

Abstract
There is increasing evidence that prokaryotes maintain chromosome structure, which in turn impacts gene expression. We recently characterized densely occupied, multi‐kilobase regions in the E. coli genome that are transcriptionally silent, similar to eukaryotic heterochromatin. These extended protein occupancy domains (EPODs) span genomic regions containing genes encoding metabolic pathways as well as parasitic elements such as prophages. Here, we investigate the contributions of nucleoid‐associated proteins (NAPs) to the structuring of these domains, by examining the impacts of deleting NAPs on EPODs genome‐wide in E. coli and B. subtilis. We identify key NAPs contributing to the silencing of specific EPODs, whose deletion opens a chromosomal region for RNA polymerase binding at genes contained within that region. We show that changes in E. coli EPODs facilitate an extra layer of transcriptional regulation, which prepares cells for exposure to exotic carbon sources. Furthermore, we distinguish novel xenogeneic silencing roles for the NAPs Fis and Hfq, with the presence of at least one being essential for cell viability in the presence of domesticated prophages. Our findings reveal previously unrecognized mechanisms through which genomic architecture primes bacteria for changing metabolic environments and silences harmful genomic elements.
Keywords: bacterial gene regulation, chromatin, gene regulation, nucleoid‐associated proteins
Subject Categories: Chromatin, Transcription & Genomics; Microbiology, Virology & Host Pathogen Interaction
Distinct heterochromatin‐like domains promote transcriptional memory and silence parasitic genetic elements in bacteria.
Introduction
All organisms must organize immense amounts of genetic information into a relatively small physical space within the cell. The maintenance and accessibility of the DNA architecture are required for efficient DNA replication, repair, and transcription, and division, and thus for proper cell division required to sustain life. In the bacterium Escherichia coli (E. coli), chromosome structure is mediated by roughly a dozen small, basic nucleoid‐associated proteins (NAPs) (Luijsterburg et al, 2008; Dillon & Dorman, 2010; Shen & Landick, 2019) that work in concert to modulate supercoiling, DNA looping, and distant chromosomal contacts (Postow et al, 2004; Deng et al, 2005; Grainger et al, 2008; Lim et al, 2012; Boudreau et al, 2018). Despite substantial research effort, it is largely unclear how individual NAPs impact the overall structure of the chromosome, due in part to their promiscuous and overlapping binding across the genome (Ali Azam et al, 1999; Dillon & Dorman, 2010; Amemiya et al, 2021).
In order to gain a comprehensive understanding of the contribution of NAPs to global protein occupancy and gene expression, we used in vivo protein occupancy display at high resolution (IPOD‐HR). IPOD‐HR is a method that provides a global snapshot of areas in the genome where proteins are bound to DNA, thus yielding insight into genome‐wide regulation and structure (Vora et al, 2009; Freddolino et al, 2021a). IPOD‐HR revealed approximately two hundred regions of the E. coli genome that are densely packed with protein but exclude RNA polymerase, inviting striking comparisons to heterochromatin found in eukaryotes (Freddolino et al, 2021a). These regions are referred to as extended protein occupancy domains (EPODs) and identified based on the presence of high levels of protein occupancy over a length of 1 kb or more (Vora et al, 2009). While EPODs have been shown to be partially occupied by NAPs, and in some cases overlap with known binding sites for NAPs such as H‐NS (Freddolino et al, 2021a), the contributions of individual NAPs on EPOD formation remain unexplored. Our previous findings, consistent with studies in the literature, have implicated H‐NS as a dominant gene silencer in E. coli (Ueguchi & Mizuno, 1993; Deng et al, 2005; Shen & Landick, 2019; preprint: Freddolino et al, 2021b). H‐NS has the capacity to form filaments, tightly compacting dsDNA. In vitro, H‐NS has been shown that the two types of filaments, linear or bridged, which block transcription initiation, while only bridged filaments block transcription by blocking elongation (Singh et al, 2014; Kotlajich et al, 2015; Landick et al, 2015). Other proteins, such as Hha and the H‐NS paralog StpA, promote the formation of H‐NS filaments and modulate their structural properties (Valk et al, 2017; Boudreau et al, 2018). In addition to H‐NS’s role in global gene silencing, it is well documented to specifically silence horizontally acquired DNA (Baumler, 2006; Lucchini et al, 2006; Navarre, 2006; Kahramanoglou et al, 2011; Freddolino et al, 2021a). While the dominant form of filaments in vivo remains unknown, it is clear that H‐NS plays a major role in transcriptional silencing in the bacteria that carry it.
Recent data suggest that EPODs contribute to both the regulation of metabolic pathways and the silencing of horizontally acquired DNA (Scholz et al, 2019; Freddolino et al, 2021a); however, the NAPs mediating this response for individual EPODs are unknown. Additionally, evidence suggests that bacterial genomes have integration hotspots for horizontally acquired DNA (preprint: Nakamura et al, 2021). We have found that EPODs have a higher Tn5 integration frequency compared to the rest of the genome (Freddolino et al, 2021a), perhaps giving insight that EPODs may be functional units that serve as hotspots for foreign DNA integration. Additional data using different mechanisms of horizontal gene transfer would be required to further investigate the extent to which EPODs show enhanced integration rates, as opposed to simply forming on horizontally acquired DNA once it has already integrated. Understanding the key protein components of EPODs and their roles in genome organization will shed light on the relationship between the regulation of transcription and genome architecture in bacterial genomes.
Here, we investigate the contributions of the major NAPs in E. coli to maintaining the pattern of global protein occupancy, and the effects of that occupancy on gene expression. Rather than simply observing where one particular protein binds, we monitor the complete set of changes in protein occupancy caused by loss of any single NAP. We show that loss of any single NAP results in changes to the global landscape of protein occupancy, albeit often less dramatic than might be expected, as we show that NAPs are able to compensate for each other to maintain EPOD structure even after single NAP knockouts. Further, we demonstrate the function of NAP mediated silencing of rare carbon metabolism and suggest that this mechanism facilitates a transcriptional memory response. Additionally, we describe a previously unidentified prophage silencing role for NAPs Hfq and Fis. Finally, we demonstrate the presence of EPOD‐like structures in the evolutionary distinct Gram‐positive Bacillus subtilis, which function similarly to facilitate silencing of metabolic pathways and horizontally acquired genes.
Results
Large‐scale patterns of protein occupancy are highly maintained across conditions and laboratory isolates
In order to investigate the contributions of the major E. coli nucleoid‐associated proteins to the genome‐wide pattern of protein occupancy, we applied our recently developed IPOD‐HR method (Freddolino et al, 2021a) to profile the changes in global protein occupancy on the chromosome in the presence of several deletions of nucleoid‐associated protein coding genes. The IPOD‐HR procedure begins similarly to a chromatin immunoprecipitation (ChIP) experiment, in which cells are cross‐linked under a physiological condition of interest, lysed, and the DNA digested (Appendix Fig S1A). However, rather than pull down a single protein target with an antibody as in ChIP, a phenol–chloroform extraction is performed wherein an enrichment of protein–DNA complexes forms in a white disk at the aqueous‐organic interphase (Appendix Fig S1A). The disk is isolated and washed thoroughly, and DNA is then purified from the disk and compared to an input sample to reveal the locations on the genome that were bound by proteins at the time of crosslinking. The raw IPOD‐HR signal contains contributions both from RNA polymerase and from regulatory proteins. In order to visualize protein bound to DNA aside from RNA polymerase, we perform an RNA polymerase ChIP‐seq experiment in parallel and subtract the binding of RNA polymerase from our raw IPOD‐HR signal (Appendix Fig S1B). In‐depth methodological details are given in the Methods section and in Freddolino et al (2021a). In our initial applications of IPOD‐HR in E. coli, we found that the method was able to reveal the dynamics of individual transcription factor binding sites after both genetic and physiological perturbations (Freddolino et al, 2021a). We saw in contrast, however, that hundreds of regions of extended protein occupancy (termed Extended Protein Occupancy Domains or EPODs (Vora et al, 2009)) were fairly static across all tested conditions (three transcription factor deletions and three growth conditions) (Freddolino et al, 2021a). Given that EPODs appear to arise largely due to the binding of nucleoid‐associated proteins (NAPs), in the present work, we examined the contributions of different NAPs to the pattern of EPODs along the chromosome.
To identify condition‐dependent EPOD locations in the E. coli genome, we applied EPOD calling equivalent to that in preprint: Freddolino et al (2021b). There are two EPOD thresholds defined by regions that are greater than 1,024 bp in length over which the rolling mean protein occupancy in that region exceeds a specified percentile compared to the rest of the genome (see Materials and Methods). We term the two levels of stringency used here as “Loose EPODs” (75th percentile cutoff; the “relaxed” threshold) and “Strict EPODs” (90th percentile cutoff) (Fig 1A; further explained in Vora et al (2009)). Here, we will focus primarily on “Strict EPODs” unless otherwise noted. It was previously shown that the ~200 EPODs identified across the E. coli MG1655 (WT) genome show significantly enriched overlaps with loci encoding genes involved in metabolism and silencing of mobile elements (Freddolino et al, 2021a). As remodeling of nucleoid organization has been previously detected in response to environmental changes, such as media richness and growth phase (Neeli‐Venkata et al, 2016; Remesh et al, 2020; Verma et al, 2020), we tested whether EPODs were one of the functional units that mediated changes across growth conditions. To examine the robustness of these domains under various physiological conditions, we performed IPOD‐HR on an MG1655 isolate (referred to as “WT”), varying the media type and growth phase. IPOD‐HR was performed as previously described (Freddolino et al, 2021a); samples were taken at mid‐exponential growth phase or deep stationary growth phase (D.S.; described in Materials and Methods below), in both rich defined media and minimal media with glucose (see Materials and Methods). For all experiments, we used defined media to maintain consistency in physiological experiments with buffer conditions, salts, etc., and to avoid the noted variation occurring in Lysogeny broth (LB medium) (Neidhardt et al, 1974; https://schaechter.asmblog.org/schaechter/2009/11/the‐limitations‐of‐lb‐medium.html). Even within E. coli MG1655 isolates, there are genetic differences from variation in parental origin (Freddolino et al, 2012) that have led to differences in what is deemed “WT” across different laboratories. To examine the robustness of EPODs between relatively similar isolates with minor genetic differences, we used another MG1655 isolate (labeled WT (2)). In comparison with WT, WT (2) is crl+ , gatC+ , glpR+ , contains point mutations in ybhJ and mntP and has an intact dgcJ (see Methods for details). The variations between different MG1655 isolates are an important consideration when comparing across various MG1655 datasets, as our data suggest that small genetic differences may lead to wider changes in gene regulation and protein occupancy landscapes compared to changes in media sources, as we will discuss in detail below.
Figure 1. EPODs are highly robust across growth conditions.
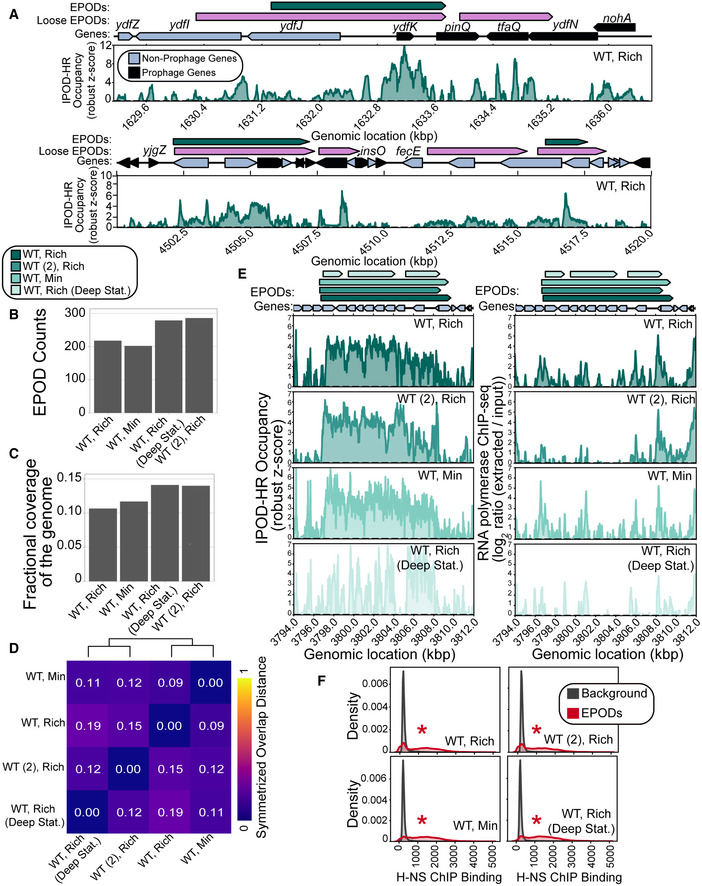
- Example regions exhibiting the IPOD‐HR occupancy where EPOD calls are made. Loose EPODs (pink) and Strict EPODs (teal) represent spans of high protein occupancy at lower and higher thresholds, respectively. Neighboring areas showing lower robust z scores are not EPODs. Note that following the definitions in (preprint: Freddolino et al, 2021b), the presence of an EPOD requires a sustained region of high average occupancy, without any regions in which occupancy drops to background levels. Thus, breaks in EPOD calls represent cases where a rolling average occupancy drops below the calling threshold (e.g., the breakpoint in the middle of pinQ in the top panel), and regions of apparent occupancy may nevertheless not be called due to being below the minimum length threshold (as is the case for nohA).
- Strict EPOD counts across WT (MG1655) E. coli cells are similar across different conditions. Cells were grown in: Rich (Rich Defined Media (see Materials and Methods)), Min (Minimal Media) and collected at log phase growth or deep stationary phase (Deep Stat.). WT (2) is a MG1655 isolate from another laboratory that differs from our WT strain by four‐point mutations and two insertion sequence integrations.
- Fractional coverage of Strict EPODs across the genome does not largely vary across conditions; however, cells grown in the Min condition have a slightly larger coverage.
- To assess the similarity between EPOD calls in each condition, we calculated the symmetrized overlap distance of EPODs. A value of 0 indicates that the set of EPODs are identical. Hierarchical clustering reveals that growth phase impacts EPOD location in this dataset.
- Specific locations across all conditions remain occupied by protein where EPODs across a representative region, the waa operon, were called (left) and unoccupied by RNA polymerase (right). The quantile‐normalized robust z scores of the protein occupancy at each 5 bp are represented by the IPOD‐HR occupancy.
- Kernel density plots displaying the normalized histograms (smoothed by a kernel density estimator) of H‐NS ChIP (Kahramanoglou et al, 2011) for regions of the genome within EPODs versus Background (i.e., protein occupancy through the rest of the genome, where contiguous regions between EPODs were treated as a single data point). H‐NS binding shows a strong overlap with EPOD locations measured in all conditions. (*) indicates FDR‐corrected P < 0.005 via permutation test indicating an enrichment in H‐NS ChIP binding in EPODs across conditions (against a null hypothesis of no difference in medians).
The number and overall genomic coverage of EPODs across conditions and genetic backgrounds remained relatively stable (Fig 1B and C), with the greatest increases in coverage and counts in cells in deep stationary phase and the WT (2) isolate. To examine the changes in the locations and boundaries of EPODs, we calculated a measure that we refer to as the symmetrized overlap distance, given by the difference between unity and the geometric mean of the EPOD overlaps:
where AB is the fraction of condition B’s EPODs overlapped by the relaxed threshold EPODs (“Loose EPODs”; example in Fig 1A) contained in condition A, and BA is the fraction of condition A EPODs overlapped by the loose threshold EPODs from condition B. A value of 0 indicates identical EPOD locations, since both overlaps would be 1. The use of loose EPODs in the comparisons as opposed to strict avoids overstating the differences between conditions based on minor thresholding differences and emphasizes large and systematic changes in occupancy. The symmetrized overlap distances between all pairs of conditions examined here are shown in Fig 1D. Values within the heatmap displayed are calculated using the fraction of EPODs contained in the relaxed threshold EPODs, defined as in preprint: Freddolino et al (2021b), for each condition. Hierarchical clustering analysis of these distances reveals that the major differences stem decreasingly from growth phase, genotype, and then media changes (Fig 1D). Notably, the EPOD location profiles for the two MG1655 isolates considered here were more similar to each other than the WT deep stationary phase condition, but in all cases a substantial majority of EPODs are conserved across conditions. These findings support ongoing research that shows major changes in nucleoid composition during later phases of growth (Ali Azam et al, 1999; Grainger et al, 2008). Previous investigation of EPODs under baseline growth conditions (exponential growth in rich defined media) demonstrated that genes contained in EPODs were enriched for several functionalities, including DNA transposition, cytolysis, and LPS biosynthesis (Freddolino et al, 2021a). To identify the pathways maintained across all conditions considered in Fig 1, we ran iPAGE (Goodarzi et al, 2009) to associate which gene ontology (GO) terms fell within EPODs relative to the rest of the genome (“Background”). We found that three GO terms remained highly enriched in EPODs under all four conditions shown here: cytolysis, LPS biosynthetic pathway, and cellular response to acid chemical, demonstrating that EPOD‐mediated regulation of these functionalities persists across both physiological condition and the selected cell lineages (Appendix Fig S2).
Important genetic differences have previously been observed between various MG1655 isolates, which include mutations expected to have regulatory consequences (Freddolino et al, 2012). In the case of the WT versus WT (2) MG1655 isolates considered here, one of the main genetic differences is the presence of an IS1 insertion sequence in the dgcJ gene (a putative diguanylate cyclase) in WT that is absent in WT (2). Cyclic di‐GMP generally regulates the switch between adhesion/biofilm formation and motility across a range of bacteria (Hengge, 2009). A dgcJ mutant exhibits increased motility (Sanchez‐Torres et al, 2011). In addition, WT (2) contains an intact crl allele compared with the IS5‐disrupted allele present in WT, and the Crl regulon likewise contains a multitude of genes involved in motility and adhesion (Santos‐Zavaleta et al, 2019). Based on these important genetic differences, we expected that the overall regulatory states of WT and WT (2) would substantially differ with regard to motility and biofilm formation, although the direction of the effect is impossible to predict a priori due to the lack of information on the precise effect of the dgcJ IS1 insertion (which could in principle cause gain or loss of function) and the complicated interaction of various GGDEF proteins in regulating motility and biofilm formation (Sanchez‐Torres et al, 2011). Indeed, we observed an enrichment of EPODs on biofilm and adhesion‐related genes in the WT (2)‐specific EPODs (Appendix Fig S2), indicating that the additional EPODs unique to WT (2) were enriched on operons involved in establishing a sessile lifestyle. As noted above, the strains considered here also differ in genotype at gatC, glpR, ybhJ, and mntP; although it appears unlikely based on the genes involved, we cannot rule out contributions from the differences at those loci in the observed changes in EPOD locations. Taken together with our deep stationary phase results, we show that IPOD‐HR is able to detect changes in global protein occupancy due to genetic and environmental differences. We also note that while most EPODs are preserved across a range of conditions, the positions of some EPODs do indeed change in response to genetic and/or environmental perturbations.
Silencing of the non‐functional LPS gene pathway is maintained across conditions
To better understand the dynamics of protein occupancy across different conditions at a locus that shows a heavily maintained EPOD, we examined the changes in occupancy at a representative region, the LPS biosynthesis locus (the waa operon) across conditions (Fig 1E). The LPS pathway was noted to be silenced by EPODs in our initial findings (Freddolino et al, 2021a). Typically, E. coli express these operons as part of O antigen biosynthesis, a highly beneficial cellular component that increases resistance to phage infection and environmental pressures (Wang & Reeves, 1998; D’Souza et al, 2002; Feng et al, 2005; Nakao et al, 2012; Linkevicius et al, 2013; Wang et al, 2016). However, MG1655 contains an insertion element in wbbL, an upstream component of the LPS pathway, rendering wbbL non‐functional (Rubirés et al, 1997). Consistent with prior observations, we observe robust protein occupancy and minimal RNA polymerase occupancy across all conditions (Fig 1E), although a qualitative change in the locations of the high‐occupancy regions is apparent in deep stationary phase, perhaps reflecting a turnover in the predominant NAPs present under that condition. Without functional genes in this pathway, there are no biological benefits to expressing downstream genes under various physiological conditions. We speculate that these components of the LPS pathway are silenced due to the insertion element in wbbL as a method to conserve resources. While the exact mechanism for the establishment and maintenance of waa operon silencing is currently unknown, we hypothesize that silencing may have been selected for over the course of the subsequent evolution of MG1655.
Nucleoid‐associated proteins are the main components of EPODs
We hypothesized that the maintenance of EPODs was largely driven by NAP occupancy. H‐NS has been widely described as a major component of the E. coli nucleoid in exponential growth, specifically inhibiting transcription and silencing mobile elements and prophages (Ueguchi & Mizuno, 1993; Lucchini et al, 2006; Navarre et al, 2007; Boudreau et al, 2018). We compared published H‐NS binding sites within EPODs compared to background (i.e., the remainder of the genome) and found significant enrichments of H‐NS binding in EPODs across all considered conditions (Fig 1F), consistent with previous observations (Vora et al, 2009). Thus, H‐NS is facilitating silencing of regions across the genome robustly in varying media, growth phase, and slight genotype differences. However, we also found previously that while the majority of EPODs overlap with known H‐NS binding regions, a substantial fraction do not (Freddolino et al, 2021a). In addition, the fact that H‐NS is present at a particular EPOD does not necessarily mean that it is the only factor (or even a necessary or sufficient factor) in forming that EPOD and silencing the genes there. We thus sought to assess whether other NAPs facilitate silencing of specific EPODs or whether H‐NS is the major silencing factor of EPODs across the E. coli genome.
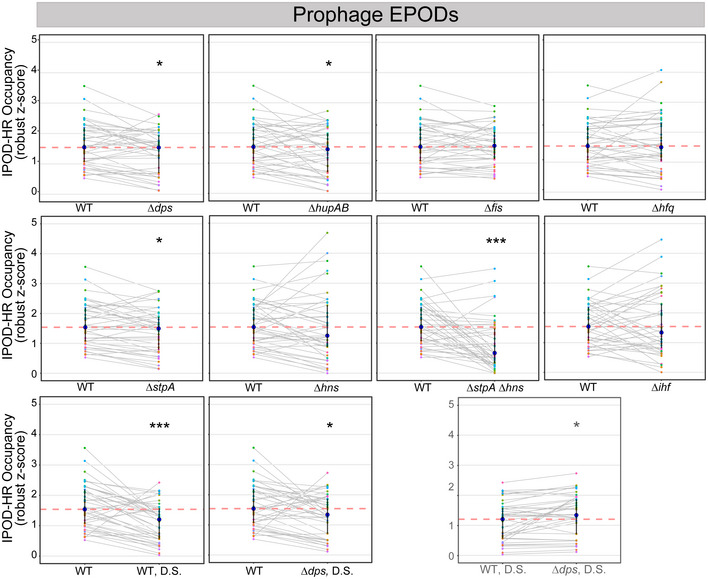
The binding locations and biological roles of NAPs have been difficult to define largely because of their promiscuous binding across the genome (Amemiya et al, 2021). Due to their propensity to bind DNA and their high abundance in the cell, we hypothesized that multiple NAPs contribute to EPODs. To examine the contributions of a range of E. coli NAPs to EPODs, we made single deletions of the most abundant E. coli NAPs (hns, stpA, fis, hfq, ihfAB, dps, and hupAB) and performed IPOD‐HR on the deletion strains. Since StpA is a known paralog of H‐NS and forms bridged filaments across DNA (Lim et al, 2012; Boudreau et al, 2018), we also created an hns/stpA double knockout and performed IPOD‐HR. As the NAP Dps is primarily expressed during stationary phase (Frenkiel‐Krispin et al, 2001; Nair & Finkel, 2004), we performed additional IPOD‐HR experiments on WT and ∆dps cells that were collected during deep stationary phase (defined in Materials and Methods). EPOD counts and coverage slightly varied across genotypes tested (Fig 2A and B), with the largest loss of coverage observed in ∆stpA∆hns. To examine shifts in EPOD locations, we again measured the symmetrized overlap distance (defined for Fig 1D) and performed hierarchical clustering (Fig 2C). The greatest changes in EPOD locations relative to the baseline condition (WT cells in exponential phase) occur within ∆stpA∆hns and ∆ihf, both of which cause profound changes in the profile of EPOD locations. The deep stationary phase samples cluster together, indicating similar shifts in EPOD locations. Interestingly, ∆hns and ∆hfq cluster together, perhaps suggesting a similar role in silencing at some EPODs. Several other NAP deletions show minimal effects on EPODs relative to WT cells, including deletions of hupAB, stpA, and dps (in exponential phase), indicating that at least under baseline conditions, these proteins do not contribute strongly to defining EPODs.
Figure 2. Loss of nucleoid‐associated proteins (NAPs) leads to changes in EPODs.
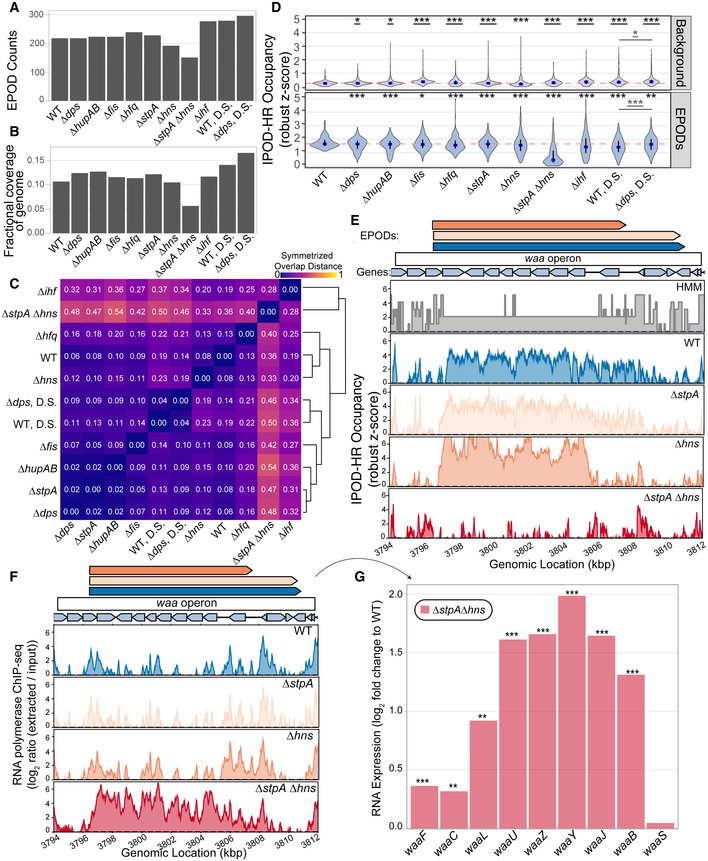
- Number of EPODs called in different NAP deletions. D.S. denotes genotypes where cells were collected in the deep stationary phase of growth.
- Fractional coverage of EPODs for each genotype across the genome.
- Symmetrized overlap statistic comparing each pair of samples in the NAP deletion dataset. The symmetrized overlap denotes similarity between EPOD locations, where a value of 0 = identical. Hierarchical clustering was performed to group like‐genotypes.
- Distributions of mean protein occupancy at WT EPOD boundaries and background. The blue dots denote the median and the black line displays the interquartile ranges in each condition. The dashed pink line represents the WT median. (*) indicate the Wilcoxon rank‐sum test P value comparing the change in pseudomedian versus WT for each condition that has been adjusted using the Benjamini and Hochberg method (against a null hypothesis of no difference in pseudomedians). The gray line denotes the same comparison between the D.S. conditions. Underlined (*)’s indicate a gain in the pseudomedian compared to baseline conditions, while no underline indicates a loss in the pseudomedian compared to baseline conditions. P value < 0.05 = *, < 0.005 = **, < 0.0005 = ***. Three biological replicates were used for the “WT, Rich” case and “WT (deep stat.)” case, and two biological replicates for the “WT, Min” and “WT (2), Rich” conditions.
- Protein occupancy over the waa operon. The quantile‐normalized robust z scores of the protein occupancy at each 5 bp are represented by the IPOD‐HR occupancy. The EPOD over the waa operon is lost in the ∆stpA∆hns condition which results in increased accessibility and RNA polymerase occupancy (Appendix Fig S3A).
- The loss in protein occupancy shown in (E) leads to increases in RNA polymerase occupancy across the waa operon.
- RNA‐seq analysis shows log fold change compared to WT of waa operon expression upon deletion of hns and stpA. (*) indicate q value < 0.05 = *, < 0.005 = **, < 0.0005 = *** (called using DeSeq2, as described in Methods).
Different EPODs are comprised of distinct combinations of NAPs
To identify the relative contribution of different NAPs in maintaining a “standard” set of EPODs present in WT cells during exponential phase growth, we calculated the average of the total protein occupancy signal across every EPOD location and other genomic regions for each genotype (Fig 2D). When comparing the WT median (pink dashed line) versus the deletion mutant medians (blue), NAPs contributing to protein occupancy at EPODs that are normally present during exponential growth phase display a statistically significant dip in occupancy. Consistent with our prior observations, the loss of occupancy at normal EPOD boundaries in ∆stpA∆hns cells is particularly profound. However, the hns single mutant shows only a minor loss of occupancy which indicates that StpA can largely compensate for its loss. Several other NAP deletions also showed significant drops in occupancy at the standard EPOD locations, often with a heavy lower tailed or even bimodal distribution of occupancy changes, suggesting that only some subset of EPODs were affected in each case. At the same time, in many cases, RNA polymerase occupancy at a subset of affected EPODs is observed to rise (Fig EV1A), indicating a de‐repression of some EPODs upon deletion of hupAB, hfq, hns, ihf, or (especially) in the stpA/hns double knockout. The impact of the double (stpA and hns) deletion is also demonstrated when looking at the association between a previously published H‐NS ChIP‐seq dataset (Kahramanoglou et al, 2011) with EPODs in each mutant, where we see that EPODs present in the double mutant lack H‐NS enrichment compared to background (Appendix Fig S3). Inspection of the patterns of both total protein occupancy and RNA polymerase occupancy across the waa operon provides an instructive example. Whereas single deletions of hns or stpA show a minor effect on the integrity of the waa EPOD, the ∆stpA∆hns cells show nearly complete loss of occupancy in this region (Fig 2E). At the same time, there is a concomitant gain in RNA polymerase occupancy and induction of RNA expression of waa operon genes (Fig 2F and G), demonstrating that H‐NS and StpA act jointly to maintain silencing of the waa operon. While either can compensate for the other, the loss of both silencers leads to substantial de‐repression of genes in this region.
Figure EV1. Loss of NAPs results in increases in RNA polymerase occupancy and decreases in overlapping EPODs.
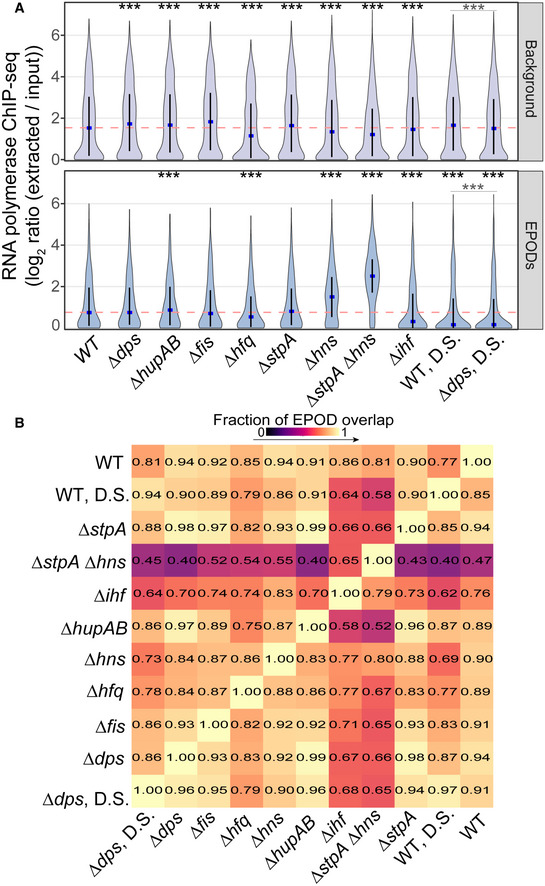
- Average RNA polymerase occupancy was calculated across intergenic regions within WT EPODs and background. Similar to Fig 2D, the blue dots denote the median and the black line displays the interquartile ranges in each condition. The dashed pink line represents the WT median. (*) indicate the Wilcoxon rank‐sum P value comparing the change in median versus WT for each condition that has been adjusted using the Benjamini and Hochberg method (against a null hypothesis of no difference in pseudomedians). The gray line denotes the same comparison between the D.S. conditions. P value < 0.0005 = ***. Data were averaged across 2 (hupAB, fis, stpA, ihf, dps), 3 (WT, hfq, WT DS, dps DS),or 4 (hns, stpA/hnS) biological replicates.
- Similar to Fig 1D, reading left to right: overlap of the relaxed set of EPOD calls (left) over the stringent set of EPOD calls (bottom).
Additionally, all NAPs exhibited changes in EPOD locations that impacted the overlap of called EPOD regions. We examined the fraction of EPODs contained in the relaxed threshold EPODs and found the overlap between EPOD locations among different genotypes dipped as low as 40% (Fig EV1B). Changes in protein occupancy compared to WT at all regions of the genome and downstream hierarchical clustering suggests a similar role of Hfq and H‐NS in silencing specific regions of the genome (Appendix Fig S4).
HMM categorization reveals distinct EPOD classes dictated by individual NAP profiles
Our data thus permit us to identify which NAPs regulate specific EPODs across the genome. To provide an automated high‐level classification of the regions across the genome specific to particular NAPs, we used our IPOD‐HR occupancy and RNA polymerase ChIP‐seq datasets across the NAP deletions and trained a six‐state hidden Markov model (HMM) that split the genome up into six classes (see Materials and Methods for details). We were able to identify three classes (2, 3, and 5) that were associated with EPODs, which exhibited significant enrichments of IHF motifs (Salgado et al, 2018), Hfq binding (see note in Materials and Methods), H‐NS binding (Kahramanoglou et al, 2011), and Fis binding (Kahramanoglou et al, 2011), as well as a low abundance of motifs for the known DNA methylases—Dam and Dcm (Salgado et al, 2018) (Table 1). In particular, HMM class 2 is especially strongly associated with H‐NS binding and likely represents H‐NS/StpA‐dependent EPODs (with IHF also apparently contributing). HMM class 5 is associated with high levels of Hfq and Fis binding and may represent EPODs comprised in part by these two factors. In addition, class 5 had a high abundance of transcription factor binding sites and promoters (Salgado et al, 2018), further implicating this class serving a regulatory role. HMM class 3 represents yet another category of EPODs that at present cannot be assigned. The utility of the HMM classification is apparent in providing a quick interpretation of the differences between EPODs. For example, the waa operon is almost entirely associated with HMM class 2 (Fig 2E), which we assign as an H‐NS filament. On the other hand, the borders of the EPOD instead fall into HMM class 3 or 5 and show loss of occupancy in an hns deletion strain even if stpA is intact (Fig 2E), whereas the class 2 region does not. Thus, we find that StpA and H‐NS together contribute to large protein regions across the genome, and are the main components silencing the O‐antigen biosynthesis pathway. At the same time, we observe the presence of two distinct classes of EPOD occupancy (class 3 and class 5) that appear largely H‐NS independent and likely represent different types of large‐scale repressive protein occupancy.
Table 1.
HMM class enrichments.
| Log2 ratio enrichment | Group‐level mean | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HMM Class | EPODs | TFs | Prom. | IHF | Dam | Dcm | H‐NS | Hfq | Fis | HU |
| 0 | −0.95 | −3.49 | −2.31 | −1.12 | 0.16 | 0.08 | 276.55 | −0.14 | 227.68 | 1.02 |
| 1 | −0.36 | −1.10 | −1.17 | −1.01 | 0.09 | −0.04 | 340.21 | 0.19 | 231.26 | 1.07 |
| 2 | 1.36 | 1.22 | −0.054 | 1.83 | −0.65 | −0.78 | 1230.42 | 0.42 | 153.19 | 0.88 |
| 3 | 1.66 | −2.52 | −4.24 | −1.42 | −0.15 | −0.19 | 608.69 | 0.06 | 196.31 | 0.92 |
| 4 | −1.53 | −4.03 | −2.41 | −3.68 | 0.14 | 0.30 | 268.33 | −0.36 | 220.40 | 0.93 |
| 5 | 0.06 | 2.14 | 1.70 | 2.21 | −0.39 | −0.35 | 437.99 | 0.77 | 238.11 | 1.08 |
| Genome average | ‐‐‐‐ | ‐‐‐‐ | ‐‐‐‐ | ‐‐‐‐ | ‐‐‐‐ | ‐‐‐‐ | 422.24 | 0.03 | 217.63 | 0.99 |
Log2 ratio enrichment: The ratio of the number of EPODs or motifs in a given HMM class to the total number of EPODs or motifs was calculated for each HMM class. A chi‐squared test was performed, and all categories were significantly associated with each class; values underlined had a P‐value < 0.05. Group‐level mean: The 500‐bp rolling mean for the binding of each NAP was used to calculate the group‐level means for across each HMM class and compared with the overall average for the genome. Permutation based P‐values were calculated comparing each class versus the background. The values underlined had a P‐value < 0.05.
It is essential to note that the HMM classes themselves are built solely off of changes in total protein occupancy and RNA polymerase occupancy in the presence of various NAP deletions. Thus, these data cannot by themselves distinguish between direct binding of the indicated NAPs, versus indirect effects on protein occupancy occurring downstream of the NAP deletions. However, the enrichment and depletion statistics shown in Table 1 reflect direct measurements of the binding profiles of the indicated factors and thus provide strong independent evidence for enrichments of binding of specific NAPs at the indicated HMM classes (i.e., H‐NS at classes 2 and 3, IHF and Hfq at classes 2 and 5, Fis at class 5).
Metabolic pressures induce changes in EPODs
Many EPODs overlap operons involved in metabolic pathways and thus silence pathways that may not be actively used in the cell under the conditions that we studied. For example, during growth in glucose rich defined media we observed an EPOD, associated with HMM class 2 (H‐NS filament), overlapping the idnDOTR operon, specifically in the promoter of the operon. The idn operon is essential for the metabolism of carbon sources such as idonate and 5‐ketogluconate, which are not present under typical laboratory conditions. In particular, the idnD gene codes for the enzyme L‐idonate 5‐dehydrogenase, which catalyzes the oxidation of L‐idonate to 5‐ketogluconate (Bausch et al, 1998, 2004; Gómez et al, 2011). The idnDOTR operon and idnK are known to be transcriptionally regulated by CRP, IdnR, GlaR, and GntR, but, to our knowledge, no connection to regulation by NAP occupancy has been described (Barh & Azevedo, 2017). Upstream of the idnDOTR operon, there is a 215‐bp regulatory region that lies in between idnK and idnD (Bausch et al, 2004). In this region, there is a single putative IdnR/GntR‐binding site, CRP binding site, and an UP element (Bausch et al, 2004). Bausch et al (2004) previously showed that induction of this pathway can occur due to exposure to L‐idonate or 5‐ketogluconate (KDG). The local positive regulator, IdnR, is activated by KDG (Bausch et al, 1998), and promotes the induction of the rest of the operon. Following exposure to KDG in the absence of glucose, IdnT enables uptake of the carbon source, IdnD (as previously mentioned) catalyzes the reversible reduction of L‐idonate to 5‐ketogluconate, IdnO catalyzes the oxidation of 5‐ketogluconate to d‐gluconate, and IdnK catalyzes the phosphorylation of d‐gluconate which then proceeds through the Entner–Doudoroff pathway to be metabolized (Bausch et al, 1998). Since the idn operon appears to be controlled by three local and one global regulators, the question arises of what additional regulatory role might be provided by the apparent silencing EPOD covering the idn promoter under standard defined media conditions used here.
To explore the mechanisms of silencing at the idnDOTR operon, we first referred to our NAP deletion dataset to see whether specific NAP(s) silenced the operon. Data from a previous H‐NS ChIP‐seq dataset (Kahramanoglou et al, 2011) plus the classification of this region as a type 2 EPOD in our HMM suggested the presence of H‐NS bound to the idn promoter region. We performed RNA‐seq in the ∆stpA∆hns background compared with the parental cells, and discovered that H‐NS and StpA indeed repress expression of the idnDOTR operon (Fig EV2A and B). We leveraged this knowledge to address whether we could induce changes in protein occupancy at the EPOD covering the idn promoter by performing a carbon source shift experiment outlined in Fig 3A (further explanation in Materials and Methods). Briefly, cells were grown in minimal media with 0.2% glucose (M9 Min Glu), shifted to minimal media with 0.2% KDG as a sole carbon source (M9 Min KDG), and shifted back to M9 Min Glu. In all conditions, cells were collected at an OD600 of ~0.1 for both IPOD‐HR to examine changes in EPODs and RNA‐seq for changes in expression. Notably, there was a severe lag for growth in KDG. We found that growth in KDG led to a reduction in protein occupancy and loss of the EPOD within the idnDOTR operon promoter (Fig 3B; raw data represented in Fig EV2B). Upon shifting the cells back to glucose, both the original pattern of protein occupancy and the EPOD were restored (Fig 3B). The consistency with the previously reported H‐NS binding (Kahramanoglou et al, 2011), coupled with our own findings that protein occupancy is reduced while there is an induction of idnDOTR operon genes in the stpA and hns double mutant (Fig EV2B), further supports our hypothesis that H‐NS is a major contributor to the suppression of the idnDOTR operon (Fig 3B). The loss of EPOD occupancy in the KDG condition was accompanied by an induction of expression of the idnDOTR operon, and repression when EPOD occupancy was restored upon the return to glucose as a carbon source (Fig 3C). Due to the long lag in growth during the transfer from glucose to KDG as a carbon source, we examined the correlation between the expression of all genes in the three conditions to see whether there were broad changes in expression when cells were forced to metabolize an exotic carbon source (Fig 3D, top panel). The Spearman correlation was extremely high when comparing all conditions (≥ 0.9), suggesting that changes are localized to the idn operon and the small set of other genes specifically regulated in response to the KDG carbon source. Similarly, the symmetrized overlap distances comparing the variation in EPOD locations across conditions were low, again supporting the notion that changes are specific to the induced operon (Fig 3D, bottom panel) and that no global rearrangement of protein occupancy occurs during growth on KDG.
Figure EV2. H‐NS and StpA mediate silencing of the idn operon.
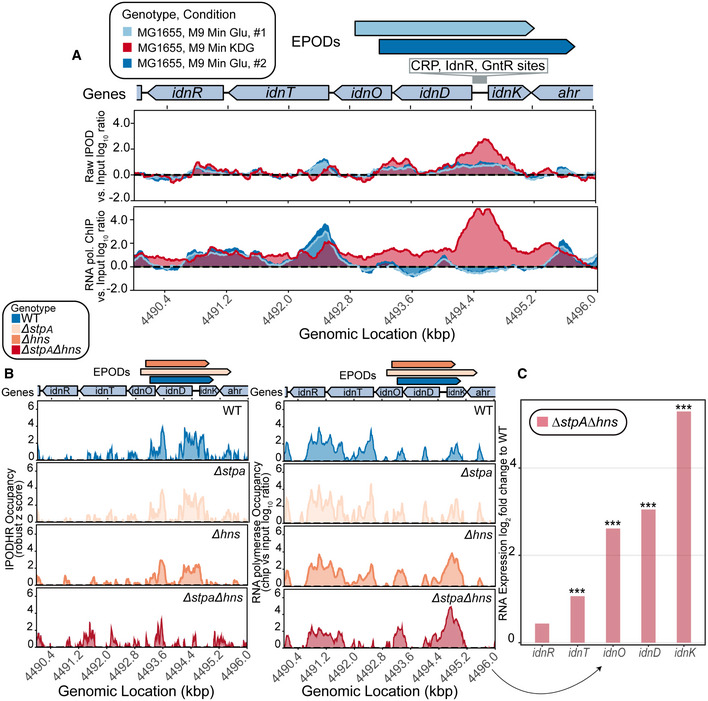
- Raw IPOD and RNA polymerase ChIP‐seq (both versus input log10 ratios) over the idn operon.
- IPOD‐HR was performed in rich defined medium (RDM) supplemented with glucose and exhibits a loss of occupancy in the deletion of both hns and stpA, as well as increased RNA polymerase occupancy.
- RNA‐seq was performed in parallel and shows log2 fold change compared to WT of idn operon expression upon deletion of hns and stpA. (*) indicate adjusted P‐value < 0.0005 = *** (calculated using DeSeq2 as described in Methods).
Figure 3. Changes in EPODs are induced in specific conditions.
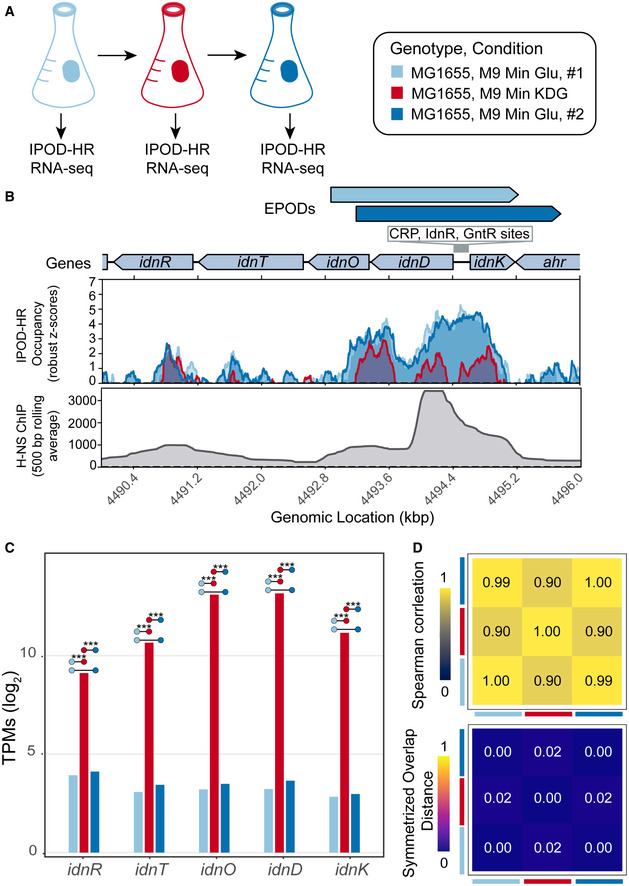
- Experimental overview. WT, MG1655 cells were grown in M9 minimal media with 0.2% glucose, samples were collected at mid log phase of growth (OD600 ~0.2) for RNA‐seq and IPOD‐HR. The cells were back diluted to an OD600 of ~0.1 in M9 minimal media with 0.2% 5‐Keto‐D‐gluconic‐acid (KDG), grown to an OD600 of ~0.2, collected for RNA‐seq and IPOD‐HR. In the final shift, the cells were back diluted to an OD600 of ~0.003 in M9 minimal media with 0.2% glucose, grown to OD600 of 0.2 and collected for RNA‐seq and IPOD‐HR. Two biological replicates were performed.
- Protein occupancy over the idn operon for each condition (colors are denoted in (A)). The quantile‐normalized robust z scores of the protein occupancy at each 5 bp are represented by the IPOD‐HR occupancy. There is a large loss in protein occupancy when cells are shifted to KDG, leading to the loss of the called EPOD. Protein occupancy is restored once cells are grown in the second glucose condition. The 500bp normalized average of previously published H‐NS ChIP‐seq (Kahramanoglou et al, 2011) exhibits high H‐NS binding on the idnD promoter region.
- To examine the expression of the idn operon at each shift, RNA‐seq was performed. RNA‐seq expression estimates log2‐scaled for idn operon genes for WT cells grown in each condition (as colored in (A)). Comparisons are denoted with colored dots with significance stars representing the adjusted P‐value where (***) signify q‐values < 0.0005 (called using DeSeq2, as described in Methods).
- Spearman correlations are represented with the heatmap comparing the whole‐transcriptome expression profiles in each condition, where identical expression values for every gene show a Spearman correlation of 1. The symmetrized overlap distance was calculated for all EPODs for each condition, where a value of 0 is identical. The colored squares on the sides of the heatmaps denote the condition (following the colors shown in (A)).
Given the sophisticated regulatory logic implemented at the idn promoter by a combination of local (GlaR, GntR, IdnR) and global (CRP) regulators, it is unclear what additional function is played by the apparently repressive EPOD covering this region. Drawing inspiration from the behavior of chromatin modifications in eukaryotes (e.g., Francis & Kingston, 2001; Kundu et al, 2007; Lagha et al, 2017; Palozola et al, 2019), we hypothesized that one function of EPODs could be to facilitate transcriptional memory. We thus performed head‐to‐head competition experiments in KDG‐containing and glucose‐containing media comparing the growth of cells that have been exposed to KDG (Fig 4A). We utilized MG1655 lacZ::cat cells as a reference strain in order to enable colony counting on MacConkey‐lactose plates as a read out for the competitions. WT (MG1655 lacZ +) cells were exposed to M9 minimal media + KDG for 48 h, back diluted and grown in M9 minimal media + glucose. During these growth periods, WT cells and lacZ::cat cells that had been grown in + glucose overnight were back diluted and mixed together into a tube containing KDG or glucose as carbon sources. Serial dilutions were plated onto MacConkey‐lactose plates immediately after mixing and after 48 h of growth for each carbon source condition (Fig 4A).
Figure 4. EPODs mediate transcriptional memory.
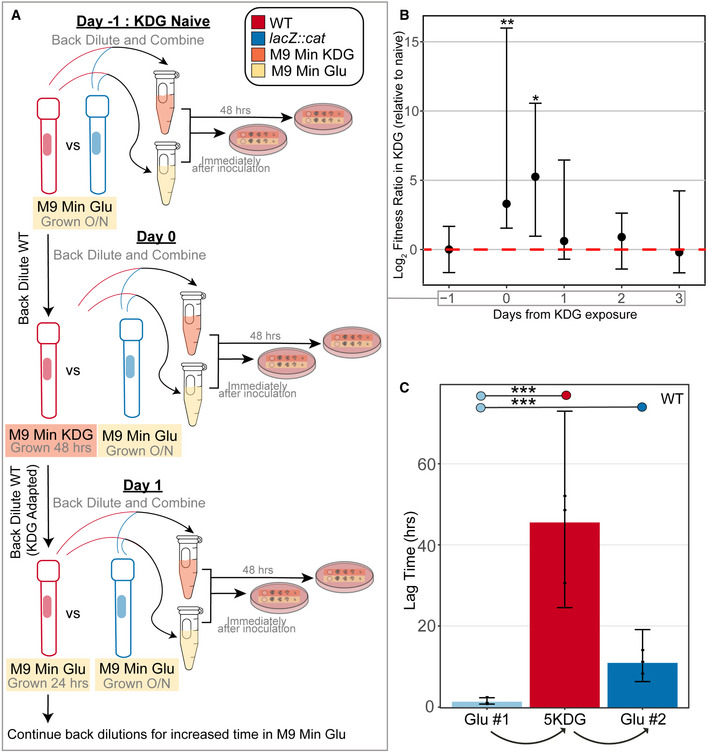
- Schematic of KDG competition experiments. Day −1: both the WT (red) and lacZ::cat (blue) are grown overnight (O/N) in M9 Min Glu. They are back diluted together in M9 Min Glu (yellow) and M9 Min KDG (orange). Serial dilutions of each media condition were spotted both immediately after inoculation and after 48 h of growth at 37°C. Day 0: The WT cells grown O/N in glucose from Day 0 were back diluted and grown in M9 Min KDG for 48 h—these cells are now “KDG Adapted”. These cells were then competed against fresh lacZ::cat cells that were grown in M9 Min Glu O/N. A mixture of both of these cells were placed in M9 Min Glu and M9 Min KDG, and spotted as previously described in Day 0. Day 1: The WT “KDG Adapted” cells grown in M9 Min KDG for 48 h were back diluted into M9 Min Glu and grown for 12 or 24 h (in separate cultures). As with Day 0, these cells were then competed against fresh lacZ::cat cells that were grown in M9 Min Glu O/N: A mixture of these cells was placed in M9 Min Glu and M9 Min KDG, and spotted as previously described in Day 0, and recorded as Day 0.5 (12 h outgrowth in M9 Min Glu) and Day 1 (24 h outgrowth in M9 Min Glu). The subsequent time points shown in (B) indicate the time KDG‐adapted cells were grown in M9 Min Glu after they were KDG adapted (i.e., Day 2 = 48 h, Day 3 = 72 h; fresh back‐dilutions were performed every 24 h from the previous M9 Min Glu culture). All later time points follow the same workflow as that shown for Day 1, beginning from cells that had been passaged additional times in M9 Min Glu.
- Difference in Log2 ratio of fitnesses of the KDG‐exposed cells with unexposed lacZ::cat (Δλ; see Materials and Methods) relative to that observed prior to KDG exposure, given as a function of time since exposure to KDG. Day −1 corresponds to the day before KDG exposure, Day 0 is cells taken immediately after growth in KDG, and subsequent time points reflect different durations of growth in M9 Min Glu prior to competition (see (A)). Points show medians across replicates (3 biological replicates for 0.5 days, 6 biological replicates for all other time points); error bars show 95% confidence intervals for the difference relative to the Day −1 time point, calculated using the R wilcox.test function. Significance was assessed using a Wilcoxon rank‐sum test comparing the distribution at each time point to the −1 day (naive) time point: *P < 0.05; **P < 0.005. Individual data points are shown in Appendix Fig S5.
- Lag times for cells grown in glucose, shifted to 5KDG, and then shifted back to glucose. (***) is defined as where the mean posterior probability of difference > 0.999, assessed using a Bayesian model (see Materials and Methods for details); data points show the values obtained for three biological replicates. Errors bars show 95% credible intervals.
As shown in Fig 4B, we found that immediately after growth in KDG media, the KDG‐exposed cells showed strongly increased fitness during competition in KDG relative to the KDG‐naive cells (Day 0). The advantage of the KDG‐exposed cells was lessened, but still significant, after 12 h of outgrowth in glucose minimal media (Day 0.5) and then strongly diminished by 24 h (Day 1) and subsequently absent. We interpret the very strong initial competitive advantage of cells taken immediately from KDG as reflecting a cellular state directly adapted to growth in KDG (including accumulations of the IdnO/IdnT proteins), whereas the persistent advantage of the previously KDG‐exposed cells after outgrowth in glucose demonstrates a memory effect where the cells more readily respond to the presence of KDG as a sole carbon source upon repeated exposure despite the transcriptional regulatory state having reset to be virtually indistinguishable from the original round of growth in glucose minimal media prior to the second 5KDG challenge (Fig 3). Given the long outgrowth between the 0 and 0.5 day 5KDG challenges (the KDG‐exposed cells used in the competitions underwent no fewer than 8.0 doublings in M9 Glu between the 0 and 0.5 day time points; count data are given in Appendix Table S1), it is expected that essentially all of the Idn proteins would have been diluted to irrelevant levels. While additional direct evidence is needed, our findings are consistent with the possibility that the structure of the EPOD in this region is such that transcriptional initiation is faster upon second induction within some time window after an initial induction, providing a transcriptional memory that facilitates responses to repeated stresses. Given that EPOD occupancy on the idn promoter re‐forms rapidly after transfer back to M9 Glu media (Fig 3), any “memory” must be implemented by molecular information beyond the simple presence or absence of protein binding. This response may be mediated by formation of bridged versus unbridged H‐NS filaments in this region, by post‐translational modification of H‐NS comprising the EPOD (analogous to the histone code (Moazed, 2011)), or binding of unknown accessory factors. Further investigation of the kinetics of EPOD re‐formation after induction is lifted, and indeed the identities of proteins that bind to the idn region immediately after EPOD re‐formation, will be required to distinguish between the possible mechanisms noted above.
We also note that the competitive fitness in glucose of cells immediately after KDG exposure was particularly poor, and indeed, in a separate set of monoculture experiments, we observed that the lag time for cells transferred from stationary phase in 5KDG media to glucose media was significantly longer than for the transfer from glucose media to glucose media (Fig 4C; note that the pregrowth procedures and growth conditions differ between the competition experiments and the monocultures in Fig 4C, likely accounting for the even more pronounced post‐KDG lag observed in the latter case). One possible explanation for these findings would be that some of the gene products induced to metabolize 5KDG may themselves be detrimental under normal growth conditions, and thus, another role of the EPOD at the idn promoter may be to ensure tight silencing of these genes until they are needed. It is also possible, however, that some other aspect of global physiological state (e.g., stress‐induced changes in transcription or translation), rather than idn gene expression, are responsible for the observed lag.
Multiple NAPs contribute to the silencing of prophages
In addition to metabolic processes, we also found that genes within EPODs were overrepresented in Gene Ontology (GO) terms associated with annotated prophages (Freddolino et al, 2021a). Across the E. coli genome, there are a number of xenogeneic elements that have been integrated and can be potentially toxic to the cell, although maintenance of these elements can also be beneficial, as they can promote resistance in the face of antibiotics (Wang et al, 2010). H‐NS is known to silence cryptic prophages and is likely to contribute to silencing the majority of horizontally acquired DNA in E. coli (Hong et al, 2010; Wang et al, 2010). IPOD‐HR successfully resolved known H‐NS silenced prophages (Fig 5A), where reductions in protein occupancy and corresponding increases in accessibility to RNA polymerase are observed in an hns knockout strain. For example, we observed a decrease in protein occupancy along the promoter and gene body regions of sieB, a gene in the Rac prophage that controls phage superinfection (Faubladier & Bouché, 1994), and corresponding increases in RNA polymerase occupancy, in the hns knockout strain. This indicates that while EPOD binding is not completely absent in the hns mutant across this particular prophage, there is still a quantitative reduction in silencing protein occupancy and corresponding transcriptional de‐repression. As noted above, not all EPODs correspond to H‐NS binding; therefore, it is likely that not all silenced prophages are located in H‐NS repressed regions. To examine the role of other nucleoid‐associated proteins in silencing prophages, we calculated the mean protein occupancy across WT EPODs that overlap prophages, and determined how those occupancies changed upon deletion of different NAPs. Overall median decreases in occupancy across prophage‐containing EPODs are observed upon deletion of dps, hupAB, hfq, stpA, hns, stpA/hns, ihf, and in both deep stationary phase samples compared to WT (occupancy distributions and statistical significance calling are shown in Fig 5B). In addition, as shown in Fig EV3, many individual EPODs show substantial changes in occupancy even in cases where the median of the distribution is unchanged.
Figure 5. Nucleoid‐associated proteins contribute to protein occupancy at EPODs that contain prophages.
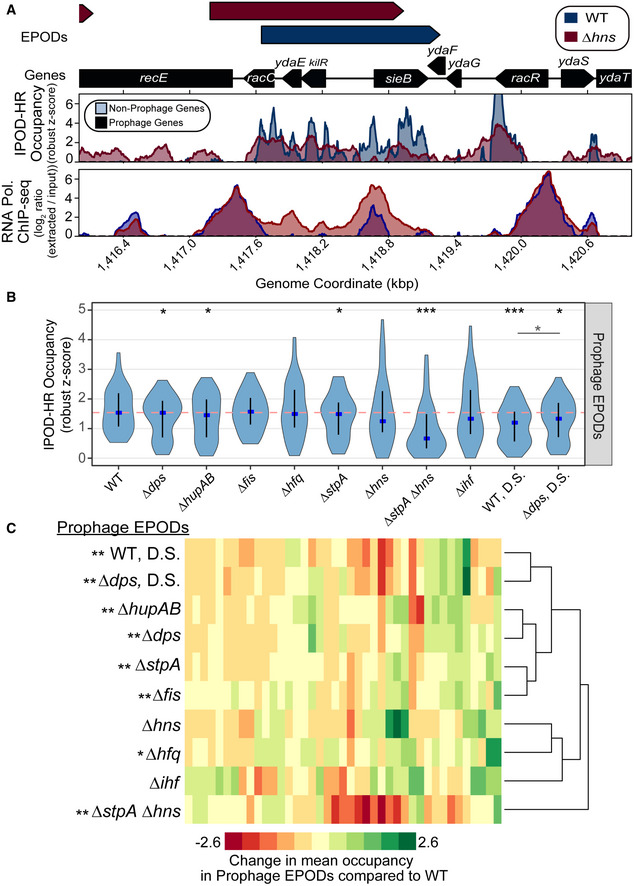
- IPOD‐HR occupancy and RNA polymerase occupancy over a known H‐NS silenced prophage in WT (blue) and ∆hns (red) cells. The quantile‐normalized robust z scores of the protein occupancy at each 5 bp are represented by the IPOD‐HR occupancy.
- The mean protein occupancy (IPOD‐HR occupancy) was calculated over WT EPOD locations that contain prophages (41 EPOD locations). As in Fig 2D, the blue dots denote the median and the black line displays the interquartile ranges in each condition. The dashed pink line represents the WT median. (*) indicate significance assessed via a Wilcoxon rank‐sum test comparing the change in pseudomedian versus WT for each condition (against a null hypothesis of no difference in medians), after application of a Benjamini–Hochberg correction for multiple hypothesis testing. The smaller horizontal line denotes the same comparison between the D.S. conditions. Q value < 0.05 = *, < 0.005=**, < 0.0005=***. Data were averaged across 2 (hupAB, fis, stpA, ihf, dps), 3 (WT, hfq, WT DS, dps DS), or 4 (hns, stpA/hns) biological replicates.
- Mean protein occupancy was calculated across all WT EPOD locations that contain annotated prophages. The change in mean protein occupancy compared to WT was calculated for each condition, where anything negative is a loss in occupancy compared to WT. Hierarchical clustering was performed to examine which genotypes clustered together and were more similar. Permutation tests comparing the change in occupancy over EPODs containing prophages were performed against the rest of the genome. P value < 0.05 = *, < 0.005 = ** indicate a negative mean change in occupancy compared to WT.
Figure EV3. Global protein occupancy across prophage EPODs changes with deletion of NAPs.
The average IPOD‐HR occupancy (robust z scores) across each of the 41 prophage EPODs of WT and NAP deletions. Each colored dot represents an individual EPOD. Gray lines connect the same EPOD in WT (or WT D.S.) versus NAP deletion to appreciate the change in occupancy for that particular EPOD. The pink dashed line indicates the median of the WT comparison. The dark blue dot indicates the median of the genotype and transparent lines display the interquartile ranges. The summary of these data is displayed as violins in Fig 5B. As shown in Fig 5B, (*) indicate the Wilcoxon rank‐sum P value comparing the change in pseudomedian versus WT for each condition that has been adjusted using the Benjamini and Hochberg method (against a null hypothesis of no difference in pseudomedians). The gray line denotes the same comparison between the D.S. conditions. P value < 0.05 = *, < 0.0005 = ***.
Inspired by our findings above that only the combined deletion of hns and stpA can reveal large changes in occupancy of many H‐NS silenced EPODs (Fig 2E–G), the minor loss of occupancy in some genetic backgrounds led us to investigate whether there are certain NAPs that work together to silence specific toxic elements. We examined the change in occupancy from each condition versus WT at WT EPODs that overlap prophages and performed hierarchical clustering analysis (Fig 5C) to identify regulators that play similar roles. Interestingly, ∆hns and ∆hfq are clustered together, as are ∆stpA and ∆fis, suggesting prophages are silenced by multiple factors and NAPs. The association of multiple NAPs to specific prophage EPODs may indicate that they act cooperatively, for instance in the case of H‐NS and StpA, or independently, which in many cases remains to be determined. Our findings implicate the well‐documented RNA chaperone Hfq (Nagai, 2002; Valentin‐Hansen et al, 2004) (which has also been shown to bind and compact dsDNA (McQuail et al, 2020; Orans et al, 2020)) as a novel silencer of prophages at the level of protein occupancy across large genomic regions. Since H‐NS and StpA are paralogs that bind to similar regions of the chromosome, we speculated that Hfq and Fis might play similarly complementary roles to each other in terms of silencing some prophages despite their lack of homology or structural similarity. In addition, from our HMM analysis, both Fis and Hfq binding are enriched in HMM class 5 (Table 1), again suggesting a link between the silencing roles and binding locations of Fis and Hfq.
Fis and Hfq are required for cell viability in a prophage‐dependent manner
Building off of the noted co‐occurrence of Fis and Hfq binding in HMM class 5 EPODs, we screened the genome for EPODs that contained prophages that lost global protein occupancy upon deletion of fis or hfq individually (e.g., Fig 6A). RNA‐seq analysis revealed that the genes within the prophage region depicted in Fig 6A were de‐repressed in both a ∆hfq and ∆fis background, despite only minimal losses in total protein occupancy (Fig 6B). While Fis and Hfq are both NAPs, highly expressed, and bind promiscuously across the genome, they are not known to silence genes via the formation of densely occupied large‐scale binding regions. Fis, while known more in the literature as a transcriptional activator (Bokal et al, 1995; Appleman et al, 1998), can inhibit transcription and act as a repressor in certain contexts (Cho et al, 2008; Chintakayala et al, 2013). Hfq is well known as a RNA chaperone (Updegrove et al, 2016) and can bind nucleic acids (including DNA) across the faces of the wheel‐like homohexamer (Orans et al, 2020). Therefore, we were not surprised to find that comparing the log fold change of expression (relative to WT) of ∆hfq and ∆fis cells were not highly correlated among all genes (Fig 6C). However, when examining only prophage genes, most genes were induced in one or both of the knockout genotypes (Fig 6D). We quantified the number of genes that were up or down in each genotype or both using the quadrant map in Fig 6E. We applied this map and counted the rate ratios comparing all genes versus prophage genes in each quadrant. The rate ratios of all genes versus prophage genes in the quadrant which represented induced expression in both ∆hfq and ∆fis was significantly higher in prophage genes (Fig 6F). Thus, prophage genes are specifically and significantly enriched among the set of genomic loci that are repressed by both Fis and Hfq, suggesting that Fis and Hfq bind and silence similar prophages, and also that a substantial fraction of all prophages in fact appear to have strongly Fis/Hfq‐dependent occupancy.
Figure 6. Loss of Fis and Hfq is lethal in a prophage‐dependent manner.
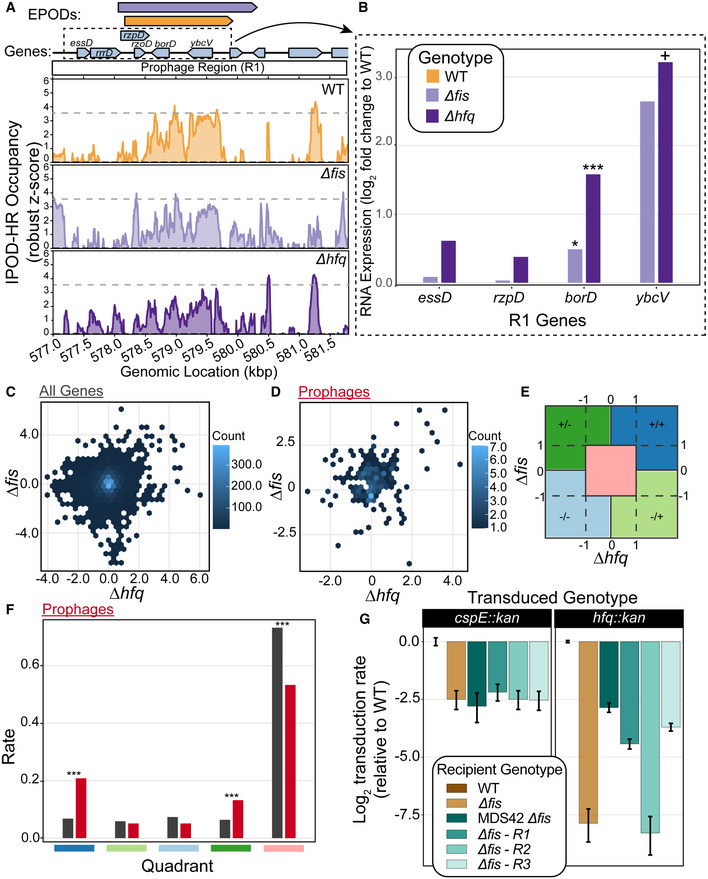
- Example prophage region that is annotated with the Fis‐ and Hfq‐associated HMM class 5 in our genome‐wide HMM classification (Table 1). The quantile‐normalized robust z scores of the protein occupancy at each 5 bp are represented by the IPOD‐HR occupancy. Prophage genes are highlighted with a red box. The major peak associated with WT IPOD‐HR occupancy is represented in a gray dashed line as a reference to compare IPOD‐HR occupancy in the other genotypes. Modest loss of protein occupancy was observed at the same prophage‐containing EPOD for ∆fis (light purple; see color key in (B)) and ∆hfq (dark purple; see color key in (B)) conditions compared to WT (gold).
- RNA‐seq of WT, ∆fis, and ∆hfq were performed. The log fold change compared to WT was calculated at prophage genes contained in the dashed box in (A). Induction of prophages across the region where loss in occupancy is observed. (*) indicate the adjusted P‐value: < 0.10 = +, < 0.05 = *, < 0.005 = **, < 0.0005 = *** (calculated using DeSeq2 as described in Methods).
- The log fold change of all genes for ∆fis and ∆hfq are shown in a hexbin plot. Counts for each gene transcript contained in one bin are denoted with the counts bar.
- The log fold change of all prophage genes for ∆fis and ∆hfq are shown in a hexbin plot.
- Outline of quadrant map to calculate the number of genes that fall within each quadrant for (F). The symbols represent log fold changes compared to WT in ∆fis / ∆hfq. For instance, +/+ denotes a positive log fold change in ∆fis and ∆hfq, −/+ denotes negative log fold change in ∆fis and positive in ∆hfq.
- Rate ratios of all genes (black) and prophage genes (red) in each quadrant outline in (E), showing a higher rate of genes that resided in the +/+ category, indicating that many prophages are de‐repressed in both ∆fis and ∆hfq. (*) indicate the P‐value calculated from testing the null that the rate ratios are the same. P‐value < 0.05 = *, < 0.005 = **, < 0.0005 = *** (calculated using DeSeq2 as described in Methods).
- P1 vir transduction experiment to test the viability of ∆fis and ∆hfq. Strain identities are indicated in the box. Number of transductions was counted on LB + Kan plates; all efficiencies are relative to WT, and thus the log‐scaled relative transduction rate for the WT itself is 0 by definition. ‐R1 indicates that the prophage region in (A) was deleted to test whether the loss of prophages silenced by Fis and Hfq restored viability of a ∆fis∆hfq genotype. R2 and R3 were other regions in the genome that contained prophages that appeared to have Fis/Hfq‐dependent EPODs. Plotted values are mean efficiencies across replicates, with error bars showing a 95% credible interval obtained via Bayesian inference, assuming the replicate‐level colony counts are Poisson distributed with a (conjugate) Gamma(0,0) prior; all log ratios (including error bounds) are plotted relative to the mean WT value. Data obtained from 5 biological replicates for hfq::kan transductions and 4 biological replicates for cspE::kan transductions.
Since the expression of genes from lysogenized bacteriophages can be toxic to the cell even if they are no longer able to form replication‐competent virions (particularly if a lytic operon is induced), we asked whether the combined loss of hfq and fis would more strongly impact cell physiology. We performed P1 transduction experiments in a WT, MG1655 background and a ∆fis background, where we attempted to delete the genomic copy of hfq. Interestingly, there was a dramatic loss of transduction efficiency in the ∆fis background, and we were not able to create the ∆hfq ∆fis mutant (Fig 6G). The transduction efficiency for hfq::kan dropped more than 100‐fold in the ∆fis background, and the very small number transductants that did form colonies could not be propagated upon restreaking. To test whether the deletion of fis impacts transduction efficiency as a whole, we attempted the same experiment deleting cspE, another gene that has not been associated with prophage silencing, and did not observe a similarly dramatic loss in transductants, with simple loss of fis leading to only a ~4‐fold loss in transduction efficiency; thus, we infer that loss of hfq and fis is synthetic lethal. To further test our hypothesis of synthetic lethality between fis and hfq, we attempted to complement the combined loss of fis and hfq using a copy of hfq on a temperature sensitive plasmid. We thus cloned hfq and its native promoters on a plasmid with a temperature sensitive origin of replication (Link et al, 1997). The plasmid was placed into WT and ∆fis cells, and the genomic copy of hfq was deleted using P1 vir transduction (see Materials and Methods). Cells were grown at permissive temperature (30°C), and spot titers were performed on LB and LB + chloramphenicol plates to measure CFUs of the culture and presence of the plasmid. Cultures were then shifted to 42°C, which prevents plasmid replication, and thus induces dropping of the plasmid containing hfq. After 8 h of growth at 42°C, spot titers were performed to assess CFUs. We found again that the combination of ∆hfq∆fis was not viable (Fig EV4), supporting our conclusion of synthetic lethality. We hypothesized that the expression of prophages silenced by Fis and Hfq led to the inviability phenotype. To test this hypothesis, we utilized the strain MDS42 (Posfai, 2006), which lacks the mobile elements and prophages in the E. coli K12 genome. We placed the temperature sensitive plasmid containing hfq in MDS42 and MDS42 ∆fis cells and performed the same temperature shift experiment as described above. Removal of the prophages from the genome restored viability (Fig EV4). We also observed a substantial rescue of transduction efficiency, which reached the same level in the ∆fis background as that of the control transduction with cspE::kan (Fig 6G); thus, the lethality of the ∆hfq∆fis is dependent on the presence of at least one of the regions lost in MDS42. Since 42 large regions are deleted from MDS42, we asked whether we could identify specific prophage regions that led to inviability. Since there is loss in occupancy in both ∆hfq and ∆fis in the Fig 6A region (R1:564815‐585633, within the DLP12 prophage; Table 2), we deleted only R1:564815‐585633, and found a partial rescue of transduction efficiency (Fig 6G). We found another region that met the same criteria (R3, within the Qin prophage; Table 2) and also saw rescuing effects (Fig 6G). Another region that contained prophages, but did not dip in occupancy in both genotypes, did not impact viability (R2, within the Rac prophage; Table 2; Fig 6G). Thus, we were able to define regions that contribute to viability, and hone in on specific prophages that are silenced by Fis and Hfq and, in the absence of repression by those two NAPs, prevent cell growth. In particular, loss of either of two prophages (DLP12 or Qin) was sufficient to restore the viability of a ∆hfq ∆fis double mutant. This novel interaction defines a new role for NAPs in regulating the expression of prophages, implicating additional E. coli NAPs in the establishment of defense mechanisms against horizontally acquired DNA. It is especially notable that the NAPs apparently involved in this case are not the classic xenogeneic silencers (H‐NS/StpA), but rather, appear to represent a separate and complementary system for silencing potential lethal prophage gene expression.
Figure EV4. Growth deficiency of Δfis Δhfq cells.
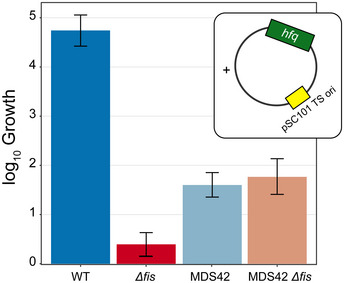
WT, MDS42, Δfis, and MDS42 Δfis cells containing a temperature sensitive plasmid with hfq had their genomic copy of hfq deleted. Cells were grown in a permissive temperature (30°C), and then shifted to a non‐permissive temperature for plasmid replication (42°C), thus removing hfq as the plasmid is dropped. The log10 fold change in CFU is displayed. Δfis cells were unable to grow with the loss of the temperature sensitive hfq plasmid, however, upon deletion of mobile elements and prophages from the genome (MDS42 strain background), viability was restored. Data show fitted values plus 95% credible intervals for a Bayesian analysis, in which the plate counts themselves were treated as Poisson random variables, with rate parameters arising from an initial cell concentration in cells/ml (at the permissive temperature) and then a post‐treatment scaling factor applied to that initial concentration. Both parameters were fitted on a log10 scale, with a Uniform (0,15) prior on the initial cell concentrations and Uniform (−10,10) prior on the treatment effect. The treatment effect parameter is plotted as “log10 growth”, reflecting the observed growth at 42°C. Fits were based on three biological replicates for each genotype and were performed using pymc3 (Salvatier et al, 2016).
Table 2.
MDS42 regions containing prophages.
| Region # | Coordinates | Genes | Gene functions |
|---|---|---|---|
| 1 | 564815‐585633 | essD, ybcS, rzpD, rzoD, borD, ybcV, ybcW, nohB | DLP12 prophage, putative prophage endopeptidase, lysozyme |
| 2 | 1400247‐1482201 | lar, recT, ydaQ, ydaC, intR | Rac prophage, recombinase, DNA renaturation |
| 3 | 1627517‐1652838 | ynfO, ydfO, gnsB, ynfN, cspI, ydfP, hokD | Qin prophage, cold shock, toxin anti‐toxin system |
Extended protein occupancy domains silence horizontally acquired DNA across diverse species
Because it relies only on elementary physico‐chemical principles rather than specific affinity reagents, IPOD‐HR is an approach that could be implemented in a wide variety of bacterial species. To further our understanding of conserved features that regulate bacterial genome architecture, we investigated whether a distantly related bacterial species contained EPODs. We performed IPOD‐HR on the Gram‐positive Firmicute Bacillus subtilis (B. subtilis)—a soil‐dwelling bacterium that has the ability to enter a number of developmental platforms upon nutrient deprivation or other environmental stressors, including the formation of desiccation resistant endospores, biofilm formation, genetic competence, and swimming/swarming motility phenotypes (Albano et al, 2005). We performed IPOD‐HR in B. subtilis strain PY79 and found multi‐kb regions of protein occupancy (EPODs) spanning genes that function in a number of metabolic pathways, suggesting a feature conserved with E. coli (Fig 7A). Many of these pathways are activated in times of nutrient limitation and stress, similarly to the silenced pathways we observe in E. coli. As regions of protein occupancy were observed in horizontally acquired DNA in E. coli, we proposed that regions of protein occupancy may play a role in horizontally acquired DNA in B. subtilis. As B. subtilis is naturally competent, the tight regulation of competence development is especially important to regulate and protect against the acquisition of harmful exogenous DNA elements. Surprisingly, we found that many large negative occupancy peaks overlapped annotated prophage genes (Fig 7B), appearing similar to EPODs but inverted in sign.
Figure 7. IPOD‐HR in Bacillus subtilis reveals Rok‐bound and SMC‐bound domains.
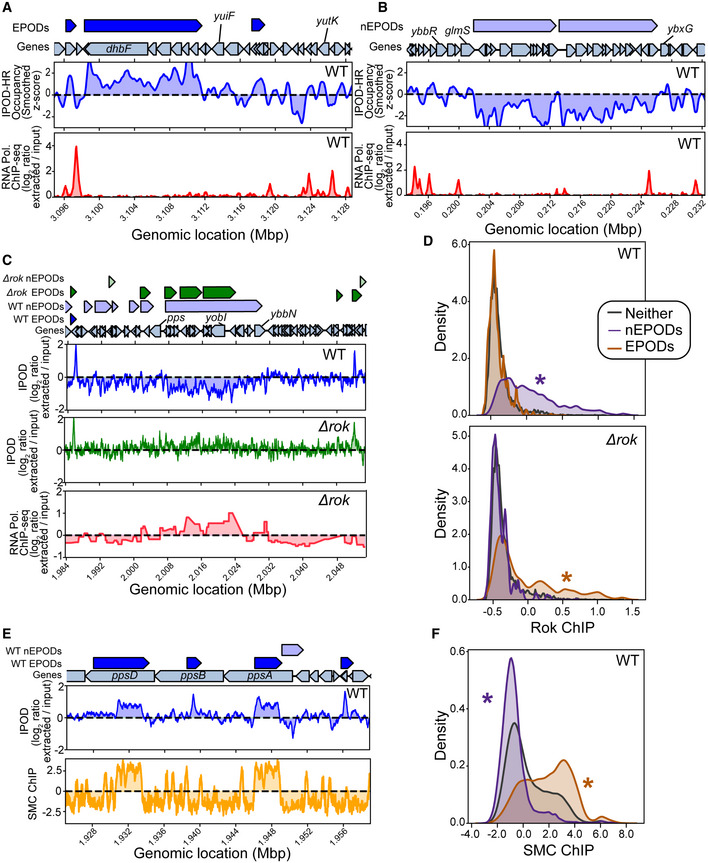
-
AIPOD‐HR ChIP‐subtracted z scores and RNA polymerase ChIP in the vicinity of an extended protein occupancy domain (EPOD). ChIP tracks are shown as log2 extracted/input ratios; the z score is the ChIP‐subtracted robust z score smoothed with a 512‐bp rolling median.
-
BIPOD‐HR and RNA polymerase ChIP tracks in the vicinity of a negative EPOD (nEPOD).
-
CEffects of deletion of rok in the vicinity of an nEPOD; Rok ChIP data from (Smits & Grossman, 2010) shows a strong overlap with the nEPOD boundary, whereas that occupancy region is lost in Δrok cells.
-
DDistributions of Rok ChIP occupancies (see Materials and Methods) in the EPODs and nEPODs called in WT (top) or Δrok (bottom) cells; note that the Rok ChIP occupancy was taken only in WT cells. (*) indicates a significant difference from the “neither” distribution (that is, genomic sites that are not in an EPOD or nEPOD); P < 0.05, permutation test.
-
EComparison of IPOD occupancy and SMC ChIP occupancy (see Materials and Methods) in the vicinity of several typical EPODs.
-
FGenome‐wide distributions of SMC binding in EPODs versus nEPODs as assessed in WT cells; (*) indicates a significant difference from the background distribution as in panel D.
One of the main regulators of competence in B. subtilis is Rok, which acts as a direct repressor of competency genes, regulates several secreted proteins and is involved in repression of mobile genetic elements (Hoa et al, 2002; Albano et al, 2005; Smits & Grossman, 2010). Due to the impact of Rok on gene regulation, coupled with the promiscuous binding activity to A + T‐rich DNA, we propose that Rok may be a main component of protein occupancy in B. subtilis. Surprisingly, while we did not see an enrichment of Rok binding in EPODs using available rok‐myc ChIP‐chip data (Smits & Grossman, 2010), Rok binding was highly correlated with the negative occupancy regions (Fig 7C and D). To investigate this further, we performed IPOD‐HR in ∆rok cells and found that indeed, the loss of Rok resulted in an increase in RNA polymerase binding at sites correlated with Rok binding and loss of negative occupancy regions found in the WT condition (Fig 7C). We examined whether negative peaks were associated with discrepancies with read mapping efficiency and found that there were no changes in the distribution of reads in background, EPODs, or negative peaks (Fig EV5A). We subsequently performed our analysis pipeline to incorporate negative occupancy and found that these negative EPODs, termed nEPODs, correlated with Rok binding and were enriched for genes known to be regulated by Rok, such as sporulation genes and genes involved in competence activation. Importantly, we did not observe correlations with any suspected nucleoid‐associated proteins or global regulators in negative peaks in E. coli, where instead negative peaks are associated with highly transcribed genes (Fig EV5). Thus, whereas in B. subtilis regions of extended negative IPOD signal appear indicative of gene silencing due to Rok binding, large regions of negative measured IPOD occupancy in E. coli are areas of active transcription. These assessments are critical when understanding global protein occupancy in new species. The most likely explanation for our observations is that Rok‐DNA complexes are likely depleted rather than enriched from the interphase during the IPOD‐HR phenol–chloroform extraction, which would give rise to a large region of apparent depletion in the resulting signal. It is possible that other proteins will be encountered in other species that have similar properties, although as noted above, we found no evidence for such behavior among E. coli proteins.
Figure EV5. nEPODs are a biological feature in Bacillus subtilis .
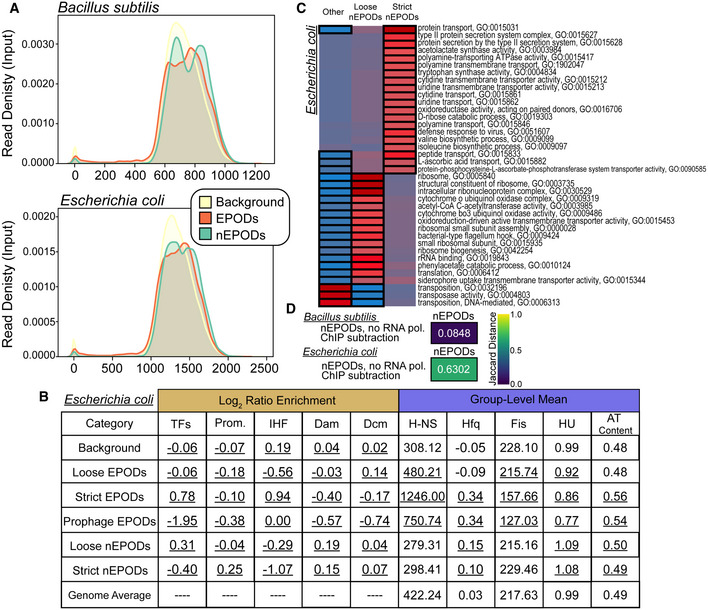
- Kernel density plots displaying the input read density Bacillus subtilis and Escherichia coli in regions called as EPODs (orange), nEPODs (green), and Background (yellow).
- Assessment of Escherichia coli nEPODs compared to other regions. Log2 ratio enrichment: The ratio of the number of TFs, promoters, or motifs in a given category to the total number of TFs, promoters, or motifs. A chi‐squared test was performed, and all categories were significantly associated with each class; values underlined had a P‐value < 0.05. Group‐level mean: The 500‐bp rolling mean for the binding of each NAP was used to calculate the group‐level means for across each category and compared with the overall average for the genome. Permutation based P‐values were calculated comparing each class versus the background. The values underlined had a P‐value < 0.05.
- Escherichia coli nEPODs overlap highly transcribed gene categories.
- Jaccard distance comparing nEPODs called with and without RNA polymerase ChIP‐seq subtraction. The high Jaccard distance in Escherichia coli indicates that nEPODs change dramatically with and without the inclusion RNA polymerase binding, and therefore RNA polymerase associated. However, locations of nEPODs in Bacillus subtilis are unchanged with RNA polymerase binding and are distinct, protein occupied regions.
Positive (standard) EPODs were also apparent in the B. subtilis data, and we found them to be highly correlated with SMC binding (Fig 7E and F), which is known to compact the genome in preparation for chromosomal segregation (Graumann, 2000; Sullivan et al, 2009; Wilhelm et al, 2015). Our findings align with known datasets (preprint: Al‐Bassam et al, 2021), and further our understanding of the role of nucleoid binding proteins in defining the genome landscape across species. Without the dependency on antibody‐based methods, we can explore unknown protein functions across a variety of developmental platforms and species. These findings highlight the broad utility of IPOD‐HR, with the ability to detect both conserved and novel genome architecture features in a variety of distantly related bacterial species. In addition, we see that the general pattern of large regions of high protein occupancy apparently silencing horizontally acquired DNA is an extremely widespread feature, occurring in at least two bacterial species separated by more than 2 billion years of evolution.
Discussion
There is increasing evidence of a regulated genome architecture in E. coli, both in terms of its three dimensional structure (Lioy et al, 2018; Verma et al, 2020; Walker et al, 2020; preprint: Wasim et al, 2021) and of the landscape of protein occupancy on the genome. Both of these classes of features are largely supported by binding of NAPs (Lioy et al, 2018; Freddolino et al, 2021a). IPOD‐HR enabled us to study global changes in chromosomal architecture—here defined as highly protein occupied regions of the genome—across species and implicated NAPs as the main component of EPODs. Here, we show the robustness of these large protein domains across media conditions, growth phase, and small genotype differences (Fig 1). The maintenance of EPODs across conditions and ancestral strains aligns with the idea that EPODs serve an important regulatory silencing role. A variety of questions emerge: How are EPODs maintained through replication and growth? What recruits proteins to these regions? Further studies are being performed to examine the role of methylation in maintenance and recruitment of protein to EPOD regions. As horizontally acquired DNA and methylation have been shown to be intertwined in E. coli (Shin et al, 2016), and we found that dam and dcm sites are depleted in EPODs, we suspect that DNA methylation may play a role in regulation of EPODs containing prophages.
Our study shows that EPODs are partly composed by NAPs in E. coli (Fig 2), with the largest contribution clearly made by the major transcriptional silencers H‐NS and StpA (consistent with an abundance of prior evidence on the silencing roles of H‐NS (Dorman, 2004; Singh et al, 2016), but with other NAPs or pairs of NAPs making important contributions at a subset of loci. Due to the wide binding capacity of NAPs across the genome, the question regarding recruitment to EPODs emerges again. IPOD‐HR successfully shows losses in occupancy upon deletion of NAPs; however, there may be accessory proteins that facilitate recruitment and maintenance. IPOD‐HR may miss subtle changes in proteins that are not as abundant in the cell, so we have begun to design proteomic analysis of EPODs to define the exact composition of EPODs.
We were able to define the key proteins involved in EPOD regulation of metabolic pathways (Figs 3 and 4) and silencing of prophages across the genome (Figs 5 and 6). EPODs appear likely to mediate the formation of transcriptional memory (Fig 4), which allows for strong repression of a rarely‐used metabolic operon when it has not been transcribed in recent memory, but aids in faster induction of genes important for metabolic response after a single exposure to a relevant nutrient. This type of regulation poses exciting ideas for understanding how architectural proteins facilitate a genome architecture regulation across the genome. Importantly, understanding bacterial genome regulation can be incredibly useful for biotechnology purposes, especially in the case where cells can be grown in a number of conditions that may induce changes in their overall genome architecture that can impact induction of particular genes.
We have also identified a novel silencing mechanism for prophages and toxic elements across the genome. Together, Hfq and Fis are required to silence some prophages, most notably DLP12 (contained in R1 of Fig 6) and Qin (R3 of Fig 6) prophages. What defines particular prophages to recruit Hfq and Fis remains to be explored. However, these findings contribute to an overarching theme of the genome structure serving as an immune response to a variety of horizontally acquired DNA. As we have previously shown, reporter constructs integrate with higher frequency in EPODs and are efficiently silenced (Scholz et al, 2019; Freddolino et al, 2021a). We propose that EPODs serve as DNA sinks for foreign DNA, and quickly silence potentially harmful elements. Further investigation into the mechanisms underlying this response will bolster our view of the defenses of bacterial species against potentially harmful genetic elements.
Many NAPs and their functions are conserved across bacterial species, and the overall roles of NAPs in establishing large regions of silencing protein occupancy appear to be conserved even beyond the reach of recognizable homologs of any given NAP. We found that the use of genome architecture as a mode of immunity may also be conserved in a distantly related species to E. coli, B. subtilis (Fig 7). The exploitation of such a system has a number of promising outcomes. For instance, the work established here could inform new antibiotic approaches for pathogenic bacteria by targeting proteins required to suppress toxic elements already in existence in the genome. In addition, understanding how bacteria recruit and build their genome architecture around foreign DNA can inform on how bacteria interact with their environment, and how they may evolve over time as they acquire new genetic elements.
Materials and Methods
Strain construction
The MG1655 “WT” isolate used in all figures was obtained from Hani Goodarzi (Tavazoie Lab, then at Princeton University) in 2009 and is isogenic with ATCC 700926, with the exception of an IS1 insertion at position 991 in dgcJ (Freddolino & Tavazoie, 2012). The MDS42 strain was obtained from Alison Hottes in 2009 (Tavazoie Lab, then at Princeton University) and contains a C‐>T mutation in ribD and missing coverage for lysV (Posfai, 2006). These modifications may be ancestral to MDS42, or specifically present in our MDS42 parental strain. MDS42 deletions were validated using PCR. The MG1655 (2) isolate was obtained from the Jakob Lab at the University of Michigan in 2018. The strain appears to arise from the ATCC 47076 lineage of MG1655 (Freddolino et al, 2012) and thus is crl+ , gatC+ , and glpR+ ; relative to the reference sequence for this lineage, however, the MG1655(2) strain also contains point mutations in ybhJ (L54I) and mntP (G25D), with unknown functional implications (genotypes obtained via analysis with breseq (Deatherage & Barrick, 2014)). The MG1655 lacZ::cml‐gfp cells used as a reference strain in the competition experiments were separately obtained from Hani Goodarzi (Tavazoie Lab, then at Princeton University).
NAP deletion strains
All nucleoid‐associated protein (NAP) gene deletions were performed in the same MG1655 “WT” base strain stock discussed above. All NAP deletions were obtained by P1 transduction of the FRT‐flanked kanR marker from the corresponding knockout strain of the Keio collection (Baba et al, 2006; Thomason et al, 2007). With the exception of ΔihfA::Km, ΔihfB::Clm(ΔihfAB), and ΔhupA ΔhupB::Km (ΔhupAB), the pCP20 plasmid (Cherepanov & Wackernagel, 1995) containing Flp recombinase was used to excise the kanR marker, leaving a small scar in the place of the original open reading frame. Once candidates were isolated for each deletion and contained the pCP20 plasmid, they were grown overnight at 42°C to drop the temperature sensitive pCP20. The overnight cultures were streaked onto LB plates and grown overnight at 37°C. Individual colonies were replica plated onto appropriate selective plates to ensure the loss of both the marker and pCP20 plasmid. The ΔihfAB and ΔhupAB strains were not cured due to incredibly low efficiency to excise markers via pCP20, and markers were retained to avoid potential suppressor mutations.
Bacillus subtilis strains
The B. subtilis PY79 and rok::kan strains came from the Simmons lab at the University of Michigan. Details of the genome sequence can be found in Schroeder and Simmons (2013).
Media/culture conditions
LB (Lennox) media (10 g/l tryptone, 5 g/l yeast extract, 5 g/l NaCl) was used for cloning and recovery of cryogenically preserved cells, with addition of 15 g/l bacteriological agar for plating.
In the case of physiological experiments, we used appropriately supplemented versions of M9 defined minimal medium (6 g/l Na2HPO4, 3 g/l KH2PO4, 1 g/l NH4Cl, 0.5 g/l NaCl, 1 mM MgSO4). Minimal M9 medium (M9/min) contained includes 0.2% (w/v) carbon source (glucose or glutamine or 5‐Keto‐D‐gluconic acid potassium salt), 0.4 mM CaCl2, 40 μM ferric citrate, and the micronutrient mixture typically incorporated in MOPS minimal media (Neidhardt et al, 1974). For all of the remaining IPOD‐HR experiments, we used our M9‐rich defined medium (M9/rdm) incorporated with 0.4% (w/v) glucose, MOPS micronutrients, 4 μM CaCl2, 40 μM ferric citrate, and 1x supplements ACGU and EZ as used in MOPS rich defined medium (Neidhardt et al, 1974).
B. subtilis cell growth and harvest for IPOD‐HR
For Bacillus subtilis, strains were struck from frozen stocks and grown on LB plates overnight at 37°C. WT and Δrok (rok::kanR) strains were inoculated into LB and LB supplemented with 5 μg/ml kanamycin, respectively, from a plate wash at a starting an OD600 of 0.025 and grown at 37°C with shaking to an OD600 between 0.65 and 0.85. Rifampicin was added to a final concentration of 150 μg/ml and cultures were incubated for an additional 10 min at 37°C with shaking. Sodium phosphate (final concentration 0.01 M) and formaldehyde (1% v/v) were added to 30 ml aliquots of culture and cross‐linked at room temperature for 5 min with shaking. Reactions were quenched by the addition of 0.333 M glycine for 10 min at room temperature. Cells were collected via centrifugation and watched twice with ice‐cold PBS and cell pellets were subsequently flash‐frozen in liquid nitrogen and stored at −80°C.
E. coli cell growth and harvest for IPOD‐HR
Cryogenically preserved cells were streaked onto an LB plate and grown in the media of interest with 1/10th of the carbon source indicated overnight at a temperature of 37°C and shaking at 200 rpm. The culture was back diluted into fresh, prewarmed media to an OD600 of 0.003 the next day. The culture was grown to the target OD600 (0.2, except in the case of deep stationary phase samples, described below) and treated with a final concentration of 150 μg/ml of rifampicin and incubated for 10 min under the same growth conditions previously described. The cultures were rapidly poured into a 50‐ml conical tube and mixed with concentrated formaldehyde/sodium phosphate (pH 7.4) buffer sufficient to yield a final concentration of 10 mM NaPO4 and 1% v/v formaldehyde. Crosslinking proceeded for 5 min at room temperature and quenched with an excess of glycine (final concentration 0.333 M) for 10 min with shaking at room temperature. The cross‐linked cells were chilled on ice for 10 min and washed twice with 10 ml ice‐cold phosphate‐buffered saline (PBS). The resulting pellets were carefully dried, remaining media pipetted and discarded, and snap‐frozen in a dry ice‐ethanol bath and stored at −80°C for no longer than 1 month.
Deep stationary samples followed the same process of being grown in the appropriate media overnight and back diluted to an OD600 0.003. Once the cells reached an OD600 of 0.2, they were grown for an additional 24 h, treated with rifampicin for 20 min, and proceeded through the same treatment for crosslinking as described above.
Cell lysis and DNA preparation
IPOD‐HR interface extraction, RNA polymerase chromatin immunoprecipitation, and crosslinking reversal and recovery of DNA was performed as previously described (Freddolino et al, 2021a). Due to the high biomass of deep stationary phase cells, cells were diluted 10× prior to lysis. In the case of the B. subtilis, samples were sonicated four times for 5s at 25% power with 15s between pulses.
Preparation of next‐generation sequencing (NGS) libraries
All DNA samples were prepared for Illumina sequencing using the NEBNext Ultra (or Ultra II) Library Prep Kit (NEB product #E7370 or #E7103, respectively). The NEBNext Ultra II Library Prep Kit was used on the Δhns ΔstpA samples and biological replicate 2 of the deep stationary phase samples (Δdps and corresponding WT). We consulted with NEB to confirm that there are no differences between the kits that would impact our results. Single index or dual index primers from NEB were used in the prep. The manufacturer’s instructions were followed with the same modifications as listed in Freddolino et al (2021a).
All libraries were sequenced on an Illumina NextSeq instrument.
Analysis of NGS data, read quality control and preprocessing, DNA sequencing and protein occupancy calling, and feature calling was performed as previously described (preprint: Freddolino et al, 2021b). In brief, after removal of technical sequences (adapters) and trimming of low‐quality read ends, reads were aligned to the U00096.3 reference genome (E. coli samples) or py79 reference genome (B. subtilis samples) using bowtie2 with “very sensitive, end to end” presets. Read densities were then calculated at 5‐bp resolution across the chromosome, and quantile normalized. Overall effects on fragment abundance due to distance from the origin of replication were removed by dividing each dataset by a spline‐smoothed version of the corresponding input data, and then, the resulting occupancy tracks were rescaled to each have the same mean value. Separate IPOD and RNA polymerase ChIP occupancy tracks were calculated based on the log2 ratio of normalized fragment abundances in each pulldown sample and the corresponding input sample, and the RNA polymerase subtracted IPOD‐HR signal was then calculated based on a LOESS fit between the observed IPOD and ChIP occupancies [as described in detail in preprint: Freddolino et al (2021b)—n.b. the data analysis methods here follow precisely the description in that reference, except as noted below].
Rescaling of IPOD‐HR occupancy tracks and subsequent EPOD calling
While we have previously shown the robustness of the IPOD‐HR analysis pipelines for both deletions of local regulators and substantial changes in physiological conditions (preprint: Freddolino et al, 2021b), we found that for the nucleoid‐associated protein deletions considered here, in many cases the assumptions underlying the IPOD‐HR normalization methods (that the overall shape of the distribution of occupancy values across the genome would not change substantively between conditions) was violated. We thus modified the EPOD calling scheme to be able to compare EPOD count, coverage, and occupancy across NAP deletion datasets, where some NAP deletions are sufficient to substantively shift the overall score distribution, due to the large changes in overall protein binding caused by deletion of any one NAP. We rescaled the IPOD‐HR occupancy tracks, beginning with the robust z score values indicating occupancy at every five base pairs of the genome, using the following procedure: We found the intersection of EPODs between each NAP deletion and WT with a minimum fractional overlap of 0.2, and plotted the mean occupancy of each overlapped EPOD (NAP deletion versus WT). We then used a robust linear model with Huber’s T for M estimation to estimate the slope of the NAP deletion values as a function of the WT values for the same EPODs and used the slope to rescale the NAP deletion dataset by dividing the robust z score values by the slope across the genome— thus, we rescaled the overall protein occupancy in each condition to bring into register the observed occupancies at regions that were highly occupied both in that deletion and in the wild type cells. These new values were used as input to call EPODs using the algorithm described in preprint: Freddolino et al (2021b): we calculated 512‐bp and 256‐bp rolling means across the genome, and set as a threshold score the 90th percentile (for normal EPODs) or 75th percentile (for loose EPODs) from the 256‐bp rolling mean signal. Then, any region of the genome at least 1,024 bp in length with a median occupancy score from the 512‐bp smoothed signal above the threshold score was considered a potential EPOD. Potential EPODs were expanded in both directions on the genome as far as possible while maintaining an median occupancy score above the specified threshold, and without ever reaching an occupancy score below zero. To call EPODs in the NAP deletion datasets considered here, we set the cutoff of what would be counted as an EPOD using the WT thresholds and applied this to all datasets (after the rescaling described above). In summary, these rescaling and thresholding enabled us to make more accurate comparisons between EPODs and occupancy across the NAP deletion datasets. By bringing the observed occupancies at conserved EPODs into register with each other across the different genotypes in our dataset. The procedure described here represents the best normalization across datasets that can be achieved in the absence of a global standard (e.g., a spike‐in sample), which would provide a simpler adjustment with fewer assumptions and may be advisable for future work.
KDG growth experiments
Experimental design for IPOD‐HR collection and RNA‐seq experiments on KDG exposure (Fig 3)
Cells were grown from a cryogenic stock on LB plates at 37°C and inoculated into our M9 minimal medium including 0.02% glucose at 37°C. In the morning, cultures were back diluted to an OD600 0.003 in fresh, prewarmed M9 minimal medium including 0.2% glucose at 37°C. Once cells reached a target OD600 of 0.2, cells were pelleted and washed twice with 5 ml of warmed PBS. 1 ml of sample was mixed with 1 ml DNA / RNA shield and flash‐frozen in a dry ice‐ethanol bath and stored at −80°C. The remainder of the cells were placed in our M9 minimal medium including 0.2% 5‐Keto‐D‐gluconic acid (KDG; Sigma‐Aldrich: Catalog #K4125) to an estimated OD600 of 0.1 and placed at 37°C. Cells were grown to an OD ~0.2 and collected for IPOD‐HR (with 1 ml for RNA‐seq) as described above, and some of the remaining cells back diluted to an estimated OD600 of 0.003 in M9 minimal medium with 0.2%§ glucose. Once cells were grown to an OD600 ~0.2 in 0.2%§ glucose, cells were collected for IPOD‐HR (with 1 ml for RNA‐seq) as described above.
Experimental design for KDG competition experiments (Fig 4B)
Our competition experiments were conducted in head‐to‐head mixed growth between a strain of interest and an otherwise‐isogenic lacZ::cat strain. For each competition, the two competing strains were pregrown under appropriate prior conditions (see below), and then, 10 µl of the pregrowth culture for each of the two competing strains was inoculated into prewarmed destination media (separate tubes of M9+glucose and M9+KDG, 2 ml per tube) and allowed to grow for approximately 48 h at 37°C with shaking at 200 rpm. Both immediately after inoculation, and again after 48 h, a dilution series of each culture was spotted on MacConkey/lactose plates to enable counting of the lac+ and lac− colonies present.
At the beginning of each experimental series, both the test (lacZ+) and reference (lacZ−) strains were pregrown overnight in M9 + 0.2% glucose (starting from a single colony taken from an LB plate); this is referred to as “Day −1” in our experimental timelines, and indicates that both the test and reference strains are KDG‐naive. At the same time as assembly of the initial competitions, the test strain for each experimental series was then back diluted 200× into 2 ml of M9 minimal media + 0.2% KDG (M9 Min KDG), and grown for 48 h. The KDG‐adapted test cells were then competed against a fresh culture of the reference cells that had been grown overnight in M9 minimal media + glucose (M9 Min Glu)—we refer to this as “Day 0” in our experimental timelines. At the same time as the Day 0 competition, some of the KDG‐adapted test cells were also diluted 200× into fresh M9 Min Glu and allowed to grow overnight. These glucose‐recovered test cells were competed the following day (in both M9 Min Glu and M9 Min KDG) against reference cells that had likewise been grown for 12 and 24 h in M9 Min Glu, resulting in our “Day 0.5” and “Day 1” competitions, respectively (that is, the test cells are 0.5 or 1.0 day removed from their KDG exposure). We continued several additional cycles of passaging the test cells in M9 Min Glu followed by competition with naive reference cells, resulting in competitions referred to as “Day 2”, “Day 3”, etc.—in all cases the stated duration is the number of days (and passages in M9 Min Glu) that the test cells are removed from their growth in KDG.
For each competition, we established competitive indices λ for the test cells relative to reference cells separately in KDG and in glucose media, using the following equation:
Where m denotes a media type (either M9 Min Glu or M9 Min KDG) and the specified cell counts (cfu) are taken for that media type. A pseudocount of 0.1 was added to all counts, as in a small number of cases zero colonies of one type or the other were observed due to the lopsidedness of the competition. One quantity of particular interest is the difference in the competitive fitness of the test strain (relative to its reference) in KDG versus that in glucose; thus, we define the difference in fitnesses, Δλ, as.
Here, more positive values of Δλ indicate that the test strain is (relative to the reference) more fit during competition in KDG media, after normalization by their competition growth in glucose media; these values are used for the analysis. The quantity Δλ is referred to as the “Log2 Fitness Ratio in KDG (relative to naive)” in Fig 4B.
Experimental design for Fig 4C
Similar to the experiments performed in Fig 3, cells were grown from a cryogenic stock on LB plates at 37°C and inoculated into our M9 minimal medium including 0.02% glucose at 37°C. In the morning, cultures were back diluted to an OD600 0.003 in fresh, prewarmed M9 minimal medium including 0.2% glucose at 37°C in a total volume 150 µl with 100 µl of mineral oil in a plate reader. Measurements were taken every 10 min at 37°C with shaking to calculate lag times. Once cells reached an OD600 ~0.2, they were back diluted to an OD600 of 0.01 in M9 minimal medium with 0.2% KDG. Cells were grown to an OD600 ~0.2, then diluted to an OD600 0.003 in M9 minimal medium with 0.2% glucose and grown to an OD600 ~0.2.
Growth curves (using log2‐scaled optical densities [ODs]) were smoothed using a cubic spline with one knot per 5 h (or fraction thereof) in the data; we then identified the maximum value of the slope of the resulting spline as the growth rate. The lag time was calculated by projecting a line with a slope equal to the growth rate through the point at which the most rapid growth was observed and took the time at which that line intercepted a horizontal line at the initially observed log2 OD for that culture. Summary statistics were calculated via Bayesian regression as implemented in the brms R package, with population‐level effects for the experiment under consideration and default brm values for all other arguments.
RNA isolation and sequencing preparation
All RNA‐sequencing samples were collected and prepared for sequencing as follows. Once cells were grown to the appropriate OD600, 2.5 ml of culture was mixed with 5 ml of RNAprotect (Qiagen: Catalog #76506), vortexed and incubated at room temperature for 5 min. Cells were spun at 4°C for 10 min at 5,000g in a fixed‐angle rotor. The supernatant was removed, and the pellet was flash‐frozen in a dry ice‐ethanol bath and stored at −80°C. For RNA extraction, the pellet was resuspended in 100 µl of TE and treated with 177kU (1 µl) Ready‐lyse lysozyme solution (Lucigen: Catalog #R1804) and 0.2 mg (10 µl) proteinase K (Thermo Fisher Scientific: Catalog #EO0492), incubated for 10 min at room temperature with vortexing every 2 min. The RNA was purified using RNA Clean and Concentrator kit‐5 (Zymo: Catalog #R1014), treated with 5 units of Baseline‐ZERO DNase (Epicentre: Catalog #DB0715K) in the presence of RNAse inhibitor (NEB: Catalog #M0314L) for 30 min at 37°C. RNA was purified again using RNA Clean and Concentrator kit‐5 (Zymo: Catalog #R1014). RNA was flash‐frozen in a dry ice‐ethanol bath and stored at −80°C.
rRNA depletion was performed using the bacterial rRNA depletion kit following manufacturer instructions (New England Biolabs (NEB): Catalog #E7850L). The only modification performed was the last step, where instead of a bead clean up, we used the RNA Clean and Concentrator kit‐5 (Zymo: Catalog #R1014).
Sequencing preparation was performed using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina following manufacturer instructions (NEB: Catalog #E7420L) for rRNA‐depleted RNA. Random primers were used, and RNA was considered intact for the NEBNext Ultra protocol. Slight modifications to the protocol are as follows. To purify cDNA, the Oligo clean & concentrator was used (Zymo: Catalog #D4061). Following adapter ligation, DNA was purified using DNA Clean & Concentrator‐5 (Zymo: Catalog #D4014). Dual index primers from NEB were used in the prep. Libraries were sequenced on an Illumina NextSeq.
RNA‐seq analysis began with read preprocessing identical to that described in preprint: Freddolino et al (2021b), through the end of the bowtie2 alignment stage. After alignment to the U00096.3 genome, we performed gene‐level quantitation with htseq‐count (Anders et al, 2015), using union mode for scoring and all mode for non‐unique assignments (appropriate for polycistronic mRNAs where a single read is often expected to span multiple genes). Differential expression calling was then performed using deseq2 with default settings; separate models were fitted for comparison of WT/ihf/hns stpA cells, WT/fis/hfq cells, and the time course for 5‐KDG exposure.
HMM classes
HMM fits were performed using the hmmlearn python package (version 0.2.4) with Gaussian emissions. As input features we used the IPOD‐HR robust z scores and RNA polymerase log2(extracted/input) ratios, for a total of 2 features per condition at each of 928,330 sites on the genome (5‐bp resolution). We trained a series of HMMs using 20‐fold cross‐validation (dividing the genome into 20 evenly sized blocks), in which we assessed the log‐likelihoods for the withheld folds based on an HMM trained on the rest of the genome. After training and evaluating models from 2 to 20 components, we found that the predictive performance increased sharply with component count up to six components, and after that increased much more gradually. We thus used a six‐component model to provide a balance of interpretability and predictive performance. We fitted 20 final models using the entire genome and selected the one with the highest likelihood to provide the final HMM; state assignments were then obtained using the Viterbi algorithm. Default parameters for hmmlearn were used unless otherwise noted.
Hfq binding was measured by cloning Hfq‐PAmCherry from McQuail et al (2020) into MG1655 (2) and performing ChIP‐seq with a monoclonal mCherry antibody (Thermo Fisher M11217) (manuscript in press). A 500‐bp rolling mean of the log2 extracted/input ratio was calculated and used for comparison in this paper.
Data visualization and analysis
For data analysis as previously described (preprint: Freddolino et al, 2021b), we made heavy use of numpy (Harris et al, 2020; preprint: Freddolino et al, 2021b), R version 3.6.3 (Verzani, 2011; RStudio, 2020), https://www.Tidyverse.org/, and ggplot2 (Wickham, 2009).
Author contributions
HMA and PLF contributed to conceptualization; HMA, TJG, and PLF contributed to methodology; HMA, TJG, TMN, LAS, RLH, and PLF contributed to investigation; HMA and PLF contributed to data analysis and curation; HMA, TMN, LAS, and PLF contributed to writing—original draft; and LS and PLF contributed to funding acquisition.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Acknowledgements
This work was supported by National Institutes of Health grants R00‐GM097033 and R35‐GM128637 (to PLF) and National Science Foundation grant MCB 1714539 (to LS).
The EMBO Journal (2022) 41: e108708
Footnotes
Correction added on 1 of February 2022, after first online publication: Twice “0.02% glucose” has been corrected to “0.2% glucose”.
Data availability
RNA‐Seq, IPOD‐HR, DNA Sequencing, and ChIP‐seq data: Gene Expression Omnibus GSE164796 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164796).
References
- Albano M, Smits WK, Ho LTY, Kraigher B, Mandic‐Mulec I, Kuipers OP, Dubnau D (2005) The Rok protein of Bacillus subtilis represses genes for cell surface and extracellular functions. J Bacteriol 187: 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Bassam MM, Moyne O, Chapin N, Zengler K (2021) Nucleoid openness profiling links bacterial genome structure to phenotype. bioRxiv 10.1101/2020.05.07.082990 [PREPRINT] [DOI]
- Ali Azam T, Iwata A, Nishimura A, Ueda S, Ishihama A (1999) Growth phase‐dependent variation in protein composition of the Escherichia coli nucleoid. J Bacteriol 181: 6361–6370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amemiya HM, Schroeder J, Freddolino PL (2021) Nucleoid‐associated proteins shape chromatin structure and transcriptional regulation across the bacterial kingdom. Transcription 12: 182–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W (2015) HTSeq–a Python framework to work with high‐throughput sequencing data. Bioinformatics 31: 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleman JA, Ross W, Salomon J, Gourse RL (1998) Activation of Escherichia coli rRNA transcription by FIS during a growth cycle. J Bacteriol 180: 1525–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H (2006) Construction of Escherichia coli K‐12 in‐frame, single‐gene knockout mutants: the Keio collection. Mol Syst Biol 2: 2006.0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barh D, Azevedo V (2017) Omics technologies and bio‐engineering: volume 1: towards improving quality of life. Academic Press; [Google Scholar]
- Baumler A (2006) Faculty Opinions recommendation of Selective silencing of foreign DNA with low GC content by the H‐NS protein in Salmonella. Faculty Opinions – Post‐Publication Peer Review of the Biomedical Literature. 10.3410/f.1032929.492954 [DOI]
- Bausch C, Peekhaus N, Utz C, Blais T, Murray E, Lowary T, Conway T (1998) Sequence analysis of the GntII (subsidiary) system for gluconate metabolism reveals a novel pathway for L‐idonic acid catabolism in Escherichia coli . J Bacteriol 180: 3704–3710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bausch C, Ramsey M, Conway T (2004) Transcriptional organization and regulation of the L‐idonic acid pathway (GntII system) in Escherichia coli . J Bacteriol 186: 1388–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokal AJ 4th, Ross W, Gourse RL (1995) The transcriptional activator protein FIS: DNA interactions and cooperative interactions with RNA polymerase at the Escherichia coli rrnB P1 promoter. J Mol Biol 245: 197–207 [DOI] [PubMed] [Google Scholar]
- Boudreau BA, Hron DR, Qin L, van der Valk RA, Kotlajich MV, Dame RT, Landick R (2018) StpA and Hha stimulate pausing by RNA polymerase by promoting DNA‐DNA bridging of H‐NS filaments. Nucleic Acids Res 46: 5525–5546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov PP, Wackernagel W (1995) Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp‐catalyzed excision of the antibiotic‐resistance determinant. Gene 158: 9–14 [DOI] [PubMed] [Google Scholar]
- Chintakayala K, Singh SS, Rossiter AE, Shahapure R, Dame RT, Grainger DCE (2013) coli Fis protein insulates the cbpA gene from uncontrolled transcription. PLoS Genet 9: e1003152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho B‐K, Knight EM, Barrett CL, Palsson BØ (2008) Genome‐wide analysis of Fis binding in Escherichia coli indicates a causative role for A‐/AT‐tracts. Genome Res 18: 900–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorman CJ (2004) H‐NS: a universal regulator for a dynamic genome. Nat Rev Microbiol 2: 391–400 [DOI] [PubMed] [Google Scholar]
- D’Souza JM, Wang L, Reeves P (2002) Sequence of the Escherichia coli O26 O antigen gene cluster and identification of O26 specific genes. Gene 297: 123–127 [DOI] [PubMed] [Google Scholar]
- Deatherage DE, Barrick JE (2014) Identification of mutations in laboratory‐evolved microbes from next‐generation sequencing data using breseq. Methods Mol Biol 1151: 165–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng S, Stein RA, Higgins NP (2005) Organization of supercoil domains and their reorganization by transcription. Mol Microbiol 57: 1511–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon SC, Dorman CJ (2010) Bacterial nucleoid‐associated proteins, nucleoid structure and gene expression. Nat Rev Microbiol 8: 185–195 [DOI] [PubMed] [Google Scholar]
- Faubladier M, Bouché JP (1994) Division inhibition gene dicF of Escherichia coli reveals a widespread group of prophage sequences in bacterial genomes. J Bacteriol 176: 1150–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Han W, Wang Q, Bastin DA, Wang L (2005) Characterization of Escherichia coli O86 O‐antigen gene cluster and identification of O86‐specific genes. Vet Microbiol 106: 241–248 [DOI] [PubMed] [Google Scholar]
- Francis NJ, Kingston RE (2001) Mechanisms of transcriptional memory. Nat Rev Mol Cell Biol 2: 409–421 [DOI] [PubMed] [Google Scholar]
- Freddolino PL, Amemiya HM, Goss TJ, Tavazoie S (2021a) Dynamic landscape of protein occupancy across the Escherichia coli chromosome. PLoS Biol 19: e3001306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freddolino PL, Amini S, Tavazoie S (2012) Newly identified genetic variations in common Escherichia coli MG1655 stock cultures. J Bacteriol 194: 303–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freddolino PL, Goss TJ, Amemiya HM, Tavazoie S (2021b) Dynamic landscape of protein occupancy across the Escherichia coli chromosome. bioRxiv 10.1101/2020.01.29.924811 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Freddolino PL, Tavazoie S (2012) Beyond homeostasis: a predictive‐dynamic framework for understanding cellular behavior. Annu Rev Cell Dev Biol 28: 363–384 [DOI] [PubMed] [Google Scholar]
- Frenkiel‐Krispin D, Levin‐Zaidman S, Shimoni E, Wolf SG, Wachtel EJ, Arad T et al (2001) Regulated phase transitions of bacterial chromatin: a non‐enzymatic pathway for generic DNA protection. EMBO J 20: 1184–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez KM, Rodríguez A, Rodriguez Y, Ramírez AH, Istúriz T (2011) The subsidiary GntII system for gluconate metabolism in Escherichia coli: alternative induction of the gntV gene. Biol Res 44: 269–275 [PubMed] [Google Scholar]
- Goodarzi H, Elemento O, Tavazoie S (2009) Revealing global regulatory perturbations across human cancers. Mol Cell 36: 900–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger DC, Goldberg MD, Lee DJ, Busby SJW (2008) Selective repression by Fis and H‐NS at the Escherichia coli dps promoter. Mol Microbiol 68: 1366–1377 [DOI] [PubMed] [Google Scholar]
- Graumann PL (2000) Bacillus subtilis SMC is required for proper arrangement of the chromosome and for efficient segregation of replication termini but not for bipolar movement of newly duplicated origin regions. J Bacteriol 182: 6463–6471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CR, Millman KJ, van der Walt SJ, Gommers R, Virtanen P, Cournapeau D et al (2020) Array programming with NumPy. Nature 585: 357–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengge R (2009) Principles of c‐di‐GMP signalling in bacteria. Nat Rev Microbiol 7: 263–273 [DOI] [PubMed] [Google Scholar]
- Hoa TT, Tortosa P, Albano M, Dubnau D (2002) Rok (YkuW) regulates genetic competence in Bacillus subtilis by directly repressing comK. Mol Microbiol 43: 15–26 [DOI] [PubMed] [Google Scholar]
- Hong SH, Wang X, Wood TK (2010) Controlling biofilm formation, prophage excision and cell death by rewiring global regulator H‐NS of Escherichia coli . Microb Biotechnol 3: 344–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahramanoglou C, Seshasayee ASN, Prieto AI, Ibberson D, Schmidt S, Zimmermann J, Benes V, Fraser GM, Luscombe NM (2011) Direct and indirect effects of H‐NS and Fis on global gene expression control in Escherichia coli . Nucleic Acids Res 39: 2073–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlajich MV, Hron DR, Boudreau BA, Sun Z, Lyubchenko YL, Landick R (2015) Bridged filaments of histone‐like nucleoid structuring protein pause RNA polymerase and aid termination in bacteria. Elife 4: e4970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu S, Horn PJ, Peterson CL (2007) SWI/SNF is required for transcriptional memory at the yeast GAL gene cluster. Genes Dev 21: 997–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagha M, Ferraro T, Dufourt J, Radulescu O, Mantovani M (2017) Transcriptional memory in the Drosophila embryo. Mech Dev 145: S137 [Google Scholar]
- Landick R, Wade JT, Grainger DC (2015) H‐NS and RNA polymerase: a love‐hate relationship? Curr Opin Microbiol 24: 53–59 [DOI] [PubMed] [Google Scholar]
- Lim CJ, Whang YR, Kenney LJ, Yan J (2012) Gene silencing H‐NS paralogue StpA forms a rigid protein filament along DNA that blocks DNA accessibility. Nucleic Acids Res 40: 3316–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link AJ, Phillips D, Church GM (1997) Methods for generating precise deletions and insertions in the genome of wild‐type Escherichia coli: application to open reading frame characterization. J Bacteriol 179: 6228–6237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkevicius M, Sandegren L, Andersson DI (2013) Mechanisms and fitness costs of tigecycline resistance in Escherichia coli . J Antimicrob Chemother 68: 2809–2819 [DOI] [PubMed] [Google Scholar]
- Lioy VS, Cournac A, Marbouty M, Duigou S, Mozziconacci J, Espéli O, Boccard F, Koszul R (2018) Multiscale structuring of the E.coli chromosome by nucleoid‐associated and condensin proteins. Cell 172: 771–783.e18 [DOI] [PubMed] [Google Scholar]
- Lucchini S, Rowley G, Goldberg MD, Hurd D, Harrison M, Hinton JCD (2006) H‐NS mediates the silencing of laterally acquired genes in bacteria. PLoS Pathog 2: e81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg MS, White MF, van Driel R, Dame RT (2008) The major architects of chromatin: architectural proteins in bacteria, archaea and eukaryotes. Crit Rev Biochem Mol Biol 43: 393–418 [DOI] [PubMed] [Google Scholar]
- McQuail J, Switzer A, Burchell L, Wigneshweraraj S (2020) The RNA‐binding protein Hfq assembles into foci‐like structures in nitrogen starved. J Biol Chem 295: 12355–12367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moazed D (2011) Mechanisms for the inheritance of chromatin states. Cell 146: 510–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai K (2002) Faculty Opinions recommendation of Hfq: a bacterial Sm‐like protein that mediates RNA‐RNA interaction. Faculty Opinions – Post‐Publication Peer Review of the Biomedical Literature. 10.3410/f.1003709.40104 [DOI]
- Nair S, Finkel SE (2004) Dps protects cells against multiple stresses during stationary phase. J Bacteriol 186: 4192–4198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Ogura Y, Gotoh Y, Hayashi T (2021) Prophages integrating into prophages: a mechanism to accumulate type III secretion effector genes and duplicate Shiga toxin‐encoding prophages in Escherichia coli . bioRxiv 10.1101/2020.11.04.367953 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Nakao R, Ramstedt M, Wai SN, Uhlin BE (2012) Enhanced biofilm formation by Escherichia coli LPS mutants defective in Hep biosynthesis. PLoS One 7: e51241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarre WW (2006) Selective silencing of foreign DNA with low GC content by the H‐NS protein in Salmonella. Science 313: 236–238 [DOI] [PubMed] [Google Scholar]
- Navarre WW, McClelland M, Libby SJ, Fang FC (2007) Silencing of xenogeneic DNA by H‐NS—facilitation of lateral gene transfer in bacteria by a defense system that recognizes foreign DNA. Genes Dev 21: 1456–1471 [DOI] [PubMed] [Google Scholar]
- Neeli‐Venkata R, Martikainen A, Gupta A, Gonçalves N, Fonseca J, Ribeiro AS (2016) Robustness of the process of nucleoid exclusion of protein aggregates in Escherichia coli . J Bacteriol 198: 898–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neidhardt FC, Bloch PL, Smith DF (1974) Culture medium for enterobacteria. J Bacteriol 119: 736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orans J, Kovach AR, Hoff KE, Horstmann NM, Brennan RG (2020) Crystal structure of an Escherichia coli Hfq Core (residues 2–69)–DNA complex reveals multifunctional nucleic acid binding sites. Nucleic Acids Res 48: 3987–3997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palozola KC, Lerner J, Zaret KS (2019) A changing paradigm of transcriptional memory propagation through mitosis. Nat Rev Mol Cell Biol 20: 55–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posfai G (2006) Emergent properties of reduced‐genome Escherichia coli . Science 312: 1044–1046 [DOI] [PubMed] [Google Scholar]
- Postow L, Hardy CD, Arsuaga J, Cozzarelli NR (2004) Topological domain structure of the Escherichia coli chromosome. Genes Dev 18: 1766–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remesh SG, Verma SC, Chen J‐H, Ekman AA, Larabell CA, Adhya S, Hammel M (2020) Nucleoid remodeling during environmental adaptation is regulated by HU‐dependent DNA bundling. Nat Commun 11: 2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- RStudio (2020) https://rstudio.com/
- Rubirés X, Saigi F, Piqué N, Climent N, Merino S, Albertí S, Tomás JM, Regué M (1997) A gene (wbbL) from Serratia marcescens N28b (O4) complements the rfb‐50 mutation of Escherichia coli K‐12 derivatives. J Bacteriol 179: 7581–7586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado H, Martínez‐Flores I, Bustamante VH, Alquicira‐Hernández K, García‐Sotelo JS, García‐Alonso D, Collado‐Vides J (2018) Using RegulonDB, the Escherichia coli K‐12 gene regulatory transcriptional network database. Curr Protoc Bioinformatics 61: 1.32.1–1.32.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatier J, Wiecki TV, Fonnesbeck C (2016) Probabilistic programming in Python using PyMC3. PeerJ Compu Sci 2: e55 [Google Scholar]
- Sanchez‐Torres V, Hu H, Wood TK (2011) GGDEF proteins YeaI, YedQ, and YfiN reduce early biofilm formation and swimming motility in Escherichia coli . Appl Microbiol Biotechnol 90: 651–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos‐Zavaleta A, Pérez‐Rueda E, Sánchez‐Pérez M, Velázquez‐Ramírez DA, Collado‐Vides J (2019) Tracing the phylogenetic history of the Crl regulon through the bacteria and archaea genomes. BMC Genom 20: 299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz SA, Diao R, Wolfe MB, Fivenson EM, Lin XN, Freddolino PL (2019) High‐resolution mapping of the Escherichia coli chromosome reveals positions of high and low transcription. Cell Syst 8: 212–225.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder JW, Simmons LA (2013) Complete genome sequence of Bacillus subtilis strain PY79. Genome Announc 1: e01085‐13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen BA, Landick R (2019) Transcription of bacterial chromatin. J Mol Biol 431: 4040–4066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J‐E, Lin C, Lim HN (2016) Horizontal transfer of DNA methylation patterns into bacterial chromosomes. Nucleic Acids Res 44: 4460–4471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K, Milstein JN, Navarre WW (2016) Xenogeneic silencing and its impact on bacterial genomes. Annu Rev Microbiol 70: 199–213 [DOI] [PubMed] [Google Scholar]
- Singh SS, Singh N, Bonocora RP, Fitzgerald DM, Wade JT, Grainger DC (2014) Widespread suppression of intragenic transcription initiation by H‐NS. Genes Dev 28: 214–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits WK, Grossman AD (2010) The transcriptional regulator Rok binds A+T‐Rich DNA and is involved in repression of a mobile genetic element in Bacillus subtilis . PLoS Genet 6: e1001207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan NL, Marquis KA, Rudner DZ (2009) Recruitment of SMC to the origin by ParB‐parS organizes the origin and promotes efficient chromosome segregation. Cell 137: 697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason LC, Costantino N, Court DL (2007) E. coli genome manipulation by P1 transduction. Curr Protoc Mol Biol 1: 1.17.1–1.17.8 [DOI] [PubMed] [Google Scholar]
- Ueguchi C, Mizuno T (1993) The Escherichia coli nucleoid protein H‐NS functions directly as a transcriptional repressor. EMBO J 12: 1039–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Updegrove TB, Zhang A, Storz G (2016) Hfq: the flexible RNA matchmaker. Curr Opin Microbiol 30: 133–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin‐Hansen P, Eriksen M, Udesen C (2004) The bacterial Sm‐like protein Hfq: a key player in RNA transactions. Mol Microbiol 51: 1525–1533 [DOI] [PubMed] [Google Scholar]
- van der Valk RA, Vreede J, Qin L, Moolenaar GF, Hofmann A, Goosen N, Dame RT (2017) Mechanism of environmentally driven conformational changes that modulate H‐NS DNA‐bridging activity. Elife 6: e27369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma SC, Qian Z, Adhya SL (2020) Correction: architecture of the Escherichia coli nucleoid. PLoS Genet 16: e1009148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzani J (2011) Getting started with RStudio: an integrated development environment for R. Sebastopol, CA: O'Reilly Media, Inc; [Google Scholar]
- Vora T, Hottes AK, Tavazoie S (2009) Protein occupancy landscape of a bacterial genome. Mol Cell 35: 247–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DM, Freddolino PL, Harshey RM (2020) A well‐mixed E. coli genome: widespread contacts revealed by tracking mu transposition. Cell 180: 703–716.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Reeves PR (1998) Organization of Escherichia coli O157 O antigen gene cluster and identification of its specific genes. Infect Immun 66: 3545–3551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Kim Y, Ma Q, Hong SH, Pokusaeva K, Sturino JM, Wood TK (2010) Cryptic prophages help bacteria cope with adverse environments. Nat Commun 1: 147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wang J, Ren G, Li Y, Wang X (2016) Deletion of the genes waaC, waaF, or waaG in Escherichia coli W3110 disables the flagella biosynthesis. J Basic Microbiol 56: 1021–1035 [DOI] [PubMed] [Google Scholar]
- Wasim A, Gupta A, Mondal J (2021) Mapping the multiscale organisation of Escherichia coli chromosome in a Hi‐C‐integrated model. bioRxiv 10.1101/2020.06.29.178194 [PREPRINT] [DOI]
- Wickham H (2009) ggplot2: Elegant graphics for data analysis. Springer Science & Business Media; [Google Scholar]
- Wilhelm L, Bürmann F, Minnen A, Shin H‐C, Toseland CP, Oh B‐H, Gruber S (2015) SMC condensin entraps chromosomal DNA by an ATP hydrolysis dependent loading mechanism in Bacillus subtilis . Elife 4: e6659 [DOI] [PMC free article] [PubMed] [Google Scholar]