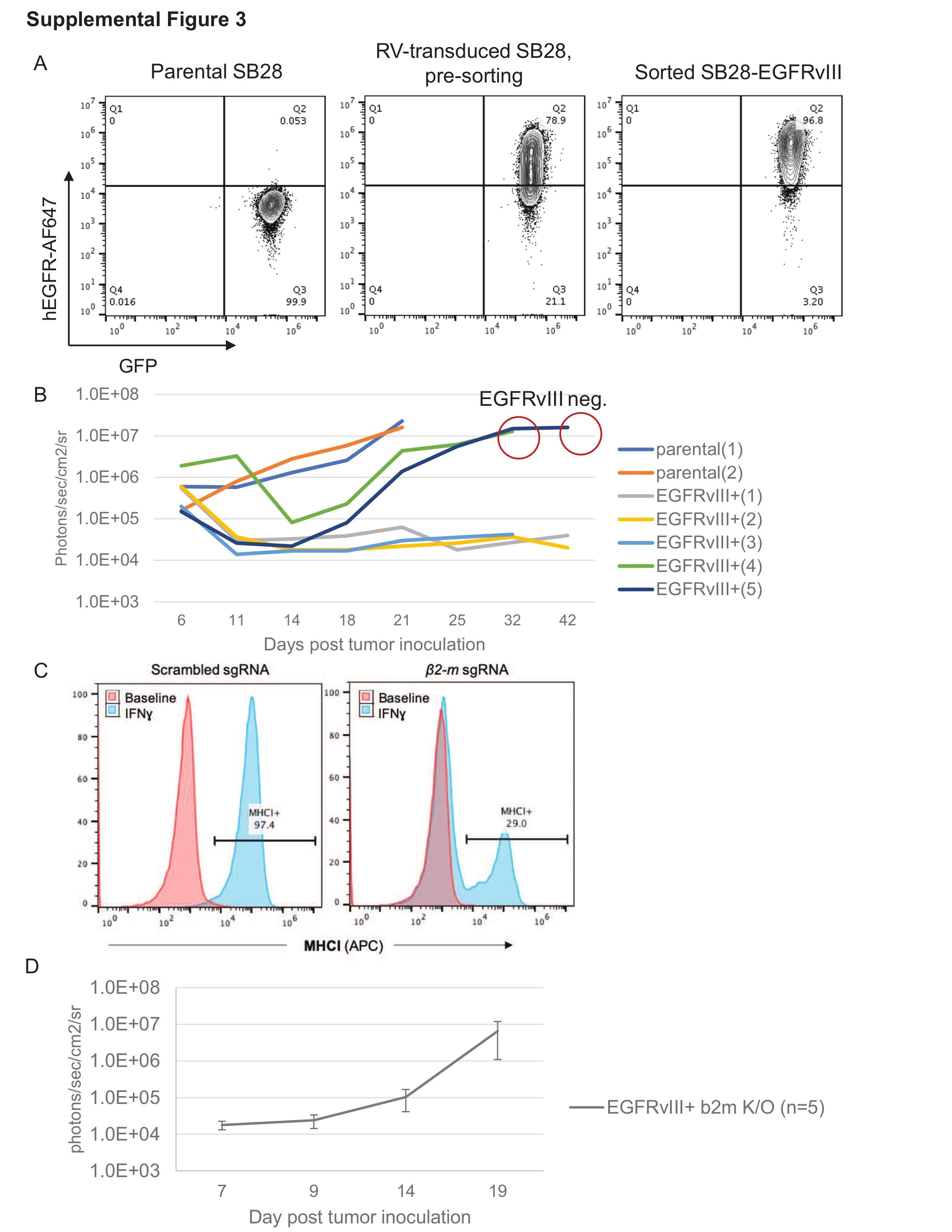
A detailed description of the cell line is available in Supplementary Materials and Methods and Supplementary Figure 3.
Real-Time Cytotoxicity Assay (xCELLigence)
CD8+ CART cells were added when the target’ cell index (CI) value reached 1, and impedance measurements were performed every 15 minutes. CI values were used to calculate the percentage of tumor cell lysis (experimental) relative to cells in the absence of CART cells (spontaneous) using the formula [1–(experimental/spontaneous)] × 100.
Orthotopic Glioma Models
All experiments used 6- to 10-week-old female C57BL/6J mice (JAX). Animals were handled in the Animal Facility at UCSF, per an Institutional Animal Care and Use Committee-approved protocol. Anesthetized mice received stereotactic inoculations of 5 × 103 cells as previously described.16
Isolation of Tissue-Infiltrating Leukocytes
The procedure for the isolation of brain-infiltrating leukocytes (BILs) using Percoll (GE Healthcare Life Sciences) was previously described.17 To isolate tumor-infiltrating leukocytes (TILs), tumors were macro-dissected from surrounding brain tissue first.
In Vitro Differentiation and Analysis of Mouse Bone Marrow
A detailed description is available in the Supplementary Methods. Briefly, bone marrow from C57BL/6J mice was cultured in 50% cRPMI/50% SB28 conditioned media (CM) either in the presence of solvent control or ONO-AE3-208 at indicated concentrations for 10 days. Adherent cells were analyzed by flow cytometry (FC) or subjected to RNA isolation using the RNeasy Mini Kit (Qiagen #74106).
Flow Cytometry
Single-cell suspensions were stained with fluorescently labeled antibodies using concentrations recommended by the manufacturers. A list of the antibodies used is available in Supplementary Table 2. A detailed description of the FC procedure is provided in the Supplementary Materials and Methods.
PCR-Based Genotyping
Details on the PCR-based genotyping are described in the Supplementary Materials and Methods.
Reagents
Lymphodepletion: combination of cyclophosphamide (4 mg/mouse) and fludarabine (1 mg/mouse) administered i.p.
EP4 inhibition in vivo: ONO-AE3-208 (Sigma, SML2076) was resuspended at 1 mg/ml in water, and 10 mg/kg body weight was administered daily p.o.
Statistical Analyses
Log-rank (Mantel-Cox) test by GraphPad Prism software (v8.1) was used to determine significance between Kaplan-Meier survival plots. Results from experiments with only 2 groups were analyzed using Student t test. For experiments with more than 2 groups, results were analyzed by 1-way ANOVA followed by Tukey posttest. All data are shown as mean ± SEM unless otherwise indicated. In vivo treatment groups were repeated as described in Supplementary Table 6. Symbols indicate statistical significance as follows: *P < .05; **P < .002; ***P < .0002.
Results
Murinized EGFRvIII-CART Cells Display EGFRvIII-Specific Cytotoxic Effects and Prolong Survival of Syngeneic Mice Bearing EGFRvIII+ Glioma
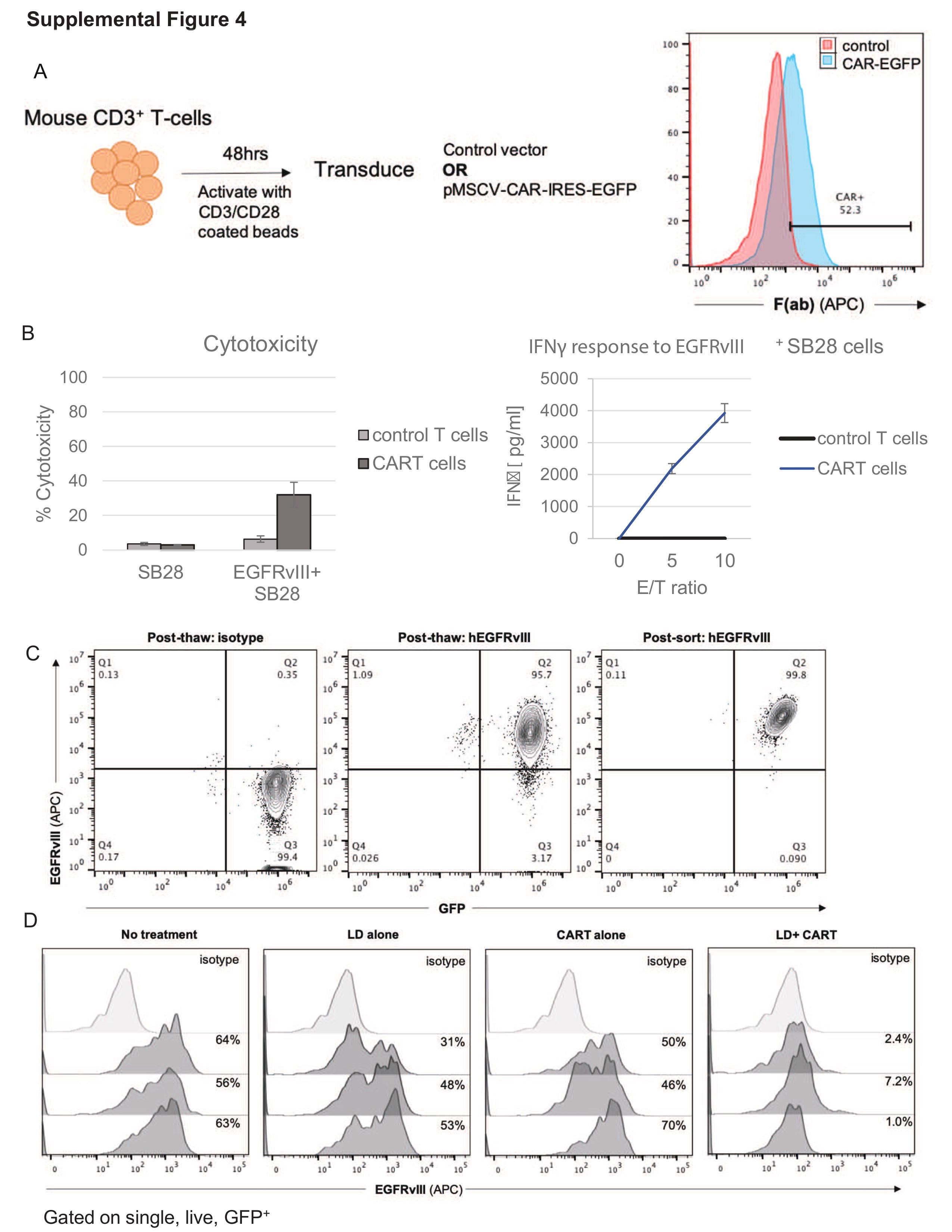
As a first step in establishing a preclinical immunocompetent model, we evaluated the therapeutic efficacy of T cells transduced with an RV encoding a murinized anti-EGFRvIII-CAR (RV-CART; Figure 1A and Supplementary Figure 1A) in syngeneic mice bearing SB28-EGFRvIII+ gliomas. The SB28-EGFRvIII tumors grew aggressively in vivo and resulted in median survival (MS) of 19 days if untreated (Figure 1B). RV-CART cells showed specific cytotoxicity against EGFRvIII+ SB28 tumor cells in vitro (Supplementary Figure 4A and B). In vivo (Figure 1B), RV-CART treatment alone showed no therapeutic benefits compared to untreated mice, underscoring the need for lymphodepletion (LD) prior to systemic adoptive cell therapy (i.v. ACT).18 LD alone significantly extended the MS of tumor-bearing mice (24 days vs No tx P = .04). Combining LD and CART, however, led to the longest MS (28 days vs CART alone P = .0004). Upon necropsy (Figure 1C), while tumors from untreated or single therapy-treated mice retained some expression of the target antigen (range 31%-70% positive; Supplementary Figure 4D), tumor cells from combination-treated animals displayed a significant loss of EGFRvIII expression (3.5 ± 1.9% vs CART alone P = .0006), strongly reminiscent of human trial observations.6 In the TME, we observed that a high proportion of each tumor was composed of CD11b+ cells (Figure 1D). Combination treatment did not influence this observation. However, XCR1+CD11b− dendritic cells, which can cross-present intracellular-derived antigens,19 significantly increased in the animals treated with both LD and RV-CART cells (Figure 1D, right). In a smaller, prospective cohort, we examined the levels of programmed death-ligand 1 (PD-L1) expression 3 days after treatment with LD + CART (Figure 1E). We detected a small population of CD45.1+ RV-CART cells within the tumors. Similar to clinical trial observations,6 PD-L1 expression on tumor cells was variable. However, 2 of 3 tumors derived from RV-CART-infused mice showed upregulation of PD-L1 signal and an increased number of positive cells (Figure 1E).
Generation of Transgenic Mice
To overcome challenges inherent to producing large numbers of consistent and high-quality CART cells needed for multi-arm in vivo studies, we generated a novel mouse strain using targeted genetic insertion of the EGFRvIII-specific CAR. We cloned the mCAR expression cassette (Figure 1A) into the Rosa26 targeting vector CTV (Figure 2A) between the Lox-Stop-Lox (LSL) cassette and the frt-flanked IRES-EGFP sequence (ie, Rosa-LSL-mCAR-EGFP vector). Following the electroporation of C57BL/6J ESC with the Rosa-LSL-mCAR-EGFP vector, 4 clones (26, 27, 32, and 36) had the Ki cassette in the targeted Rosa26 locus (Figure 2B, Supplementary Figure 4). We selected clones #26 and #32 for further expansion and blastocyst microinjection, resulting in 8 chimeric male mice. After breeding 6 of the chimeras to B6-albino females, 4 out of 6 chimeras demonstrated germline transmission of the injected ESCs by producing offspring with black coats. Founder 26-4 was selected to establish the CAR Ki colony (summarized in Figure 2C).
Tg-CART Cells Demonstrate EGFRvIII-Specific Cytotoxicity In Vitro
To validate the functionality of the transgene, we isolated T cells from the spleens of CAR Ki mice and transduced them with a Cre-mCherry RV (Figure 3A). CAR Ki T cells were readily transduced with the control mCherry virus (middle panels) and showed no background expression of EGFP (readout for transgene cassette expression). Specifically, after transduction with Cre, we detected expression of EGFP in both CD4+ and CD8+ T cells (bottom panels).
Fig. 3.

The transgene CAR is expressed following Cre recombinase expression. (A) CD3+ T cells, isolated from the spleens of CAR Ki mice, were transduced with a Cre-mCherry RV, a control RV (“empty virus”), or sham control “no virus” media. Five days after transduction, T cells were analyzed by FC for the expression of mCherry and EGFP. (B) CAR Ki mice were bred with CD4-Cre mice to induce the expression of the Ki cassette in vivo. Representative flow plots are shown of spleen-derived CD4+ and CD8+ T cells. (C) CD8+ and CD4+ T cells from CAR Ki+; Cre− and CAR Ki+; Cre+ were analyzed for the expression of EGFP and mouse F(ab). (D) RV- and Tg-CART cells induce cytotoxicity against EGFRvIII+ SB28 cells. CD8+ CART cells were co-cultured with SB28-EGFRvIII+ or parental SB28 (control) cells at different effector to target (E/T) ratios. Data from 2 separate experiments show variability in RV-CART performance. Abbreviations: CAR, chimeric antigen receptor; RV, retroviral vector.
To induce expression of the CAR in T cells in vivo, we bred the CAR Ki mice to the CD4-Cre transgenic mice. CARki/wt; CD4-Cre+ (referred to as Tg mice hereafter) mice appeared anatomically healthy, without observable differences in size or weight compared to WT or CARki/wt; CD4-Cre− controls (data not shown). Spleens from Tg mice displayed normal cellularity (Figure 3B, Supplementary Figure 5A). As the CD4-Cre mice express Cre in all T cells during the double-positive stage of thymic development,20 the CAR should be expressed in both CD4+ and CD8+ mature T-cell subsets (Tg-CART cells). In splenocytes, we observed EGFP signal in all CD4+ and CD8+ T cells (Figure 3C, top panel) but not NK cells, B cells, or myeloid cells (Supplementary Figure 5E). Flow staining with anti-mouse F(ab) antibody directly detected CAR expression in splenic Tg-CART cells, with no observable differences between male and female mice (Supplementary Figure 5B). The absence of gross pathological phenotype suggests that the CAR reactivity is specific to the human EGFRvIII neoantigen, thereby sparing the host mice from off-target toxicities.
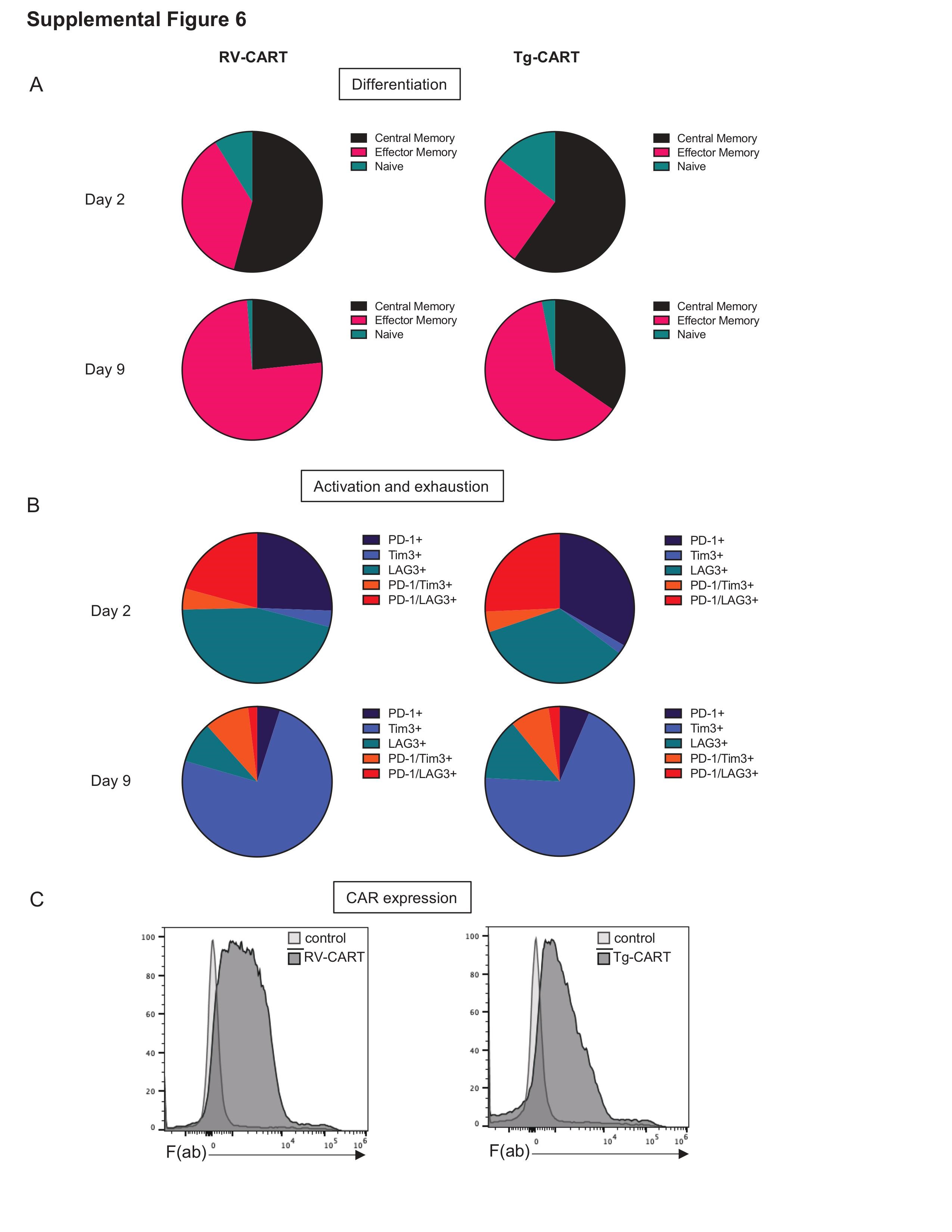
Next, we tested the functionality of the Tg-CART cells in vitro. We generated CD8+ CART cells through either retroviral transduction of WT C57BL/6 T cells (RV-CART) or by isolating them from age-matched Tg mice (Tg-CART). In both groups, we activated and expanded the cells under the same conditions. Differentiation and activation markers showed no significant differences between the 2 groups (Supplementary Figure 6). Cytotoxicity assays against SB28-EGFRvIII+ cells or parental SB28 cells (control cells) demonstrated both RV-CART and Tg-CART cells had specific cytotoxicity against EGFRvIII+ cells but not parental SB28 (Figure 3D). We also tested control T cells (sham-transduced or isolated from CARki/wt; CD4-Cre− mice) and saw no effect on the viability of parental or EGFRvIII+ cells (data not shown). It is noteworthy that cytotoxicity by Tg-CART cells at all E/T ratios remained comparable in 2 separate experiments, while RV-CART cells showed variable cytotoxicity levels between experiments (Figure 3D). Overall, these data reveal that T cells from the Tg mouse strain are able to express the EGFRvIII-CAR gene specifically and effectively upon Cre expression, thereby serving as a reliable source of EGFRvIII-specific murine CART cells.
Potent Anti-EGFRvIII+ Glioma Effects of Tg-CART Cells In Vivo
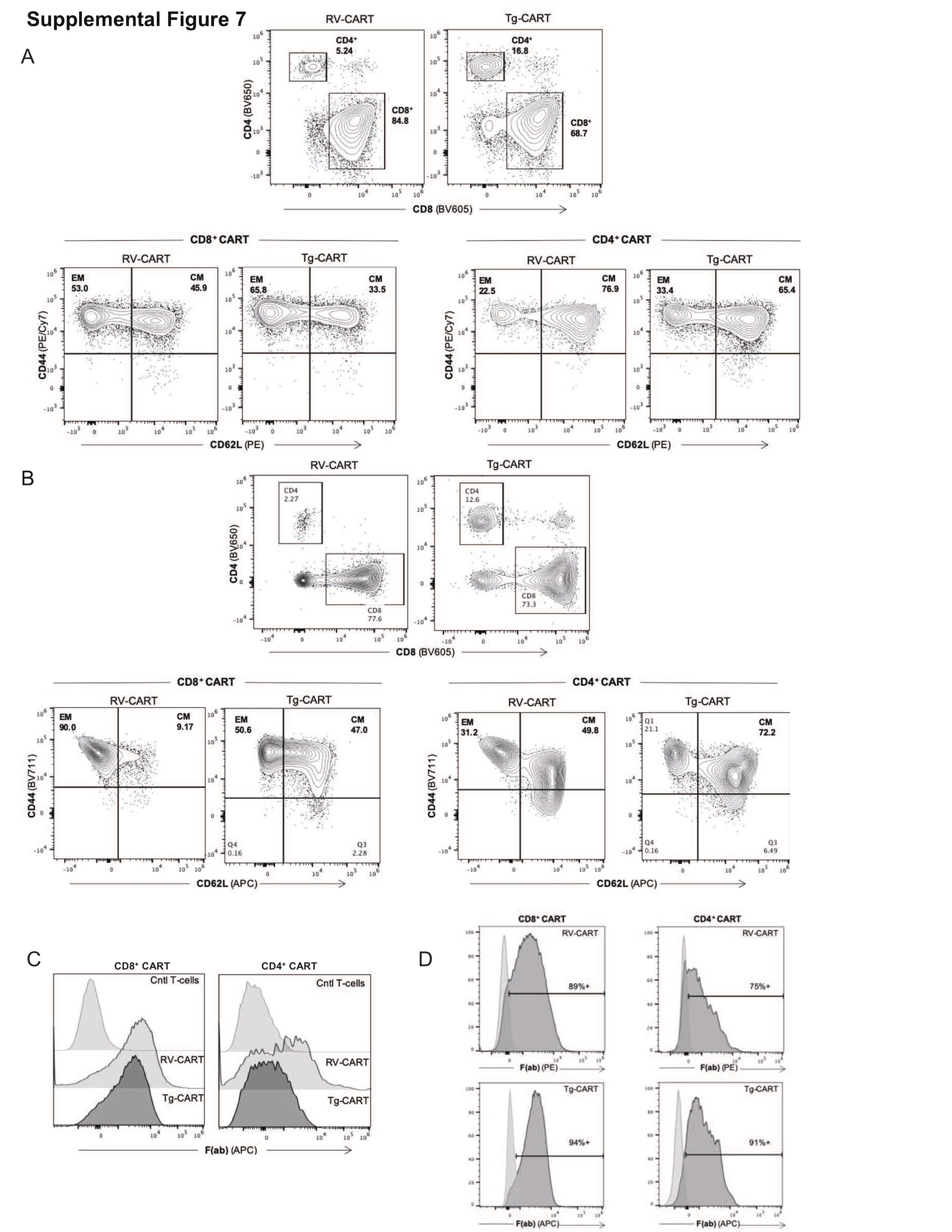
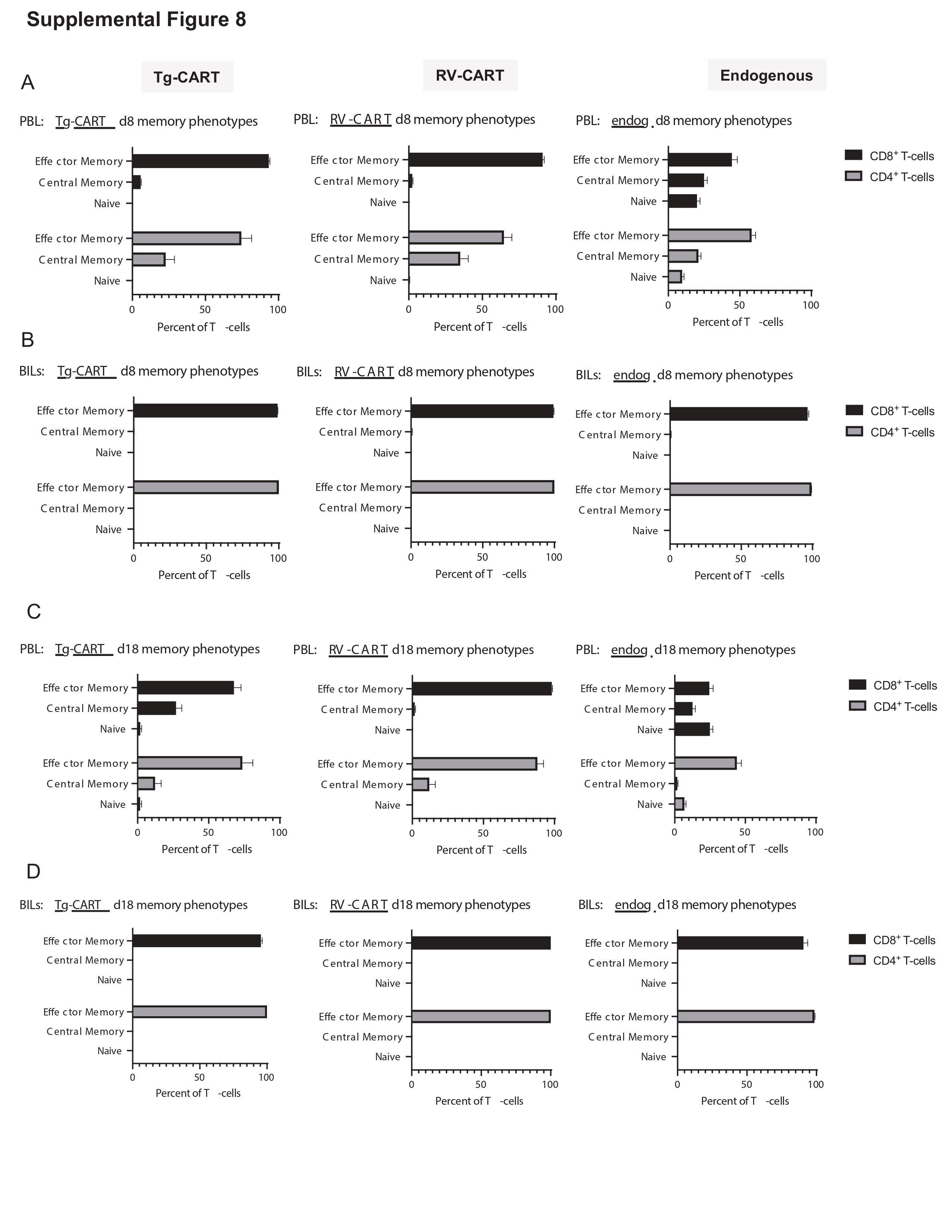
We then evaluated the engraftment and anti-glioma efficacy of Tg-CART cells in comparison to RV-CART cells in vivo. We analyzed lymphoid and nonlymphoid tissues from animals sacrificed 8 days (Figure 4A) and 18 days (Figure 4B) after the i.v. ACT. The FC analysis showed no significant differences in the pre-infusion differentiation status between the Tg- and RV-CART cells (Supplementary Figure 7). As shown in Figure 4B, higher numbers of CD8+ Tg-CART cells were detected compared with CD8+ RV-CART cells in the peripheral blood, the spleen, and the tumor-bearing brain on both days 8 and 18 after ACT except for the spleen on day 8. No significant difference in the levels of CD4+ cells was seen on day 8. However, CD4+ Tg-CART cells were significantly more abundant than RV-CART in the periphery on day 18 (Figure 4B). On day 8 post-infusion, we did not observe significant differences between the Tg-CART and RV-CART populations as to the proportion of central or effector memory cells (Supplementary Figure 8A and B). We noted a measurable population of central memory CD8+ CART cells among the Tg-CART cell population in the peripheral blood at day 18 (Supplementary Figure 8C), whereas the majority of CD8+ RV-CART cells in the blood at day 18 were differentiated effector memory cells. All detected CART cells in the TME appeared to be effector memory T cells. These results indicate that, despite the similar profiles at the time of infusion, Tg-CART cells engrafted better in the periphery and persisted longer in the TME than RV-CART cells.
Fig. 4.

Tg-CART cells persist better following IV infusion in tumor-bearing animals and prolong mouse survival further than RV-CART cells. Tumor-bearing mice were treated as in Figure 1B. Tissues were analyzed by FC for the presence of CART cells 8 days (A) and 18 days (B) after infusion. CART cell abundance is shown as the percentage of all CD3+ lymphocytes. Bars represent the mean of 6 biological replicates for (A) and 3-6 pooled biological replicates for (B). (C) Schematic of the treatment protocol. (D) Kaplan-Meier curves: LD group (MS = 24 days, n = 7), control T cells (MS = 27 days, n = 7), RV-CART cells (MS = 29 days, n = 14), and Tg-CART cells (MS = 34 days, n = 7). Abbreviations: CART, chimeric antigen receptor-transduced T cells; FC, flow cytometry; IV, intravenous; LD, lymphodepletion; MS, median survival.
To evaluate the therapeutic efficacy of Tg-CART cells, mice bearing day 7 SB28-EGFRvIII+ gliomas received LD and i.v. T cells (CAR− T cells [control), RV-CART cells, or Tg-CART cells) on the following day (Figure 4C). While RV-CART cells prolonged survival of treated mice (29 days vs LD P = .03) including 1 long-term survivor, treatment with Tg-CART cells resulted in a longer MS (34 days vs LD P = .004) and long-term survival of 3/7 treated animals (Figure 4D), although the difference did not reach a statistical significance when comparing the RV-CART and Tg-CART cohorts (P = .05).
Inhibition of the PGE2 Pathway Enhances the Efficacy of Anti-EGFRvIII-CART Therapy
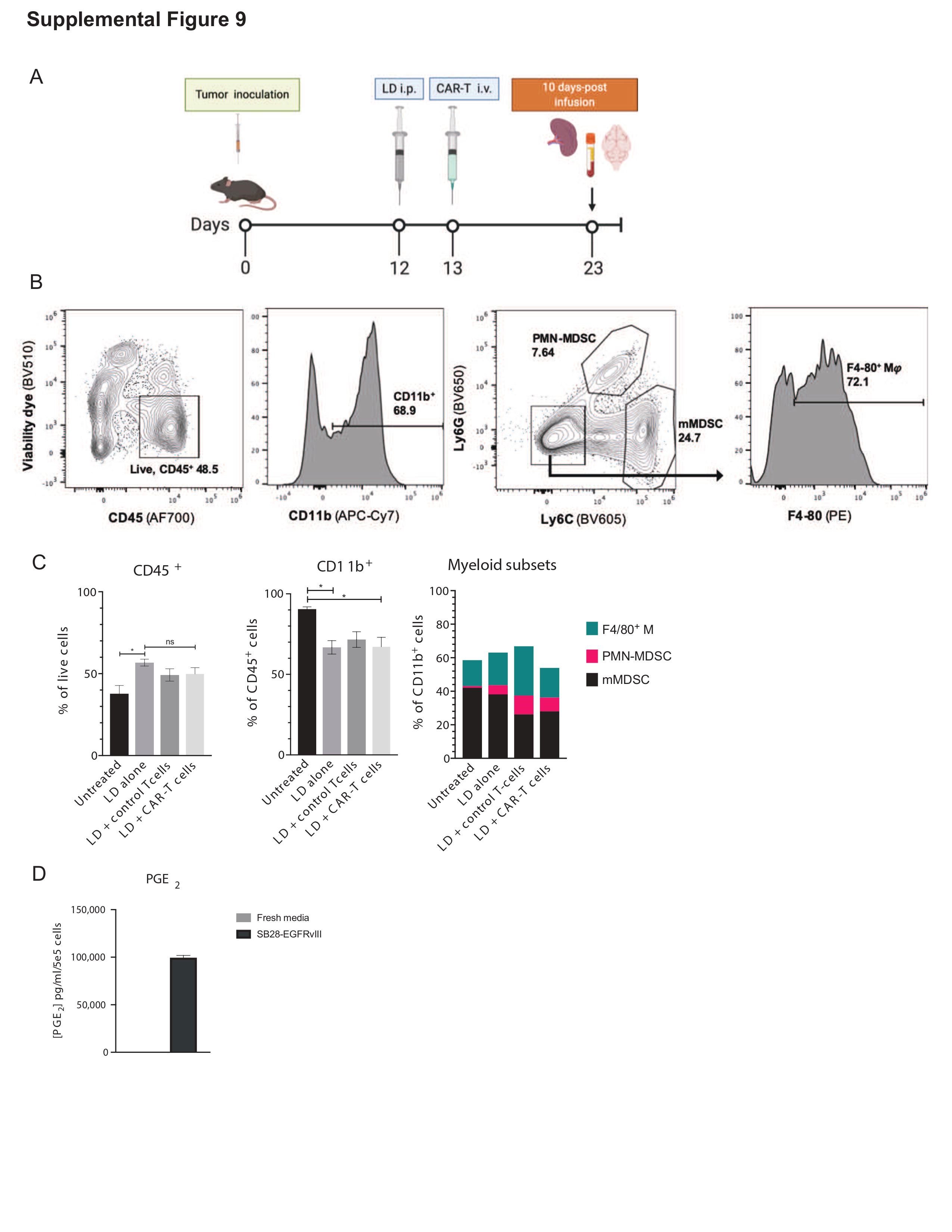
Similar to human GBM, a large component of the immune TME in SB28-EGFRvIII tumors consists of MDSC (Supplementary Figure 9A and B) which were not significantly altered by the CART infusion (Supplementary Figure 9C). We evaluated the MDSC phenotypes in female tumor-bearing hosts in order to be consistent with earlier in vivo experiments. However, as recent findings described significant differences in the response to MDSC-targeted therapy between male and female tumor-bearing mice,21 future studies are necessary to assess any potential differences in MDSC composition in response CART therapy between male and female hosts. Both SB28 and SB28-EGFRvIII cells produced high levels of PGE2 (Figure 5A, Supplementary Figure 9D). PGE2 acts by binding to its receptors EP2 and EP4 on myeloid cells.22 Mouse bone marrow-derived CD11b+ cells cultured in the presence of SB28 CM demonstrated the expression of EP4, but only background levels of EP2 (Figure 5B). A small-molecule antagonist of EP4, ONO-AE3-208,14 significantly decreased the SB28-CM induced expression of Arginase I (ArgI), a known mediator of MDSC-mediated immunosuppression (Figure 5C).23 Furthermore, ONO-AE3-208 treatment effectively decreased the expression of the MDSC marker Gr1 in CM-cultured CD11b+ cells (Figure 5D), suggesting a shift away from their immunosuppressive phenotype.24 In mice bearing SB28 glioma, treatment with ONO-AE3-208 treatment significantly suppressed the expression of immunoregulatory molecules in the TME, such as ArgI, Tgfb1, and FoxP3, and upregulated Ifng in CD3+ lymphocytes (Figure 5E). These data point to the PGE2/EP4-signaling axis as a major contributor to MDSC-induced immunosuppression in SB28 tumors and led us to test the therapeutic efficacy of ONO-AE3-208 when combined with anti-EGFRvIII-CART cells.
Fig. 5.

Inhibition of the PGE2 pathway suppresses myeloid-derived immunosuppression and improves tumor-infiltrating lymphocyte phenotype in vivo. (A) PGE2 production by SB28 glioma cells by ELISA. (B) FC analysis of EP2 and EP4 in bone marrow-derived CD11b+ cells following 6-day culture in the presence of SB28-CM; black histogram: unstained cells, red histogram: cells stained with anti-EP2 or anti-EP4 antibody. (C) ONO-AE3-208 inhibits Arg1 mRNA expression in CD11b+ cells cultured to SB28-CM. (D) ONO-AE3-208 inhibits Gr1 expression in CD11b+ cells cultured in SB28-CM. (E) mRNA levels in glioma-infiltrating leukocytes were evaluated by qRT-PCR. Lymphocytes, CD11b+Ly6G+, and CD11b+Ly6C+ MDSCs were sorted from SB28 glioma-bearing mice treated for 14 days with either vehicle or 10 mg/kg ONO-AE3-208. Bars depict mean values and error bars represent SD of 3 biological replicates. Abbreviations: PGE2, prostaglandin-E2; qRT-PCR, quantitative reverse transcription polymerase chain reaction.
Mice bearing day 7 SB28-EGFRvIII tumors were stratified to receive no treatment (n = 7) or LD (n = 35) (Figure 6A). The following day, LD-treated mice received an i.v. infusion of either control- (n = 7) or RV-CART cells (n = 28). In addition, we began daily p.o. treatment of the RV-CART-treated cohort with either vehicle (n = 14) or 10 mg/kg ONO-AE3-208 (n = 14) for up to 35 days. As in earlier experiments, both LD (MS = 27 days vs Untreated P = .0003) and LD + RV-CART (MS = 29 days vs Untreated P < .0001) extended the survival of tumor-bearing mice significantly. Although ONO-AE3-208 did not extend the MS of CART-treated mice (MS = 28 days), we observed long-term survival in 3/14 (21%) animals treated with the inhibitor, whereas only 1/14 (7%) mouse treated with RV-CART alone remained tumor-free on day 73 (Figure 6A). On day 74, we re-challenged the ONO-AE3-208-treated long-term survivors with s.c. injections of parental SB28 cells (EGFRvIII−; left flank) and SB28-EGFRvIII+ cells (right flank). As a control group, we injected age-matched treatment-naïve mice (n = 3) and measured tumor volumes every 3 days (Figure 6B). EGFRvIII+ tumors in naïve mice grew significantly faster than those in the previously treated mice (Figure 6B). Furthermore, the growth of the parental SB28 cells was inhibited in the treated mice compared with the naïve cohort (P < .05 at all time points, Figure 6B). These data suggest that the treatment regimen of RV-CART + ONO-AE3-208 in mice bearing EGFRvIII+ gliomas induced a memory response to antigens present in both parental and EGFRvIII+ cells; ie, “antigen-spreading” following the initial targeting of EGFRvIII.
Fig. 6.

The use of Tg-CART cells allows for the evaluation of long-term anti-tumor memory response. (A) Treatment protocol for the combination regimen of CART therapy and ONO-AE3-208. Mice receiving control T cells, RV-CART (in A) or Tg-CART (in C) therapy were randomized to receive daily p.o. treatment of either vehicle or 10 mg/kg ONO-AE3-208 for up to 35 consecutive days. Kaplan-Meier curves in (A) Untreated (MS = 20 days, n = 7), LD + Control T cells (MS = 27 days, n = 7), LD + RV-CART cells (MS = 29 days, n = 14), and LD + RV-CART cells + ONO-AE3-208 (MS = 28 days, n = 14); Kaplan-Meier curves in C: LD + Control T cells (MS = 34 days, n = 5), LD + Control T cells + ONO-AE3-208 (MS = 50 days, n = 5), LD + Tg-CART cells (MS not reached, n = 7), LD + Tg-CART + ONO-AE3-208 (MS not reached, n = 7). (B) Long-term survivors from the LD + CART + ONO-AE3 group received s.c. injections of 4 × 105 parental SB28 cells (EGFRvIII−, left flank) and 4 × 105 SB28-EGFRvIII cells (EGFRvIII+, right flank). Tumor size (mm3) was recorded every 3 days. Treatment-naïve, age-matched C57BL6/J mice received identical s.c. injections as controls. (D, E) Long-term survivors treated with Tg-CART cells in (C) were re-challenged with s.c. tumors as described in (B). Sizes of EGFRvIII+ (D) or parental EGFRvIII− tumors (E) in mice treated with LD + Tg-CART cells (pink lines; n = 6), LD + Tg-CART + ONO-AE3-208 (green lines; n = 5), or control treatment-naïve mice (black lines; n = 5) were recorded every 3 days. Mean tumor size values of naïve vs previously treated mice were analyzed by Student t test. Abbreviations: CART, chimeric antigen receptor-transduced T cells; LD, lymphodepletion; MS, median survival; s.c., subcutaneous.
As our transgenic animals provided a reliable source of EGFRvIII-CART cells for in vivo experiments, we evaluated Tg-CART cells in another experiment with the same experimental design outlined in Figure 6A. As shown in Figure 6C, the treatment with Tg-CART cells resulted in 6/7 mice remaining tumor-free 70-day post-CART ACT (MS not reached; n = 7). We observed a similar outcome for mice treated with a combination of Tg-CART cells and ONO-AE3-208 (MS not reached; n = 6). Both Tg-CART cell-treated groups had a significantly longer MS than those treated with control T cells (Figure 6C). As Tg-CART cell therapy alone led to enhanced long-term survival, the addition of ONO-AE3-208 did not demonstrate an additional benefit. All surviving mice were re-challenged with s.c. tumors (as in Figure 6A), and the size of tumors was recorded every 3 days (Figure D and E). As in the earlier cohort, both EGFRvIII+ and parental SB28 tumors grew more slowly in mice that had previously received CART cells, while tumors in control T-cell-treated mice (n = 3) grew similarly to those in the treatment-naïve group (data not shown).
Discussion
To our knowledge, this is the first development of Rosa26-targeted CAR Tg mice. The field has greatly appreciated the availability of mice with transgenic T-cell receptors (TCR), such as ones against gp100 (Pmel)25 or ovalbumin (OT-126 and OT-227). The consistent quality and availability of a large number of T cells derived from these mice allow us to perform experiments with adequate scientific rigor and to compare results between experiments within a project and across multiple studies that test different strategies. As there have been few CAR-based preclinical models of GBM using syngeneic models,28–31 our EGFRvIII-CAR Tg mice will likely be highly valuable for much-needed future preclinical studies in the neuro-oncology field. Because EGFRvIII is a tumor-specific antigen, we hypothesized that despite the 90% homology between human and mouse EGFR, constitutive expression of the CAR on T cells in CARKi CD4-Cre mice would not cause any adverse effects. Our data show that the new transgenic mice are viable, fertile, and display no aberrant phenotype. Additionally, the Tg-CART cells offer the possibility to easily access therapeutic cells that can be tracked in vivo using EGFP or a congenic marker like CD45.1+.
With regard to CAR Tg mice, the Kershaw lab created a Tg mouse strain expressing a CAR against Her2 under the control of a pan-hematopoietic promoter, Vav.32 Subsequently, Kershaw and colleagues generated dual-specific T cells expressing both a Her2-specific CAR and a gp100-specific TCR and demonstrated potent anti-tumor effects against a variety of Her2+ tumors.33 While the Her2-CAR Tg mice have random chromosomal insertions of the CAR transgene,34 our model is the first to target the CAR insertion to a chromosomal safe harbor locus.
The Rosa26-targeted feature may have contributed to the observed superior in vivo functions of Tg-CART cells compared to RV-CART cells. Recent studies with human T cells showed that TRAC locus-targeted expression of a CAR results in more uniform CAR expression and superior anti-tumor efficacy compared to CART cells generated via traditional viral integration methods.35 Furthermore, Roth et al demonstrated that nonviral locus-targeted insertion of a TCR36 transgene can be performed safely in human T cells and at a scale allowing for in vivo therapeutic evaluation. We noted that in vitro Tg-CART cells displayed more uniform but lower CAR expression levels based on F(ab) staining, compared with RV-CART cells. Increased uniformity of CAR expression on Tg-CART cells likely owes to a single copy of the cassette in the Rosa26 locus. Although the optimal level of CAR density is unknown, too high a density may result in early CART cell exhaustion due to ligand-independent tonic signaling due to CAR molecule clustering.37 Our mCAR vector design includes the 4-1BB co-stimulation molecule which has been shown to reduce the exhaustion effects of persistent CAR signaling.37 Together, our data suggest that CAR expression from a single allele in a known locus may provide CART cells with enhanced efficacy and support the development of locus-targeted engineering of human T cells.35,36
We and others have studied and reviewed the important contributions of the myeloid components within the glioma microenvironment (reviewed in Chuntova et al4). Our syngeneic model allowed us to evaluate the effects of the EP4 antagonist ONO-AE3-208 in combination with CART cells. The in vitro data suggested that EP4 inhibition prevents MDSC development and suppresses their ability to produce ArgI. When tested in vivo, the combination of ONO-AE3-208 and RV-CART cells resulted in long-term survival in a fraction of mice (Figure 6A). This suggests that TME remodeling during treatment may have contributed to long-lasting anti-tumor effects (Figure 6C). Using both RV-CART and Tg-CART cells, our re-challenge experiments demonstrated an anti-tumor memory response to not only EGFRvIII+ tumors but also SB28 parental tumors. The use of Tg-CART cells presents a valuable new model for future studies of the critical question of how antigen-spreading is initiated and how it can be further enhanced.
GBM possesses substantial degrees of inter- and intratumor heterogeneity, which contributes to the failure of specific antigen-targeted immunotherapeutic approaches.38,39 Despite sorting the SB28-EGFRvIII cells for EGFRvIII expression prior to intracerebral inoculation (Supplementary Figure 3C), the mice treated with LD + CART eventually recurred with tumors which lacked EGFRvIII expression. This strongly suggests that the CART-cell activity results in tumor immunoediting, allowing for the regrowth of glioma cell subpopulations that have undergone either EGFRvIII cDNA silencing or genetic loss of the transgene.
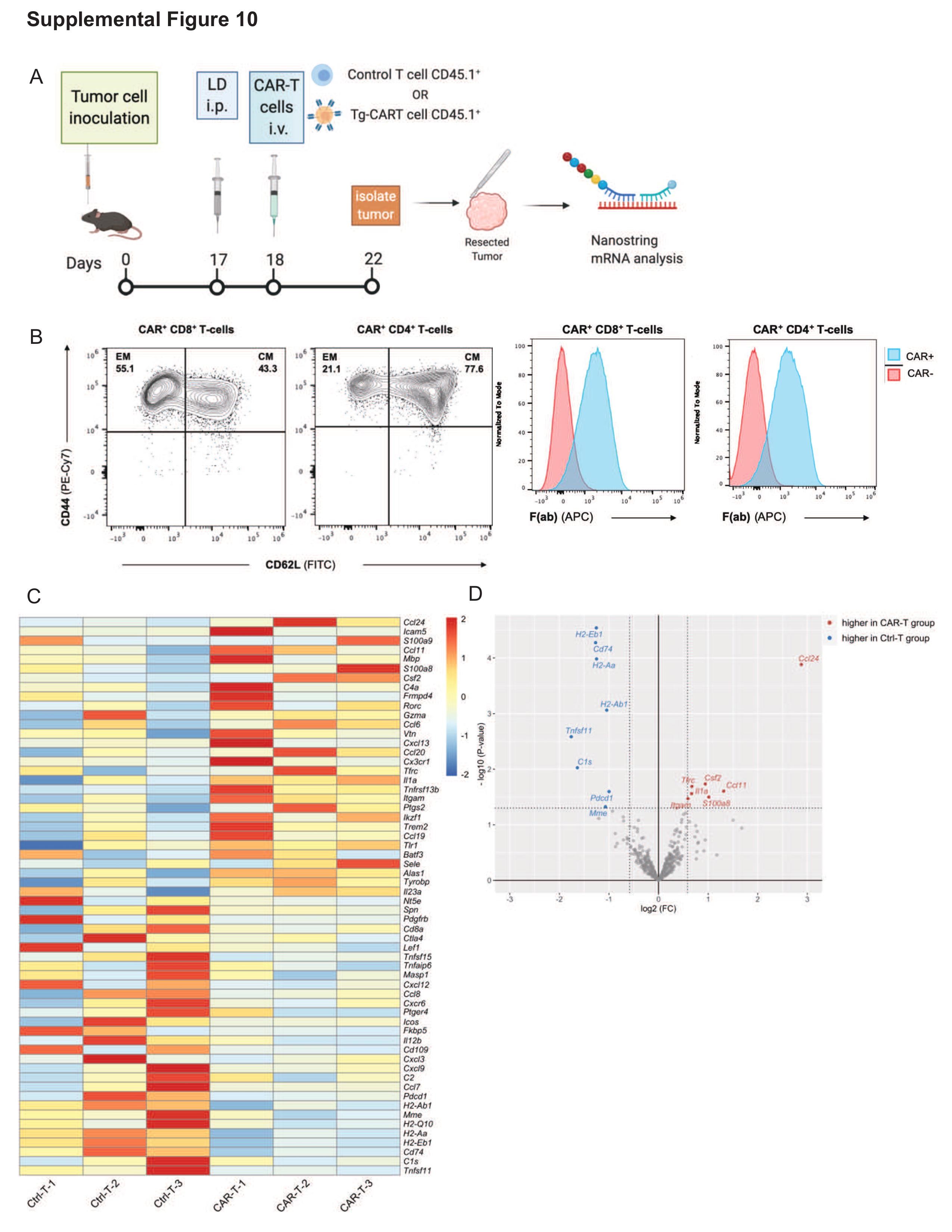
To understand the effects of CART treatment on the TME in our model, we evaluated the expression of immune-related genes in the tumor via Nanostring following Tg-CART infusion (Supplementary Figure 10A and B). As differential expression analyses showed a great level of inter-individual heterogeneity (Supplementary Figure 10C and D; Supplementary Tables 4 and 5), further studies are warranted to validate the observed trends and understand mechanisms for efficacy and resistance.
Based on preliminary safety data in recurrent GBM patients receiving CART therapy, there exists a strong basis to move this form of therapy forward to newly diagnosed patients who completed the standard-of-care, adjuvant chemoradiation therapy. Preclinical studies by us (Ohkuri, Kosaka et al, in press)40 and others29 showed that temozolomide could enhance the post-infusion expansion of adoptively transferred T cells.
As successful immunotherapy for primary GBM faces numerous challenges,4 our new Tg mouse strain allows access to high number of EGFRvIII-CART cells with a reproducible phenotype within a flexible preclinical in vivo system. Coupled with our syngeneic tumor model, the Tg-CART mice will be a valuable new tool to address issues including antigen immune escape, the highly immunosuppressive TME, and whether approaches combining separate therapies can improve outcomes for GBM patients.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Tohru Kotani, Payal Watchmaker, Bindu Hegde, Ryan Gilbert, Davide Ruggero, and Ruggero lab members; UCSF Core Facilities: Laboratory for Cell Analysis, Genome Analysis Core, Preclinical Therapeutics Core, and Gladstone Transgenic Gene Targeting Core. All experimental diagrams were created with BioRender.
Funding
H.O. was supported by Parker Institute for Cancer Immunotherapy and National Institutes of Health/National Institute of Neurological Disorders and Stroke [1R35 NS105068].
Conflict of interest statement. None.
Authorship statement. Concept and design: P.C. and H.O. Methodology: P.C., Y.H., R.N., T.N., H.E.L., B.S., and H.O. Data acquisition: P.C., Y.H., R.N., Y.G., A.Y., R.H., T.N., G.K., T.C., A.L.M., M.M., K.M.D., D.D., J.S., and H.E.L. Analysis and data interpretation: P.C., Y.H., R.N., Y.G., T.N., G.K., B.S., and H.O. Manuscript preparation: P.C., A.L.M., K.M.D., B.S., and H.O. Study supervision: H.O.
References
- 1. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379(1):64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chuntova P, Downey KM, Hegde B, Almeida ND, Okada H. Genetically engineered T-cells for malignant glioma: overcoming the barriers to effective immunotherapy. Front Immunol. 2018;9:3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kwok D, Okada H. T-cell based therapies for overcoming neuroanatomical and immunosuppressive challenges within the glioma microenvironment. J Neurooncol. 2020;147(2):281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chuntova P, Chow F, Watchmaker P, et al. . Unique challenges for glioblastoma immunotherapy – discussions across neuro-oncology and non-neuro-oncology experts in cancer immunology. Neuro Oncol. 2020;23(3):356–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thorne AH, Zanca C, Furnari F. Epidermal growth factor receptor targeting and challenges in glioblastoma. Neuro Oncol. 2016;18(7):914–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. O’Rourke DM, Nasrallah MP, Desai A, et al. . A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399): eaaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kosaka A, Ohkuri T, Okada H. Combination of an agonistic anti-CD40 monoclonal antibody and the COX-2 inhibitor celecoxib induces anti-glioma effects by promotion of type-1 immunity in myeloid cells and T-cells. Cancer Immunol Immunother. 2014;63(8):847–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Genoud V, Marinari E, Nikolaev SI, et al. . Responsiveness to anti-PD-1 and anti-CTLA-4 immune checkpoint blockade in SB28 and GL261 mouse glioma models. Oncoimmunology. 2018;7(12):e1501137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Simonds EF, Lu ED, Badillo O, et al. . Deep immune profiling reveals targetable mechanisms of immune evasion in immune checkpoint inhibitor-refractory glioblastoma. J Immunother Cancer. 2021;9(6): e002181. doi: 10.1136/jitc-2020-002181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Müller S, Kohanbash G, Liu SJ, et al. . Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017;18(1):234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kohanbash G, Okada H. Myeloid-derived suppressor cells (MDSCs) in gliomas and glioma-development. Immunol Invest. 2012;41(6–7):658–679. [DOI] [PubMed] [Google Scholar]
- 12. Fujita M, Kohanbash G, Fellows-Mayle W, et al. . COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res. 2011;71(7):2664–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Obermajer N, Kalinski P. Generation of myeloid-derived suppressor cells using prostaglandin E2. Transplant Res. 2012;1(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xin X, Majumder M, Girish GV, Mohindra V, Maruyama T, Lala PK. Targeting COX-2 and EP4 to control tumor growth, angiogenesis, lymphangiogenesis and metastasis to the lungs and lymph nodes in a breast cancer model. Lab Invest. 2012;92(8):1115–1128. [DOI] [PubMed] [Google Scholar]
- 15. Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood. 2011;118(20):5498–5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Okada H, Tahara H, Shurin MR, et al. . Bone marrow-derived dendritic cells pulsed with a tumor-specific peptide elicit effective anti-tumor immunity against intracranial neoplasms. Int J Cancer. 1998;78(2):196–201. [DOI] [PubMed] [Google Scholar]
- 17. Nishimura F, Dusak JE, Eguchi J, et al. . Adoptive transfer of type 1 CTL mediates effective anti-central nervous system tumor response: critical roles of IFN-inducible protein-10. Cancer Res. 2006;66(8):4478–4487. [DOI] [PubMed] [Google Scholar]
- 18. Gattinoni L, Finkelstein SE, Klebanoff CA, et al. . Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202(7):907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bottcher JP, Bonavita E, Chakravarty P, et al. . NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018;172(5):1022–1037.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee PP, Fitzpatrick DR, Beard C, et al. . A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15(5):763–774. [DOI] [PubMed] [Google Scholar]
- 21. Bayik D, Zhou Y, Park C, et al. . Myeloid-derived suppressor cell subsets drive glioblastoma growth in a sex-specific manner. Cancer Discov. 2020;10(8):1210–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Obermajer N, Kalinski P. Key role of the positive feedback between PGE(2) and COX2 in the biology of myeloid-derived suppressor cells. Oncoimmunology. 2012;1(5):762–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gielen PR, Schulte BM, Kers-Rebel ED, et al. . Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100A8/9 and arginase and suppress T cell function. Neuro Oncol. 2016;18(9):1253–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gabrusiewicz K, Rodriguez B, Wei J, et al. . Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. 2016;1(2): e85841. doi: 10.1172/jci.insight.85841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Overwijk WW, Tsung A, Irvine KR, et al. . gp100/Pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med. 1998;188(2):277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clarke SR, Barnden M, Kurts C, Carbone FR, Miller JF, Heath WR. Characterization of the ovalbumin-specific TCR transgenic line OT-I: MHC elements for positive and negative selection. Immunol Cell Biol. 2000;78(2):110–117. [DOI] [PubMed] [Google Scholar]
- 27. Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76(1):34–40. [DOI] [PubMed] [Google Scholar]
- 28. Jin L, Ge H, Long Y, et al. . CD70, a novel target of CAR T-cell therapy for gliomas. Neuro Oncol. 2018;20(1):55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Suryadevara CM, Desai R, Abel ML, et al. . Temozolomide lymphodepletion enhances CAR abundance and correlates with antitumor efficacy against established glioblastoma. Oncoimmunology. 2018;7(6):e1434464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murty S, Haile ST, Beinat C, et al. . Intravital imaging reveals synergistic effect of CAR T-cells and radiation therapy in a preclinical immunocompetent glioblastoma model. Oncoimmunology. 2020;9(1):1757360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alizadeh D, Wong RA, Gholamin S, et al. . IFNgamma is Critical for CAR T Cell-mediated Myeloid Activation and Induction of Endogenous Immunity. Cancer Discov. 2021;11:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yong CS, John LB, Devaud C, et al. . A role for multiple chimeric antigen receptor-expressing leukocytes in antigen-specific responses to cancer. Oncotarget. 2016;7(23):34582–34598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Slaney CY, von Scheidt B, Davenport AJ, et al. . Dual-specific chimeric antigen receptor T cells and an indirect vaccine eradicate a variety of large solid tumors in an immunocompetent, self-antigen setting. Clin Cancer Res. 2017;23(10):2478–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yong CS, Westwood JA, Schröder J, et al. . Expression of a chimeric antigen receptor in multiple leukocyte lineages in transgenic mice. PLoS One. 2015;10(10):e0140543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eyquem J, Mansilla-Soto J, Giavridis T, et al. . Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roth TL, Puig-Saus C, Yu R, et al. . Reprogramming human T cell function and specificity with non-viral genome targeting. Nature. 2018;559(7714):405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Long AH, Haso WM, Shern JF, et al. . 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Patel AP, Tirosh I, Trombetta JJ, et al. . Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Choe JH, Watchmaker PB, Simic MS, et al. . SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med. 2021;13(591): eabe7378. doi: 10.1126/scitranslmed.abe7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ohkuri T, Kosaka A, Ikeura M, Salazar AM, Okada H. IFN-γ- and IL-17-producing CD8+ T (Tc17-1) cells in combination with poly-ICLC and peptide vaccine exhibit antiglioma activity. J Immunother Cancer. 2021; 9: e002426. doi: 10.1136/jitc-2021-002426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.