

Abstract
Introduction
Fasting glucose increases with age and is linked to modifiable Alzheimer's disease risk factors such as cardiovascular disease and Type 2 diabetes (T2D).
Methods
We leveraged available biospecimens and neuroimaging measures collected during the Alzheimer's Prevention Through Exercise (APEx) trial (n = 105) to examine the longitudinal relationship between change in blood glucose metabolism and change in regional cerebral amyloid deposition and gray and white matter (WM) neurodegeneration in older adults over 1 year of follow‐up.
Results
Individuals with improving fasting glucose (n = 61) exhibited less atrophy and regional amyloid accumulation compared to those whose fasting glucose worsened over 1 year (n = 44). Specifically, while individuals with increasing fasting glucose did not yet show cognitive decline, they did have regional atrophy in the hippocampus and inferior parietal cortex, and increased amyloid accumulation in the precuneus cortex. Signs of early dementia pathology occurred in the absence of significant group differences in insulin or body composition, and was not modified by apolipoprotein E ε4 carrier status.
Discussion
Dysregulation of glucose in late life may signal preclinical brain change prior to clinically relevant cognitive decline. Additional work is needed to determine whether treatments specifically targeting fasting glucose levels may impact change in brain structure or cerebral amyloid in older adults.
Keywords: Alzheimer's disease, amyloid, glucose, hippocampus, imaging, metabolism
1. INTRODUCTION
Approximately 50 million individuals worldwide suffer from dementia, the most common type being Alzheimer's disease (AD). Fasting glucose increases with age 1 and is linked to potentially modifiable AD risk factors such as cardiovascular disease and Type 2 diabetes (T2D). 2 Both prediabetic fasting glucose levels and history of dietary sugar intake track positively with increased regional cerebral amyloid cross sectionally, 3 , 4 and higher fasting glucose has been causally associated with increased AD risk. 5 Preclinically, blood glucose tracks positively with interstitial amyloid, 6 and in tissues, elevated glucose is associated with increased amyloid beta (Aβ). 7 This suggests that change in fasting glucose may be a window into early metabolic decline related to AD. However, longitudinal multi‐modal brain imaging studies of fasting glucose in cognitively healthy older adults are lacking.
A multi‐faceted neuroimaging approach is necessary to comprehensively quantify the relationship of glucose metabolism with measures of brain health. Here, we present a secondary analysis of data from the Alzheimer's Prevention Through Exercise trial (APEx; NCT02000583), which found that a 1‐year exercise intervention improved fitness but did not significantly affect cerebral amyloid or hippocampal volume. 8 Fasting glucose is often taken during clinical visits and when acquired over time, can index metabolic changes either associated with or preceding dementia. We hypothesized that positive change in fasting glucose levels over 1 year would be associated with decreased limbic gray matter (GM) volume, decreased white matter (WM) integrity, and increased cerebral amyloid deposition in highly metabolic regions, indicative of early stages of dementia‐related pathology.
2. METHODS
2.1. Standard protocol approvals, registrations, and patient consents
APEx was registered in ClinicalTrials.gov (NCT02000583) and was approved by the Institutional Review Board at the University of Kansas Medical Center. All participants provided written informed consent.
2.2. Participants
Participants were at least 65 years old, sedentary or underactive, 9 and on stable medications for at least 30 days. Participants were excluded if they had insulin‐dependent diabetes, uncontrolled hypertension, or cognitive impairment. 10 , 11 Participants either completed 150 minutes per week of moderate intensity (AEx) or standard of care education only (Control) in a 2:1 ratio. Additional APEx inclusion and exclusion criteria can be found in the primary publication. 8 Our analyses included individuals with both pre‐ and post‐intervention blood glucose and amyloid positron emission tomography (PET; n = 105). We classified individuals whose fasting glucose decreased over the longitudinal follow‐up into a “glucose improves” (GI) group (n = 61). Individuals whose fasting glucose increased were included in the “glucose worsens” (GW) group (n = 44).
RESEARCH IN CONTEXT
Systematic Review: The literature was reviewed for glucose/metabolism, anatomical brain imaging/diffusion tensor imaging (DTI)/white matter hyperintensities (WMH), and Alzheimer's disease (AD), mainly in PubMed, and all studies are appropriately cited.
Interpretation: Change in fasting glucose over 1 year was related to multi‐modal brain imaging biomarkers associated with dementia. Our findings indicate that the group of individuals with improving fasting glucose exhibited less atrophy and regional amyloid accumulation compared to the group whose fasting glucose worsened over 1 year.
Future Directions: Identifying individuals at risk of AD is essential for early prevention. Targeting systemic glucose metabolism for intervention may modulate these outcomes in cognitively healthy older adults at risk for AD.
All APEx participants underwent a baseline 18F‐AV45 PET scan and were adjudicated to have either “elevated” cerebral amyloid or levels that fell into the “subthreshold” range, as previously described. 8 , 12 Briefly, all images were interpreted independently by three experienced clinicians, without reference to any clinical information, to determine amyloid status. During this evaluation, raters combined both visual and quantitative information to determine status as “elevated” versus “non‐elevated,” with final status determined by majority of the raters 8 , 13 , 14 The MIMneuro Amyloid Workflow (version 6.8.7, MIM Software Inc.) was used to view and analyze images with florbetapir templates as the target for a two‐phase registration: first rigid registration, then deformable registration to a common template space. First, raters reviewed raw PET images visually. Then raters examined the cerebellum‐normalized standardized uptake value ratios (SUVRs) in six regions of interest (ROIs; anterior cingulate, posterior cingulate, precuneus, inferior medial frontal, lateral temporal, and superior parietal cortex) and projection maps comparing SUVRs to an atlas of amyloid‐negative scans. 46 Participants were eligible for the study if they had an elevated scan or were in the subthreshold range. We defined subthreshold as a mean cortical SUVR for the six ROIs >1.0, which represented the upper half of non‐elevated scans (mean cortical SUVR for non‐elevated scans [n = 166] 0.99 [0.06 standard deviation (SD)]).
2.3. Apolipoprotein E genotyping and blood glucose measures
Individuals reported to the University of Kansas Clinical and Translational Science Unit for metabolic analyses. Two blood pressure measures were taken after 5 minutes of seated rest, and averaged to determine the mean systolic and diastolic values. Fasting whole blood was collected for all individuals at baseline and 1‐year follow‐up in acid citrate dextrose (ACD) tubes, from which plasma was subsequently generated. Plasma samples were stored at –80 °C until analysis. We used a Taqman single nucleotide polymorphism allelic discrimination assay (ThermoFisher) to determine apolipoprotein E (APOE) ε4 carrier status (carrier vs. noncarrier). APOE genotyping failed on one individual. Fasting plasma glucose was measured using a YSI 2300 Glucose and Lactate analyzer (Yellow Springs Instruments). Fasting plasma insulin was measured using enzyme‐linked immunosorbent assay (ALPCO). We also calculated homeostasis model assessment of insulin resistance (HOMA‐IR). 15
2.4. Anthropometric measures
Participants donned standardized gowns and were asked to void. Total body mass was measured using a digital scale accurate to 0.1 kg (Seca Platform Scale, model 707). Body composition was estimated using dual energy x‐ray absorptiometry (DEXA; Lunar Prodigy, version 11.2068) to determine lean mass, fat mass, and bone mineral density.
2.5. Cognitive outcomes
Participants were characterized with nine cognitive tests that are part of the Uniform Data Set version 2.0 (UDS 2.0), 16 and z‐scores for these tests were calculated using a normative calculator as previously described. 17 Test tests included the Mini Mental State Examination, Logical Memory (Immediate and Delayed), Category Fluency (Animals and Vegetables), Trail Making Test Parts A and B, Boston Naming Test, and Digit Symbol Substitution. The z‐scores for each individual test are given in Table S1 in supporting information, and were averaged to generate a “global” normed score. All UDS tests were administered except Digit Span tests due to time constraints for the study visit.
2.6. Magnetic resonance imaging
Anatomic brain imaging (magnetization prepared rapid gradient echo [MP‐RAGE]) and diffusion tensor imaging (DTI) magnetic resonance imaging (MRI) were also performed at both baseline and 1‐year timepoints using a Siemens 3.0 Tesla Skyra scanner. Full sequence details are described in a previous publication. 8 Both high‐resolution T1 (MP‐RAGE; 1 × 1 × 1.2 mm voxels; TR = 2300 milliseconds, TE = 2.98 milliseconds, TI = 900 milliseconds, FOV 256 × 256 mm, 9° flip angle) and DTI (TR = 10,000 milliseconds; TE = 90 milliseconds; 75 slices; voxel size, 2.3 × 2.3 × 2.0 mm2 in plane; slice thickness = 2.0 mm; FOV = 300 mm; 64 gradient directions, b‐value, 1000 s/mm2) were acquired for detailed anatomical assessment.
Every MP‐RAGE was checked for image and motion artifacts and gross anatomical abnormalities, resulting in the further removal of three qualifying subjects with either poor structural image quality at one timepoint, leaving a longitudinal MP‐RAGE imaging sample of 102 subjects. T1‐weighted images were evaluated using voxel‐based morphometry (VBM). Images were analyzed and pre‐processed with the Computational Anatomical Toolbox 12 (CAT12 Version 12.6, C. Gaser, Structural Brain Mapping Group, Jena University Hospital; http://dbm.neuro.uni‐jena.de/cat/) through Statistical Parametric Mapping version 12 (SPM12; Wellcome Trust Centre for Neuroimaging; http://www.fil.ion.ucl.ac.uk/spm/software/spm12/) that operate under Matlab (R2019b) (The Mathworks) on a Mac. As part of the longitudinal pipeline in CAT12, T1‐weighted images from both timepoints were registered using intra‐subject coregistration, which were then realigned across subjects and bias‐corrected with reference to the mean images computed from each subject's pre‐ and post‐intervention images. Next, images from both timepoints as well as subject's mean images were segmented into GM, WM, cerebrospinal fluid (CSF), and white matter hyperintensities (WMH). Detection of WMH volume is both outputted as a volumetric measure and also accounted for during the spatial normalization step. Baseline and 1‐year images were spatially normalized using the high‐dimensional DARTEL algorithm into Montreal Neurological Institute (MNI) space. 18 We calculated total intracranial volume (TICV) using total GM, WM, and CSF volumes. The amount of volume changes were scaled to retain the original local volumes (modulating the segmentations). 19 The modulated GM segmentations were smoothed using a 10 × 10 × 10 mm full‐width at half‐maximum Gaussian kernel prior to group level analysis. After preprocessing, a sample homogeneity check was performed in accordance with the CAT12 manual.
2.7. Florbetapir PET
We performed 18F‐AV45 PET scanning at baseline and 1‐year follow‐up on all individuals to measure amyloid neuropathology. Approximately 50 minutes after intravenous Florbetapir 18F‐AV45 (370 MBq) injection, PET scanning was performed on a GE Discovery ST‐16 PET/CT scanner. Two PET brain frames of 5 minutes in duration were acquired continuously, summed, and attenuation corrected as previously described. 8 Briefly, we analyzed scans using MIMneuro Amyloid Workflow (version 6.8.7, MIM Software Inc.), registering each image to a template, then calculating the cortical‐to‐cerebellar SUVR across the image. We calculated average SUVRs in six regions (frontal, temporal, superior parietal, anterior cingulate, posterior cingulate, and precuneus) from an established MIM atlas. 13 We also calculated a “global” mean SUVR of these regions.
In addition to mean amyloid values for atlas‐based regional analysis, we also normalized the cerebellar‐standardized (using a probability threshold of 50% for GM specificity), co‐registered PET images into MNI space as well as smoothed them using an 8 × 8 × 8 mm full‐width at half‐maximum Gaussian kernel for use in longitudinal voxel‐based analysis, which was done to verify the SUVR statistical results in imaging space, as well as provide a visual of regions tested.
2.8. DTI
DTI data were processed using FMRIB's Software Library (FSL) version 6.0.3. 20 FSL's workflow (http://fsl.fmrib.ox.ac.uk/fsl/fslwiki/TBSS/UserGuide) for tract‐based spatial statistics (TBSS 21 ) was used to extract fractional anisotropy (FA) values from atlas‐based WM pathways for statistical analysis in SPSS. Reconstructed directional and B0 scans were checked for motion artifact and image quality before processing. For DTI analyses, one additional subject was excluded due to motion artifacts in the baseline diffusion scan. Standard TBSS preprocessing included correction for eddy currents and head motion (ECC), extraction of brain tissue from the skull using brain extraction tool (BET), followed by fitting a tensor model to the data using DTIFIT to generate spatial maps of FA values for each subject at each timepoint. We focused on FA values over other diffusion measures of integrity due to it being the predominant DTI abnormality in prediabetic patients. 22 We used an a priori tract‐of‐interest method to measure WM integrity in tracts associated with early decline in AD: the cingulum (two sections, the middle cingulate portion and the hippocampal extension), and the uncinate fasciculus (UF). After the initial steps of the TBSS processing pipeline, individual FA maps were non‐linearly registered, aligned, and transformed into a common 1 × 1 × 1mm standard MNI space template (FMRIB58), followed by skeletonization. 21 During skeletonization, all subjects’ FA data were thinned to create a mean FA skeleton that represents the center of all the tracts common to the group. Each subject's diffusivity metrics were projected into the group mean skeleton and thresholded to include only voxels with FA values higher than 0.3. The accuracy of the registration was visually checked for each participant.
To determine AD‐related a priori tracts, we used each subject's skeleton and ran FSLMATHS to extract the FA value in a given region using binary WM masks from the Johns Hopkins University (JHU) probabilistic atlas. 23 , 24 The intersection of each subject's skeletonized FA values and binary mask for each WM tract of interest were used to calculate an average diffusivity value and percent change in FA over 1 year and compared between glucose groups using age, sex, treatment group, elevated status, and APOE ε4 status as covariates.
2.9. Statistical analyses
For all analyses performed outside of imaging space, we used multiple regression to determine the relationship between continuous variables. Covariates included age, sex, treatment group (aerobic exercise vs. education only) and cerebral amyloid status (elevated vs. subthreshold). We used analysis of covariance to determine group differences in our continuous outcome measures, again including age, sex, treatment group, and cerebral amyloid status as covariates. Further correction for APOE ε4 carrier status did not affect the significance of our findings. Relationships between categorical outcomes were assessed using Chi‐square analyses. Findings were considered significant at P < .05.
2.9.1. Neuroimaging statistics
To determine brain regions that reflected greater changes in GM volume over time between glucose change groups, we performed a 2 × 2 mixed analysis of variance (ANOVA) on the normalized GM images using the “flexible factorial” module in CAT12. The two factors were Session (baseline and 1 year; within‐subjects) and Group (GI or GW; between‐subjects; determined using the elevated/subthreshold cutpoint). A follow‐up one‐tailed t‐test was used to determine the directionality of the effect. We included the following covariates: age, sex, baseline amyloid elevated status, treatment, and TICV. We did an additional analysis adding APOE ε4 as a covariate. For all analyses, voxels are reported with reference to the MNI standard space within SPM12. To avoid possible edge effects at the border between GM and WM and to include only relatively homogeneous voxels, we used an absolute threshold masking of 0.10 for each analysis. Results for F‐tests and t‐tests were considered significant at P < .05 after correction for multiple comparisons (family‐wise error [FWE]), and results at P < .001 (uncorrected) for t‐tests are shown in supplemental data with a minimum cluster size of 100 voxels (k >100).
After whole brain analysis, we used a small volume correction (SVC) to test for differences between glucose‐change group status and change in GM volume over 1 year in the medial temporal cortex and inferior parietal/precuneus cortex, where we have previously shown associations with baseline insulin sensitivity. 25 , 26 The SVC's included left and right hippocampus, and also the left and right inferior parietal cortex and precuneus from the integrated automatic anatomic labeling (AAL) tool. 27 Significant clusters were extracted from the hippocampus (mean voxel value of the cluster) and used to calculate the percentage of change of GM over 1 year for purposes of plotting the results visually.
For amyloid PET analyses, we calculated mean cerebral amyloid values from the cortical‐to‐cerebellar SUVRs in a priori determined regions, including the frontal, temporal, superior parietal, anterior cingulate, posterior cingulate, and precuneus, which were used for region‐specific analyses. To verify the cluster size and location of the PET SUVR statistical analysis and to visualize brain regions that reflected differences in amyloid deposition over time between glucose change groups, we performed a 2 × 2 mixed ANOVA on the normalized, smoothed cerebellar standardized PET images using the “flexible factorial” model in CAT12. We used the same model as on VBM above with age, sex, baseline amyloid elevated status, original exercise intervention status, and TICV. We used a SVC using the left and right precuneus and cuneus from the integrated AAL tool 27 in each glucose change group to create the statistical map of amyloid change.
3. RESULTS
3.1. Baseline group characteristics
Age, sex, education, treatment group (aerobic exercise vs. education only), baseline cerebral amyloid status (not elevated vs. elevated), APOE ε4 carrier status, and baseline fasting glucose levels were not different between individuals whose fasting glucose improved (GI) longitudinally compared to those whose fasting glucose worsened (GW; Table 1). Individuals whose glucose worsened tended to have higher systolic blood pressure compared to those whose glucose improved, despite higher frequency of diagnosed hypertension in those whose glucose improved. Approximately equal numbers of individuals with other comorbiditites, including T2D, were represented in each group (Table 1).
TABLE 1.
Baseline characteristics of groups
| Measure | FG improves (n = 61) | FG worsens (n = 44) | P‐value | |
|---|---|---|---|---|
| Demographics | Age (years) | 71.6 [4.9] | 71.7 [5.6] | .889 |
| Sex (#, % male) | 21 [34] | 14 [32] | .780 | |
| Elevated (#, %) | 46 [75] | 27 [61] | .123 | |
| AEx group (#, %) | 44 [72] | 28 [64] | .355 | |
| Education (years) | 15.9 [2.3] | 16.5 [2.2] | .180 | |
| APOE ε4 (#,% carrier) | 32 [53] | 16 [36] | .086 | |
| Metabolic/vascular | Glucose (bl; mg/dL) | 100.3 [11.3] | 97.4 [14.1] | .377 |
| Body mass index | 28.3 [5.4] | 28.4 [7.1] | .972 | |
| VO2 peak (mL/kg/min) | 22.2 [4.6] | 22.4 [5.9] | .552 | |
| Systolic BP (mmHg) | 127.4 [11.8] | 132.8 [17.1] | .026* | |
| Diastolic BP (mmHg) | 75.5 [8.9] | 75.2 [9.8] | .955 | |
| White matter hyperintensities | 3.59 [3.3] | 2.90 [2.6] | .303 | |
| Resting HR (bpm) | 69.6 [10.3] | 66.8 [9.5] | .147 | |
| Comorbidities | Type 2 diabetes (%) | 6 [9.8] | 9 [20.5] | .125 |
| Hyperlipidemia (#, %) | 28 [45.9] | 25 [56.8] | .270 | |
| Hypertension (#, %) | 28 [45.9] | 11 [25] | .029* | |
| Atrial fibrillation (#,%) | 2 [3.3] | 2 [6.8] | .401 | |
| Angina | 1 [1.7] | 0 [0] | .396 | |
| Myocardial infarction | 3 [4.9] | 2 [4.5] | .930 | |
| Hyposomnia | 7 [11.7] | 5 [11.4] | .962 |
Abbreviations: AEx, aerobic exercise; APOE, apolipoprotein E; BP, blood pressure; mmHg, millimeters of mercury; bpm, beats per minute; FG, fasting glucose; SD, standard deviation.
Notes: Overall baseline characteristics for individuals included in the study. APOE ε4 genotype information was unavailable for one individual in the FG improves group. Indvidudals whose FG worsened had higher baseline systolic blood pressure than those whose FG improved, although a diagnosis of hypertension was more common in those whose FG improved. Values are given as means [SD] or number [percent].
No additional significant differences in baseline characteristics existed between groups.
P < .05.
Covariates included age, sex, elevated status, and treatment group.
3.2. Metabolic, anthropometric, and cognitive outcomes
Glucose levels declined by ≈7% in the GI group compared to an increase of >6% in the GW group (P < .001). Insulin resistance (HOMA‐IR) was significantly decreased in the GI group compared to an increase to the GW (P = .007). This was an effect driven primarily by glucose, as insulin levels were not significantly different between the groups (Table 2). Change in body composition over the course of the study was quite similar between groups, including measures of regional fat mass, lean mass, and bone mineral density. Further, there was no significant difference in change in blood pressure over 1 year between groups (Table 2). Change in cognitive performance outcomes also did not differ significantly between groups over 1 year (Table S1).
TABLE 2.
Percent change in metabolic, anthropometric, and brain markers over 1 year
| Measure | FG improves (n = 61) | FG worsens (n = 44) | P‐value |
|---|---|---|---|
| Glucose | −7.4 [5.4] | 6.3 [5.6] | .000 |
| Insulin | −3.0 [53.9] | 17.2 [66.9] | .144 |
| HOMA‐IR | −9.3 [52.9] | 25.8 [77.4] | .014 |
| VO2 | 7.43 [10.7] | 6.83 [24.2] | .921 |
| Lean mass | −0.50 [2.9] | −0.04 [2.3] | .342 |
| Fat mass | −3.7 [7.3] | −2.9 [7.8] | .629 |
| Android | −5.5 [12.3] | −3.9 [10.7] | .550 |
| Gynoid | −3.7 [7.7] | −3.3 [7.8] | .687 |
| BMD | −0.28 [1.7] | −0.14 [1.6] | .733 |
| BMC | −0.66 [1.9] | −0.67 [1.5] | .890 |
| Systolic BP | 1.75 [0.10] | 1.94 [0.11] | .934 |
| Diastolic BP | 1.35 [12.1] | 1.33 [10.7] | .829 |
| WMH | −0.68 [1.3] | −0.46 [1.2] | .358 |
Abbreviations: BMC, bone mineral composition; BMD, bone mineral density; BP, blood pressure; FG, fasting glucose; HOMA‐IR, homeostasis model assessment of insulin resistance; SD, standard deviation; VO2, peak volume of oxygen consumption during a graded exercise test; WMH, white matter hyperintensities.
Notes: Percent change in metabolic, anthropometric, and brain markers in individuals whose glucose improves compared to those whose glucose worsens.
Values are given as means [SD].
P < .05.
Covariates included age, sex, elevated status, and treatment group.
3.3. Fasting glucose groups differ in longitudinal hippocampal brain volume change
Whole brain voxel‐based analysis of the main effect of time on change in GM volume showed significant atrophy across all subjects in the bilateral caudate, putamen, hippocampus, inferior frontal gyrus, and posterior temporal cortex at P <.001 FWE voxel‐level corrected (Table 3). Interaction analysis on the overall effect of time and group on GM volume showed a significant effect on volume of the right hippocampus (P = .031, FWE voxel‐level corrected, k = 299), and t‐test directional analysis revealed that decreases in hippocampal volume were driven by the GW group. Moreover, significance remained within the ROI analysis on this directional t‐test (P = .035, FWE voxel‐level corrected). There was also a significant interaction of group and time showing increased atrophy in the GW group in the precuneus/inferior parietal region of interest (P = .003, FWE voxel‐level corrected). Hippocampal (Figure 1A‐C) and parietal (Figure 1D‐F) results were extracted and percent change was calculated and plotted against percent change in glucose to visually show directionality of results. Results remained significant in the t‐test in the ROIs when APOE ε4 was added as a covariate.
TABLE 3.
Voxel‐based morphometry analysis of effect of time and glucose groups on gray matter volume
| Peak p (FWE‐corr) | Peak T or F | Z | cluster (k) | Peak p (unc) | x,y,z (mm) | Regions | ||
|---|---|---|---|---|---|---|---|---|
| F‐test main effect of time | F | |||||||
| Whole brain | 0.000 | 127.26 | Inf | 16353 | 0.000 | −8 9 4 | Left caudate | |
| 0.000 | 83.32 | 7.63 | 5794 | 0.000 | 48 20 −9 | Right inferior frontal gyrus (BA 47) | ||
| 0.000 | 67.41 | 7.03 | 8846 | 0.000 | −46 18 −10 | Left inferior frontal gyrus (BA 47) | ||
| 0.000 | 66.22 | 6.98 | 382 | 0.000 | −26 −16 −16 | Left parahippocampal gyrus | ||
| 0.000 | 63.7 | 6.87 | 373 | 0.000 | 15 24 −9 | Right caudate | ||
| 0.000 | 58.29 | 6.63 | 668 | 0.000 | 52 15 30 | Right middle frontal gyrus (BA 9) | ||
| 0.000 | 57.01 | 6.57 | 3506 | 0.000 | 57 −24 50 | Right postcentral gyrus (BA 2) | ||
| 0.000 | 54.26 | 6.44 | 372 | 0.000 | −12 22 −9 | Left hippocampus | ||
| 0.000 | 53.86 | 6.42 | 886 | 0.000 | −40 −40 −36 | Left cerebellum | ||
| 0.000 | 49.93 | 6.22 | 181 | 0.000 | −15 9 −22 | Left orbitofrontal gyrus | ||
| 0.000 | 47.55 | 6.09 | 435 | 0.000 | 44 −26 −33 | Right fusiform gyrus | ||
| 0.000 | 44.73 | 5.93 | 373 | 0.000 | 44 −45 −28 | Right fusiform gyrus | ||
| 0.000 | 39.75 | 5.63 | 229 | 0.000 | 28 −15 −18 | Right hippocampus | ||
| 0.000 | 39.7 | 5.63 | 675 | 0.000 | −52 −34 52 | Left postcentral gyrus | ||
| 0.001 | 37.33 | 5.48 | 307 | 0.000 | −39 −14 −45 | Left inferior temporal gyrus | ||
| 0.001 | 37.08 | 5.46 | 271 | 0.000 | −14 −16 78 | Left precentral gyrus | ||
| 0.001 | 36.08 | 5.4 | 244 | 0.000 | −33 −51 48 | Left inferior parietal cortex | ||
| F‐test interaction of time and glucose group | F | |||||||
| Whole brain | n.s | |||||||
| Hippocampus | 0.031 | 11.11 | 3.03 | 81 | 0.000 | 33, −10, −26 | Right hippocampus | |
| Precuneus/inferior parietal cortex | n.s. | |||||||
| T‐test | ||||||||
| Negative interaction between time and glucose group | T | |||||||
| Whole brain | n.s. | |||||||
| Hippocampus | n.s. | |||||||
| Precuneus/inferior parietal | n.s. | |||||||
| Positive interaction between time and glucose group | T | |||||||
| Whole brain | 0.577 | 3.98 | 3.82 | 156 | 0.000 | 44, −44, 42 | Right parietal lobe (BA 40) | |
| 0.990 | 3.36 | 3.26 | 135 | 0.000 | 62, −24, −2 | Right superior temporal gyrus | ||
| Hippocampus | 0.035 | 3.33 | 3.23 | 133 | 0.000 | 33, −10, −26 | Right hippocampus | |
| Precuneus/inferior parietal cortex | 0.003 | 3.98 | 3.82 | 358 | 0.000 | 44, −42, 42 | Right inferior parietal cortex | |
Abbreviations: BA, Brodmann's area; FWE‐corr, family‐wise error corrected P‐value; n.s., not significant; p (unc), uncorrected P value; x,y,z, coordinates from Talairach and Tournoux.
Notes: Voxel‐based longitudinal gray matter results in individuals whose glucose improves compared to those whose glucose worsens over 1 year.
Regions are listed by descending cluster size.
FIGURE 1.
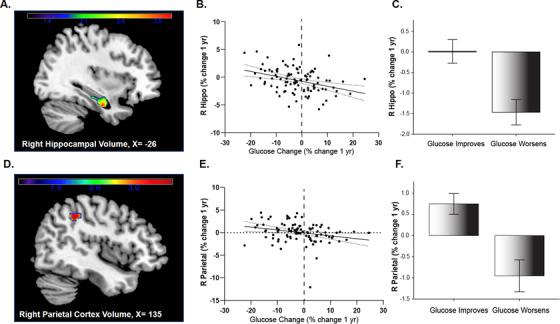
Gray matter atrophy over 1 year occurs in individuals whose fasting glucose worsens. A, Longitudinal voxel‐based results from significant cluster (at 33, –10, –26) from region of interest analysis overlaid on three‐dimensional surface demonstrating right hippocampal atrophy. B, Mean gray matter volume loss was extracted from the cluster showing maximal statistical difference at P < .05 family‐wise error (FWE) corrected and transformed to percent change value to plot against fasting glucose change (B) and group (C) for visual purposes. D, Longitudinal voxel‐based results from region of interest analysis demonstrating right parietal cortex atrophy from significant cluster (44, –42, 42), and is presented as a plot against glucose change (E) and group (F) to show the visual relationship. Covariates included age, sex, elevated status, treatment group, and total intracranial volume (TICV)
3.4. Fasting glucose groups differ in cerebral amyloid accumulation
We next characterized differences in regional and global cerebral amyloid change. We found that the GW group accumulated more global cerebral amyloid over 1 year (P = .036), a change that was driven by increases in precuneus (P = .003) and superior parietal regions (P = .029, Table 4). Figure 2 shows a visual comparison of amyloid change in the precuneus ROI, which is of particular interest due to high metabolic rate and early accumulation of amyloid, in those whose GI (A) versus worsens (B).
TABLE 4.
Regional and global cerebral amyloid increases occur in individuals whose glucose regulation worsens
| Region of interest | FG improves (n = 61) | FG worsens (n = 44) | P‐value |
|---|---|---|---|
| Precuneus | 0.472 [4.6] | 2.81 [4.3] | .003* |
| Superior parietal | −0.392 [4.9] | 1.67 [5.6] | .029* |
| Anterior cingulate | −0.698 [5.4] | 0.701 [4.8] | .087 |
| Inferior medial frontal | 0.102 [5.3] | 1.08 [4.9] | .132 |
| Posterior cingulate | −0.347 [5.5] | 0.253 [6.1] | .390 |
| Lateral temporal | 0.896 [5.4] | 1.32 [4.5] | .306 |
| Global cerebral SUVR (6 ROI) | 0.010 [4.4] | 1.32 [4.1] | .040* |
Abbreviations: FG, fasting glucose; ROI, region of interest; SD, standard deviation; SUVR, standardized uptake value ratio.
Notes: Percent change in regional and global cerebral amyloid SUVR values in glucose in glucose change groups. Global SUVR was calculated as the mean of the six regions of interest.
Values are given as means [SD].
P < .05.
FIGURE 2.
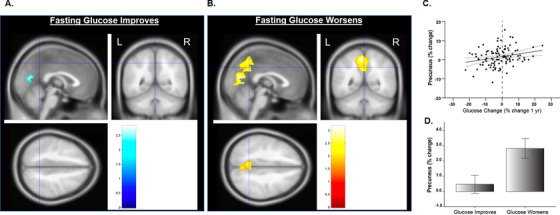
Longitudinal voxel‐based positron emission tomography (PET) standardized uptake value ratio (SUVR) shows increases in amyloid over 1 year in the precuneus/cuneus in the group whose glucose improves (A; PET amyloid increases are visible in blue) and in the group whose glucose worsens (B; PET amyloid increases are visible in yellow). Percent change in regional (precuneus) mean cerebellar standardized SUVR are also plotted percent change in glucose (C) and group (D). Covariates included age, sex, elevated status, and treatment group
3.5. Fasting glucose groups do not differ regarding change in limbic WM integrity
We did not observe significant differences in FA between groups, although trends for group differences (decreased FA in the GI group) were observed in the right cingulum bundle and right cingulum hippocampus (P = .068 and P = .089, respectively) when controlling for age, sex, and treatment group. Significance was not affected when APOE ε4 was added as a covariate.
3.6. Fasting glucose change tracks with change in cerebral amyloid
For the cerebral amyloid PET neuroimaging measure, we analyzed six a priori designated ROIs. Thus, for this measure, we also assessed glucose and cerebral amyloid change as continuous variables, in addition to the categorical analysis of “GI versus GW” as detailed in section 4.4. Across all individuals, the percent change in fasting glucose was linearly associated with percent change in cerebral amyloid in both the precuneus and posterior cingulate regions, as well as globally (Table S2).
4. DISCUSSION
We sought to test whether change in fasting glucose over 1 year was related to multi‐modal brain imaging measures of early neuropathological changes in the brain associated with dementia. Here, we present evidence that in cognitively healthy older adults, increasing fasting glucose over 1 year is associated with decreased limbic GM volume, decreased parietal cortex volume, and regional increases in brain amyloid. These initial signs of cerebral AD neuropathology across multiple modalities occurred in the absence of significant differences in insulin, body weight, body composition, cognitive change, or APOE ε4 carrier status.
There is accumulating evidence that abnormal cellular bioenergetics increases AD risk. A recent large‐scale analysis (>100,000 individuals) used Mendelian randomization to test whether measures of glucose metabolism were associated with increased risk for AD, and found that higher fasting glucose was causally associated with substantially increased AD risk. 5 Prior work suggests that higher blood glucose and markers of insulin resistance may be detrimental to brain structure and function. 25 , 26 , 28 , 29 As such, glucose regulation across the lifespan appears important for brain health; even variability in visit‐to‐visit fasting glucose during young adulthood (which may play a role in vascular complications 30 ) is associated with lower hippocampal volume and WM integrity at midlife 31 and cognitive function at midlife. 32 Recently, Ekblad et al. showed that insulin resistance in midlife was associated with greater amyloid burden regardless of APOE ε4 status. 33 In our analysis of older adults, significant differences in GM atrophy between fasting glucose change groups were seen in the right hippocampus and right parietal cortex, regions known to decline early in AD. Consistent with these findings, we also observed a weak trend for a decline in FA in the hippocampal cingulum bundle, a key component of the limbic system involved in communication with the hippocampus. Post mortem studies have shown increased tau burden early in the hippocampal entorhinal cortex in aging and AD, which may play a key role in limbic region neurodegeneration. 34 , 35 The association between impaired systemic metabolism and limbic atrophy suggests that increases in brain neuropathology were occurring concomitantly with GM and WM structural changes in individuals whose glucose worsened over time.
As mentioned, fasting glucose tracks with amyloid cross‐sectionally 3 and longitudinally in individuals considered elevated for amyloid at baseline. 36 Preclinical and biochemical evidence suggests a bidirectional link between glucose and amyloid processing. Decreased beta secretase 1 (BACE1), which regulates amyloid precursor protein processing are associated with improved glucose disposal and insulin sensitivity in animal models and cell lines. 37 Consistent with these findings, inhibition of BACE1 increases glucose uptake and basal oxygen consumption in cells. 38 In mouse hippocampal neurons, high glucose increases BACE1 expression and Aβ production, 39 and in adipocytes glucose increases Aβ secretion. 40 Finally, biochemical work suggests that the presence of glucose favors Aβ42 oligomer formation, which occurs rapidly and spontaneously. 41
Taken together, this evidence provides molecular rationale for the relationship of fasting glucose and cerebral amyloid. However, while meta‐analyses of human studies support T2D diagnosis as a risk factor for AD, 42 most autopsy data suggests no relationship 43 or a negative relationship 44 , 45 between T2D diagnosis and traditional AD neuropathology. Yet, exacerbated neuropathology has been observed in APOE ε4 carriers with T2D. 46 , 47 It is likely that in a disease state such as T2D, additional factors such as medication use, genetics, disease duration, and survival bias may also affect these relationships.
Interestingly, the changes in glucose regulation observed between our groups occurred independently of significant changes in body weight or body composition. This is important, as others have shown changes in glucose regulation can occur through changes in light physical activity or breaking up sitting time, 48 lifestyle changes that may not be large enough to trigger significant changes in body weight or body composition. Although insulin resistance as measured by HOMA‐IR was different between groups, this was primarily driven by glucose differences, with fasting insulin not different between the groups. We did observe higher systolic blood pressure levels at baseline in those whose glucose worsened over time, and these individuals were less likely to have a diagnosis of hypertension, suggesting potential differences in diagnosis and treatment between groups. However, the groups were similar in terms of hypertensive medication use, and had similar change in blood pressure over the 1‐year follow‐up. We also did not observe longitudinal differences in cognitive performance, suggesting that the neuropathological changes occur early prior to clinically detectable cognitive change. Importantly, we did not quantify changes in tau neuropathology in this study. Others have recently shown that increasing tau neuropathology (measured by PET) tracks with memory decline in cognitively healthy older adults, 49 and tau neuropathology may be of particular importance in individuals with impaired glucose metabolism and diabetes. 50 , 51 Future work is needed to elucidate the relationship between change in glucose and tau neuropathology in older adults at risk for AD.
There are limitations to consider in our study. We included individuals with elevated amyloid and those who fell in what is considered the “subthreshold” range. While this is of importance because these individuals may be at greatest risk for developing AD, individuals with varying cerebral amyloid at baseline may accumulate amyloid longitudinally at varying rates. Our inclusion of sedentary individuals with relatively high quantitative levels of amyloid may limit the generalizability of the study to more active individuals or populations with lower cerebral amyloid levels. Additionally, this is a longitudinal study over 1 year, and studies of greater duration are needed to determine whether these patterns are consistent and associated with eventual cognitive decline or AD diagnosis. Additional studies are also needed to investigate the relationship between glucose change and tau brain neuropathology, which was not assessed in our study. Finally, APEx was a clinical trial of exercise. Although there were no differences in fasting glucose change between exercise groups (data not shown), and we included “exercise group” as a covariate, this is a limitation.
Overall, this work suggests that cognitively healthy older adults who have worsening glucose regulation over time are potentially at risk for worsening neuropathological and structural brain outcomes. A shift in fasting glucose from normal to impaired, which could occur with the magnitude of change we observed in the current study, has been associated with all‐cause mortality, 52 and increased rates of fasting glucose over time are linked to cardiovascular markers such as increased arterial stiffness 53 and cerebrovascular disease incidence. 54 However, there are more clinically relevant outcomes of glycemic control than fasting glucose, such as HbA1c. Future studies of longitudinal change in brain structure and neuropathology with additional clinically meaningful outcomes of glycemic control are also needed. Nonetheless, our findings suggest that changes in fasting glucose, controlling for age, sex, exercise treatment group, and baseline cerebral amyloid level, associate positively with longitudinal amyloid accumulation in highly metabolic regions. Additional work to explore the effects of targeting systemic glucose metabolism as a potential way to modulate these outcomes in cognitively healthy older adults at risk for AD is warranted.
CONFLICTS OF INTEREST
Dr. Robyn A. Honea reports no disclosures. Casey John reports no disclosures. Zachary Green reports no disclosures. Paul Kueck reports no disclosures. Dr. Matthew K. Taylor receives research support from the National Institutes of Health (NIH). Dr. Rebecca J. Lepping receives research support from the NIH, Novartis, and Veloxis. Dr. Ryan Townley has received honoraria from the University of Kentucky and the University of Kansas and the law firm Tenopir and Huerter. Dr. Eric D. Vidoni is named on patent 18KU028M and receives research support from the NIH. Dr. Jeffery M. Burns receives research support from the NIH and for clinical trials from the Alzheimer's Association, Eli Lilly, Avid Radiopharmaceuticals, Novartis, Amylyx, Eisai, Merck, Biogen, AbbVie, vTv Therapeutics, Genentech, Janssen, Astra‐Zeneca, and Roche. Dr. Jeffery M. Burns has also received consulting fees from Biogen, Stage 3 Innovations, Avanir Pharmaceuticals, and Sage Therapeutics. Dr. Jeffery M. Burns has received honoraria from USC and the University of Washington and has an unpaid advisory role in the Albert Trust Institute for White Matter and Cogntion. Dr. Jill K. Morris receives research support from the NIH.
AUTHOR CONTRIBUTIONS
All authors and contributors agreed to the conditions outlined in the Authorship and Contributorship section of the Information for Authors. All authors take full responsibility for the data, the analyses and interpretation, and the conduct of the research; have full access to all data; and the right to publish all data.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
This project is supported by funding from the National Institute on Aging, including R01AG052954 (Jeffery M. Burns), R01AG062548 and R00AG050490 (Jill K. Morris), R21AG061548 (Eric D. Vidoni), and P30AG035982 (KU Alzheimer's Disease Research Center). Additional support is provided through the Margaret “Peg” McLaughlin and Lydia A. Walker Opportunity Fund and the Leo and Anne Albert charitable trust. We would also like to thank Riley Kemna for technical support for this manuscript.
Honea RA, John CS, Green ZD, et al. Relationship of fasting glucose and longitudinal Alzheimer's disease imaging markers. Alzheimer's Dement. 2022;8:e12239. 10.1002/trc2.12239
DATA AVAILABILITY STATEMENT
De‐identified data will be shared at the request of qualified investigators for purposes of replicating procedures and results.
REFERENCES
- 1. O'Sullivan JB. Age gradient in blood glucose levels. Magnitude and clinical implications. Diabetes. 1974;23:713‐715. [DOI] [PubMed] [Google Scholar]
- 2. Morris JK, Honea RA, Vidoni ED, Swerdlow RH, Burns JM. Is Alzheimer's disease a systemic disease? Biochim Biophys Acta. 2014;1842(9):1340‐1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Morris JK, Vidoni ED, Wilkins HM, et al. Impaired fasting glucose is associated with increased regional cerebral amyloid. Neurobiol Aging. 2016;44:138‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Taylor MK, Sullivan DK, Swerdlow RH, et al. High glycemic diet is associated with cerebral amyloid in cognitively normal elderly. Am J Clin Nutr. 2017;106(6):1463‐1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pan Y, Chen W, Yan H, Wang M, Xiang X. Glycemic traits and Alzheimer's disease: a Mendelian Randomization Study. Aging (Albany NY). 2020;12:22688‐22699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Macauley SL, Stanley M, Caesar EE, et al. Hyperglycemia modulates extracellular amyloid‐beta concentrations and neuronal activity in vivo. J Clin Investig. 2015;125:2463‐2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Akhtar MW, Sanz‐Blasco S, Dolatabadi N, et al. Elevated glucose and oligomeric beta‐amyloid disrupt synapses via a common pathway of aberrant protein S‐nitrosylation. Nat Commun. 2016;7:10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vidoni ED, Morris JK, Watts A, et al. Effect of aerobic exercise on amyloid accumulation in preclinical Alzheimer's: a 1‐year randomized controlled trial. PloS One. 2021;16:e0244893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mayer CJ, Steinman L, Williams B, Topolski TD, LoGerfo J. Developing a Telephone Assessment of Physical Activity (TAPA) questionnaire for older adults. Prev Chronic Dis. 2008;5:A24. [PMC free article] [PubMed] [Google Scholar]
- 10. Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412b‐2414b. [DOI] [PubMed] [Google Scholar]
- 11. Weintraub S, Besser L, Dodge HH, et al. Version 3 of the Alzheimer disease centers' neuropsychological test battery in the Uniform Data Set (UDS). Alzheimer Dis Assoc Disord. 2018;32:10‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harn NR, Hunt SL, Hill J, Vidoni E, Perry M, Burns JM. Augmenting amyloid PET interpretations with quantitative information improves consistency of early amyloid detection. Clin Nucl Med. 2017;42:577‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clark CM, Pontecorvo MJ, Beach TG, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid‐beta plaques: a Prospective Cohort Study. Lancet Neurol. 2012;11:669‐678. [DOI] [PubMed] [Google Scholar]
- 14. Joshi AD, Pontecorvo MJ, Clark CM, et al. Performance characteristics of amyloid PET with florbetapir F 18 in patients with Alzheimer's disease and cognitively normal subjects. J Nucl Med. 2012;53:378‐384. [DOI] [PubMed] [Google Scholar]
- 15. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta‐cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412‐419. [DOI] [PubMed] [Google Scholar]
- 16. Weintraub S, Salmon D, Mercaldo N, et al. The Alzheimer's disease centers' Uniform Data Set (UDS): the neuropsychologic test battery. Alzheimer Dis Assoc Disord. 2009;23:91‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shirk SD, Mitchell MB, Shaughnessy LW, et al. A web‐based normative calculator for the uniform data set (UDS) neuropsychological test battery. Alzheimers Res Ther. 2011;3:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ashburner J. A fast diffeomorphic image registration algorithm. NeuroImage. 2007;38:95‐113. [DOI] [PubMed] [Google Scholar]
- 19. Good CD, Ashburner J, Frackowiak RS. Computational neuroanatomy: new perspectives for neuroradiology. Rev Neurol (Paris). 2001;157:797‐806. [PubMed] [Google Scholar]
- 20. Smith SM, Jenkinson M, Woolrich MW, et al. Advances in functional and structural MR image analysis and implementation as FSL. NeuroImage 2004;23(Suppl 1):S208‐S219. [DOI] [PubMed] [Google Scholar]
- 21. Smith SM, Jenkinson M, Johansen‐Berg H, et al. Tract‐based spatial statistics: voxelwise analysis of multi‐subject diffusion data. NeuroImage. 2006;31:1487‐1505. [DOI] [PubMed] [Google Scholar]
- 22. Wang DQ, Wang L, Wei MM, et al. Relationship between type 2 diabetes and white matter hyperintensity: a systematic review. Front Endocrinol (Lausanne). 2020;11:595962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mori S, Wakana S, Van Zijl PC, Nagae‐Poetscher L. MRI Atlas of Human White Matter. Amsterdam, Netherlands: Elsevier; 2005. [Google Scholar]
- 24. Hua K, Zhang J, Wakana S, et al. Tract probability maps in stereotaxic spaces: analyses of white matter anatomy and tract‐specific quantification. NeuroImage. 2008;39:336‐347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Morris JK, Vidoni ED, Perea RD, et al. Insulin resistance and gray matter volume in neurodegenerative disease. Neuroscience. 2014;270:139‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morris JK, Vidoni ED, Honea RA, Burns JM. Alzheimer's Disease Neuroimaging I . Impaired glycemia increases disease progression in mild cognitive impairment. Neurobiol Aging. 2014;35:585‐589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tzourio‐Mazoyer N, Landeau B, Papathanassiou D, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single‐subject brain. NeuroImage. 2002;15:273‐289. [DOI] [PubMed] [Google Scholar]
- 28. Burns JM, Donnelly JE, Anderson HS, et al. Peripheral insulin and brain structure in early Alzheimer disease. Neurology. 2007;69:1094‐1104. [DOI] [PubMed] [Google Scholar]
- 29. Burns JM, Honea RA, Vidoni ED, Hutfles LJ, Brooks WM, Swerdlow RH. Insulin is differentially related to cognitive decline and atrophy in Alzheimer's disease and aging. Biochim Biophys Acta. 2012;1822:333‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Su G, Mi S, Tao H, et al. Association of glycemic variability and the presence and severity of coronary artery disease in patients with type 2 diabetes. Cardiovasc Diabetol. 2011;10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xiong Z, Li J, Zhong X, et al. Visit‐to‐visit fasting glucose variability in young adulthood and hippocampal integrity and volume at midlife. Diabetes Care. 2019;42:2334‐2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bancks MP, Carnethon MR, Jacobs DR, Jr. , et al. Fasting glucose variability in young adulthood and cognitive function in middle age: the Coronary Artery Risk Development in Young Adults (CARDIA) Study. Diabetes Care. 2018;41:2579‐2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ekblad LL, Johansson J, Helin S, et al. Midlife insulin resistance, APOE genotype, and late‐life brain amyloid accumulation. Neurology 2018;90:e1150‐e1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Puladi B, Dinekov M, Arzberger T, Taubert M, Kohler C. The relation between tau pathology and granulovacuolar degeneration of neurons. Neurobiol Dis. 2021;147:105138. [DOI] [PubMed] [Google Scholar]
- 35. Xie L, Wisse LEM, Das SR, et al. Longitudinal atrophy in early Braak regions in preclinical Alzheimer's disease. Hum Brain Mapp. 2020;41:4704‐4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gomez G, Beason‐Held LL, Bilgel M, et al. Metabolic syndrome and amyloid accumulation in the aging brain. J Alzheimer's Dis. 2018;65:629‐639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Meakin PJ, Harper AJ, Hamilton DL, et al. Reduction in BACE1 decreases body weight, protects against diet‐induced obesity and enhances insulin sensitivity in mice. Biochem J. 2012;441:285‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hamilton DL, Findlay JA, Montagut G, et al. Altered amyloid precursor protein processing regulates glucose uptake and oxidation in cultured rodent myotubes. Diabetologia. 2014;57:1684‐1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee HJ, Ryu JM, Jung YH, et al. High glucose upregulates BACE1‐mediated abeta production through ROS‐dependent HIF‐1alpha and LXRalpha/ABCA1‐regulated lipid raft reorganization in SK‐N‐MC cells. Sci Rep. 2016;6:36746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tharp WG, Gupta D, Smith J, Jones KP, Jones AM, Pratley RE. Effects of glucose and insulin on secretion of amyloid‐beta by human adipose tissue cells. Obesity (Silver Spring). 2016;24:1471‐1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kedia N, Almisry M, Bieschke J. Glucose directs amyloid‐beta into membrane‐active oligomers. Phys Chem Chem Phys. 2017;19:18036‐18046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bellou V, Belbasis L, Tzoulaki I, Middleton LT, Ioannidis JPA, Evangelou E. Systematic evaluation of the associations between environmental risk factors and dementia: an umbrella review of systematic reviews and meta‐analyses. Alzheimers Dement. 2017;13:406‐418. [DOI] [PubMed] [Google Scholar]
- 43. Arvanitakis Z, Schneider JA, Wilson RS, et al. Diabetes is related to cerebral infarction but not to AD pathology in older persons. Neurology. 2006;67:1960‐1965. [DOI] [PubMed] [Google Scholar]
- 44. Beeri MS, Silverman JM, Davis KL, et al. Type 2 diabetes is negatively associated with Alzheimer's disease neuropathology. J Gerontol A Biol Sci Med Sci. 2005;60:471‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ahtiluoto S, Polvikoski T, Peltonen M, et al. Diabetes, Alzheimer disease, and vascular dementia. Neurology. 2010;75:1195‐1202. [DOI] [PubMed] [Google Scholar]
- 46. Malek‐Ahmadi M, Beach T, Obradov A, et al. Increased Alzheimer's disease neuropathology is associated with type 2 diabetes and ApoE epsilon.4 carrier status. Curr Alzheimer Res. 2013;10:654‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dos Santos Matioli MNP, Suemoto CK, Rodriguez RD, et al. Diabetes is not associated with Alzheimer's disease neuropathology. J Alzheimer's Dis. 2017;60:1035‐1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Colberg SR, Sigal RJ, Yardley JE, et al. Physical activity/exercise and diabetes: a position statement of the American Diabetes Association. Diabetes Care. 2016;39:2065‐2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen X, Cassady KE, Adams JN, Harrison TM, Baker SL, Jagust WJ. Regional tau effects on prospective cognitive change in cognitively normal older adults. J Neurosci. 2021;41:366‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moran C, Beare R, Phan TG, et al. Type 2 diabetes mellitus and biomarkers of neurodegeneration. Neurology. 2015;85(13):1123‐1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McIntosh EC, Nation DA. Alzheimer's Disease Neuroimaging I . Importance of treatment status in links between Type 2 diabetes and Alzheimer's disease. Diabetes Care. 2019;42:972‐979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee G, Kim SM, Choi S, et al. The effect of change in fasting glucose on the risk of myocardial infarction, stroke, and all‐cause mortality: a Nationwide Cohort Study. Cardiovasc Diabetol. 2018;17:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wu Y, Yu J, Jin C, et al. Longitudinal fasting blood glucose patterns and arterial stiffness risk in a population without diabetes. PloS One. 2017;12:e0188423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ogata S, Watanabe M, Kokubo Y, et al. Longitudinal trajectories of fasting plasma glucose and risks of cardiovascular diseases in middle age to elderly people within the general Japanese population: the Suita Study. J Am Heart Assoc. 2019;8:e010628. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
De‐identified data will be shared at the request of qualified investigators for purposes of replicating procedures and results.