

Abstract
The vacuolar ATPase (V‐ATPase) is a rotary motor proton pump that is regulated by an assembly equilibrium between active holoenzyme and autoinhibited V1‐ATPase and Vo proton channel subcomplexes. Here, we report cryo‐EM structures of yeast V‐ATPase assembled in vitro from lipid nanodisc reconstituted Vo and mutant V1. Our analysis identified holoenzymes in three active rotary states, indicating that binding of V1 to Vo provides sufficient free energy to overcome Vo autoinhibition. Moreover, the structures suggest that the unequal spacing of Vo’s proton‐carrying glutamic acid residues serves to alleviate the symmetry mismatch between V1 and Vo motors, a notion that is supported by mutagenesis experiments. We also uncover a structure of free V1 bound to Oxr1, a conserved but poorly characterized factor involved in the oxidative stress response. Biochemical experiments show that Oxr1 inhibits V1‐ATPase and causes disassembly of the holoenzyme, suggesting that Oxr1 plays a direct role in V‐ATPase regulation.
Keywords: Cryo‐electron microscopy, Oxr1p, reversible disassembly, TLDc domain, vacuolar ATPase
Subject Categories: Membranes & Trafficking, Organelles, Structural Biology
Comprehensive Cryo‐EM and biochemical analysis identifies Oxr1 as novel binding partner of V‐ATPase, controlling reversible enzyme assembly.

Introduction
The vacuolar H+‐ATPase (V‐ATPase; V1Vo ATPase) is a large, multisubunit protein complex that acidifies the lumen of intracellular organelles, including the Golgi apparatus, early and late endosomes, lysosomes, and secretory vesicles (Maxson & Grinstein, 2014; Kane, 2016; Futai et al, 2019; Collins & Forgac, 2020; Eaton et al, 2021). V‐ATPase’s proton pumping activity is essential for many cellular processes such as pH and ion homeostasis, protein trafficking and degradation, endocytosis, autophagy, phagocytosis, signaling, hormone secretion, and neurotransmitter release. In some specialized tissues of higher organisms, V‐ATPase is targeted to the plasma membrane to pump protons out of the cell. V‐ATPase‐mediated extracellular acidification serves to facilitate processes such as bone remodeling, sperm maturation, and maintaining blood pH. V‐ATPases are associated with several widespread diseases, including distal renal tubular acidosis (dRTA) (Smith et al, 2000), osteopetrosis (Frattini et al, 2000), sensorineural deafness (Karet et al, 1999), male infertility (Breton et al, 1996), diabetes (Sun‐Wada et al, 2006), neurodegeneration (Bagh et al, 2017), and cancer (Stransky et al, 2016). Moreover, V‐ATPase‐mediated acidification is required for efficient infection by viruses such as influenza or Ebola (Perez & Carrasco, 1994; Lindstrom et al, 2018), or targeted for inhibition by microbial pathogens including Mycobacterium tuberculosis, Legionella pneumophila, or Vibrio parahaemolyticus (Xu et al, 2010; Wong et al, 2011; Sreelatha et al, 2015). Thus, V‐ATPase is essential for normal cellular processes and plays an important role in the pathogenesis of a wide spectrum of diseases.
Eukaryotic V‐ATPases are highly conserved across species. The relative ease of yeast genetics and the ability to purify milligram amounts of yeast V‐ATPase and its subcomplexes make Saccharomyces cerevisiae a powerful model system to study the structure, catalytic mechanism, and regulation of the enzyme from higher organisms, including mammals. The yeast V‐ATPase is composed of 16 different polypeptides that are organized into two subcomplexes – a cytosolic V1‐ATPase, composed of A3B3(C)DE3FG3H, and a membrane integral Vo proton channel, made of ac 8 c′c′′defVoa1 (Harrison & Muench, 2018; Vasanthakumar & Rubinstein, 2020; Wilkens et al, 2021) (Appendix Fig S1A). V‐ATPase is a member of the family of rotary ATPases, which also includes F1Fo‐ATP synthase found in bacteria, mitochondria, and chloroplasts, A1Ao‐ATPase/synthase found in archaea, and A/V‐like ATPases found in eubacteria (Hilario & Gogarten, 1998). In V‐ATPase, ATP is hydrolyzed in three catalytic sites that are located on the A3B3 catalytic hexamer at alternating A‐B interfaces. The resulting free energy drives rotation of a central rotor stalk formed by subunit D, which is embedded in the central cavity of the A3B3 hexamer and, together with subunit F, protrudes ~45 Å towards the membrane. Rotation of the central stalk is coupled to the Vo via subunit d, which forms a structural link between the DF rotor of the V1 and the ten‐membered proteolipid‐ or c‐ring formed by the hydrophobic c subunits (c, c′, c′′) of the Vo. Proton translocation occurs at the interface of the c‐ring and the C‐terminal transmembrane domain of subunit a (a CT), which contains two half‐channels lined by polar residues that provide access of water molecules to the c‐ring from opposite sides of the membrane. During ATP hydrolysis‐driven c‐ring rotation, conserved glutamates present in each of the proteolipid subunits become protonated at the cytoplasmic half‐channel, and as the c‐ring rotates, the protons are carried through the lipid bilayer before being released into the lumen or extracellular space. Three heterodimers of subunits E and G (peripheral stalks EG1‐3) together with subunits H and C, and the N‐terminal domain of subunit a (a NT) keep the catalytic hexamer static during rotation of the central stalk for efficient energy coupling.
V‐ATPase activity is regulated by a unique process called reversible disassembly, wherein V1 detaches from the membrane integral Vo domain with concomitant release of subunit C into the cytosol (Parra et al, 2014; Oot et al, 2017) (Appendix Fig S1B). This process is reversible in vivo, and thus dissociated components can reassemble into a holoenzyme under favorable conditions. V‐ATPase regulation by reversible disassembly has been extensively characterized on a cellular level in yeast (Parra et al, 2014), and while the process is conserved in mammals (Trombetta et al, 2003; Sautin et al, 2005; Stransky & Forgac, 2015; Bodzeta et al, 2017), much less is known about the mechanism and signaling pathways in higher organisms. From studies in yeast, we know that disassembly due to, e.g., glucose withdrawal requires both ATP hydrolysis by the enzyme, as well as an intact microtubule network, and that (re)assembly is facilitated by the heterotrimeric chaperone complex “Regulator of ATPases of Vacuolar and Endosomal membranes” (RAVE), along with glycolytic enzymes aldolase and phosphofructokinase‐I (Parra & Kane, 1998; Xu & Forgac, 2001; Lu et al, 2007; Chan & Parra, 2014; Smardon et al, 2015). Recent structural studies conducted with the yeast enzyme showed that V‐ATPase disassembly is accompanied by dramatic conformational changes of the V1 and Vo subunits that form the interface in the holoenzyme (Couoh‐Cardel et al, 2015; Oot et al, 2016). These structural changes result in autoinhibition of disassembled V1 and Vo subcomplexes, in that V1 can no longer hydrolyze MgATP, and Vo becomes sealed to protons (Zhang et al, 1992; Graf et al, 1996; Parra et al, 2000; Couoh‐Cardel et al, 2015). In Vo, autoinhibition is largely due to an association of a NT (part of the “stator” of the Vo motor) with subunit d (part of the rotor), an interaction not present in the holoenzyme (Couoh‐Cardel et al, 2015; Mazhab‐Jafari et al, 2016; Roh et al, 2018). Another structural change that accompanies enzyme dissociation occurs in V1 subunit H: upon V1 detachment from Vo, subunit H’s C‐terminal domain (HCT) rotates and moves from a binding site on a NT, which is required for activity of the holoenzyme (Ho et al, 1993), to a binding site on A3B3, which leads to autoinhibition of V1 (Oot et al, 2016). HCT binding to V1 stabilizes an open catalytic site, which results in entrapment of MgADP at an adjacent closed catalytic site (Oot et al, 2016). During in vivo reassembly, HCT and inhibitory MgADP are released from V1 by an as yet unknown mechanism (Oot et al, 2017). Thus, HCT‐mediated inhibition is a major obstacle in the reassembly of V1 and Vo into holoenzyme in vitro. However, amino acid sequence analysis revealed that mammalian HCT lacks the eleven‐residue loop found in yeast HCT that is responsible for autoinhibition of yeast V1 (Oot et al, 2016). We recently showed that replacing wild‐type H (Hwt) in yeast with a chimeric form of H consisting of yeast HNT and human HCT (Hchim) activates V1’s MgATPase activity and allows for in vitro assembly of coupled holo V‐ATPase (Sharma et al, 2019). Moreover, biophysical experiments indicated that V‐ATPase containing Hchim is more stable (less prone to disassembly) than the wild‐type yeast enzyme since human HCT binds well to yeast a NT, but has little to no affinity for yeast V1. However, despite recent progress with structure determination of holo V‐ATPases from a variety of sources (Abbas et al, 2020; Wang et al, 2020a, 2020b), key aspects of the molecular mechanism of V‐ATPase regulation by reversible disassembly remain largely elusive.
Here, we characterize in vitro assembled and lipid nanodisc reconstituted yeast V‐ATPase (V1HchimVoND) using cryo‐electron microscopy (cryoEM) and biochemical experiments. Previous cryoEM and crystal structures of yeast holoenzyme and autoinhibited V1 and Vo subcomplexes revealed that while active V‐ATPases populate three distinct rotary states (states 1–3) (Zhao et al, 2015), autoinhibited V1 and Vo subcomplexes are halted in states 2 and 3, respectively (Mazhab‐Jafari et al, 2016; Oot et al, 2016; Stam & Wilkens, 2017; Roh et al, 2018). In the current study, we found that V‐ATPases that were assembled in vitro from state 2 V1Hchim and lipid nanodisc reconstituted state 3 Vo populated all three rotary states, with the majority of the particles in state 1, similar to what had been observed for the wild‐type holoenzyme purified from yeast. Since in vitro assembly was carried out in absence of ATP, this means that activation of the Vo and subsequent rotation of the DFd:c‐ring central rotor subcomplex is driven solely by the formation of protein‐protein interactions at the V1‐Vo interface. Another peculiar feature of the eukaryotic V‐ATPase is the unequal angular spacing of the proton carrying glutamic acid residues of the c‐ring due to the presence of the single‐copy proteolipid subunit c″, a structural feature whose role in enzyme function is not understood. An analysis of the structures of the three rotary states at the level of the c‐ring suggests that this unequal spacing of the c‐ring glutamates serves to alleviate the symmetry mismatch between the three‐step V1 motor and the ten‐step c‐ring proton turbine, a hypothesis that is supported by in vivo mutagenesis experiments that show that a symmetric c‐ring results in complete loss of function. Finally, the cryoEM analysis of in vitro assembled holoenzyme revealed a population of free V1‐ATPase molecules bound to subunit C and a hitherto unseen binding partner of V1, the TLDc domain of yeast Oxr1p. Biochemical experiments show that Oxr1p binding inhibits V‐ATPase activity and promotes holoenzyme disassembly. From the data, we propose that Oxr1p provides a mechanism of V‐ATPase disassembly that is independent of the enzyme’s catalytic activity.
Results
CryoEM structures of in vitro reconstituted yeast holo V‐ATPase
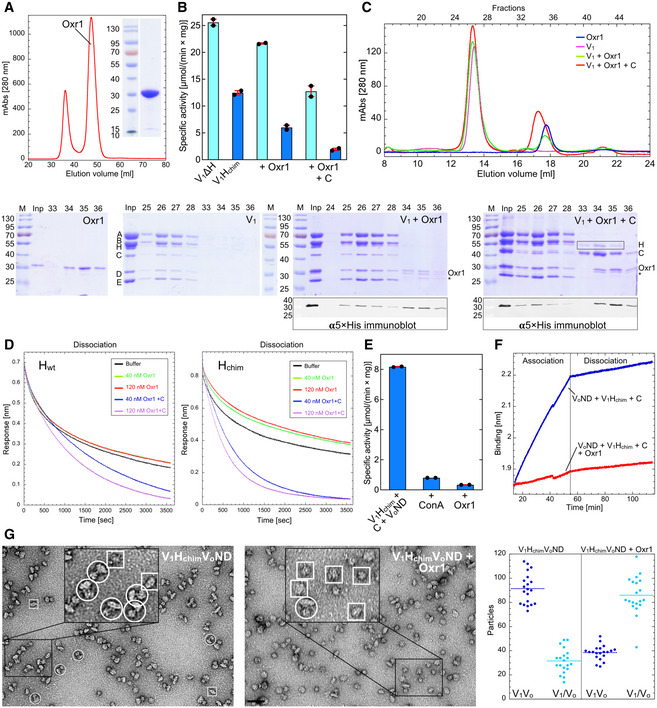
We recently described an in vitro procedure for reconstituting highly active and stable yeast holo V‐ATPase from purified Vo, subunit C, and V1‐ATPase containing a mutant form of subunit H (Hchim) composed of yeast N‐ and human C‐terminal domains (Sharma et al, 2019). Briefly, yeast V1‐ATPase is affinity captured from a strain in which the gene for subunit H is deleted, and the resulting V1ΔH is reconstituted with recombinant Hchim. Purified V1Hchim is then combined with Vo in lipid nanodisc (VoND) together with recombinant subunit C to assemble V1HchimVoND (Fig 1A and B). V1HchimVoND’s specific MgATPase activity is typically in the range of 8–12 µmol × (min × mg)−1 and > 90% sensitive to the V‐ATPase specific inhibitor Concanamycin A (ConA) (Fig 1C), indicating that the Hchim containing enzyme is tightly coupled. Purified V1HchimVoND (Fig 1D) was vitrified on holey gold grids and imaged by cryoEM, and a dataset of ~570,000 particles was subjected to 2‐D reference‐free classification and 3‐D reconstruction protocols (Fig EV1). From the analysis, we obtained maps of the holoenzyme in the previously observed rotational states 1–3 at overall resolutions of between 4.2 and 6.6 Å (Appendix Table S1). Focused refinement using V1 and Vo masks improved local resolution for some parts of the maps to ~3.2 Å, allowing visualization of side‐chain density for most of the V1 subcomplex (Appendix Fig S2).
Figure 1. Reconstitution and cryoEM analysis of yeast V‐ATPase.
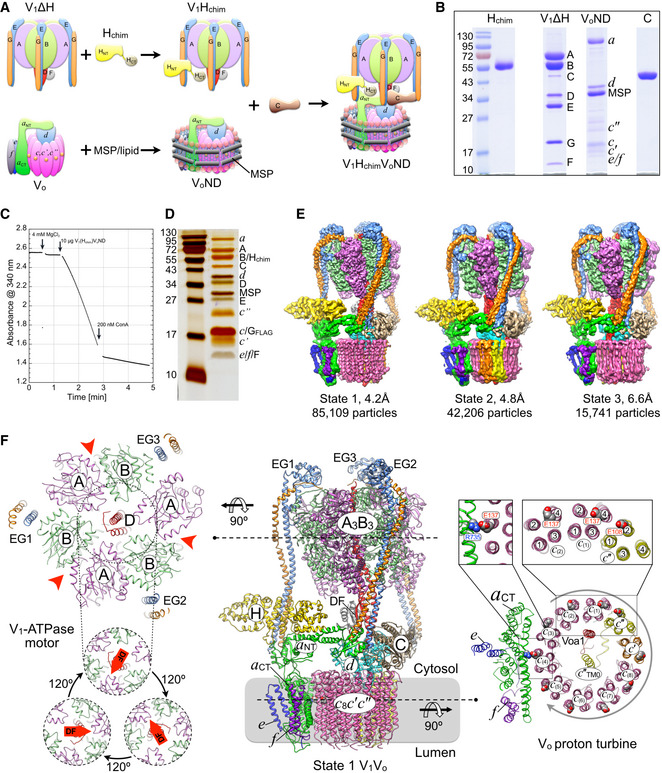
- Reconstitution of yeast V‐ATPase from Hchim containing V1 (V1Hchim), lipid nanodisc reconstituted Vo (VoND), and recombinant subunit C.
- Coomassie‐stained SDS‐PAGE (from left to right) of Hchim, V1ΔH, VoND, and C.
- Representative ATPase activity assay (ATP regenerating system). At the indicated time, 200 nM ConA is added to the assay.
- Silver stained SDS‐PAGE of V1HchimVoND.
- CryoEM maps of rotational states 1–3 of the in vitro assembled holoenzyme.
- Middle panel: Ribbon diagram of the state 1 V1HchimVoND holoenzyme. Subunit A, magenta; B, light green; C, tan; D, red; E, light blue; F, grey; G, orange; H, yellow; a, green; c 8, pink; c′, orange; c″, yellow; d, cyan; e, blue; f, purple; Voa1, red. Left panel, top: Cross section of V1‐ATPase at the level indicated. The three nucleotide‐binding sites are indicated by red arrowheads. Left panel, bottom: The central rotor of the V1 motor (subunits DF) rotates in steps of 120°. Right panels: Cross section of the c‐ring at the level indicated. The c‐ring of the Vo proton turbine rotates in ten steps, with each step stabilized by a transient salt bridge between a c‐ring glutamic acid and the essential arginine of a CT (Arg735 in yeast). Each proteolipid subunit is organized in a four α helix bundle, with helices 2 and 4 forming the outer, and helices 1 and 3 forming the inner ring of the c‐ring. Whereas the c and c′ subunits have the glutamic acid on α helix 4, c″ has the glutamate on helix 2, creating an unequal spacing of the c‐ring glutamic acid residues, a unique feature of eukaryotic V‐ATPase.
Figure EV1. CryoEM data processing workflow.

- Representative electron micrograph of reconstituted V1(Hchim)VoND with representative 2‐D class averages on the right. A few holoenzyme particles are highlighted by green circles. Bar is 50 nm.
- Image processing workflow for 3‐D classification and map refinement. The class designated “noH” resulted in a low‐resolution map with only weak density for subunit H that could not be improved by further classification, suggesting that the class represents a heterogeneous population of dynamic states and/or assembly intermediates.
- Map resolution for states 1, 2, 3, and V1(C)Oxr1 according to the gold‐standard Fourier shell correlation (FSC) at the 0.143 criterion indicated by the dashed line.
Reconstituted V1HchimVoND populates all three rotational states
Previous work by the Rubinstein group showed that wild‐type V‐ATPase purified from yeast exists in three rotary states (termed states 1‐3) that are distinguished by ~120° rotations of the central rotor (subunits DFdc 8 c′c″Voa1, or DFd:c‐ring) relative to the stator (subunits A3B3CE3G3Haef) (Zhao et al, 2015). The three states, which were not equally populated, likely resulted when MgATP was exhausted upon cell lysis and subsequent protein purification. However, since purified V1Hchim and Vo are locked in rotary states 2 and 3, respectively, and since in vitro assembly of V1Hchim, VoND, and C was carried out in absence of MgATP, our expectation was that the resulting V1HchimVoND complexes should be in state 2 and/or state 3. Surprisingly, the cryoEM analysis of the in vitro assembled V‐ATPases revealed the presence of all three rotary states, with classes representing states 1, 2, and 3 containing 59, 30, and 11% of the holoenzyme particles (Figs 1E and EV1). This distribution, with the majority of particles in state 1, is similar to the one observed for wild‐type complexes isolated from yeast (Zhao et al, 2015). The observation that most of the particles populated state 1 indicates that the free energy that is released upon the formation of protein‐protein interactions at the V1‐Vo interface (Fig 1A) is sufficient to relieve the inhibitory a NT‐d interaction present in free Vo (Couoh‐Cardel et al, 2015), and rotate the central stalk complex (DFd:c‐ring) in the absence of ATP to produce the equilibrium distribution dictated by the states’ overall energies, with state 1 having the lowest energy.
The c‐ring asymmetry is required for V‐ATPase function
V‐ATPase consists of two stepping motors, V1 and Vo, that are coupled via the central stalk (Fig 1F). In the V1‐ATPase motor, the DF subcomplex rotates in steps of 120° powered by cyclic ATP hydrolysis on the three catalytic nucleotide‐binding sites located at alternating AB interfaces (Fig 1F, left panels). The proton carrying c‐ring of the Vo, however, rotates in ten steps, with the pauses (dwell times) between each step transiently stabilized by the formation of a salt bridge between proton carrying c‐ring glutamic acid residues and the essential arginine of a CT (Fig 1F, top left of right panels). A peculiar feature of eukaryotic V‐ATPase is the asymmetry of its c‐ring due to the presence of the c″ proteolipid. Each proteolipid has four transmembrane (TM) α helices, with helices 2 and 4 forming the outer, and helices 1 and 3 the inner ring of the c‐ring. The c″ proteolipid contains an extra N‐terminal α helix that is positioned in the center of the c‐ring cylinder and referred to as c″TM0 (Fig 1F, right panels). Whereas nine of the ten proteolipids (c 8 c′ in yeast) have the proton carrying glutamate on the outer surface of TM helix 4 (E137 and E145 for c and c′, respectively), c″ has the glutamic acid on helix 2 (E108 in yeast) (Hirata et al, 1997; Mazhab‐Jafari et al, 2016; Roh et al, 2018; Fig 1F, right panels). This arrangement creates an unequal angular spacing of the glutamic acid residues, with one full 360° rotation of the c‐ring being divided into eight steps of 36°, and each one step of 18° and 54° between the glutamate on c″ and the glutamates on the neighboring c (1) and c′ subunits (Fig 2A). We recently determined a 2.7 Å cryoEM structure of yeast Vo, and from accompanying MD simulations, we found that the unequal spacing of the glutamic acid residues contributes to the autoinhibition of passive proton translocation through free Vo (Roh et al, 2020). However, whether the asymmetry, which is unique to the eukaryotic V‐ATPase (virtually all of the related F‐, A‐ and A/V‐like ATPases/synthases have symmetric c‐rings) also plays a role in holoenzyme function, is not known. To address the question whether the c‐ring asymmetry is required for V‐ATPase function, we generated three mutants in yeast c″: (i) the essential glutamate in c″ (E108) was changed to glycine (c″E108G; Delete), (ii) the equivalent position in TM α helix 4 was changed to a glutamate (c″S192E; Addition), (iii) and a double mutant was generated to switch the essential glutamate from TM α helix 2 to helix 4 (c″E108G/S192E; Switch) to create a symmetric distribution of c‐ring glutamic acid residues (Fig 2B). We then tested the vma‐ growth phenotype of the three mutants compared to wild type (Fig 2C). The vma‐ phenotype of mutant yeast strains that lack functional V‐ATPase is characterized by varying levels of growth inhibition depending on pH of the media and the presence of calcium or zinc ions (Nelson & Nelson, 1990). From previous work it was known that c″E108 is essential for V‐ATPase function (Hirata et al, 1997), and we therefore included the c″E108G Delete mutant as a control. Growth on rich media (YPD) buffered to pH 5 does not require V‐ATPase activity, and consequently, all strains grow under this condition (Fig 2C1). Growth is also observed on synthetic dropout media (SD) containing all amino acids (Fig 2C2), but since the URA3 selection marker was used to delete the gene for c″ (VMA16), only vma16Δ grows in absence of uracil (Fig 2C3). The most stringent growth media is buffered to pH 7 and contains Zn2+ or Ca2+, conditions under which only wild type is able to grow (Fig 2C4,5). The least stringent media, YPD pH 7, allows some growth of all strains (Fig 2C6); however, the more stringent YPD + Ca2+ only shows growth for the Delete (c″E108G) and Addition (c″S192E) mutants, but not for the Switch (c″E108G/S192E) mutant strain (Fig 2C7). Moreover, the Switch mutant shows weakest growth compared to the Deletion and Addition mutants under any of the permissive conditions, indicating that the asymmetry in the angular spacing of the c‐ring due to c″ is essential for V‐ATPase activity.
Figure 2. Asymmetry in c‐ring serves to accommodate symmetry mismatch.
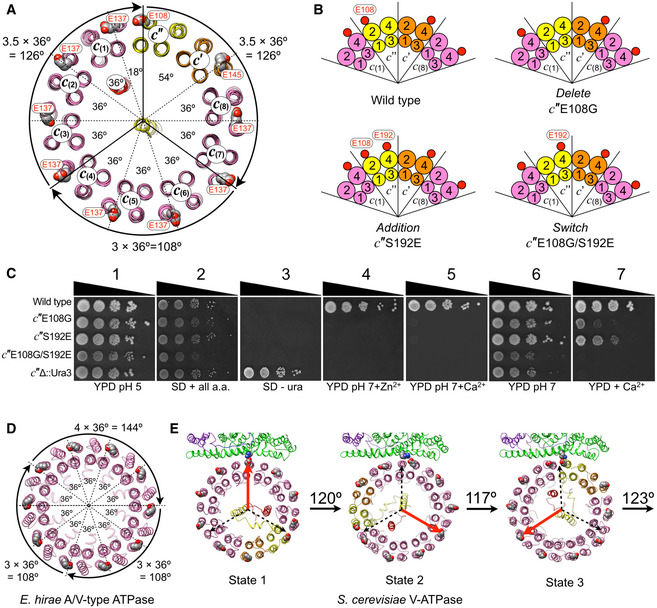
- Cross section of the yeast V‐ATPase c‐ring (as seen from the cytosol toward the membrane), highlighting the unequal angular spacing between c‐ring essential glutamic acid residues. Due to the unique position of c″E108, the angular spacings between c‐ring essential glutamates are 8 × 36°, 1 × 18°, and 1 × 54°. This asymmetry allows one full 360° rotation to be divided into two steps of 3.5 × 36° = 126°, and one step of 3 × 36° = 108°, thus causing less strain in the DF central rotor subcomplex.
- Positions of c‐ring essential glutamic acid residues in wild‐type and mutant strains c″E108G (Delete), c″S192E (Addition) and c″E108G/S192E (Switch). Small red circles indicate glutamic acid residues and numbered circles indicate the TM helix of each proteolipid chain. Only a part of the c‐ring is shown.
- Growth phenotypes of wild‐type and c″‐mutant yeast strains probed on (1) rich media buffered to pH 5 (YPD pH 5); (2) synthetic dropout (SD) plus all amino acids (SD + all a.a.); (3) SD minus uracil (SD ‐ ura); (4,5) YPD buffered to pH 7 plus 4 mM Zn2+ or 60 mM Ca2+, respectively; (6) YPD buffered to pH 7; (7) YPD plus 60 mM Ca2+.
- Cross section of the symmetric c 10 ring of the A/V‐type ATPase from Enterococcus hirae (PDB 2BL2) (Murata et al, 2005). The three steps of the symmetric c‐ring are two steps of 108°, and one step of 144°.
- Measured angles of c‐ring rotation between states 1–3 of yeast V‐ATPase. The measured rotation angles deviate slightly from the predicted angles due to a small “over rotation” of state 2, which brings the three c‐ring angles even closer to the 120° steps of the DF central rotor.
The unequal spacing of c‐ring glutamates alleviates symmetry mismatch between c‐ring and catalytic sites
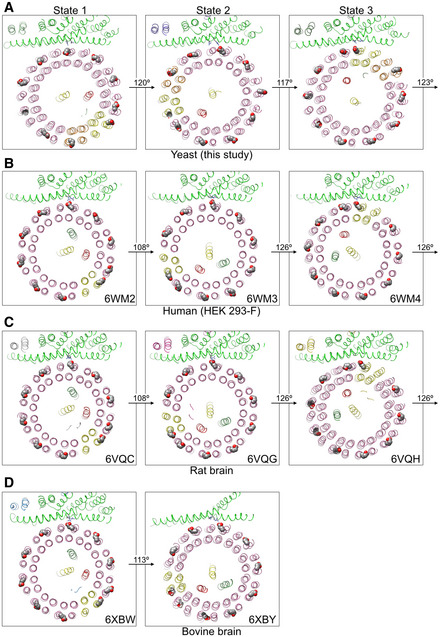
The loss of function caused by the Switch mutant is striking and can be rationalized by the following considerations: One full rotation of a symmetric c‐ring as found in, e.g., the Enterococcus hirae A/V‐like enzyme would have to be divided into two steps of 108° and one step of 144° (Fig 2D), a mismatch to the 120° steps of the ATPase motor that would require significant under‐ and overtwisting of the central rotor in at least one of the three rotary states. The situation, however, is different in the eukaryotic V‐ATPase. As a result of the asymmetry, one full 360° rotation of the c‐ring can be divided into two steps of 126° (each 3.5 × 36°) and one step of 108° (3 × 36°), which is closer to the 120° steps of the V1 (Fig 2A). Whereas the resolution of the Vo in our maps of yeast V1HchimVoND, especially in states 2 and 3, is insufficient to accurately model amino acid side chains, the rotary positions of the c‐ring in the three states are well defined and allow measurement of the rotation angles relative to a CT with high confidence. An analysis of the holoenzyme structures reveals that the stopping positions of the c‐ring in the three rotary states are even more closely aligned with the three 120° steps of the ATPase motor due to the conformational flexibility of the arginine side chain. This flexibility allows the ring to pause in state 2 a little further toward state 3 than predicted by above considerations, resulting in angles of ~120, 117, and 123° between states 1 and 2, 2 and 3, and from 3 to 1, respectively (Fig 2E). Of note, similar c‐ring rotation angles between states 1 and 3 as observed here for the yeast enzyme are also obtained for V‐ATPases from other species (Abbas et al, 2020; Wang et al, 2020a, 2020b) (Fig EV2), highlighting the conserved nature of V‐ATPases from yeast to mammals. Importantly, this evening out of the c‐ring rotation steps works only if c″E108 forms a salt bridge with a CTR735 in one of the three rotary states of the holoenzyme. This condition is exactly what is observed in state 3 (Mazhab‐Jafari et al, 2016; Roh et al, 2018), thus lending support for our hypothesis that the unequal spacing of the c‐ring glutamates serves to alleviate the symmetry mismatch between ATPase motor and c‐ring stepping.
Figure EV2. c‐ring rotation angles between states 1–3 of yeast and mammalian V‐ATPases.
Structure of V1‐ATPase in complex with subunit C and Oxr1p
3‐D classification of the ~570,000 particle dataset revealed a minor subset (~3%) of V1‐ATPase particles, which allowed reconstruction of a 3.8 Å resolution map. Importantly, while this V1 subcomplex lacked subunit Hchim, the complex contained subunit C and a hitherto unseen density of ~25 kDa that was wedged between C, EG2, and the C‐termini of the adjacent A and B subunits (Fig 3A). Upon close examination of this density, we found that none of the previously structurally characterized components (V‐ATPase subunits or subunit domains) provided a match, and since there was no prior report of a V1 or V1Vo structure with a macromolecule bound at that site, we reasoned that this extra density may belong to a separate and unknown polypeptide that is co‐purified with V1∆H used for in vitro V‐ATPase assembly. To determine the identity of this structural component, we performed mass spectrometry of V1Hchim (Fig 3B; Dataset EV1). The analysis revealed several proteins that were present at significant levels based on the peptide spectrum matches (PSM) and the overall posterior error probability (PEP) score, most notably YPL196W, the yeast homolog of Oxidation Resistance protein 1 (Oxr1p, hereafter referred to as Oxr1) (Fig 3B). Yeast Oxr1 is a 273‐residue protein with a C‐terminal Tre2/Bub2/Cdc16, LysM, domain catalytic (TLDc) domain, which is conserved in the OXR1 and nuclear receptor coactivator 7 (NCOA7) paralogs found in higher organisms (Durand et al, 2007; Finelli & Oliver, 2017; Fig EV3A and B). An interaction between NCOA7 and V‐ATPase has previously been described in the mouse brain and kidney (Merkulova et al, 2015; Castroflorio et al, 2021). To explore whether the novel density belongs to Oxr1, we generated a homology model of the yeast protein using the crystal structure of zebrafish OXR2 TLDc domain (PDB 4ACJ) (Blaise et al, 2012) as a template (Fig 3C). Fitting the model to the map showed that secondary structure elements and many bulky side chains matched the cryoEM density well (Fig 3D), providing strong evidence that the density indeed represents Oxr1. The V1 complex containing C and Oxr1 is hereafter referred to as V1(C)Oxr1 (Fig 3E). PISA analysis revealed that the interface of Oxr1’s TLDc domain with peripheral stator EG2, the C‐terminal domains of subunit A and B, and the foot domain of subunit C (Cfoot) is largely driven by electrostatic and hydrophobic interactions, with buried surface areas of 649, 250, 395, and 572 Å2, respectively (Fig 3F). The analysis also showed that many of the interacting residues on both Oxr1 and V‐ATPase subunits are strictly conserved between yeast and higher organisms (Fig EV3C and D). It should be noted that there was no clear density that could account for the N‐terminal 62 residues of Oxr1, which are predicted to be largely disordered (Appendix Fig S3).
Figure 3. Structure of yeast V1 in complex with subunit C and Oxr1’s TLDc domain.
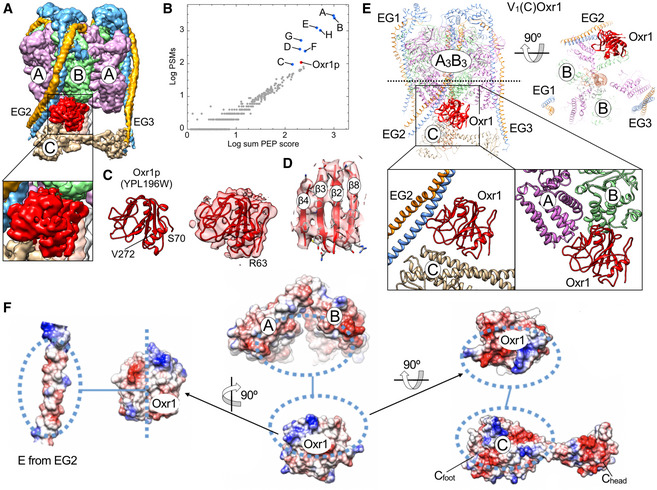
- 3.8 Å cryoEM map of V1 in complex with subunit C, highlighting the hitherto unseen density (red) wedged between subunits B and C and the N‐terminal part of peripheral stator EG2.
- Mass spectrometry analysis of V1Hchim. Protein IDs are plotted as a function of overall peptide spectrum matches (PSM) and the posterior error probability (PEP) scores.
- Left, homology model of yeast Oxr1 (residues 70–232) based on zebrafish OXR2’s TLDc domain (PDB 4ACJ) (Blaise et al, 2012). Right, fit of the real space refined yeast Oxr1 homology model into the cryoEM density.
- Enlarged view of the central β sheet comprising residues Y130‐K135 (β2), I141‐S146 (β3), F165‐K168 (β4), and A266‐R271 (β8).
- Ribbon diagram of V1(C)Oxr1 featuring the binding position of Oxr1 between EG2, the C‐terminal domains of the subunit AB pair adjacent to the open catalytic site, and subunit C.
- Electrostatic surface representations of V1 subunits E, A, B, and C involved in Oxr1 binding.
Figure EV3. Conservation of TLDc domains and the TLDc binding site on the V‐ATPase.
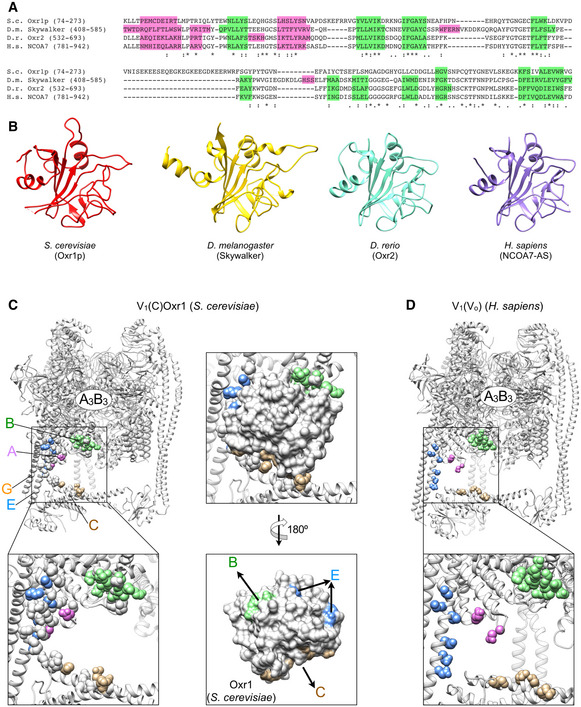
- Multiple sequence alignment of the TLDc domains from S. cerevisiae Oxr1p (Uniprot accession Q08952), Drosophila melanogaster GTPase activating Skywalker (accession Q9VIH7, 30% identical to yeast Oxr1p), Danio rerio Oxr2 (accession A9JTH8, 35% identical to yeast Oxr1p), and Homo sapiens NCOA7 (accession Q8NI08, 38% identical to yeast Oxr1p). The sequences are colored according to secondary structure from available experimental structures with α‐helices in pink and β‐strands in green.
- Binding site of Oxr1 on yeast V1(C). Residues that contact Oxr1 as determined by PDBePISA are in spacefill. Residues that are identical in yeast, fly, fish, and human are colored according to the corresponding subunit (C, tan; E, light blue, B, light green; A, magenta).
- CryoEM structure of the V1 subcomplex from human V‐ATPase (PDB 6WM2; Wang et al, 2020a), highlighting the conservation of residues that constitute the binding site of Oxr1’s TLDc domain on yeast V1(C) The residue coloring is as in panel (C).
Oxr1 inhibits V1‐ATPase and promotes V‐ATPase disassembly
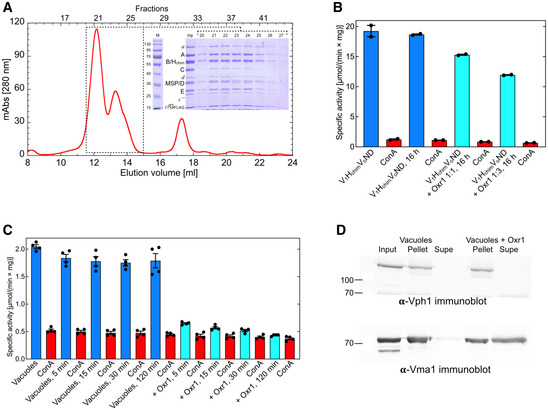
To explore the functional consequence of the interaction of Oxr1 with yeast V‐ATPase, we cloned yeast OXR1 (YPL196W) with a N‐terminal 7×His‐tag for expression in Escherichia coli. Size exclusion chromatography shows that Oxr1 elutes from the column in a single peak corresponding to a mass of ~30 kDa, indicating that the protein is a monomer in solution (Fig 4A).
Figure 4. Oxr1 inhibits V‐ATPase’s MgATPase activity and in vitro assembly.
- Size exclusion chromatography elution profile (1.6 cm × 50 cm Superdex S75) and SDS‐PAGE of full‐length N‐terminally 7×His tagged yeast Oxr1.
- Inhibition of V1ΔH and V1Hchim’s MgATPase activity by Oxr1 in absence and presence of subunit C. The data from one representative of two experiments using two different preparations are shown. Individual data points (n = 2) and mean from one representative of two biological replicates are shown with error bars representing SEM.
- Oxr1 binds wild‐type V1. Samples of V1, Oxr1, and mixtures of V1 with a 2‐fold molar ratio of Oxr1 in absence or presence of a 3‐fold molar ratio of subunit C were subjected to size exclusion chromatography on a 1 cm × 30 cm Superose 6 Increase column. SDS‐PAGE is shown for all four samples and immunoblot analysis of column fractions using a penta‐His antibody is shown for samples containing Oxr1 and Oxr1 plus C. Note that incubation of V1 with Oxr1 in presence of excess subunit C leads to partial release of subunit H (highlighted by the rectangular box in the V1 + Oxr1 + C gel). Also, some degradation of Oxr1’s unstructured N‐terminus is observed as indicated by the asterisk (only the full‐length protein is detected by the αHis‐tag antibody). The data from one representative of two experiments using two different preparations are shown.
- Dissociation kinetics of Hwt and Hchim from V1ΔH in presence of Oxr1 and subunit C as probed by BLI. Recombinant wild‐type (left) and chimeric subunit H (right) with an N‐terminal fusion to maltose‐binding protein (MBP) was immobilized on anti‐mouse Fc (AMC) BLI sensors via an αMBP monoclonal antibody. The sensors were then dipped into wells with V1ΔH followed by wells with buffer in absence or presence of Oxr1 and C to monitor the kinetics of V1ΔH dissociation. Only the dissociation phase is shown. The data from one representative of three experiments using two different preparations are shown.
- Oxr1 mediated inhibition of assembly of holo V‐ATPase from V1Hchim, subunit C and VoND. The data from one representative of two experiments using two different preparations are shown. Individual data points (n = 2) and mean from one representative of two biological replicates are shown with error bars representing SEM.
- Oxr1 prevents binding of V1Hchim to VoND as probed by BLI. Streptavidin‐coated BLI sensors were loaded with Vo in lipid nanodiscs containing biotinylated MSP, and sensors were then dipped into wells containing recombinant C and V1Hchim in absence and presence of Oxr1. Only the V1Hchim association and dissociation phase from one representative of two experiments using two different preparations is shown.
- Oxr1 mediated dissociation of holo V‐ATPase as analyzed by negative stain electron microscopy. V1HchimVoND was incubated without and with a 3‐fold molar ratio of Oxr1 for 16 h. Samples were then spotted on carbon‐coated copper grids, stained with 1% uranyl acetate, and observed by transmission electron microscopy. Number of assembled V1Vo and disassembled V1 and Vo particles were counted on a total of 20 micrographs each. The horizontal dotted lines indicate the mean. A few holoenzyme and free V1 and VoND particles are highlighted by white circles and boxes, respectively. Bar = 50 nm. The data from one representative of two experiments using two different preparations are shown.
Oxr1 inhibits V1‐ATPase
The structural model of V1(C)Oxr1 shows Oxr1’s TLDc domain wedged between the N‐termini of EG2, Cfoot, and the C‐terminal domains of the subunit AB pair that faces the open catalytic site in rotary state 1, respectively (Fig 3E), suggesting that binding of Oxr1 may interfere with the activity of the complex. We therefore first tested whether Oxr1 has an effect on V1’s ATPase activity. While V1 purified from yeast containing Hwt has no ATPase activity in presence of Mg2+ (Parra et al, 2000; Zhang et al, 2003), V1∆H and V1Hchim possess robust MgATPase activity, which, however, quickly decays due to trapping of inhibitory MgADP in a catalytic site (Parra et al, 2000; Oot et al, 2016). Preincubation of V1 mutant complexes with equimolar amounts of Oxr1 resulted in ~10 and ~50% inhibition of the MgATPase activity of V1∆H and V1Hchim, respectively, as measured in an ATP regenerating assay (Fig 4B). Addition of recombinant subunit C increased Oxr1’s inhibitory effect for V1∆H and V1Hchim to ~50 and ~80%, respectively. Together with our structural observations (Fig 3), this suggests that Oxr1 interferes with cooperative catalysis, and while subunit C enhances the inhibitory effect, it is not strictly required for binding of Oxr1 to V1.
Oxr1 binding to V1‐ATPase is cooperative with subunits C and H
We next tested whether Oxr1 also interacts with wild‐type V1 (containing Hwt) using size exclusion chromatography. We incubated V1 with Oxr1 in absence or presence of excess subunit C and applied the mixtures onto a Superose 6 Increase column. SDS‐PAGE and immunoblot analysis of elution fractions indicated that Oxr1 co‐eluted with V1, irrespective of whether subunit C was added (Fig 4C). Surprisingly, the size exclusion chromatography analysis of the mixture of wild‐type V1 with Oxr1 and subunit C indicated that Oxr1 and C binding leads to partial release of subunit H, suggesting that stable binding of H or C/Oxr1 is mutually exclusive. To test this hypothesis, we immobilized maltose‐binding protein (MBP) tagged Hwt or Hchim on BLI sensors to determine off‐rates of V1∆H dissociation in presence and absence of Oxr1 and Oxr1 + C. While Oxr1 alone had little to no effect on V1∆H dissociation, dipping BLI sensors into wells with Oxr1 in presence of recombinant C resulted in a significantly faster off‐rate, with the effect more pronounced for the dissociation of V1∆H from Hchim compared to Hwt (Fig 4D). The data thus confirms that stable binding of H, or Oxr1 and C, is mutually exclusive, and suggests that V1(C)Oxr1 is overall more stable (has a lower energy) than V1Hwt or V1Hchim.
Oxr1 prevents in vitro assembly of holo V‐ATPase
Given that Oxr1 binds and inhibits V1Hchim in a subunit C‐dependent manner, we wished to determine whether Oxr1 interfered with in vitro assembly of holo V‐ATPase. We mixed V1Hchim with VoND and subunit C with and without Oxr1 and incubated the mixture for 16 h at room temperature. In absence of Oxr1, we measured a specific MgATPase activity of ~8 µmol × (min × mg)−1. Incubation of V1Hchim, VoND, and C in presence of Oxr1, however, did not result in significant MgATPase activity (Fig 4E), indicating that binding of Oxr1 and C to V1Hchim interfered with the formation of active holoenzyme. However, this result did not exclude the possibility that assembly took place, but that the assembled complex was inactive due to bound Oxr1. To answer this question, we performed a biolayer interferometry (BLI)‐based assay that we had developed previously to study in vitro V‐ATPase assembly (Sharma et al, 2019). Here, VoND is immobilized on streptavidin‐coated BLI sensors via biotinylated MSP and sensors are then dipped into wells containing V1Hchim and C in presence or absence of Oxr1. In absence of Oxr1, robust binding was observed, indicating binding of V1Hchim to immobilized Vo. However, very little BLI signal was produced in presence of Oxr1, indicating that assembly does not take place under these conditions (Fig 4F). The results thus suggest that binding of Oxr1 and C to V1Hchim prevented assembly of holo V‐ATPase.
Oxr1 causes disassembly of V‐ATPase holoenzyme
To test whether Oxr1 has an effect on the assembled enzyme, reconstituted V1HchimVoND was first subjected to size exclusion chromatography on a Superose 6 Increase column to remove unassembled components (Fig EV4A). V1HchimVoND was then incubated with and without Oxr1 for 16 h before the assembly state was determined using negative stain electron microscopy (Fig 4G). The analysis shows that in absence of Oxr1, ~85% of the complexes were assembled, whereas in presence of Oxr1, fewer than 40% of the complexes remained intact (Fig 4G). Consistent with this analysis, the ATPase activity was reduced by about 35% from ~18.7 to ~12 µmol × (min × mg)−1 after the 16 h incubation in presence of Oxr1 (Fig EV4B). To rule out the possibility that Oxr1‐induced dissociation is due to the presence of Hchim, we isolated vacuoles with Hwt containing V‐ATPases to test the effect of Oxr1 binding. As with in vitro reconstituted V1HchimVoND, Oxr1 led to inhibition and dissociation of the wild‐type enzyme (Fig EV4C). Interestingly, inhibition of the Hwt containing enzyme in vacuoles was significantly faster compared to the Hchim containing complex, consistent with our earlier BLI‐based observations that showed a greater stability of V1HchimVoND compared to wild‐type V1VoND (Sharma et al, 2019). In summary, Oxr1 (i) binds and inhibits mutant V1 subcomplexes in a subunit C‐dependent manner, (ii) destabilizes the interaction of V1∆H with Hwt and Hchim, (iii) prevents assembly of holoenzyme, and (iv) interacts with holo V‐ATPase to cause disassembly into V1 and Vo subcomplexes.
Figure EV4. Inactivation of V1HchimVoND and V1Vo on vacuoles by Oxr1.
- In vitro assembled V1(Hchim)VoND was applied to a 1 cm × 30 cm Superose 6 Increase column and eluted with 20 mM Tris‐HCl, pH 7.2, 100 mM NaCl, 0.5 mM EDTA in 0.5 ml fractions. Inset, SDS‐PAGE of column elution fractions. The MgATPase activity of fractions 21 and 22 was 18 and 17 µmol × (min × mg)−1, respectively.
- MgATPase activity assays using fraction 21 were performed in absence and presence of ConA and Oxr1 after a 16 h incubation at room temperature. The data from one representative of two experiments using two different preparations are shown. Individual data points (n = 2) and mean from one representative of two biological replicates are shown with error bars representing SEM.
- Vacuoles isolated by flotation on Ficoll gradients from a yeast strain expressing FLAG‐tagged G subunit were incubated without (blue bars) and with Oxr1 (cyan bars) for the indicated times followed by MgATPase activity assays. MgATPase activity was then measured in the presence of 200 nM ConA (red bars). Individual data points (n = 4) and mean from each two measurements using two biological replicates are shown with error bars representing SEM.
- Vacuoles were incubated in absence or presence of Oxr1. Vacuoles were collected by ultracentrifugation (37,000 g for 20 min) and resuspended pellets and supernatant were resolved on SDS‐PAGE gels, blotted on PVDF membranes, and probed for presence of V1 subunit A (Vma1) and Vo subunit a (Vph1). The data from one representative of two experiments using two different vacuole preparations are shown.
Conformational changes in V1‐ATPase due to Oxr1 and subunit C binding
We previously reported a crystal structure of yeast V1 that showed HNT attached to peripheral stator EG1 and HCT bound at the bottom of the A3B3 hexamer (Oot et al, 2016) (Fig 5A). The binding of HCT stabilized an open catalytic site (see arrow in Fig 5A, lower panel), which in turn stabilized inhibitory MgADP at an adjacent site, resulting in the autoinhibited conformation of V1. A comparison to the three rotary states of the holoenzyme revealed that autoinhibited V1 is halted in state 2 (Oot et al, 2016). In contrast, the map of V1(C)Oxr1 does not have density for Hchim (Figs 3A and 5B). Based on the positions of subunit C and the central DF rotor, the complex is in rotary state 1, with the open catalytic site next to Oxr1 (see arrow in Fig 5B, lower panel). However, a comparison to rotary state 1 of the holoenzyme (Fig 5C) reveals significant changes in the conformations of the subunits involved in Oxr1 binding, including the N‐terminal domains of EG2 and EG3, which are displaced by 30 and 20 Å, respectively, and subunit C, which is displaced towards the A3B3 hexamer by 22–26 Å (Fig 5C and Movie EV1). It is evident that especially the conformation of EG2 in the V1(C)Oxr1 complex is no longer positioned correctly for supporting the interface between a NT and Cfoot, a structural change that may provide a rationale why Oxr1 inhibits Hchim mediated assembly of the holoenzyme (Figs 4F and 5D). In summary, the structural and biochemical data indicate that Oxr1 and subunit C binding to V1 is highly cooperative, and that the resulting V1(C)Oxr1 complex has higher stability than V1 or V1Vo.
Figure 5. Conformational changes in V1‐ATPase due to Oxr1 and subunit C binding.
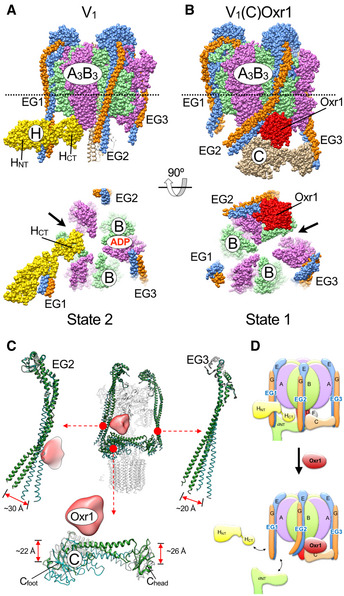
- Crystal structure of V1 in the autoinhibited state 2 conformation (PDB 5D80) (Oot et al, 2016). HCT of wild‐type H stabilizes an open catalytic site (arrow) with inhibitory MgADP bound to an adjacent site.
- Model of V1 in complex with subunit C and Oxr1p. In this complex, V1 is in state 1, with C bound between EG2 and EG3. Oxr1 (red) is wedged between the N‐terminal domain of EG2, the subunit AB pair next to the open catalytic site, and Cfoot.
- Structural changes in EG2, EG3, and subunit C upon Oxr1 binding.
- Oxr1 causes disassembly of holo V‐ATPase. In this model, Oxr1 binding destabilizes the interaction between EG2, Cfoot, and the distal domain of a NT, which leads to release of V1 from the membrane.
Discussion
We have previously shown that autoinhibited wild‐type V1 and Vo do not reassemble in vitro with a measurable rate, despite the process being thermodynamically favored (Sharma et al, 2019). We reasoned that the disassembled subcomplexes do not reassociate because both are stabilized by interactions not present in the holoenzyme, resulting in a conformational mismatch: autoinhibited V1 is locked in rotary state 2 by inhibitory MgADP and HCT binding to the open catalytic site, whereas Vo is arrested in state 3, which is stabilized by the interaction between a NT and d. Thus, breaking one or both of these interactions must represent the rate‐limiting kinetic barrier(s) that prevent spontaneous reassembly.
The crystal structure of autoinhibited V1 showed that the inhibitory interaction between HCT and the open catalytic site is mediated by an eleven‐residue loop that is not found in higher organisms (Oot et al, 2016). We therefore introduced a chimeric subunit H construct with yeast HNT and human HCT (Hchim). Importantly, human HCT lacks the inhibitory loop found in yeast HCT, but retains the binding site for a NT, which is conserved from yeast to human (Oot et al, 2016). Unlike wild type yeast V1, which is catalytically inactive, V1∆H reconstituted with Hchim has robust MgATPase activity and is able to bind C and Vo to form coupled holo V‐ATPase (Sharma et al, 2019). Our cryoEM analysis of the in vitro reconstituted holoenzyme revealed a similar distribution of rotary states 1–3 as had been observed for the enzyme purified from yeast (Zhao et al, 2015). This suggests that the main kinetic barriers for (re)assembly of the wild‐type enzyme are the release of (i) MgADP from a closed catalytic site and (ii) HCT from its inhibitory binding site on V1. However, the mechanisms by which HCT and inhibitory MgADP are released from wild‐type V1 in vivo are currently not known.
A unique feature of the eukaryotic V‐ATPase is the unequal spacing of its ten c‐ring glutamic acid residues due to the presence of the proteolipid subunit c″, which carries its essential glutamate on TM helix 2 instead of helix 4 as for the other nine proteolipids. From molecular dynamics simulations, we recently argued that the asymmetry created by the unequal glutamate spacing contributes to the autoinhibition of passive proton transport across free Vo (Roh et al, 2020). However, whether the asymmetry also functions in ATP hydrolysis‐driven proton transport remained unknown. The mutagenesis experiments presented here show that a V‐ATPase c‐ring with equally spaced glutamic acid residues is inactive, suggesting that the asymmetry is indeed required for a functional enzyme. Based on the holoenzyme structures of rotary states 1‐3, we propose that the unequal glutamate spacings around c″ serve to alleviate the symmetry mismatch between the three 120° rotary steps of V1 and the ten steps of the Vo by allowing a full 360° turn of the c‐ring to be divided into two steps of 126° (3.5 × 36°) and one step of 108° (3 × 36°). In contrast, a symmetric c‐ring would have to step 108° twice and 144° once, which would require a too large under‐ and/or overtwist of the central DFd rotor. Of note, the c‐ring steps measured for the yeast enzyme are even closer to 120° due to the conformational flexibility of the side chain of Arg735. It is unclear at this point why the E. hirae enzyme can tolerate the larger 144° step of a symmetric c‐ring. However, unlike eukaryotic V‐ATPase, the bacterial enzyme has only two peripheral stators, a structural feature that may allow more flexibility and storage of elastic energy during ATP hydrolysis‐driven rotation of the central stalk.
Importantly, our cryoEM analysis of the reconstituted holoenzyme also revealed a structure of unassembled V1‐ATPase in complex with subunit C and Oxr1, a poorly characterized factor that has hitherto not been described as being associated with the yeast V‐ATPase. Our experiments showed that recombinant Oxr1 not only binds and inhibits V1’s MgATPase activity in a subunit C‐dependent manner, the factor also leads to inhibition and disassembly of the in vitro reconstituted and vacuole associated holoenzyme. The inhibitory activity can be rationalized from the V1(C)Oxr1 structure, which shows Oxr1 bound to the C‐terminal domain of the B subunit of the open catalytic site. This mode of interaction between Oxr1 and state 1 V1, which, together with C, provides significant asymmetry to V1ΔH subcomplex in rotational state 1, is reminiscent of the interaction of wild‐type HCT with the open catalytic site as seen in the crystal structure of state 2 wild‐type V1 (Oot et al, 2016). Therefore, it appears that both H (via HCT) and Oxr1 inhibit V1 rotary catalysis by stabilizing an open catalytic site, with concomitant binding of inhibitory MgADP in an adjacent site as shown for wild‐‐type V1. It is tempting to speculate that subunit H and Oxr1 compete for the binding interface formed at an open catalytic site. Moreover, subunit H and Oxr1 appear to stabilize different rotary states, with H and Oxr1 stabilizing state 2 and 1, respectively. This differential stabilization by H or Oxr1 suggests that formation of a stable V1H or V1(C)Oxr1 complex is mutually exclusive, which may explain why the V1(C)Oxr1 complex here visualized by cryoEM does not contain Hchim, and why binding of Oxr1 to wild‐type V1 in presence of C leads to release of H. Thus, aside from the well‐characterized regulatory mechanism of reversible disassembly in response to nutrient availability, the work presented here uncovers an alternative pathway of V‐ATPase regulation, which is induced by binding of Oxr1 (Fig 6). Our biochemical experiments show that Oxr1‐induced disassembly, unlike the disassembly caused by glucose withdrawal, does not require ATP hydrolysis. We previously provided evidence that the ATP‐dependent step of reversible disassembly is subunit C release (Sharma et al, 2019). Here we showed that subunit C remains bound during Oxr1‐induced dissociation, an observation that provides a possible explanation why Oxr1‐dependent disassembly does not require input of energy, and suggests that the process is solely driven by the formation of new protein‐protein interactions.
Figure 6. Regulation of V‐ATPase assembly by nutrients and Oxr1.
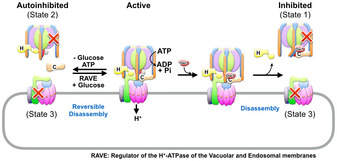
Left side: V‐ATPase activity is regulated by reversible disassembly in a nutrient‐dependent manner. Upon glucose withdrawal, active V1Vo dissociates into subunit C and autoinhibited V1‐ATPase and Vo proton channel subcomplexes in an ATP‐dependent manner. Reassembly of active V1Vo from autoinhibited V1 and Vo and C subunit requires the “Regulator of the H+‐ATPase of the Vacuolar and Endosomal membranes” (RAVE) complex. Right side: The here presented data show that disassembly of V1Vo can also be induced by presence of Oxr1. The physiological role of Oxr1‐induced disassembly, and whether there is a mechanism in vivo to release Oxr1 and reactivate V1 for reassembly, is currently not known.
Early reports suggested that Oxr1 in yeast is targeted to mitochondria, and that strains lacking the protein have increased sensitivity to oxidative stress (Volkert et al, 2000). More recent reports on the mammalian homologs OXR1 and NCOA7 provided direct evidence for an interaction with V‐ATPase, most likely via their conserved C‐terminal TLDc domain (Merkulova et al, 2015; Castroflorio et al, 2021). Several studies indicated that OXR1/NCOA7 protects cells from oxidative stress in neuronal tissues, and that lack of, e.g., NCOA7 is correlated with reduced V‐ATPase activity on lysosomal membranes. Reduced activity upon NCOA7 knockout was also observed for plasma membrane V‐ATPases in mouse kidney, resulting in a partial dRTA phenotype (Merkulova et al, 2018). However, these latter findings appear to be at odds with the in vitro experiments presented here, showing that yeast Oxr1 inhibits and disassembles the holoenzyme. One possible explanation for the conflicting observations is that Oxr1 may play a role in the quality control of V‐ATPase activity (Fig EV5). As mentioned in the previous paragraph, only active V‐ATPases have the ability to disassemble (Parra & Kane, 1998), which means that V1‐ATPase subcomplexes that have lost the ability to hydrolyze ATP (due to, e.g., oxidative stress) would remain on the membrane, thereby occupying and preventing Vo complexes from associating with undamaged, active V1‐ATPases. Binding of Oxr1 to inactive holoenzyme complexes and releasing the damaged V1 subcomplexes would free up intact Vo complexes to allow for (re)assembly of active holoenzymes with the help of assembly factors such as the RAVE complex. However, whether mechanisms exist to release Oxr1 and repair and reactivate damaged V1 to allow reassembly of active holo V‐ATPases is currently unknown. Recently, the TLDc domain‐containing protein mEAK7 was found to bind mammalian V‐ATPase at a site equivalent to the site where Oxr1 interacts with the complex from yeast (preprint: Tan et al, 2021). However, unlike Oxr1, binding of mEAK7 was found to have no effect on V‐ATPase’s MgATPase activity or assembly state, indicating that while TLDc domain‐containing proteins share V‐ATPase as a common binding partner, their interactions with the complex are involved in diverse cellular functions. Further experiments will be required to uncover the physiological role of the interaction of Oxr1 with the V‐ATPase, and whether the observations obtained here for the yeast system are conserved in higher organisms including humans.
Figure EV5. Model of Oxr1 mediated V‐ATPase regulation under oxidative stress.
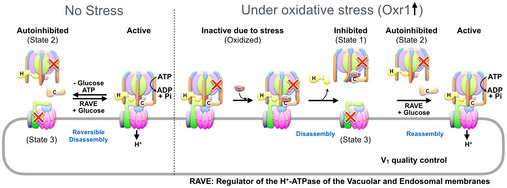
Left: Under normal conditions (no stress), V‐ATPase activity is regulated by reversible disassembly in a nutrient‐dependent manner. Under these conditions, holoenzyme dissociation results in autoinhibited V1 and Vo that can reassemble catalyzed by the RAVE complex and other cellular factors. Right: Oxidative stress conditions lead to inactive holoenzyme complexes (due to e.g. intramolecular disulfide bond formation) that are no longer able to dissociate. Under these conditions, Oxr1 binds and dissociates inactive complexes. The resulting free Vo can then be reassembled with undamaged V1 to form active holoenzyme complexes.
Materials and Methods
Purification of Vo and reconstitution into lipid nanodiscs
Cell growth, membrane isolation, Vo purification, and reconstitution were performed as described previously (Couoh‐Cardel et al, 2016; Stam & Wilkens, 2017). Briefly, yeast cells containing calmodulin‐binding peptide (CBP) tag fused to the C‐terminus of Vph1 (the vacuolar isoform of subunit a) were harvested after 16 h of growth (OD600 = 13.0) by centrifugation at 3,500 g, washed in water, and resuspended in a lysis buffer (25 mM Tris‐HCl, pH 7.4, 500 mM sorbitol, 2 mM CDTA). Cells were broken using zirconium beads in an inverted bead‐beater. Cellular debris and mitochondria were removed by low‐ (3,500 g) and medium‐speed (13,000 g) centrifugation, respectively. Total membranes were isolated by high‐speed centrifugation (200,000 g) for 2 h, washed once in lysis buffer, pelleted again (100,000 g) for 1 h, and resuspended in a Dounce homogenizer. The protein concentration of the preparation was measured by a modified Pierce TCA‐BCA assay (Couoh‐Cardel et al, 2015) and membranes were stored at −80°C until use.
Membranes were thawed and diluted in lysis buffer to reach a concentration of 10 mg/ml. Membrane protein was extracted with undecyl maltoside (UnDM) at a final concentration of 0.6 mg/mg in presence of protease inhibitors (2 µg/ml leupeptin, 2 µg/ml pepstatin, 5 µg/ml aprotinin, 0.5 µg/ml chymostatin, and 1 mM PMSF) at 4°C with gentle agitation. After 45 min, 4 mM CaCl2 was added and extraction was continued for another 15 min. Extracted membranes were cleared by ultracentrifugation (100,000 g) for 1 h and passed over a 15 ml calmodulin sepharose (CaM) column pre‐equilibrated with calmodulin washing buffer (10 mM Tris‐HCl, pH 8, 150 mM NaCl, 10 mM beta‐mercaptoethanol (β‐ME), 2 mM CaCl2, 0.06% UnDM) to capture CBP‐tagged Vo. The column was washed with each 100 ml of calmodulin washing buffer with and without NaCl. Captured Vo from the column was then eluted with elution buffer (10 mM Tris‐HCl, pH 8, 10 mM β‐ME, 10 mM CDTA, 0.06% UnDM) in fractions of 8 ml. Vo containing fractions were concentrated using 100 kDa MWCO centrifugal concentrators (VivaSpin) and the final concentration was measured using the modified Pierce TCA‐BCA assay (Couoh‐Cardel et al, 2015).
To reconstitute Vo into lipid nanodisc (VoND), detergent‐solubilized Vo, membrane scaffold protein (MSP1E3D1) and E. coli polar lipids were mixed in disc forming buffer (20 mM Tris‐HCl, pH 7.4, 0.5 mM CDTA, 100 mM NaCl, 1 mM DTT) at a molar ratio of 0.02:1:25. Reconstitution was carried out at room temperature for 1 h in presence of protease inhibitors and 1.5% UnDM with gentle mixing followed by detergent removal with 0.4 g/ml Bio‐Beads SM2 (Bio‐Rad). After separation from Bio‐Beads, the reconstitution mixture was carefully supplemented with 5 mM CaCl2 and passed over a 3 ml CaM column to remove the empty nanodiscs. The column was eluted as before and the VoND containing fraction applied to a 1.6 cm × 50 cm Sephadex‐200 size‐exclusion chromatography column equilibrated in disc forming buffer. VoND containing fractions were pooled, mixed with 20% glycerol, concentrated, flash‐frozen in liquid nitrogen, and stored at −80°C until use.
Expression and purification of subunits Hchim, MBP‐Hchim, MBP‐H, and C
Expression and purification of subunits (Hchim, C) and subunit fusions with maltose‐binding protein (MBP) (MBP‐Hchim, MBP‐Hwt) was done as described (Oot & Wilkens, 2010; Sharma et al, 2019). Briefly, E. coli Rosetta2 cells expressing N‐terminally MBP‐tagged Hchim, ‐Hwt, and ‐C subunits were grown to an OD600 of 0.6 in RB media (LB + 0.2% glucose) supplemented with 50 µg/ml of carbenicillin and 34 µg/ml of chloramphenicol. Expression was then induced with 0.5 mM isopropyl‐β‐D‐1‐thiogalacto‐pyranoside (IPTG) at 30°C for 5.5 h except for MBP‐Hwt, which was induced at 20°C for 6 h. Cells were harvested by centrifugation at 3,500 g, resuspended in amylose column buffer (ACB) (20 mM Tris‐HCl, pH 7.4, 200 mM NaCl, 1 mM EDTA), and stored at −20°C until use. Cells were thawed, treated with lysozyme (25 mg/l cells) and DNase (2 mg/l cells), and lysed by sonication. Cellular debris was removed by centrifugation at 13,000 g for 30 min at 4°C, and the supernatant was passed through a 20 ml amylose column pre‐equilibrated with ACB. The column was washed with 10 CV of ACB, and protein was eluted with 25 ml of ACB supplemented with 10 mM maltose and 1 mM DTT. For further purification, MBP‐Hchim and MBP‐Hwt were applied to a 1.6 cm × 50 cm Superdex‐200 size‐exclusion chromatography column equilibrated in ACB without salt. For purification of Hchim and C, the MBP tag was cleaved with Prescission protease for 2 h in presence of 5 mM DTT. For Hchim, the buffer was changed by dialysis overnight to 25 mM sodium phosphate, pH 7, 5 mM β‐ME, 0.5 mM EDTA. The dialyzed protein was then passed over a carboxymethyl (CM) column to remove MBP. Bound protein was eluted, concentrated, and applied to a Superdex‐200 size‐exclusion chromatography column for further purification. For subunit C, the buffer was changed by overnight dialysis to 20 mM Bis‐Tris, pH 6.5, 0.5 mM EDTA, and the protein was passed over a diethylaminoethyl (DEAE) column to remove MBP. The flowthrough containing C was dialyzed to adjust the pH away from the isoelectric point, concentrated, and applied to a Superdex‐200 size‐exclusion chromatography column attached to an ÄKTA FPLC (GE Healthcare). Purified MBP‐H was immediately used, and other proteins were stored at −80°C in 20% glycerol until use.
Purification of wild‐type and mutant V1
Cell growth and V1 purifications were performed as described (Sharma et al, 2019). Briefly, yeast cells expressing N‐terminally FLAG‐tagged subunit G were grown to an OD600 of ~3.5 in synthetic dropout medium (SD minus leucine). Cells were harvested by centrifugation at 4,000 g, resuspended in TBSE (20 mM Tris, pH 7.2, 150 mM NaCl, 0.5 mM EDTA) and stored at −80°C until use. For purification, cells were broken by ~16 passes through a microfluidizer with intermittent cooling on ice. The cell lysate was initially centrifuged at 4,000 g for 30 min at 4°C, and the supernatant was centrifuged again at 13,000 g for 40 min at 4°C. Cleared cell lysate was passed through a 5 ml αFLAG column pre‐equilibrated with TBSE. The column was washed with 10 CV of TBSE, and bound protein was eluted with 5 CV of TBSE plus 0.1 mg/ml FLAG peptide. V1 containing fractions were pooled, concentrated and applied to a Superdex‐200 (1.6 cm × 50 cm) size‐exclusion chromatography column equilibrated in 20 mM Tris, pH 7.2, 0.5 mM EDTA, 1 mM DTT or TCEP. Fractions were analyzed by SDS‐PAGE, pooled, and concentrated using a 50 kDa MWCO centrifugal concentrator. To reconstitute V1Hchim, V1∆H elution fractions from the αFLAG column were pooled and mixed with an estimated 3‐fold molar excess of Hchim. Reconstituted V1Hchim was then concentrated and subjected to Superdex‐200 size‐exclusion chromatography as for V1. Fractions were analyzed by SDS‐PAGE and V1Hchim containing fractions were pooled and concentrated.
Reconstitution of V1HchimVoND
To reconstitute functional V‐ATPase, the three components V1Hchim, VoND, and subunit C were mixed in a 1:1:3 molar ratio and incubated for 2–18 h at room temperature to complete the reassembly. Reconstituted V‐ATPase was then used as is, or passed over a 1 cm × 30 cm Superose 6 Increase size‐exclusion chromatography column in disc forming buffer plus 1 mM TCEP to separate assembled V1HchimVoND from unassembled components.
Mutagenesis of c″ glutamates
The plasmids pRS316 containing the genes for subunit c (VMA3) and c″ (VMA16) were a generous gift from Dr. Tom Stevens, University of Oregon. The yeast strain 5Aα in which VMA3 was replaced with URA3 was a gift from Dr. Patricia Kane, SUNY Upstate Medical University.
Parent yeast strain generation
To generate the parent strain for c″ mutagenesis, pRS316 containing VMA3 was digested with EcoR1 and BceA1 and the resulting ~1.6 kb fragment was used for homologous recombination by transformation of SF838‐1D vma3Δ:URA3 using a standard lithium acetate protocol (Gietz & Woods, 2002). Transformants were selected against Ura3 by their ability to grow on FOA plates and screened for their inability to grow on SD‐Ura pH 5 plates. Colonies with successful insertion of VMA3 were verified using PCR amplification and DNA sequencing. VMA11 and VMA16 were PCR amplified to corroborate their unaltered sequence.
Mutagenesis of c″
A pRS316 plasmid containing VMA16 gene was subjected to site directed mutagenesis by the QuikChange protocol (Stratagene). The single mutant c″E108G (Delete) was generated using the following primers: vma16_E108G: CCA CCA AGA ATT TAA TTT CCA TTA TTT TCT GTG GAG TGG TTG CCA TTT AC, vma16_E108G_r: CAT TAT TTT CTG TGG AGT GGT TGC CAT TTA CGG TCT GAT TAT TGC. The single mutant c″S192E (Addition) was generated using the following primers: vma16_S192E: GCA TTG TTT GTT AAA ATT TTG GTC ATT GAA ATT TTC GGG GAA ATT TTA GGT TTA TTA G, vma16_S192E_r: CAT TGA AAT TTT CGG GGA AAT TTT AGG TTT ATT AGG TTT GAT TGT TGG TTT ATT G. The double mutant c″E108G/S192E (Switch) was generated using sequential mutagenesis steps. Sequences were confirmed by DNA sequencing (Eurofins) using custom primers: ORF_vma16: CTC ACC GGA AGG CGA ATA AAA TAC or ORF_vma16_2: CGT TTT TAC GAG CTA GAT AAG CGC A. Resulting mutant plasmid was digested using restriction enzymes EcoRI and NcoI to generate a ~2 kb fragment that was gel purified and transformed for homologous recombination in SF838‐ID (MATa, ade6, leu2‐3, leu2‐112, ura3‐52, pep4‐3, gal2; tfpl‐D8), which was deleted for VMA16 using a Ura3 cassette. Transformants were counterselected against Ura3 by their ability to grow on FOA plates and screened for inability to grow on SD‐Ura pH 5 plates. False positives were discarded by PCR and final colonies were DNA sequenced using ORF_vma16 primers. VMA3 and VMA11 were also PCR amplified to validate their sequence.
Spot test
To test the growth phenotype of c″ single and double mutants, cells were grown in YPD pH 5 liquid media to an OD600 between 0.7 and 1.1 and 1 OD of cells was pelleted, washed, and resuspended to the same density. 1:10 serial dilutions were sequentially spotted onto restrictive media (YPD, pH 7, 60 mM CaCl2; YPD, 4 mM ZnCl2; SD‐Ura), semi‐restrictive media (YPD, 60 mM CaCl2 and YPD, pH 7), and non‐restrictive media (YPD, pH 5, and SD + all amino acids) and plates were incubated at 30°C for 3 days and scanned from day 3 to day 7. The mutations were subsequently verified by performing colony PCR and DNA sequencing in a blind experiment using colonies from a one‐month‐old plate.
Cloning, expression, and purification of Oxr1
The open reading frame of the OXR1 gene (YPL196W) was amplified by PCR from genomic DNA and inserted into the pET28a expression vector using restriction‐free cloning (Bond & Naus, 2012). In the primary PCR reaction, the YPL196W gene was amplified from yeast genomic DNA using a pair of hybrid primers (Oxr1_ETF: 5´‐GAA GGA GAT ATA CCA TGG GTC ATC ATC ATC ATC ATC ATC ACT TTG GAG TCA AGG ATG CTA TAT TCA AG‐3´ and Oxr1_ETR: 5´‐GGT GCT CGA GTG CGG CCG CAA GCC TAT CCT ACA CGC CAT ACT TCC AAA G‐3´) that contain complementary sequences for both insert and vector. The amplified sequence from the primary PCR was then used as a megaprimer in the secondary PCR to insert the gene into pET28a. Insertion of YPL196W was confirmed by restriction digest and DNA sequencing (Eurofins). The inserted sequence is in‐frame with an N‐terminal 7×His‐tag. For expression of Oxr1, E. coli BL21DE3 cells were grown to OD600 of 0.6–0.8 in RB medium in presence of 50 μg/ml kanamycin. Expression was induced with 0.5 mM IPTG for 16 h at 20°C. Cells were harvested by centrifugation at 3,500 g, resuspended in Buffer A (20 mM Tris, 250 mM NaCl, 10 mM Imidazole, 2 mM DTT, pH 8), and stored at −20°C until use. Cells were thawed, supplemented with 1 mM PMSF and 2 mM DTT, treated with lysozyme (25 mg/l cells) and DNase (2 mg/l cells), and lysed by sonication. Cellular debris was removed by centrifugation at 13,000 g for 30 min at 4°C, and the supernatant was passed through a 10 ml Ni‐NTA column pre‐equilibrated with Buffer A. The column was washed with 5 CV of Buffer A, and protein was eluted using a 0–60% gradient of Buffer B (20 mM Tris, 250 mM NaCl, 500 mM Imidazole, 2 mM DTT, pH 8). Fractions containing Oxr1 were pooled, concentrated, and applied to a 1.6 cm × 50 cm Superdex‐75 size‐exclusion chromatography column in 20 mM Tris, 100 mM NaCl, 1.5 mM DTT, pH 7. Fractions were analyzed by SDS‐PAGE and Oxr1 containing fractions were pooled and concentrated. Purified Oxr1 was either used immediately, or stored in 20% glycerol at −80°C.
Isolation and purification of yeast vacuoles
Vacuoles were isolated by flotation on Ficoll gradients as described (Sharma & Wilkens, 2017). Briefly, yeast grown to an OD600 of ~1 was converted to spheroplasts with zymolyase 100T, lysed in a Dounce homogenizer in presence of 12% Ficoll 400. After ultracentrifugation (71,000 g, 4°C, 40 min), vacuoles were recovered from the top of the gradients, overlaid with 8% Ficoll 400, and centrifuged a second time. Vacuoles were recovered and stored at −80°C until use.
Western blot analysis
SDS‐PAGE gels of column fractions were transferred to a low fluorescence polyvinylidene fluoride (LF PVDF) membrane. The membrane was blocked with 5% milk in TBST (20 mM Tris, pH 7.5, 150 mM NaCl, 0.05% Tween‐20) for 1 h at room temperature with gentle shaking. Primary antibody was then added and incubated overnight at 4°C. To study Oxr1 binding to wild‐type V1 subcomplex, mouse monoclonal IgG Penta‐His antibody (Qiagen catalog no. 34660) was used at 1:5,000 dilution. To analyze the dissociation of V1‐subcomplex from Vo, the blot was probed for presence of V1 subunit A (Vma1) and Vo subunit a (Vph1) using mouse monoclonal antibodies 8B1‐F3 and 10D7 (Molecular Probes), respectively. Blots were washed three times with TBST for a total of 30–45 min. After washing, goat anti‐mouse IgG Alexa Fluor Plus 488 secondary antibody (Invitrogen, catalog no. A32723) was added at 1:2,000 dilution and incubated for 1–1.5 h at room temperature in the dark. After washing three more times with TBST, the blot was dried and fluorescence images were captured on a Sapphire Biomolecular Imager (Azure Biosystems, Inc.).
ATPase assays
ATPase assays were carried out as described (Oot et al, 2016). Briefly, 1 ml of the assay containing 50 mM HEPES, pH 7.5, 25 mM KCl, 0.5 mM NADH, 2 mM phosphoenolpyruvate, 5 mM ATP, 30 units each of lactate dehydrogenase and pyruvate kinase was preheated to 37°C and supplemented with 4 mM MgCl2. The assay was started by adding 10 μg V‐ATPase or V1 subcomplexes or vacuoles and the change in absorbance at 340 nm was monitored using a temperature‐controlled cuvette holder in a Varian CARY 100 Bio UV‐Visible Spectrometer in kinetics mode. Enough time was provided after adding the protein to establish a linear rate of ATP hydrolysis. In case of measuring the activity of holoenzyme, 200 nM of the V‐ATPase specific inhibitor Concanamycin A was added into the assay.
Biolayer interferometry (BLI)
All BLI experiments were carried out in the Octet RED384 system as previously described (Sharma et al, 2019) with the exception that 1 mM β‐ME was included in all buffers and samples. All wells of the BLI plate contained 200 μl of buffer or specific protein(s) diluted into the same buffer. 0.5 mg/ml BSA was included in all samples to reduce non‐specific binding of assay components to the sensors. To study the effect of Oxr1 on V‐ATPase assembly, streptavidin biosensors (ForteBio, SA biosensors catalog no. 18‐5019) were pre‐equilibrated in a buffer for 10 min before loading with VoND (3 µg/ml). The biosensors were then dipped into wells containing V1Hchim and subunit C in presence and absence of Oxr1 and then into wells containing buffer. A control for non‐specific binding of V1Hchim and subunit C to empty biosensors was included. To assess the effect of Oxr1 on the dissociation of V1∆H from wild‐type H or Hchim, MBP‐tagged Hwt or Hchim (5 µg/ml) were loaded onto Anti‐Mouse IgG Fc Capture biosensors (ForteBio, AMC biosensors catalog no. 18‐5088) that were initially dipped into wells with 1 µg/ml anti‐MBP monoclonal antibodies (New England BioLabs, catalog no. E8032S). MBP‐Hwt or ‐Hchim loaded biosensors were then dipped into wells containing V1∆H (association step) followed by wells containing buffer, Oxr1, or Oxr1 plus subunit C (dissociation step). An appropriate control was included to monitor the slow dissociation of MBP‐Hwt or ‐Hchim from the biosensors. Experiments were carried out at 23°C, with a stir speed of 1,000 rpm and a standard measurement rate of 5 s−1. The data were processed and analyzed by the Octet Data Analysis v10.0 software.
Mass spectrometry analysis of V1Hchim
In‐solution digestion of 40 µg V1Hchim with trypsin was carried out using filter‐assisted sample preparation (FASP; Wisniewski et al, 2009) and peptides desalted using mixed‐mode cation exchange (MCX) stage tips (Rappsilber et al, 2003). For LC‐MS/MS, samples were dissolved in water containing 2% acetonitrile (ACN) and 0.5% formic acid. Samples (0.5 µg) were injected onto a nano‐LC (C18) column connected inline to an Orbitrap Lumos mass spectrometer via a nanoelectrospray source operating at 2.2 kV. The MS data was searched using SequestHT in Proteome Discoverer (version 2.4, Thermo Scientific) against the yeast proteome in Uniprot.
Negative stain EM analysis
For analysis of the assembly state of the holoenzyme using TEM, 5 μl of V1HchimVoND and V1HchimVoND + Oxr1 at ~20 μg/ml were applied to glow discharged carbon‐coated copper grids for 1 min, washed with water for 10 s, and then stained with 1% (w/v) uranyl acetate for another minute. Grids were visualized using a JEOL JEM‐1400 transmission electron microscope operating at 80 keV and images were captured at a magnification of 200,000× using a Gatan Orius SC1000 CCD camera. Assembled and disassembled particles from equal numbers of micrographs from both groups were counted manually for comparative analysis.
Cryo‐EM grid preparation and data collection
V1HchimVoND was vitrified at 0.5–1 mg/ml on UltrAuFoil 2/2 grids (Quantifoil) at 4°C in a cold room using an in‐house built cryo‐plunger. 4,684 Movies were recorded on a FEI Titan Krios 300 keV G3i at Stanford SLAC, equipped with a Gatan K2 Summit detector mounted behind a Bioquantum 20 eV slit energy filter. Images were collected automatically using EPU (Thermo Fisher) with dose fractionated into 25 movie frames at defocus ranges of 0.5–3 μm. The electron exposure per frame was ~2.0 e/Å2, and the pixel size at the specimen level was 1.08 Å.
Data processing and 3D refinement
Image processing was started in RELION‐3 (Zivanov et al, 2018) (Fig EV1). Movies were aligned in 5 × 5 patches in MotionCor2 (Zheng et al, 2017), and CTF parameters were estimated with GCTF (Zhang, 2016). Utilizing template‐based autopicking, 571,609 particles were initially picked. Holoenzymes in three rotational states with 165,044 particles were selected after several rounds of 2D and/or 3D classification using RELION. Each particle stack in three rotational states (state 1: 85,109, state 2: 42,206, state 3: 15,741) was then imported to cryoSPARC (Punjani et al, 2017). Global non‐uniform refinement resulted in maps at 4.2, 4.8, and 6.6 Å resolution for states 1‐3, respectively. Local refinement focused on V1 and Vo improved map features and resolution up to ~3.5 Å. While processing the holoenzyme structures, we noticed that a significant number of particles (52,594) resembled V1‐shaped molecules without Vo sectors. We then initiated another round of image processing starting from micrographs using cryoSPARC and performed template‐based particle picking with the V1 map from RELION as a template. We manually removed clear views of holoenzyme from 2D class averages and then 178,379 particles were selected and subjected to 3D classification. Due to the presence of structural heterogeneity in the V1 particles, we used non‐uniform refinement protocols to obtain the final 3.8 Å resolution map of V1(C)Oxr1.
Model building
Building of the model of state 1 V1HchimVoND started from the model of the A3B3DFH subcomplex of the cryoEM structure of state 1 V1Vo (PDB 3J9T) (Zhao et al, 2015), the cryoEM structure of Vo (PDB 6C6L) (Roh et al, 2018), and the crystal structures of the EG heterodimer (PDB 4DL0, 4EFA) (Oot et al, 2012) and subunit C (PDB 1U7L) (Drory et al, 2004). Atomic models were placed into the cryoEM density using rigid body fitting in Chimera (Pettersen et al, 2004) followed by manual model building in Coot (Emsley et al, 2010) and real‐space refinement in Phenix (Adams et al, 2010). Progress in refinement was monitored using validation tools as implemented in Coot, Phenix, and the Molprobity server (Williams et al, 2018). For placing yeast Oxr1, a homology model was generated in Phyre2 using the TLDc domain of zebrafish Oxr2 (PDB 4ACJ) (Blaise et al, 2012) as template. The interface between Oxr1 and V‐ATPase subunits was analyzed using the PDBePISA server (Krissinel & Henrick, 2007). CryoEM data collection and 3‐D refinement parameters as well as model statistics are given in Appendix Table S1.
Author contributions
S‐HR and SW conceived the project and designed experiments. S‐HR and SW wrote the manuscript with input from authors. S‐HR, SL, and HK performed the cryoEM study, MMK, RAO, SC‐C, and SW carried out biochemical experiments, and S‐HR and SW validated the models and maps.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Movie EV1
Acknowledgements
We thank Dr. Ebbing de Jong from the SUNY Upstate Medical University Mass Spectrometry Core Facility for the mass spectrometry analysis of V1Hchim and for help with data analysis and Maureen Tarsio for assistance with the vacuole preparation. This work has been supported by NIH grants GM058600, GM141908, and CA228340 to S.W., and Korean National Research Foundation grants NRF‐2019M3E5D6063871, 2019R1C1C1004598, 2020R1A5A1018081, 2021M3A9I4021220, 2020R1A6C101A183, SUHF foundation, and the Creative‐Pioneering Researchers Program of Seoul National UniversityS to S.‐H.R. The cryoEM data was collected at the Stanford‐SLAC CryoEM Facilities (P41GM103832, R01GM079429, and S10OD021600).
Conflict of interest
The authors declare that they have no conflict of interest.
The EMBO Journal (2022) 41: e109360
Contributor Information
Stephan Wilkens, Email: wilkenss@upstate.edu.
Soung‐Hun Roh, Email: shroh@snu.ac.kr.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data are available from the authors upon request. The cryoEM maps of the three states of V1HchimVoND and V1(C)Oxr1 are deposited in the Electron Microscopy Data Bank under accession numbers EMD‐31538 (http://www.ebi.ac.uk/pdbe/entry/EMD‐31538), EMD‐31539 (http://www.ebi.ac.uk/pdbe/entry/EMD‐31539), EMD‐31540 (http://www.ebi.ac.uk/pdbe/entry/EMD‐31540), and EMD‐31541 (http://www.ebi.ac.uk/pdbe/entry/EMD‐31541), respectively. The associated coordinate models are deposited in the Protein Data Bank with accession numbers 7FDA (http://www.rcsb.org/pdb/explore/explore.do?structureId=7FDA), 7FDB (http://www.rcsb.org/pdb/explore/explore.do?structureId=7FDB), 7FDC (http://www.rcsb.org/pdb/explore/explore.do?structureId=7FDC), and 7FDE (http://www.rcsb.org/pdb/explore/explore.do?structureId=7FDE), respectively.
References
- Abbas YM, Wu D, Bueler SA, Robinson CV, Rubinstein JL (2020) Structure of V‐ATPase from the mammalian brain. Science 367: 1240–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW et al (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud‐Arnould M, Tauziet M, Moncorge O, Goujon C, Blaise M (2021) Crystal structure of the TLDc domain of human NCOA7‐AS. Acta Crystallogr F Struct Biol Commun 77: 230–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagh MB, Peng S, Chandra G, Zhang Z, Singh SP, Pattabiraman N, Liu A, Mukherjee AB (2017) Misrouting of v‐ATPase subunit V0a1 dysregulates lysosomal acidification in a neurodegenerative lysosomal storage disease model. Nat Commun 8: 14612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaise M, Alsarraf HM, Wong JE, Midtgaard SR, Laroche F, Schack L, Spaink H, Stougaard J, Thirup S (2012) Crystal structure of the TLDc domain of oxidation resistance protein 2 from zebrafish. Proteins 80: 1694–1698 [DOI] [PubMed] [Google Scholar]
- Bodzeta A, Kahms M, Klingauf J (2017) The Presynaptic v‐ATPase reversibly disassembles and thereby modulates exocytosis but is not part of the fusion machinery. Cell Rep 20: 1348–1359 [DOI] [PubMed] [Google Scholar]
- Bond SR, Naus CC (2012) RF‐Cloning.org: an online tool for the design of restriction‐free cloning projects. Nucleic Acids Res 40: W209–W213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton S, Smith PJ, Lui B, Brown D (1996) Acidification of the male reproductive tract by a proton pumping (H+)‐ATPase. Nat Med 2: 470–472 [DOI] [PubMed] [Google Scholar]
- Castroflorio E, den Hoed J, Svistunova D, Finelli MJ, Cebrian‐Serrano A, Corrochano S, Bassett AR, Davies B, Oliver PL (2021) The Ncoa7 locus regulates V‐ATPase formation and function, neurodevelopment and behaviour. Cell Mol Life Sci 78: 3503–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CY, Parra KJ (2014) Yeast phosphofructokinase‐1 subunit Pfk2p is necessary for pH homeostasis and glucose‐dependent vacuolar ATPase reassembly. J Biol Chem 289: 19448–19457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MP, Forgac M (2020) Regulation and function of V‐ATPases in physiology and disease. Biochim Biophys Acta Biomembr 1862: 183341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couoh‐Cardel S, Hsueh YC, Wilkens S, Movileanu L (2016) Yeast V‐ATPase proteolipid ring acts as a large‐conductance transmembrane protein pore. Sci Rep 6: 24774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couoh‐Cardel S, Milgrom E, Wilkens S (2015) Affinity purification and structural features of the yeast vacuolar ATPase Vo membrane sector. J Biol Chem 290: 27959–27971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drory O, Frolow F, Nelson N (2004) Crystal structure of yeast V‐ATPase subunit C reveals its stator function. EMBO Rep 5: 1148–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand M, Kolpak A, Farrell T, Elliott NA, Shao W, Brown M, Volkert MR (2007) The OXR domain defines a conserved family of eukaryotic oxidation resistance proteins. BMC Cell Biol 8: 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton AF, Merkulova M, Brown D (2021) The H(+)‐ATPase (V‐ATPase): from proton pump to signaling complex in health and disease. Am J Physiol Cell Physiol 320: C392–C414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finelli MJ, Oliver PL (2017) TLDc proteins: new players in the oxidative stress response and neurological disease. Mamm Genome 28: 395–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frattini A, Orchard PJ, Sobacchi C, Giliani S, Abinun M, Mattsson JP, Keeling DJ, Andersson A‐K, Wallbrandt P, Zecca L et al (2000) Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat Genet 25: 343–346 [DOI] [PubMed] [Google Scholar]
- Futai M, Sun‐Wada GH, Wada Y, Matsumoto N, Nakanishi‐Matsui M (2019) Vacuolar‐type ATPase: a proton pump to lysosomal trafficking. Proc Jpn Acad Ser B Phys Biol Sci 95: 261–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Woods RA (2002) Transformation of yeast by lithium acetate/single‐stranded carrier DNA/polyethylene glycol method. Methods Enzymol 350: 87–96 [DOI] [PubMed] [Google Scholar]
- Graf R, Harvey WR, Wieczorek H (1996) Purification and properties of a cytosolic V1‐ATPase. J Biol Chem 271: 20908–20913 [DOI] [PubMed] [Google Scholar]
- Harrison MA, Muench SP (2018) The vacuolar ATPase – A nano‐scale motor that drives cell biology. Subcell Biochem 87: 409–459 [DOI] [PubMed] [Google Scholar]
- Hilario E, Gogarten JP (1998) The prokaryote‐to‐eukaryote transition reflected in the evolution of the V/F/A‐ATPase catalytic and proteolipid subunits. J Mol Evol 46: 703–715 [DOI] [PubMed] [Google Scholar]
- Hirata R, Graham LA, Takatsuki A, Stevens TH, Anraku Y (1997) VMA11 and VMA16 encode second and third proteolipid subunits of the Saccharomyces cerevisiae vacuolar membrane H+‐ATPase. J Biol Chem 272: 4795–4803 [DOI] [PubMed] [Google Scholar]
- Ho MN, Hirata R, Umemoto N, Ohya Y, Takatsuki A, Stevens TH, Anraku Y (1993) VMA13 encodes a 54‐kDa vacuolar H(+)‐ATPase subunit required for activity but not assembly of the enzyme complex in Saccharomyces cerevisiae. J Biol Chem 268: 18286–18292 [PubMed] [Google Scholar]
- Kane PM (2016) Proton transport and pH control in fungi. Adv Exp Med Biol 892: 33–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez‐Soriano J, Santos F, Cremers CWRJ, Pietro AD et al (1999) Mutations in the gene encoding B1 subunit of H+‐ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 21: 84–90 [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372: 774–797 [DOI] [PubMed] [Google Scholar]
- Lindstrom A, Anantpadma M, Baker L, Raghavendra NM, Davey R, Davisson VJ (2018) Phenotypic prioritization of diphyllin derivatives that block filoviral cell entry by vacuolar (H(+) )‐ATPase inhibition. ChemMedChem 13: 2664–2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Ammar D, Ives H, Albrecht F, Gluck SL (2007) Physical interaction between aldolase and vacuolar H+‐ATPase is essential for the assembly and activity of the proton pump. J Biol Chem 282: 24495–24503 [DOI] [PubMed] [Google Scholar]
- Lüthy K, Mei D, Fischer B, De Fusco M, Swerts J, Paesmans J, Parrini E, Lubarr N, Meijer IA, Mackenzie KM et al (2019) TBC1D24‐TLDc‐related epilepsy exercise‐induced dystonia: rescue by antioxidants in a disease model. Brain 142: 2319–2335 [DOI] [PubMed] [Google Scholar]
- Maxson ME, Grinstein S (2014) The vacuolar‐type H(+)‐ATPase at a glance – more than a proton pump. J Cell Sci 127: 4987–4993 [DOI] [PubMed] [Google Scholar]
- Mazhab‐Jafari MT, Rohou A, Schmidt C, Bueler SA, Benlekbir S, Robinson CV, Rubinstein JL (2016) Atomic model for the membrane‐embedded VO motor of a eukaryotic V‐ATPase. Nature 539: 118–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkulova M, Păunescu TG, Azroyan A, Marshansky V, Breton S, Brown D (2015) Mapping the H(+) (V)‐ATPase interactome: identification of proteins involved in trafficking, folding, assembly and phosphorylation. Sci Rep 5: 14827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkulova M, Păunescu TG, Nair AV, Wang CY, Capen DE, Oliver PL, Breton S, Brown D (2018) Targeted deletion of the Ncoa7 gene results in incomplete distal renal tubular acidosis in mice. Am J Physiol Renal Physiol 315: F173–F185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata T, Yamato I, Kakinuma Y, Leslie AG, Walker JE (2005) Structure of the rotor of the V‐Type Na+‐ATPase from Enterococcus hirae. Science 308: 654–659 [DOI] [PubMed] [Google Scholar]
- Nelson H, Nelson N (1990) Disruption of genes encoding subunits of yeast vacuolar H(+)‐ATPase causes conditional lethality. Proc Natl Acad Sci USA 87: 3503–3507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oot RA, Couoh‐Cardel S, Sharma S, Stam NJ, Wilkens S (2017) Breaking up and making up: the secret life of the vacuolar H+ ‐ATPase. Protein Sci 26: 896–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oot RA, Huang LS, Berry EA, Wilkens S (2012) Crystal structure of the yeast vacuolar ATPase heterotrimeric EGC(head) peripheral stalk complex. Structure 20: 1881–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oot RA, Kane PM, Berry EA, Wilkens S (2016) Crystal structure of yeast V1‐ATPase in the autoinhibited state. EMBO J 35: 1694–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oot RA, Wilkens S (2010) Domain characterization and interaction of the yeast vacuolar ATPase subunit C with the peripheral stator stalk subunits E and G. J Biol Chem 285: 24654–24664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra KJ, Chan CY, Chen J (2014) Saccharomyces cerevisiae vacuolar H+‐ATPase regulation by disassembly and reassembly: one structure and multiple signals. Eukaryot Cell 13: 706–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra KJ, Kane PM (1998) Reversible association between the V1 and V0 domains of yeast vacuolar H+‐ATPase is an unconventional glucose‐induced effect. Mol Cell Biol 18: 7064–7074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra KJ, Keenan KL, Kane PM (2000) The H subunit (Vma13p) of the yeast V‐ATPase inhibits the ATPase activity of cytosolic V1 complexes. J Biol Chem 275: 21761–21767 [DOI] [PubMed] [Google Scholar]
- Perez L, Carrasco L (1994) Involvement of the vacuolar H(+)‐ATPase in animal virus entry. J Gen Virol 75(Pt 10): 2595–2606 [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA (2017) cryoSPARC: algorithms for rapid unsupervised cryo‐EM structure determination. Nat Methods 14: 290–296 [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, Mann M (2003) Stop and go extraction tips for matrix‐assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75: 663–670 [DOI] [PubMed] [Google Scholar]
- Roh SH, Shekhar M, Pintilie G, Chipot C, Wilkens S, Singharoy A, Chiu W (2020) Cryo‐EM and MD infer water‐mediated proton transport and autoinhibition mechanisms of V(o) complex. Sci Adv 6: eabb9605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh SH, Stam NJ, Hryc CF, Couoh‐Cardel S, Pintilie G, Chiu W, Wilkens S (2018) The 3.5‐A CryoEM structure of nanodisc‐reconstituted yeast vacuolar ATPase Vo proton channel. Mol Cell 69: 993–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sautin YY, Lu M, Gaugler A, Zhang L, Gluck SL (2005) Phosphatidylinositol 3‐kinase‐mediated effects of glucose on vacuolar H+‐ATPase assembly, translocation, and acidification of intracellular compartments in renal epithelial cells. Mol Cell Biol 25: 575–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Oot RA, Khan MM, Wilkens S (2019) Functional reconstitution of vacuolar H+‐ATPase from Vo proton channel and mutant V1‐ATPase provides insight into the mechanism of reversible disassembly. J Biol Chem 294: 6439–6449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Wilkens S (2017) Biolayer interferometry of lipid nanodisc‐reconstituted yeast vacuolar H+ ‐ATPase. Protein Sci 26: 1070–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smardon AM, Nasab ND, Tarsio M, Diakov TT, Kane PM (2015) Molecular interactions and cellular itinerary of the yeast RAVE (Regulator of the H+‐ATPase of Vacuolar and Endosomal Membranes) complex. J Biol Chem 290: 27511–27523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, Hulton SA, Sanjad SA, Al‐Sabban EA, Lifton RP et al (2000) Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116‐kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet 26: 71–75 [DOI] [PubMed] [Google Scholar]
- Sreelatha A, Bennett TL, Carpinone EM, O'Brien KM, Jordan KD, Burdette DL, Orth K, Starai VJ (2015) Vibrio effector protein VopQ inhibits fusion of V‐ATPase‐containing membranes. Proc Natl Acad Sci USA 112: 100–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam NJ, Wilkens S (2017) Structure of the Lipid Nanodisc‐reconstituted Vacuolar ATPase Proton Channel: definition of the interaction of rotor and stator and implications for enzyme regulation by reversible dissociation. J Biol Chem 292: 1749–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stransky L, Cotter K, Forgac M (2016) The function of V‐ATPases in cancer. Physiol Rev 96: 1071–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stransky LA, Forgac M (2015) Amino acid availability modulates vacuolar H+‐ATPase assembly. J Biol Chem 290: 27360–27369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun‐Wada GH, Toyomura T, Murata Y, Yamamoto A, Futai M, Wada Y (2006) The a3 isoform of V‐ATPase regulates insulin secretion from pancreatic beta‐cells. J Cell Sci 119: 4531–4540 [DOI] [PubMed] [Google Scholar]
- Tan YZ, Abbas YM, Wu JZ, Wu D, Hesketh GGB, Gingras SA, Robinson AC, Grinstein CV, Rubinstein JL (2021) Structure of mammalian V‐ATPase with the TLDc domain protein mEAK7 bound. bioRxiv 10.1101/2021.11.03.466369 [PREPRINT] [DOI] [Google Scholar]
- Trombetta ES, Ebersold M, Garrett W, Pypaert M, Mellman I (2003) Activation of lysosomal function during dendritic cell maturation. Science 299: 1400–1403 [DOI] [PubMed] [Google Scholar]
- Vasanthakumar T, Rubinstein JL (2020) Structure and Roles of V‐type ATPases. Trends Biochem Sci 45: 295–307 [DOI] [PubMed] [Google Scholar]
- Volkert MR, Elliott NA, Housman DE (2000) Functional genomics reveals a family of eukaryotic oxidation protection genes. Proc Natl Acad Sci USA 97: 14530–14535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wu D, Robinson CV, Wu H, Fu TM (2020a) Structures of a complete human V‐ATPase reveal mechanisms of its assembly. Mol Cell 80: 501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Long T, Hassan A, Wang J, Sun Y, Xie XS, Li X (2020b) Cryo‐EM structures of intact V‐ATPase from bovine brain. Nat Commun 11: 3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkens S, Oot RA, Khan MM, Zimmer K (2021) Structure and mechanism of the vacuolar H+‐ATPase. In Encyclopedia of Biological Chemistry, Joseph J (ed), 3rd edn, pp 581–593. Oxford: Elsevier; [Google Scholar]
- Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB et al (2018) MolProbity: More and better reference data for improved all‐atom structure validation. Protein Sci 27: 293–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj N, Mann M (2009) Universal sample preparation method for proteome analysis. Nat Methods 6: 359–362 [DOI] [PubMed] [Google Scholar]
- Wong D, Bach H, Sun J, Hmama Z, Av‐Gay Y (2011) Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar‐H+‐ATPase to inhibit phagosome acidification. Proc Natl Acad Sci USA 108: 19371–19376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Shen X, Bryan A, Banga S, Swanson MS, Luo ZQ (2010) Inhibition of host vacuolar H+‐ATPase activity by a Legionella pneumophila effector. PLoS Pathog 6: e1000822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Forgac M (2001) Microtubules are involved in glucose‐dependent dissociation of the yeast vacuolar [H+]‐ATPase in vivo . J Biol Chem 276: 24855–24861 [DOI] [PubMed] [Google Scholar]
- Zhang J, Myers M, Forgac M (1992) Characterization of the V0 domain of the coated vesicle (H+)‐ATPase. J Biol Chem 267: 9773–9778 [PubMed] [Google Scholar]
- Zhang K (2016) Gctf: Real‐time CTF determination and correction. J Struct Biol 193: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Charsky C, Kane PM, Wilkens S (2003) Yeast V1‐ATPase: affinity purification and structural features by electron microscopy. J Biol Chem 278: 47299–47306 [DOI] [PubMed] [Google Scholar]
- Zhao J, Benlekbir S, Rubinstein JL (2015) Electron cryomicroscopy observation of rotational states in a eukaryotic V‐ATPase. Nature 521: 241–245 [DOI] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, Agard DA (2017) MotionCor2: anisotropic correction of beam‐induced motion for improved cryo‐electron microscopy. Nat Methods 14: 331–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, Scheres SH (2018) New tools for automated high‐resolution cryo‐EM structure determination in RELION‐3. Elife 7 [DOI] [PMC free article] [PubMed]