

Abstract
Polyubiquitination by E2 and E3 enzymes is crucial to cell cycle control, epigenetic regulation, and development. The hallmark of the E2 family is the ubiquitin (Ub)‐conjugating (UBC) domain that forms a dynamic thioester conjugate with ubiquitin (E2~Ub). Numerous studies have focused on E2 surfaces, such as the N‐terminal and crossover helices, that directly interact with an E3 or the conjugated ubiquitin to stabilize the active, "closed" state of the E2~Ub. However, it remains unclear how other E2 surfaces regulate ubiquitin transfer. Here, we demonstrate the helix–turn–helix (HTH) motif of the UBC tunes the intrinsic polyubiquitination activity through distinct functions in different E2s. Interestingly, the E2HTH motif is repurposed in UBE2S and UBE2R2 to interact with the conjugated or acceptor ubiquitin, respectively, modulating ubiquitin transfer. Furthermore, we propose that Anaphase‐Promoting Complex/Cyclosome binding to the UBE2SHTH reduces the conformational space of the flexible E2~Ub, demonstrating an atypical E3‐dependent activation mechanism. Altogether, we postulate the E2HTH motif evolved to provide new functionalities that can be harnessed by E3s and permits additional regulation to facilitate specific E2‐E3‐mediated polyubiquitination.
Keywords: Anaphase‐Promoting Complex/Cyclosome, E2 ubiquitin‐conjugating enzyme, RING E3 ubiquitin ligase, UBE2R, UBE2S
Subject Categories: Post-translational Modifications & Proteolysis, Structural Biology
Surfaces not involved in direct E3 ligase binding contribute differentially to polyubiquitination by distinct donor/acceptor ubiquitin interactions in different E2 enzymes.
Introduction
The covalent attachment of ubiquitin (Ub) to proteins is fundamental to a wide range of cell signaling processes in eukaryotes. This reversible modification can dictate the fate of the substrate by altering the half‐life, enzyme activity, and subcellular localization of a Ub‐modified target. Post‐translational modification of a protein through Ub‐tagging is not a simple or binary code. A variety of outcomes for a given substrate are achieved through a complex array of Ub topologies. In addition to the many lysine residues on the protein substrate, Ub itself also contains several residues (Met1, Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, and Lys63) that are used to form polyubiquitin chains, giving rise to a multitude of possible homotypic and heterotypic chains (Komander & Rape, 2012; Haakonsen & Rape, 2019). A sophisticated and complex set of enzymes in the Ub system generate the diversity in Ub modification types (Buetow & Huang, 2016; Clague et al, 2019).
For Ub conjugation to occur, Ub is passed through an E1‐E2‐E3 cascade where the Ub‐activating enzyme (E1) is charged with Ub in a Mg‐ATP‐dependent manner to form E1~Ub (~ denotes a covalent intermediate). The Ub is then passed from the E1~Ub to the active site cysteine of a Ub‐conjugating enzyme (E2) to form an E2~Ub conjugate. E3 Ub ligases harness the E2~Ub to transfer Ub to the target substrate. The specificity for different ubiquitination products is largely controlled by E2 and E3 enzymes. RING E3s (the largest family of E3 ligases with ~600 members) co‐recruit the substrate and the E2~Ub onto the same scaffold (Deshaies & Joazeiro, 2009). Broadly stated, the role of the RING E3 ligases is to promote Ub transfer from the E2~Ub thioester to the substrate by activating the E2 (Reverter & Lima, 2005; Dou et al, 2012; Plechanovova et al, 2012; Pruneda et al, 2012; McGinty et al, 2014). In the absence of a RING, the E2~Ub complex is highly flexible, and the mobile Ub samples a large degree of conformational space around the E2 active site (Pruneda et al, 2012; Metzger et al, 2014; Khago et al, 2020). RINGs have been shown to activate the E2~Ub conjugate through direct interactions with the E2 N terminus (Reverter & Lima, 2005; Dou et al, 2012; Plechanovova et al, 2012; Pruneda et al, 2012; McGinty et al, 2014). This interaction stabilizes a “closed” form of the E2~Ub where Ub interacts with the crossover helix of the E2, leaving the thioester linkage susceptible to nucleophilic attack (Fig 1A).
Figure 1. Specific charge‐swap variants of the UBE2SHTH increase enzymatic activity and retain Ub linkage specificity.
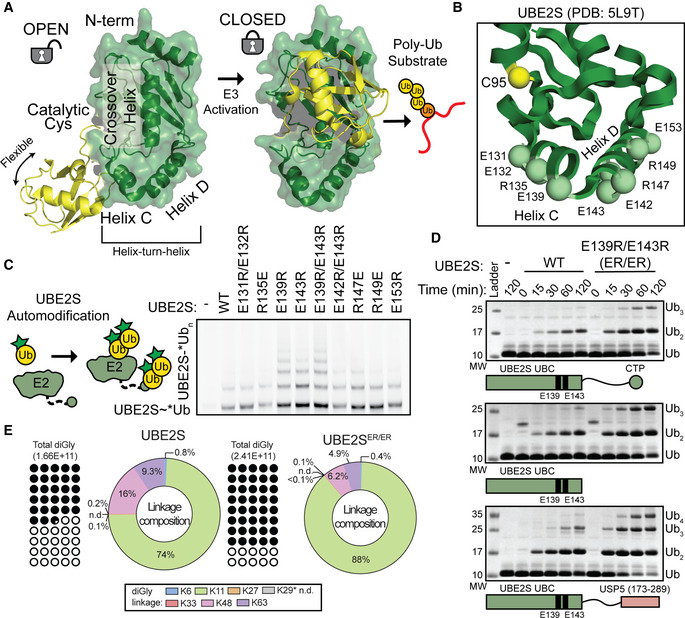
- UBE2SHTH (green, from PDB: 5L9T) charged residues (spheres) that were mutated and tested in autoubiquitination assays.
- E139R, E143R, and the combination ER/ER mutant substitutions in UBE2S increase the amount of automodification, monitored by the presence of *Ub (* denotes fluorescently labeled protein) and using SDS–PAGE and fluorescent scanning.
- ER/ER substitutions increase Ub chain formation in multiple UBE2S constructs (WT, UBC only, and UBC‐UBD fusion), monitored by Coomassie staining of non‐reduced SDS–PAGE gels.
- UBE2S WT and ER/ER form primarily K11‐linked Ub chains, determined by quantifying the total detected diGly linkages (left) and calculating the percentage of specific chain topologies (right) using mass spectrometry.
Source data are available online for this figure.
There are greater than 30 different E2s, which are identified by the hallmark Ub‐conjugating (UBC) domain (Stewart et al, 2016; Gundogdu & Walden, 2019). E2s can contain unique insertions within the UBC domain (e.g., the K48 chain former UBE2R/CDC34/UBCH3), as well as N‐ and/or C‐terminal extensions (Kleiger et al, 2009; Pierce et al, 2009; Wu et al, 2010a; Ziemba et al, 2013; Stewart et al, 2016). Currently, there are ~30 RING‐E2 structures containing a nonspecific E2 (e.g., UBE2D) and truncated E3 (Gundogdu & Walden, 2019). However, E3s often contain additional domains to recruit specific E2s (e.g., E3 binding the backside of the E2 UBC domain), and some E2s have been shown to utilize noncatalytic partners to improve activity (Reverter & Lima, 2005; Das et al, 2009; Brown et al, 2015; Haakonsen & Rape, 2019; Baek et al, 2020a). The interplay between the specificity of chain elongation, E3 domains, and intrinsic E2 activity remains unclear. E2s are highly conserved at the N‐terminal helix, crossover helix, and backside. In contrast, the C‐terminal helix–turn–helix (HTH) motif of the UBC (hereafter referred to as the E2HTH) is evolutionally divergent (Gundogdu & Walden, 2019). Therefore, it is interesting to speculate that the E2HTH has evolved to accommodate distinctive modes of E2‐E3 interactions and regulate E2 activity.
To understand how the E2HTH can be harnessed by an E3 for chain elongation, we examined the tandem polyubiquitination mechanisms of the Anaphase‐Promoting Complex/Cyclosome (APC/C). The APC/C is a member of the cullin‐RING family of E3 ligases (e.g., SCFs) and is responsible for the timely destruction of key cell cycle regulator proteins (e.g., Securin, Cyclin B; King et al, 1996; Baek et al, 2020b; Bodrug et al, 2021). The APC/C uses a dual E2 mechanism where UBE2C/UBCH10 primes the substrate with Ub and builds short polyubiquitin chains with mixed Ub linkages along with a second E2, UBE2S, to generate K11‐linked poly‐Ub and branched K11/K48 chains on the substrate‐linked Ub (Aristarkhov et al, 1996; Yu et al, 1996; Kirkpatrick et al, 2006; Garnett et al, 2009; Williamson et al, 2009b; Wu et al, 2010b; Meyer & Rape, 2014). UBE2C~Ub is recruited and activated by the cullin‐like subunit (APC2 winged‐helix B (WHB)) and the APC11 RING to facilitate the “closed” conformation of UBE2C~Ub (Tang et al, 2001; Brown et al, 2015). However, the APC/C‐UBE2S mechanism is distinct. First, UBE2S does not have the standard RING‐binding residues present in other E2s (Brown et al, 2014; Kelly et al, 2014). Second, a groove formed by the subunits APC2 and APC4 binds to a C‐terminal extension of UBE2S (Brown et al, 2016; Yamaguchi et al, 2016). Third, the APC11 RING domain has been proposed to position the substrate‐linked Ub for diUb synthesis by UBE2S, rather than binding UBE2S directly (Brown et al, 2014, 2016; Kelly et al, 2014; Yamaguchi et al, 2016). Critically, APC2 helices bind to the UBE2SHTH to allow UBE2S~Ub transfer (Brown et al, 2016; Yamaguchi et al, 2016). Currently, the “closed” form of UBE2S~Ub is the only known catalytically competent state as mutations to the crossover helix (C118A) or to the canonical ubiquitin‐binding surface (I44A) impair ubiquitination (Wickliffe et al, 2011). However, it remains unknown how the APC/C interaction with the UBE2SHTH modulates Ub chain elongation.
Here, we performed a series of intricate enzyme and binding assays, phylogenetic analysis, molecular modeling with molecular dynamics and Rosetta, and NMR experiments to understand how the E2HTH functions in chain elongation and E3‐dependent activation. We reveal that the E2HTH can play different roles in Ub chain formation depending on the E2 tested. Charge‐swapping the conserved, negatively charged residues in the UBE2SHTH results in both E3‐dependent and E3‐independent hyperactivity. The reciprocal E2HTH mutations, from positive to negative, in UBE2C and UBE2D2 reduce their enzymatic activity. Computational modeling reveals that the negatively charged UBE2SHTH can interact with the conjugated donor Ub in an extended configuration. Disrupting this interaction through positively charged substitutions or APC2 binding to the UBE2SHTH reduces the prevalence of this relatively inactive conformation and promotes the formation of the “closed” state. In contrast, we show that the mutation of a conserved, positively charged residue in the UBE2R2HTH can reduce the binding of the acceptor Ub, rather than the donor Ub, and impair catalytic activity. Overall, we demonstrate that the E2HTH impacts the intrinsic activity of multiple E2s, which can be modulated by an E3, and postulate that the diversity in the E2HTH can be important for specialization of the E2s.
Results
Specific substitutions at the helix–turn–helix (HTH) motif of UBE2S stimulate Ub chain formation
To understand how the HTH modulates E2 function, we first created a panel of charge‐swapped UBE2S variants in this region (Fig 1B). Using UBE2S autoubiquitination assays where the fluorescent *Ub (* denotes the position of the fluorescent label) is monitored, we see most substitutions had no effect on activity (Fig 1C). Surprisingly, changing the charged residues from Glu to Arg at positions 139 and 143 noticeably increased the intrinsic UBE2S autoubiquitination activity (Fig 1C). To further demonstrate that the E139R/E143R (hereafter referred to as ER/ER) substitutions enhance the activity of UBE2S, we examined if these mutations were dependent on the C‐terminal extension of UBE2S. We introduced the ER/ER mutations in constructs containing only the UBC core (residues 1–156) or a UBE2S chimera where a Ub‐binding domain (UBD) from USP5 replaces the UBE2S C‐terminal peptide (CTP, residues 196–222) that interacts with the APC/C (Bremm et al, 2010). These substitutions increased Ub chain synthesis for all of the UBE2S constructs tested (Fig 1D and Appendix Fig S1G). To confirm that a change in Ub‐linkage specificity was not responsible for the enhanced E2 activity, we performed quantitative mass spectrometry analysis on these reactions. The results revealed an increase in both percentage and amount of K11‐linked Ub chains, indicating that the intrinsic specificity of UBE2S to form K11‐linked chains remained intact (Figs 1E and EV1A).
Figure EV1. Charge‐swaps at the E2HTH impact activity across several E2 family members.
- A
-
B–DFluorescent scans monitoring the diUb production by UBE2S (B), UBE2D2 (C), and UBE2R2 (D) WT and charge‐swapped variants. The UBE2S, UBE2D2, and UBE2R2 WT panels used in Fig 2E,G,H are shown for comparison.
Taken together, substitutions in the UBE2SHTH influence the intrinsic chain elongation activity independently of the C‐terminal extension, but also retain K11‐linkage specificity in the absence of an E3. Furthermore, the positively charged nature of these substitutions and their location on the UBE2SHTH surface suggests a role for electrostatics at the E2HTH in the regulation of Ub transfer.
Helix–turn–helix motif of the UBC modulates activity of multiple E2s
To understand how the E2HTH impacts Ub chain elongation, we tested other E2s that have different charges in the HTH in diUb synthesis assays (Fig 2A). In contrast to UBE2S, UBE2C (Fig 2B), UBE2D2 (Fig 2C), and UBE2R2 (Fig 2D) have positively charged residues at analogous positions in the E2HTH. Therefore, we expected that changes from positively to negatively charged residues would reduce the Ub‐transfer activity of these E2s. To pinpoint the charged residue(s) responsible for the modulation of E2 function, substituted variants of UBE2S, UBE2C, UBE2D2, and UBE2R2 were tested in a pulse‐chase analysis of diUb synthesis, allowing us to sensitively monitor the E2~Ub conjugate during a single turnover. Briefly, the E2s are loaded with equimolar donor *Ub by the addition of E1 and Mg‐ATP, forming E2~*Ub. These reactions were then quenched with EDTA to prevent subsequent formation of E2~*Ub. In the chase reaction, a free, unlabeled acceptor Ub, missing the terminal glycine residues (residues 75–76, UbΔGG), is added and diUb formation is monitored. UBE2S will automodify on a lysine (K100) within its active site (Liess et al, 2019); therefore, we substituted K100 for Arg100 (K100R) to mitigate possible artifacts due to autoubiquitination.
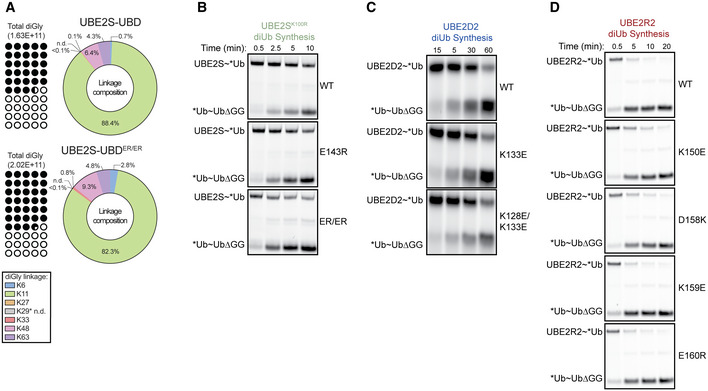
Figure 2. The charge at the E2HTH modulates the intrinsic activity of the E2.
-
ACartoon of pulse‐chase diUb synthesis assays, related to (E–H), where the donor Ub is fluorescently labeled and transferred to unlabeled acceptor UbΔGG.
- B
- C
- D
-
E–HPulse‐chase diUb synthesis assays using different E2s, UBE2S (E), UBE2C (F), UBE2D2 (G), or UBE2R2 (H). DiUb formation was monitored by fluorescent scanning (top), quantitated (bottom), and assessed for statistical significance by either an unpaired Welch’s t‐test (UBE2C and UBE2R2) or one‐way ANOVA (UBE2S and UBE2D2) (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, n = 3 independent, technical replicates for all E2s). Error bars: SEM. Data normalized to the maximal WT diUb formation. # denotes a contaminant band.
-
IWeblogos of the HTH motifs of UBE2S, UBE2C, UBE2D, and UBE2R. Residues colored according to their average net negative (red) or positive (blue) charge. Residues of interest on UBE2S, UBE2C, UBE2D, and UBE2R are labeled. UBE2R is extended to show conservation of D143, used in Fig 4.
-
JNegative charges at the E2HTH weaken the diUb synthesis activity of the E2 family. Electrostatic models of the UBE2S, UBE2C, UBE2D2, and UBE2R2 HTH motifs were generated using the PBEQ solver (Jo et al, 2008). Active site cysteine is indicated as a yellow sphere. Negative to positive charge represented as a red to blue gradient.
Source data are available online for this figure.
Interestingly, the charge‐swapped variants of these E2s had different effects depending on the E2 that was examined. First, UBE2S showed an increase in diUb formation when the charge‐swapped substitutions were added (Figs 2E and EV1B). Significantly, the E139R substitution had the most dramatic effect and enhanced diUb synthesis by ~3.5‐fold. Second, the positive to negative charge‐swaps at similar positions in UBE2C and UBE2D2 (K157E and K128E, respectively) significantly reduced the formation of diUb compared to the wild‐type versions of these E2s (Figs 2F and G, and EV1C), consistent with the results for UBE2S. Another chain‐elongating E2, UBE2R, presents a slightly different E2HTH and functions with SCF E3 ligase complexes. Human UBE2R has an additional helical turn in the HTH and a significantly more positive surface than the other E2s tested. Several charge‐swaps were created on UBE2R2 at residues R149, K150, D158, K159, and E160. Of those, R149 and K150 roughly aligned with position 139 of UBE2S. Interestingly, while most substitutions had little to no effect on UBE2R2 activity, the R149E mutation resulted in a drastic reduction in diUb formation by ~85% (Figs 2H and EV1D). Taken together, our results show that electrostatic interactions at the E2HTH impact chain elongation in several different E2s, suggesting a potential role for the E2HTH that is conserved across the E2 family.
To explore the electrostatic variability in the E2HTH, we constructed a deep sequence alignment and consensus sequence for each E2, aligned them using Promals3D, and created a phylogenetic tree. This phylogenetic reconstruction resembled others and indicated that the E2s we tested were sampled broadly across the tree (Fig EV2A; Winn et al, 2004; Osborne et al, 2021). Based on this consensus alignment, we also analyzed the Shannon entropy across E2 families. E2HTH residues were significantly more variable than the average position in the E2, and several E2s lacked an HTH region altogether (Fig EV2A and B). Through our deep sequence alignment for each E2, we found that several of our mutated positions are highly conserved. A negatively charged residue at UBE2S position 139 is strongly conserved (> 90%), while positively charged residues at UBE2D position 128 and UBE2R positions 149 and 150 are weakly conserved (~65%), strongly conserved (> 90%), and not conserved, respectively (Figs 2I and EV2C). These results mirror the direction and magnitude of activity changes due to substitutions at these positions in our assays, suggesting that there is a functional significance to highly conserved residues in the E2HTH. More broadly, UBE2S has the most overall negatively charged HTH compared to all other E2s (Figs 2I and J, and EV2A and C). Interestingly, UBE2A is the only other E2 with a highly conserved, negatively charged residue at position 139. This position is of particular interest because it displayed the greatest effect on diUb synthesis across our tested E2s, UBE2S, UBE2C, UBE2D2, and UBE2R2 (Figs 2E–H and EV2A and C). Other E2s display less conservation or contain positively charged residues at this position, including UBE2C, UBE2D, and UBE2R (Figs 2I and EV2C). Overall, we observe differences between the HTH regions of the E2 family, but residue electrostatics within specific E2s remain conserved.
Figure EV2. The HTH is a variable region across E2 family members but retains some charge conservation for individual E2s.

- Maximum likelihood phylogenetic tree of the E2 family, including charge conservation models at the E2HTH.
- Shannon entropy plot of residue conservation across the E2 family. The majority of residues in the E2HTH (pink stars) have increased Shannon entropy compared to the E2 mean represented by the horizontal line.
- Weblogos of E2HTH residue conservation for each E2 family member. UBE2S, UBE2C, UBE2D, and UBE2R are repeated from Fig 2I for comparison.
UBE2SHTH and UBE2RHTH motifs have distinct functions during chain elongation
To examine how different charges at the E2HTH function during diUb synthesis, we tested the chain‐elongating E2s UBE2S and UBE2R in E2~Ub transfer assays that eliminate the bulk of free Ub during catalysis. In these experiments, either the thioester‐linked donor Ub or the noncovalent acceptor Ub were substituted during chain elongation (Fig 3A and B). First, the E2 was subjected to the single‐turnover lysine discharge assay where the E2 was loaded with equimolar *Ub, similar to the diUb synthesis assay (Fig 3A). Then, the E2~*Ub loading reaction was quenched and free lysine was added in the place of an acceptor Ub to facilitate *Ub discharge from the E2. The lysine discharge assay is particularly sensitive and monitors the disappearance of the E2~*Ub conjugate to track product formation. In addition to the Lys100 residue, UBE2S may automodify on its C‐terminal extension (Liess et al, 2019). Therefore, we introduced the K100R substitution into a UBE2S core (1–156) construct to further limit confounding activity due to autoubiquitination. In this assay, the UBE2S charge‐swapped variants retained their enhanced activity compared to wild type (Figs 3C and EV3A). In contrast, the UBE2R2 R149E variant performed nearly identically to wild type (Figs 3D and EV3B). These results indicate that the markedly reduced activity for the UBE2R variant is due to the body of the acceptor Ub.
Figure 3. UBE2SHTH and UBE2R2HTH charged residues contribute to different steps during Ub chain elongation.

- Schematic of a lysine discharge assay. The acceptor Ub is replaced with free lysine to monitor the stability of the E2~Ub.
- Schematic of a *LRLRGG (C terminus of Ub) peptide transfer to an acceptor UbΔGG, eliminating interactions with the donor Ub.
- The E139R substitution decreases the stability of UBE2S~Ub compared to WT, revealed by fluorescent scanning of a lysine discharge assay (top) and its quantification (bottom). Statistical comparison was performed using one‐way ANOVA (*P ≤ 0.05, **P ≤ 0.01, n = 4 independent, technical replicates). Error bars: SEM. Data normalized to the initial time point of each respective E2~Ub band.
- UBE2R2 R149E has a similar E2~Ub stability as WT, monitored by fluorescent scanning (top) of the lysine discharge assay and its quantification through an unpaired Welch’s t‐test (bottom) (n = 3 independent, technical replicates). Error bars: SEM. Data normalized to the initial time point of each respective E2~Ub band.
- Fluorescent scans (top) and quantification (bottom) of a *LRLRGG peptide transfer assay comparing UBE2S WT and ER/ER reveals no significant difference through an unpaired Welch’s t‐test (n = 3 independent, technical replicates). Error bars: SEM. Data normalized to the maximal *LRLRGG‐Ub formation by WT UBE2S.
- UBE2R2 R149E is still defective in the transfer of the *LRLRGG peptide to an acceptor Ub compared to WT, probed by fluorescent scanning (top) and its quantitation (bottom). Statistical comparison was performed using an unpaired Welch’s t‐test (**P ≤ 0.01, n = 5 independent, technical replicates). Error bars: SEM. Data normalized to the maximal *LRLRGG‐Ub formation by WT UBE2R2.
Source data are available online for this figure.
Figure EV3. UBE2R2HTH facilitates acceptor Ub binding for diUb synthesis.

- Fluorescent scan of the lysine discharge assay monitoring the stability of the UBE2S~*Ub. The panel of the UBE2S WT from Fig 3C is shown for comparison.
- Fluorescent scans monitoring UBE2R2~*Ub in lysine discharge assays. UBE2R2 WT panel from Fig 3D is shown for comparison.
- Equilibrium data fitting of Ub titrations in BLI experiments to detect interactions between Ub and immobilized UBE2R2 variants. Mean data points of n ≥ 3 independent, technical replicates are shown. Error bars: SD.
- Corresponding raw BLI traces used for equilibrium data fitting in (C).
- Fitting of the METRIS roll parameter data from BLI data.
Next, we assayed the transfer of a fluorescent peptide consisting of the Ub C terminus (*LRLRGG), instead of the donor Ub, to an acceptor UbΔGG (Fig 3B) (Chaugule et al, 2020). In this *LRLRGG transfer assay, the UBE2S ER/ER variant no longer displayed enhanced activity when compared to the wild‐type enzyme, suggesting that its enhanced activity is dependent on the conjugated donor Ub (Fig 3E). In contrast, the activity of UBE2R2 harboring the R149E substitution was still significantly reduced, further suggesting that the UBE2R2HTH impacts acceptor Ub recruitment during catalysis (Fig 3F).
In summary, the lysine discharge and *LRLRGG transfer assay cleanly delineated contributions of the donor and acceptor Ub interactions with UBE2S and UBE2R2 to their intrinsic activity. These experiments revealed that UBE2S and UBE2R2 charge‐swap variants impact distinct stages of Ub transfer. For UBE2S, the HTH substitutions modulate UBE2S activity through the donor Ub. For UBE2R2, the HTH variant perturbs interactions with the acceptor Ub, indicating that the E2HTH has divergent functions in UBE2S and UBE2R2.
UBE2R2HTH is used to bind to the acceptor ubiquitin
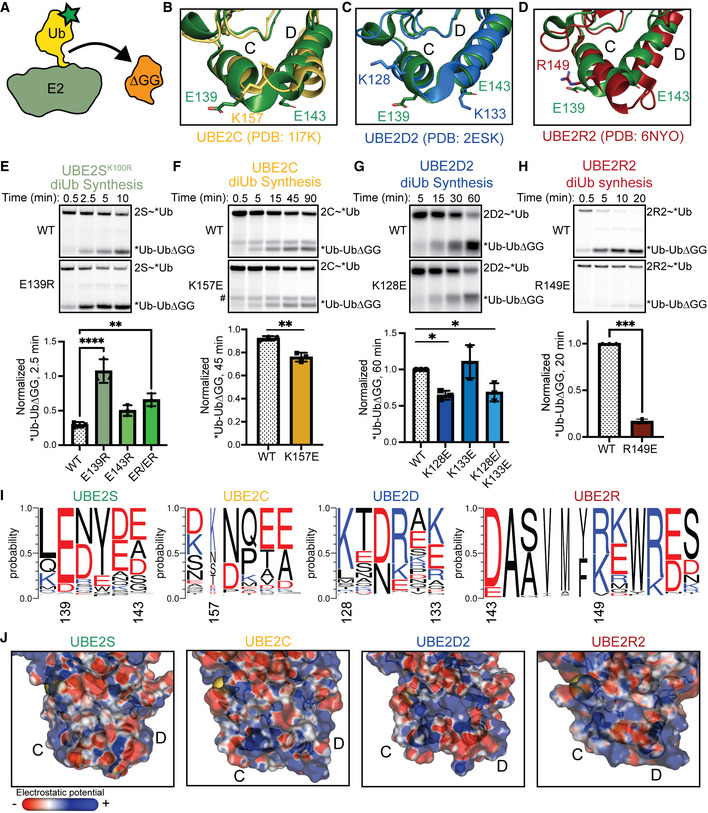
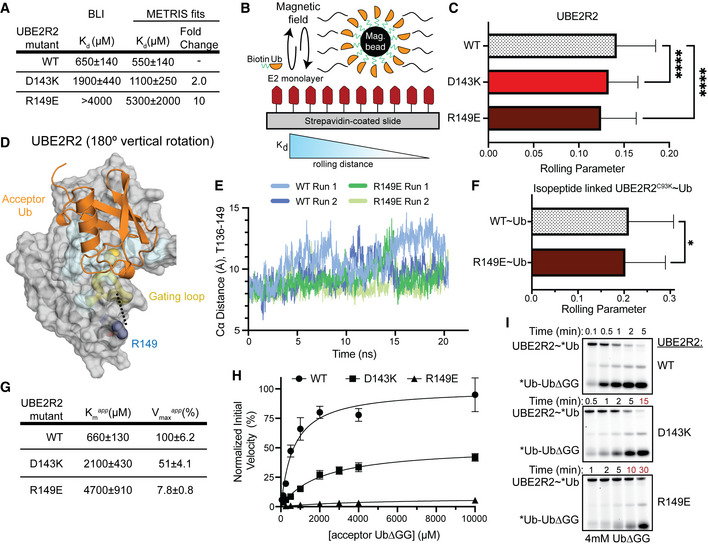
Previous studies have examined how UBE2R2 recruits the acceptor Ub. For example, an early study replaced a patch of residues (143, 147, 149, 150, and 153) with alanines to reveal a defect in activity (Gazdoiu et al, 2007). A later kinetic study characterized a specific charge‐swap mutation at the N terminus of helix 3 of the UBE2RHTH (D143K) that decreased the rate of diUb formation by 7‐fold but rescued a defective acceptor Ub R54E mutation (Hill et al, 2016). Thus, we suspected that the R149E substitution reduced Ub binding to UBE2R2. To test this hypothesis, we first measured the binding affinity for each of these substituted variants using biolayer interferometry (BLI). Previous studies found a Km for the acceptor Ub to UBE2R2 to be ~500 μM (Liwocha et al, 2021), within error of our observed K d value for wild‐type UBE2R2 of 650 μM ± 140 μM (Fig 4A). Both D143K and R149E variants had significantly reduced binding with K d values of ~2 mM and > 4 mM, respectively (Figs 4A and EV3C and D). However, the BLI curves are not saturating as a result of thermodynamic non‐ideality at high concentrations of Ub, likely due to dimerization of the Ub (Saluja & Kalonia, 2008; White et al, 2010). To overcome the weak binding of these variants and technical limitations of BLI, we used a recently developed technique called METRIS (mechanically transduced immunosorbent assay). METRIS can measure ultra‐weak interactions, uses a very small quantity of protein, and is a single particle method, meaning it can accurately measure small differences in binding affinity and has very high statistical power. The METRIS assay measures the distance that a protein‐functionalized magnetic bead will roll on a protein‐functionalized surface when subjected to a rotating magnetic field (Petell et al, 2021). The distance each bead travels is dependent on the strength of protein–protein interactions and is measured by the dimensionless rolling parameter (RP) (Fig 4B). To generate our METRIS system, we biotinylated native cysteines on UBE2R2 wild‐type, D143K, and R149E, and attached them to a streptavidin‐coated surface. Ub was attached to magnetic beads via an N‐terminal biotin. The trend of the rolling parameters matched the BLI assay: wild‐type (0.143 ± 0.001) > D143K (0.135 ± 0.001) > R149E (0.121 ± 0.001) (Fig 4C). By fitting the RP to the BLI data, we predict that the R149E variant binds the acceptor Ub 10‐fold weaker than wild‐type UBE2R2 (Fig 4A and Fig EV3E). Collectively, these data provide the first direct measurement of the acceptor Ub binding to the UBE2R2HTH.
Figure 4. The UBE2R2 R149E variant disrupts acceptor Ub binding and gating loop flexibility.
-
ADissociation constants of UBE2R2 and its variants binding to acceptor Ub as determined by BLI and fitted data from METRIS. Data from n ≥ 3 independent experiments.
-
BSchematic of METRIS assay set‐up and parameter analysis to assess the binding between UBE2R2 and Ub. In brief, biotinylated Ub is linked to a magnetic bead that is introduced to a magnetic field while on a UBE2R2 functionalized coverslip. The movement of each bead is quantified, and the rolling parameter correlates to binding affinities between Ub and E2.
-
CPlot of the METRIS rolling parameters of UBE2R2 WT, D143K, and R149E compared using one‐way ANOVA (****P ≤ 0.0001, WT: 63 beads × 17 rolls n = 1071, R149E: 46 beads × 17 rolls n = 782, D143K: 47 beads × 17 rolls n = 799; technical replicates), indicating a weaker interaction with Ub between the UBE2R2 variants compared to WT. Error bars: SEM.
- D
-
EUBE2R2 R149E restricts the movement of the gating loop compared to WT as shown by the distances of the Cα atoms from T136 and R149 in molecular dynamics simulations.
-
FPlot of METRIS rolling parameters of WT and the R149E substituted variant of UBE2R2 isopeptide‐linked to the donor Ub compared using one‐way ANOVA (*P ≤ 0.05, WT‐Ub: 47 beads × 17 rolls n = 799, R149E‐Ub: 114 beads × 17 rolls n = 1938; technical replicates). Error bars: SEM.
-
G–IRole of the UBE2RHTH in diUb formation is determined from kinetic parameters (G, H) for mutants upon titrating the acceptor Ub in pulse‐chase reactions following UBE2R2~*Ub, monitored by fluorescent scanning of nonreducing SDS–PAGE gels (I). Apparent K m and V max were determined using the Michaelis–Menten equation (n ≥ 3 independent, technical replicates). Error bars: SEM. Data normalized to the WT apparent V max.
Source data are available online for this figure.
To determine the rationale for the effects of the R149E mutation, we examined a previous model of UBE2R‐mediated diUb synthesis (Fig 4D; Hill et al, 2016). This model does not have R149 directly contacting the acceptor Ub. Furthermore, it would be geometrically unfeasible for the acceptor Ub to contact this portion of the UBE2R2HTH. Therefore, we used computational modeling to study the impact of this mutation on UBE2R. First, we used the Rosetta molecular modeling suite to insert the R149E mutation into the human UBE2R protein. The best fit rotamer allowed the formation of a hydrogen bond with the side chain of T136, which is not possible with the native R149. T136 is found in the “gate loop” adjacent to a highly conserved N135 that can form hydrogen bonds with G75 based on other E2 crystal structures (Plechanovova et al, 2012). The gate loop forms the active site cleft and interacts with both the donor Ub tail and the acceptor Ub. Dynamics of this gate loop have been broadly implicated in E2 catalysis (Rout et al, 2014; Jones et al, 2019). Therefore, we used molecular dynamics (MD) to further interrogate the stability of the R149E mutant. We ran two 20 ns simulations for the UBE2R2 wild‐type and R149E variant to investigate local impacts of the R149E mutation. Overall, the R149E mutation led to a shorter distance between the gate loop and UBE2R2HTH, on average a 1 Å shorter Cα‐Cα distance between the R149E substitution and T136 (Fig 4E). Although this change is relatively small, we suspect this substitution impacts the acceptor Ub‐binding site, including its central residues (A139 and P140), based on our model and other mechanistic data (Sadowski et al, 2010). Furthermore, we expect that this mutation could impact the enzymatic activity of the E2, as the gate loop is thought to aid in catalysis by partially stabilizing the tetrahedral oxyanion intermediate (Jones et al, 2019). A recent study showed that a similar subtle change resulted in a drastic K m and k cat defect when the Ub K48 side chain was synthetically increased in length by a single methylene (Liwocha et al, 2021).
To provide biochemical support for our gating loop hypothesis, we performed METRIS to further characterize the effect of R149E in the context of the UBE2R2~Ub conjugate. Because both the R149E substitution and donor Ub are expected to modulate the conformation and dynamics of the gating loop, we tested whether adding a donor Ub to UBE2R2 would reduce the binding defect caused by R149E. We used METRIS to study this interaction, because generating large quantities of UBE2R~Ub is prohibitive for many binding methods. After forming a UBE2R2~Ub isopeptide conjugate through a C93K mutation, we found only a small reduction in rolling parameter for the UBE2R2~Ub conjugate harboring the R149E mutation (< 2‐fold defect) in binding to the acceptor Ub (Fig 4F). This result indicates that the donor Ub can partially rescue the 10‐fold defect observed with native UBE2R2. Additionally, we performed a pulse‐chase diUb synthesis assay titrating UbΔGG to determine the apparent kinetic parameters, K m app and V max app, between the UBE2R2~Ub conjugate and acceptor Ub (Fig 4G–I). UBE2R2 D143K and R149E displayed a 3‐fold and 7‐fold increase in the K m app compared to wild type, respectively. This result agreed with the BLI and METRIS results using unconjugated UBE2R2 (Fig 4A). Furthermore, the V max app was markedly reduced by ~50% and 90% for D143K and R149E, respectively. Overall, our results suggest that the HTH impacts acceptor Ub binding and catalysis during UBE2R2‐depedent Ub chain formation.
Mechanistic basis of the UBE2SHTH during diUb synthesis
In contrast to the UBE2RHTH, specific substitutions in the UBE2SHTH promote catalysis when the full body of the conjugated donor Ub is present (Fig 3). As the E139 and E143 residues of the UBE2SHTH are distant from the site the donor Ub occupies in the “closed” state, it seems implausible that substitutions at these residues could stabilize the “closed” conformation to enhance UBE2S activity. Therefore, we hypothesized that the negatively charged residues of wild‐type UBE2SHTH stabilize an “open” UBE2S~Ub conformation. Moreover, Ub has a positively charged β‐sheet surface that is also responsible for the interactions at the E2 crossover helix in the “closed” state. To evaluate this hypothesis, we conducted four 500 ns MD simulations for the wild‐type UBE2S~Ub conjugate. In these simulations, both the E2 and Ub remain well‐folded. Within the first 50 ns of the simulation, the conjugated Ub positions itself on a surface of the E2. The Ub position was split evenly between “open” and “closed” conformations. In one of the “open” states, Ub is near the UBE2SHTH and forms a hydrogen bond interaction with E139 for more than 50% of the trajectory (Fig 5A). This hydrogen bond provides a potential open‐state‐stabilizing interaction between the donor Ub and the UBE2SHTH. To examine this hypothesis, we tested *Ub R54E in UBE2S autoubiquitination assays and found that this Ub substitution slightly reduced the enhanced activity of UBE2S E139R (Fig 5B). This result supports our model that interactions between the UBE2SHTH and the donor Ub can stabilize the “open” state and impair catalysis.
Figure 5. UBE2SHTH interacts with the donor Ub, suggesting that APC2 can prevent this conformation to facilitate the “closed” conformation.
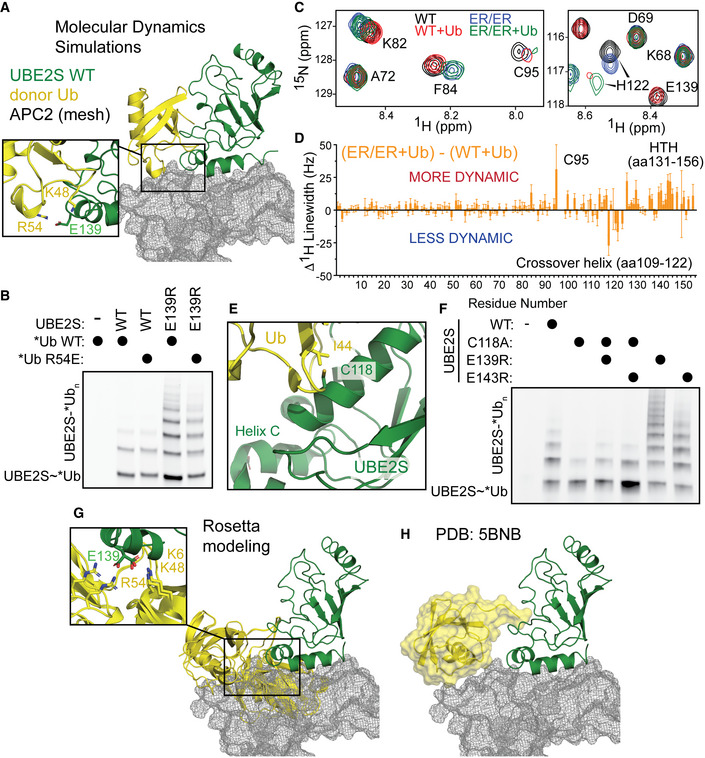
- Molecular dynamics simulation of UBE2S (green)~Ub (yellow) reveals electrostatic interactions between the positive surface of Ub and UBE2S E139 (cutout). Rosetta model of APC2 (gray mesh, from PDB: 5L9T) docked on UBE2S~Ub would sterically clash with this extended conformation (Brown et al, 2016).
- Reducing the positive charge of *Ub reduces the autoubiquitination of the E139R‐substituted UBE2S variant, monitored by fluorescent scanning of an SDS–PAGE gel.
- HSQC spectra displaying chemical shift perturbations and linewidth changes in 15N‐labeled UBE2SUBC WT apo (black) or in complex with Ub (red) and UBE2SUBC ER/ER apo (blue) or in complex with Ub (green).
- Plot of 15N‐labeled UBE2SUBC WT and ER/ER 1H linewidth changes in the presence of Ub revealing the ER/ER variant has a more dynamic active‐site cysteine and HTH, but a more stabilized crossover helix, indicative of the “closed” state. Shown are the best‐fit values and standard deviations (SD) estimated from the covariance matrix analysis of NMR spectra with n = 2048 measurement points for 16 ppm in the 1H dimension.
- C118A substitutions decrease UBE2S autoubiquitination, measured by fluorescent scanning of SDS–PAGE gels, indicating the significance of the “closed” formation of the UBE2S~Ub for activity.
- Rosetta modeling of UBE2S~Ub, in the absence of APC2, reveals multiple “open” conformations that would sterically clash with a docked Rosetta model of the APC2 (gray mesh, PDB: 5L9T; Brown et al, 2016). Cutout showing multiple possible interactions between UBE2S and Ub.
Source data are available online for this figure.
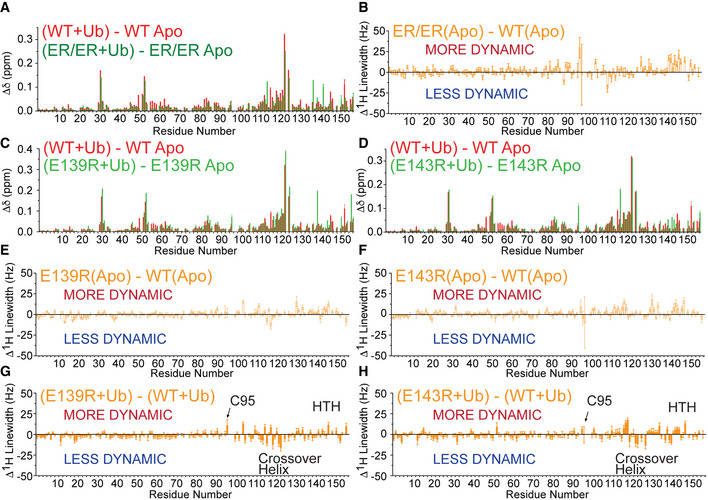
Next, we used NMR to characterize how the UBE2SHTH variants influence the interactions between UBE2S and Ub. Chemical shift analyses of 15N‐1H HSQC NMR spectra of UBE2S wild‐type and the UBE2S ER/ER variant revealed very similar perturbations upon Ub binding, where the crossover helix exhibits the largest chemical shift differences between Ub‐bound and Ub‐unbound states (Figs 5C, EV4A, and Appendix Figs S2 and S5). These findings suggest that the UBE2S ER/ER variant binds Ub in an overall “closed” conformation similar to the one shown previously for the wild‐type UBE2S (Wickliffe et al, 2011). However, differences were observed in the dynamic properties of E2‐Ub interactions when comparing the 1H linewidth data. In the free state, relative to the wild‐type enzyme, the UBE2S ER/ER variant displays broader linewidth for NMR peaks of the active site residue C95 and the HTH (Figs 5C and EV4B). This result indicates that backbones of these regions in the variant are more conformationally flexible at the micro‐to‐millisecond timescale than the wild‐type enzyme. Upon Ub binding, C95 and the HTH remain relatively more flexible in the variant, yet residues in the C terminus of the crossover helix (C118‐H122) display much sharper linewidth compared to wild type (Fig 5C and D). This observation suggests reduced flexibilities at the binding interface of the variant UBE2S‐Ub complex and further supports the formation of a more ordered, “closed” conformation, which is assumed to be the only active state of UBE2S~Ub. Furthermore, the individual E139R and E143R variants interacted with Ub in a similar manner as UBE2S harboring both substitutions but exhibited reduced effects on the 1H linewidth (Fig EV4, EV5C–H and Appendix Figs S3 and S4). These results suggest that both E139R and E143R play roles in modulating the conformational dynamics and shifting the population of the ER/ER variant UBE2S‐Ub complex to a more “closed” conformation.
Figure EV4. NMR analysis of UBE2S and its variants binding to Ub.
-
ANMR chemical‐shift perturbations due to the binding of Ub to either UBE2S WT or the ER/ER variant.
-
BNMR 1H linewidth comparisons of apo (no Ub) UBE2S WT and ER/ER variant.
-
C, DNMR chemical‐shift perturbations due to the binding of Ub to either UBE2S WT or the E139R variant (C) or E143R variant (D).
-
E, FNMR 1H linewidth comparisons of apo UBE2S WT and E139R (E) or E143R variant (F).
-
G, HNMR 1H linewidth comparisons of UBE2S WT and E139R (G) or E143R (H) variant when Ub is added.
Data information: (A–H) Shown are the best‐fit values and standard deviations (SD) estimated from the covariance matrix analysis of NMR spectra with n = 2048 measurement points for 16 ppm in the 1H dimension.
Figure EV5. APC2 competes with the donor Ub to bind to the UBE2SHTH .

-
AStructure of UBE2C bound to the APC11 RING and APC2 WHB (PDB: 5L9U; Brown et al, 2016), demonstrating that the UBE2CHTH is not interacting with the APC/C.
-
BRepresentative fluorescent scan (left) and its quantification (right) monitoring CycB* ubiquitination by APC/C and either UBE2C WT or K157E. Data were statistically analyzed using an unpaired Welch’s t‐test (*P ≤ 0.05, n = 4 independent, technical replicates). Error bars: SEM. Data normalized to the unmodified substrate band from reactions lacking an E2.
-
CQuantification of Fig 6E monitoring the polyubiquitination of Ub‐CycB* by a panel of UBE2SHTH charge‐swap variants. Data were statistically analyzed by one‐way ANOVA (*P ≤ 0.05, ****P ≤ 0.0001, n = 3 independent, technical replicates). Error bars: SEM. Data normalized to the unmodified substrate band from reactions lacking an E2.
-
D, EFluorescent scan (D) and its quantification (E) monitoring APC/C‐dependent Ub‐Securin (Ub‐Sec*) ubiquitination utilizing the panel of UBE2S variants (related to Fig 6E). Data were statistically analyzed by one‐way ANOVA (n = 3 independent, technical replicates). Error bars: SEM. Data normalized to the unmodified substrate band from reactions lacking an E2.
Source data are available online for this figure.
Both the MD and NMR results hinted that the negatively charged residues in the UBE2SHTH stabilize the catalytically inactive “open‐state” conformation and that the ER/ER substitutions promote the “closed‐state”. To further test this hypothesis, we introduced the crossover helix C118A mutation, which reduces the formation of the “closed” state, into these hyperactive UBE2S variants (Wickliffe et al, 2011; Brown et al, 2016). Indeed, the activity of the UBE2S C118A variant was markedly reduced compared to the wild‐type enzyme. However, the charge‐swapped variants of UBE2S slightly rescued this defect in autoubiquitination assays, albeit still with less activity than wild‐type UBE2S (Fig 5E and F). Together, these data support our model that the UBE2SHTH interacts with positive charges on the Ub surface and perturbing these interactions result in the formation of the active, “closed” E2~Ub state.
The concept that disfavoring the “open” states could activate an E2, as revealed by the charge‐swapped UBE2S variants, suggests that an E3 could also use this mechanism to enhance polyubiquitination. To better understand the conformational ensemble of the UBE2S~Ub conjugate, we used the E2‐thioester protocol in Rosetta to generate 100,000 structures. In the top 200 structures, we observed two different orientations where positively charged residues in the conjugated Ub interact with E139, indicating that additional “open” conformations are possible. Interestingly, the conjugated Ub would sterically clash with APC2 in both conformations (Fig 5G). These structures resemble the crystal structure of UBE2S~Ub, which would also clash with APC2 (Fig 5H; Lorenz et al, 2016). Furthermore, there are other possible “open” conformations where the Ub would clash with APC2 even though the conjugated Ub is not interacting with E139. Thus, this modeling implies that APC2 could enhance the formation of the “closed” conformation of UBE2S~Ub by reducing the possible “open” configurations of the E2~Ub conjugate.
Charge‐swapped HTH substitutions function in the context of E3‐dependent ubiquitination
To examine how an E3 can modulate E2 activity through interactions at the HTH, we interrogated three E2s (UBE2C, UBE2D2, and UBE2S) that partner with the APC/C. Previous studies have shown that substitutions at the UBE2SHTH or the UBE2S‐interacting helices of APC2 result in a catalytic defect but do not disrupt the binding of UBE2S to the APC/C (Fig 6A; Brown et al, 2014, 2016; Kelly et al, 2014). Unlike UBE2S, UBE2C and UBE2D2 are not expected to use the E2HTH to interact with the APC/C based on the available structural and mutagenesis data (Brown et al, 2014; Brown et al, 2015; Chang et al, 2015; Brown et al, 2016). Instead, they are both activated by the APC11 RING domain, while the winged‐helix B (WHB) of APC2 also binds to the backside of UBE2C (Figs 6B and EV5A; Brown et al, 2015, 2016).
Figure 6. E2HTH substitutions differentially impact substrate ubiquitination by the E3 APC/C.

-
A, BSchematic of UBE2S~Ub (A) and UBE2C~Ub interactions (B) with the APC/C. UBE2S~Ub uses its C‐terminal extension to bind to a groove formed by APC2 and APC4. The UBE2SHTH interacts with APC2 and the APC11 RING domain positions the substrate‐linked Ub for chain elongation. UBE2C~Ub is positioned by the APC11RING and APC2 winged‐helix B (WHB) domains during substrate ubiquitination (Brown et al, 2015, 2016).
-
CSecurin ubiquitination by APC/C and either UBE2C WT or K157E, resolved by SDS–PAGE and fluorescent scanning. Quantification of 4 or more ubiquitins added on the substrate (right) are calculated as a fraction of total ubiquitination and analyzed by an unpaired Welch’s t‐test (****P ≤ 0.0001, n = 5 independent, technical replicates). Error bars: SEM.
-
DSimilar to C, Ub‐securin ubiquitination by APC/C and either UBE2D2 WT or charge‐swap variants. Quantification of 4 or more ubiquitins (right) added on the substrate are calculated as a fraction of total ubiquitination and analyzed by one‐way ANOVA (****P ≤ 0.0001, n = 4 independent, technical replicates). Error bars: SEM.
-
ERepresentative fluorescent scan of an SDS–PAGE gel of APC/C‐dependent polyubiquitination of Ub‐CyclinB* by a panel of UBE2SHTH charge‐swap variants. Quantification in Fig EV5C.
-
FThe recombinant UBE2S ER/ER variant increases Cyclin B degradation in G1 extracts compared to UBE2S wild type (WT), monitored by immunoblot. G1 extracts were prepared from HeLa S3 cells treated with either control firefly luciferase siRNA (siFF) or two UBE2S siRNAs (siUBE2S).
Source data are available online for this figure.
Using a recombinantly expressed and purified system, we monitored APC/C‐E2‐dependent activity on known substrates Cyclin BNTD* (CycBNTD*) or Securin* (Sec*). Based on the quantitation of the unmodified substrate and ubiquitinated products, both charge‐swapped UBE2C and UBE2D2 variants displayed reduced APC/C‐dependent ubiquitination activity (Figs 6C and D, and EV5B), consistent with the diUb assays (Fig 2F and G). Specifically, the activities of the charged‐swapped E2 variants were decreased by ~30–60% compared to wild type. To bypass the need for UBE2C‐dependent priming of the substrate and specifically test APC/C‐UBE2S‐dependent activity, Ub was genetically linked to the substrate. As expected from previous studies, several relatively conserved (> 50% based on our sequence alignment) UBE2S substitutions (E131R/E132R, R135E, R147E) were defective in an APC/C‐dependent manner because of their interactions with APC2 (Figs 6E and EV5C–E). Consistent with the E3‐independent results, the UBE2S E139R, E143R, and ER/ER variants displayed a slight increase in activity. This enhanced APC/C‐dependent activity was then tested in a HeLa cell extract system where APC/C substrate degradation was monitored. Degradation of endogenous APC/C substrate Cyclin B was markedly reduced in UBE2S‐depleted extracts. This defect was rescued by the addition of recombinant UBE2S. When the recombinant UBE2S ER/ER variant was added, substrate degradation was further enhanced (Fig 6F). Taken together, our data suggest that the functional role of the HTH is preserved in E3 catalyzed chain elongation.
APC/C and UBE2SHTH substitutions facilitate the formation of the “closed” state of E2~Ub
To determine if both the APC/C and the UBE2SHTH substitutions enhance Ub chain elongation through a related mechanism, we tested the UBE2S variants against APC/Cs harboring mutations at APC2 that disrupt the APC2‐UBE2S interaction based on previous studies (Fig 7A; Brown et al, 2016). As expected, APC/C containing V542A/E543A and K562D substituted variants of APC2 were defective in UBE2S‐dependent substrate chain elongation assays. However, the charge‐swapped UBE2SHTH variants partially rescued these defects, suggesting that, fundamentally, both APC/C and the UBE2SHTH variants have similar effects on E2~Ub (Fig 7B). Next, we performed diUb synthesis assays using both the entire APC/C and the smaller APC/C Platform (composed of APC1, APC2, APC4, APC5, and APC11). As expected, the APC2 substitutions disrupted UBE2S‐dependent activity, but the UBE2SHTH variants partially rescued these deficiencies (Fig 7C and D). To test whether these functions are dependent on the “closed” state of E2~Ub, C118A‐substituted UBE2S variants were examined in APC/C‐dependent substrate polyubiquitination assays. Noticeably, the HTH charge‐swap substitutions were able to partially rescue the dramatic defect caused by the C118A mutation (Fig 7E). However, these C118A/E139R and C118A/E143R variants did not display the enhanced activity of their E139R and E143R counterparts. These results indicate that both APC/C‐ and substituted HTH‐dependent activation of UBE2S function by driving the UBE2S~Ub “closed” conformation.
Figure 7. UBE2SHTH substitutions partially rescue the defects in Ub chain elongation created by disrupting the UBE2S‐APC2 interaction.
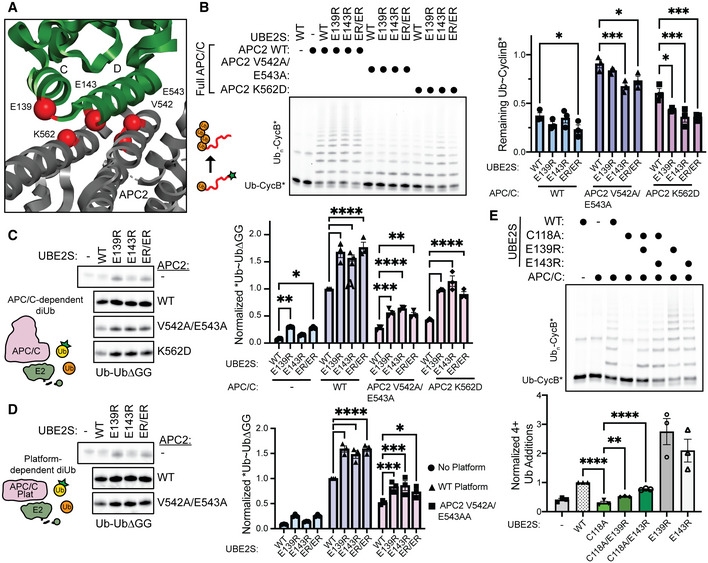
- Structural view of UBE2SHTH (green) interacting with APC2 for diUb synthesis (Brown et al, 2016). Residues of interest at the UBE2SHTH‐APC2 interface are indicated as red spheres (PDB: 5L9T).
- Representative fluorescent scan of an SDS–PAGE gel (left) and its quantification (right) where the charged‐swapped variants of the UBE2SHTH enhance substrate ubiquitination of APC/Cs harboring APC2 substitutions. Statistical significance was assessed by two‐way ANOVA (*P ≤ 0.05, ***P ≤ 0.001, n = 3 independent, technical replicates). Error bars: SEM. Data normalized to the negative control substrate band from reactions lacking an E2.
- APC/C‐UBE2S‐dependent diUb synthesis was monitored by fluorescent scanning (left), quantitated (right), and assessed for statistical significance by two‐way ANOVA (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001, n = 3 independent, technical replicates), demonstrating that the UBE2SHTH substitutions rescue the defects in diUb synthesis created by mutating APC2. Error bars: SEM. Data normalized to WT APC/C‐UBE2S diUb reactions.
- APC/C Platform‐dependent activation of UBE2S‐mediated diUb synthesis was monitored by fluorescent scanning (left), quantitated (right), and assessed for statistical significance by two‐way ANOVA (*P ≤ 0.05, ***P ≤ 0.001, ****P ≤ 0.0001, n = 3 independent, technical replicates). Error bars: SEM. Data normalized to WT APC/C Platform‐UBE2S diUb reactions.
- Similar to B, the role of the “closed” conformation in UBE2S‐dependent Ub chain elongation of the APC/C substrate Ub‐CycB*, monitored by the incorporation of C118A‐substituted UBE2S variants (top). Quantification of ubiquitinated Ub‐CycB* was assessed (bottom) and statistical significance by one‐way ANOVA between C118A and WT, C118A/E139R, and C118A/E143R (**P ≤ 0.01, ****P ≤ 0.0001, n = 3 independent, technical replicates). Error bars: SEM. Data normalized to WT UBE2S 4+ Ub additions.
Source data are available online for this figure.
Discussion
Current views of how E2 enzymes are activated to mediate polyubiquitination are largely based on simplified E2‐E3 systems using truncated versions of the E3, nonspecific E2s, and rarely include an E3‐bound substrate (Gundogdu & Walden, 2019). Therefore, additional mechanistic understandings are needed to dissect how E2‐E3 pairs achieve specific interactions to form complex Ub topologies. As the E2HTH is the most divergent region of the E2 UBC domain, it is likely that this motif has become specialized to facilitate Ub chain elongation. Collectively, our studies show that the E2HTH is important for modulating the intrinsic polyubiquitination activity of the E2 enzyme through multiple mechanisms.
UBE2S is recruited by the APC2‐APC4 groove via its C‐terminal extension, which also activates the APC/C for substrate priming with UBE2C (Brown et al, 2014, 2016; Kelly et al, 2014; Martinez‐Chacin et al, 2020). The UBE2SHTH interacts with the helices on APC2, activating UBE2S~Ub for Ub transfer (Brown et al, 2014, 2016). However, the mechanistic basis for this activation has remained unclear. By combining NMR, MD simulations, and detailed biochemical assays, we demonstrate that the negative charge present on the UBE2SHTH reduces the rate of Ub transfer. While this defect is relatively mild in single turnover reactions, it is exacerbated during the formation of an entire polyubiquitin chain because the deficiency would occur every time a Ub is added to the chain. However, why would UBE2SHTH have residues that slow down catalysis? It is possible that this mechanism reduces its activity to prevent the nonspecific discharge of UBE2S~Ub to non‐E3 bound substrates, and consequently, the APC/C facilitates specific ubiquitination of client proteins using the UBE2SHTH‐APC2 interaction to limit the conformational space of the donor Ub and enrich the “closed” E2~Ub conformation. This finding is supported by our sequence analysis, since UBE2S has the most negatively charged HTH and interacts with an E3 at this region. Moreover, since it was previously suggested that the UBE2S~Ub intermediate is not activated by the RING domain to stabilize the “closed” configuration, our proposed mechanism reveals an unexpected model for E2 activation by an E3 (Williamson et al, 2009b; Wickliffe et al, 2011; Brown et al, 2014, 2016; Kelly et al, 2014).
To expand this mechanism across E2 family members, we tested multiple E2s that are canonically activated by a RING domain and are not known to bind to the E3 through their E2HTH motifs. For these E2s, UBE2C, UBE2D2, and UBE2R2, positive to negative substitutions on the E2HTH decreased Ub chain elongation in an E3‐independent manner. UBE2R2 displayed the most striking defect and has several mechanistic features similar to UBE2S. UBE2R is the chain‐elongating E2 for SCF E3 ligases and is recruited to the E3 through the binding of its C‐terminal extension (Gazdoiu et al, 2007; Kleiger et al, 2009; Pierce et al, 2009; Wu et al, 2010a). Therefore, we performed a side‐by‐side comparison between UBE2S and UBE2R2 to decipher how interactions with Ub promote different mechanisms. Surprisingly, the results suggested that the E2HTH contributes to interactions with the donor Ub for UBE2S and the acceptor Ub for UBE2R2 (Fig 3). Therefore, Ub binding to the E2HTH has been repurposed depending on the E2 utilized. Interestingly, previous NMR analysis of the K63 chain former UBE2N suggested that the E2HTH also interacts with the donor Ub when the dynamic E2~Ub conjugate is formed (Pruneda et al, 2011). Furthermore, there are 5 structures where the donor Ub is bound to the E2HTH (PDB: 3UGB, 5IFR, 5TUT, 2KJH, 4R62) (Serniwka & Shaw, 2009; Page et al, 2012; Kumar et al, 2015; Mulder et al, 2016). These structures indicate that an “open” state where donor Ub can interact with the E2HTH may be universal and preventing this state could be a mechanism of E2 activation by additional E3 domains and adaptors.
Based on the Shannon entropy of our E2 alignment, we found that the region around helix C in the E2HTH had a high Shannon entropy, indicating that it is one of the more variable regions of the protein (Fig EV2B). While most of the E2s have a structurally homologous HTH, several E2s have unique structures, such as loop insertions, or are completely missing the E2HTH. For example, the human isoform of UBE2R has one additional helical turn in many other mammalian versions but not in Saccharomyces cerevisiae. Also, many E2s that have a divergent HTH have a specialized function. Helix C of UBE2Z is replaced with a longer loop and acts as the Fat10‐conjugating enzyme (Schelpe et al, 2016). UBE2V is the inactive adaptor that coordinates with UBE2N to form K63 chains and has only a single helical turn instead of an HTH (Eddins et al, 2006). In UBE2W, which conjugates Ub to disordered N‐terminal amines, the HTH is replaced by a dynamic loop (Vittal et al, 2015; Ye et al, 2020). UBE2K has an additional helical extension that contains a Ub binding site (Pluska et al, 2021). UBE2J and UBE2Q also do not have HTH motifs, suggesting that these E2s have acquired additional functions. Overall, it appears that the E2HTH provides a platform for diversification of E2 activity.
The variety of E2s suggests that numerous possible mechanisms exist that expand beyond the traditional view of E2 activation by an E3. In particular, the UBC domain is already known to bind to several different protein regulators to alter E2 activity and achieve specificity. Typically, these interactions occur at the N terminus or backside of the UBC domain (Buetow & Huang, 2016; Stewart et al, 2016; Gundogdu & Walden, 2019). However, the binding of Ub—either as a donor or acceptor—to the E2HTH appears to add another layer of regulation that intersects both catalysis and protein–protein interactions. Therefore, we suspect that the harnessing of the E2HTH surface by E3s for substrate polyubiquitination will be uncovered in future studies.
Materials and Methods
Protein purification
Wild‐type and mutant proteins necessary for ubiquitination assays (UBA1, UBE2S, UBE2C, APC/C, CDH1, Ub, and substrates) were expressed and purified as previously described (Brown et al, 2014, 2015, 2016; Jarvis et al, 2016). Recombinant APC/C, UBA1, and CDH1 were expressed using a baculovirus expression system, and all other proteins were purified from BL21‐Codon Plus (DE3)‐RIL cells (Weissmann et al, 2016). All purification steps were completed at 4°C unless otherwise indicated.
APC/C was expressed in baculovirus‐infected Hi‐5 insect cells, which included a twin Strep‐tag at the C terminus of APC4. After centrifugation, the cells were resuspended in lysis buffer (50 mM HEPES pH 8.0, 200 mM NaCl, 5% glycerol, 1 Roche Ultra EDTA‐free Protease Inhibitor tablet per 50 ml buffer, 10 µg/ml Leupeptin, 20 µg/ml Aprotinin, 2.5 mM Benzonase, and 2.5 mM PMSF). Cells were sonicated, and the debris was cleared by centrifugation. Clarified lysate was incubated with Strep‐Tactin Sepharose resin and eluted with desthiobiotin. APC/C was further purified with anion exchange and size exclusion chromatography (SEC) into a final buffer of 20 mM HEPES pH 8, 200 mM NaCl, and 1 mM DTT (Brown et al, 2016; Jarvis et al, 2016). All proteins were sized into this final buffer unless uniquely otherwise described. This purification protocol was used to generate phosphomimetic (pE) APC/C ΔAPC15 with APC2 VE542‐543AA or K562D substitutions, as well as APC/C Platform with APC2 VE542‐543AA substitutions (Qiao et al, 2016).
CDH1 was expressed in Hi‐5 insect cells and purified with a N‐terminal 3xMyc‐His6 tag (Qiao et al, 2016). After elution from nickel NTA agarose beads, the Myc‐His tag was removed with HRV 3C protease treatment and CDH1 was further purified by cation exchange chromatography. Lastly, CDH1 was subjected to SEC into a final buffer of 20 mM HEPES pH 7, 300 mM AmSO4, and 1 mM DTT.
UBA1 was expressed with an N‐terminal GST tag in baculovirus‐infected insect cells and purified by Glutathione Sepharose 4B (GS4B) resin. After thrombin‐dependent removal of the GST tag, UBA1 was then purified by anion exchange chromatography, passed over heparin resin to remove thrombin, and SEC. UBA1 expressed in bacteria cells contains an N‐terminal His6 tag and was purified with nickel‐affinity chromatography, anion exchange chromatography, and SEC.
UBE2S wild‐type and its variants for ubiquitination assays were expressed with an N‐terminal multipurpose tag from a customized pRSF duet vector including a His6 tag, TEV protease site, FLAG tag, then HRV 3C protease site. After expression, these proteins were purified with nickel‐affinity chromatography and treated with HRV 3C protease. The full‐length versions of UBE2S underwent further purification through cation exchange chromatography. Both the UBE2SUBC and UBE2S full‐length constructs were then subjected to SEC. UBE2S‐UBD constructs were expressed as a GST fusion. After purification by GS4B resin, the fusion protein was treated with HRV 3C protease and dialysis. Liberated GST was then removed by GS4B resin and the UBE2S‐UBD was purified by SEC.
For NMR, 15N‐labeled UBE2SUBC wild‐type and the ER/ER variant were grown to OD 0.8–1.0 in minimal media (1X M9 salts, 0.06 mM FeCl3, 2 mM MgSO4, 0.1 mM CaCl2, 2 g/l glucose) at 37°C and induced with 0.6 mM IPTG at 16°C. Isotopically labeled variants were purified as previously described above for unlabeled UBE2SUBC. Immediately prior to NMR experiments, 15N‐variants and unlabeled, wild‐type Ub were buffer‐exchanged into 20 mM HEPES pH 7, 100 mM NaCl, 10 mM DTT.
All other E2s were also expressed in BL21‐Codon Plus (DE3)‐RIL. UBE2C and mutants were purified using a C‐terminal His6 tag, nickel affinity chromatography, and SEC. UBE2D2 and mutants were expressed without an affinity tag. Cells were resuspended in 50 mM HEPES pH 7.5 and 5 mM DTT. After sonication and clarifying the lysate, UBE2D2 was isolated by cation exchange chromatography and SEC. UBE2R2 and its variants were expressed with an N‐terminal GST tag with a TEV protease cleavage site. After purification using GS4B resin, dialysis, and treatment with TEV, GS4B was used to remove the cleaved GST‐tag and TEV protease, and UBE2R2 was further purified by SEC. UBE2R2 and its mutants used in BLI experiments were not treated with TEV and therefore remained as a GST‐UBE2R fusion.
Fluorescently labeled substrates were purified as previously described (Brown et al, 2014). In short, substrates were purified with an N‐terminal GST tag with a TEV protease cleavage site and a C‐terminal Cys‐His6 uncleavable extension. Substrates were isolated on GS4B resin before elution and TEV protease treatment. After nickel affinity chromatography and SEC, the substrates were fluorescently labeled using fluorescein‐5‐maleimide (Thermo Fisher) that reacted with the single cysteine at the C terminus. These substrates were then subjected to SEC.
Unlabeled Ub and UbΔGG was purified as previously described (Brown et al, 2014). UbΔGG is missing the final C‐terminal glycine residues necessary to go through the Ub cascade. After sonication and centrifugation, the bacteria lysate was treated with acetic acid at room temperature (RT) until the lysate reached ~pH 4.5. The lysate was then centrifuged to remove precipitation and dialyzed overnight at room temperature in 25 mM Sodium Acetate pH 4.5. Ub was then further purified with cation exchange chromatography and SEC.
Ub for fluorescent labeling was expressed with an N‐terminal GST tag, TEV protease site, and a single cysteine. After purification by GS4B resin, dialysis, and protease treatment, liberated GST was removed with GS4B resin. After TEV cleavage, the amino acids of GSGGSC remain appended to the starting methionine of wild‐type Ub. Ub was then subjected to SEC, fluorescent labeling of the N‐terminal Cys by fluorescein‐5‐maleimide and purified by SEC.
Enzyme assays
All ubiquitination assays were completed by mixing reaction components on ice in assay buffer (20 mM HEPES pH 8, 200 mM NaCl) before equilibrating to room temperature (RT) and starting reactions with either Ub or MgCl2‐ATP. At the indicated times, reactions were quenched with SDS loading buffer before being separated by SDS–PAGE on either GenScript or Invitrogen 4–12% Bis‐Tris gels in non‐reducing conditions. To visualize reactions, fluorescent gels were scanned on an Amersham Typhoon 5 before being quantified on ImageQuant software and processed with GraphPad Prism. Welch’s unpaired t‐test was utilized for comparisons of two variants to account for potential differences in variances. One‐way ANOVA tests were utilized for multiple comparisons. However, two‐way ANOVA tests were utilized in Fig 7B–D to account for grouping from both UBE2S and APC/C variants. Confirmation that data met statistical test assumptions was performed with GraphPad Prism. Qualitative Coomassie gels were imaged on a Bio‐Rad ChemiDoc. All assays were performed at least 3 times (n ≥ 3).
UBE2S autoubiquitination assays (Figs 1C and 5B and F) were performed by mixing 100 nM UBA1 with 200 nM UBE2S and 4 µM *Ub. Reactions were started with 5 mM MgCl2‐ATP and quenched after 60 min.
To qualitatively test the effect of the ER/ER mutations on chain elongation, we added 5 µM of either UBE2S full‐length (FL), UBE2S‐UBD, or UBE2SUBC to 0.5 µM UBA1, 250 µM Ub, and 10 mM MgCl2‐ATP and incubated at 37°C (Fig 1D and Appendix Fig S1G). Aliquots were taken at 0, 15, 30, 60, and 120 min and resolved by non‐reductive SDS–PAGE before Coomassie staining. Similarly, to make samples for mass spectrometry analysis, 500 µl reactions were set up in triplicate with the same concentrations as above (Figs 1E and EV1A). Reactions were allowed to proceed at 37°C for 4 h before being quenched with 50 mM EDTA pH 8.0. To remove unmodified proteins, reactions were spiked with 100 µl of 1 M glacial acetic acid to pH 4.5, spun down at 18,000 g for 10 min and desalted into 20 mM HEPES pH 8, 200 mM NaCl before being snap‐frozen with liquid N2.
Pulse‐chase assays were used to monitor the diUb formation mediated by UBE2S, UBE2C, UBE2D2, and UBE2R2 (Figs 2E–H and EV1, EV2, EV3, EV4, EV5B–D). Due to low propensities to automodify, full‐length UBE2C, UBE2D2, and UBE2R2 were utilized. UBE2SUBC was used and a K100R substitution was introduced to prevent automodification (Liess et al, 2019). To load the E2, 1 µM UBA1, 5 mM MgCl2‐ATP, and equimolar E2 and *Ub (2 µM for UBE2S, 5 µM for UBE2C, UBE2D2, and UBE2R2) were incubated at RT for 10 min to generate E2~*Ub conjugates and then quenched with 50 mM EDTA pH 8.0. This mixture was combined with 1 mM UbΔGG to result in a final concentration of 0.5 µM E2~*Ub and reactions were quenched at the indicated times. Ub discharge to lysine mediated by UBE2S (Figs 3C and EV3A) or UBE2R2 (Figs 3D and EV3B) was assayed in a similar scheme. However, the E2~*Ub mixture was combined with 20 mM free lysine instead of UbΔGG for a final E2~*Ub concentration of 0.5 µM.
Fluorescent‐LRLRGG (*LRLRGG) peptide transfer assays were performed by incubating 10 µM *LRLRGG peptide, 0.5 µM UBA1, 100 µM UBΔGG, 5 mM MgCl2‐ATP, and either 1 µM UBE2S (Fig 3E) or 5 µM UBE2R2 (Fig 3F) at RT. Reactions were quenched in non‐reducing SDS buffer at the indicated time points and separated on Invitrogen 4–12% Bis‐Tris gels and imaged on the Amersham Typhoon 5.
The apparent kinetic parameters, K m (K m app) and V max (V max app), of wild‐type UBE2R2 and variants D143K and R149E were assessed in pulse‐chase diUb synthesis assays to determine mutational effects on UBE2R2 activity (Fig 4G–I). A pulse reaction of 1 μM His6‐UBA1, 5 μM *Ub, 5 μM UBE2R2, and 5 mM MgCl2‐ATP was incubated for 10 min, then quenched with 50 mM EDTA to generate the UBE2R2~*Ub conjugate. This pulse mixture was diluted 10‐fold into a secondary reaction with varying amounts of UbΔGG to allow *Ub~UbΔGG formation. Time courses suitable for each UBE2R2 variant at each UbΔGG concentration were quenched and separated by SDS–PAGE to allow for the quantification of the UBE2R2~*Ub band. Initial velocities were calculated from the resulting time courses by determining the absolute value of the slope of UBE2R2~*Ub depletion within the linear range of activity. The initial velocities were fitted into the Michaelis–Menten equation, v = V max app[X]/(K m app+[X]), where X is the acceptor UbΔGG, to estimate K m app and V max app. The wild‐type initial velocities were fitted first to determine the wild‐type V max app. This value was used to normalize the initial velocities of UBE2R2 wild‐type, D143K, and R149E as a percentage of the wild‐type V max app. All data points were performed at n ≥ 3.
APC/C‐dependent substrate ubiquitination assays for UBE2S (Figs 6E, 7B and E, and Fig EV5C–E), UBE2C (Figs 6C and EV5B), and UBE2D2 (Fig 6D) were performed with 30 nM APC/C, 500 nM CDH1, 500 nM substrate, 100 nM UBA1, 125 µM Ub, 5 mM MgCl2‐ATP, and either 200 nM UBE2S, 750 nM UBE2D2, or 1 µM UBE2C. Reactions were incubated at RT and quenched at 8 (UBE2C) or 10 (UBE2S and UBE2D2) minutes.
To determine the impact of mutations within APC2 on the activity of wild‐type UBE2S and charge‐swap mutants, diUb formation assays were performed without the pulse‐chase reaction scheme (Fig 7C). In short, 1 μM His6‐Uba1, 2 μM *Ub, 100 nM CDH1, 1 mM UbΔGG, 200 nM of the respective UBE2S variant, and 100 nM of the respective APC/C variant were combined. Reactions were started with 5 mM MgCl2‐ATP and quenched at 5 min. DiUb formation assays for UBE2S and APC/C Platform variants were performed under the same conditions but in the absence of CDH1 (Fig 7D).
Proteomics—label‐free Ub chain linkage analysis of chain elongation by UBE2S
For mass spectrometry (Figs 1E and EV1A), in vitro Ub assays were performed in triplicate as indicated (absence of E2, UBE2S, UBE2SER/ER, UBE2S‐UBD or UBE2SER/ER‐UBD) and followed by acid precipitation and then buffer exchange into 20 mM HEPES pH 8, 200 mM NaCl. Samples were subjected to reduction (5 mM tris(2‐carboxyethyl)phosphine for 10 min at room temperature) and alkylation (25 mM chloroacetamide for 20 min at room temperature) followed by Trichloroacetic acid (TCA) precipitation with 20%TCA, a wash with 10% TCA, and triplicate washes with cold acetone. Samples were briefly dried and resuspended in 30 μl digestion buffer (100 mM triethyl ammonium bicarbonate pH 8.5, 0.1% RapiGest) and digested for 2 h at 37°C with Lys‐C (0.25 μg) and then 6–8 h with trypsin (0.5 μg) at 37°C. The samples were acidified with equal volume of 1% formic acid to pH ~2, incubated for 15 min, vacuum centrifuged to near dryness and subjected to C18 StageTip desalting. Label‐free analysis was performed in biological triplicate and analyzed sequentially by LC/MS2 on a Q Exactive mass spectrometer (Thermo Fisher Scientific) coupled with a Famos Autosampler (LC Packings) and an Accela600 LC pump (Thermo Fisher Scientific). Peptides were separated on a 100 μm inner diameter microcapillary column packed in house with ~35 cm of Accucore150 resin (2.6 μm, 150 Å, Thermo Fisher Scientific, San Jose, CA) with a gradient consisting of 6–21% (15–40 min), 21–28% (40–48 min) (ACN, 0.125% FA) over a total 75 min run at ~150 nl/min. The scan sequence began with an MS1 spectrum (Orbitrap analysis; resolution 70,000 at 200 Th; mass range 350–1,400 m/z; automatic gain control (AGC) target 3 × 106; maximum injection time 100 ms). Precursors for MS2 analysis were selected using a top 10 method. MS2 analysis consisted of higher‐energy C‐trap dissociation (AGC 5.0 × 105; isolation window 1.2 Th; normalized collision energy 25; maximum injection time 100 ms). Precursors with charge state unassigned or different from 2, 3, 4, or 5 were excluded, peptide match option was set to preferred, isotopes were excluded, and previously interrogated precursors were excluded using a dynamic window (set to automatic) and minimum AGC target was 1 × 104. Mass spectra were processed using Protein Discoverer v.2.3 (Thermo Fisher Scientific) using the Minora algorithm (set to default parameters). The identification of proteins was performed using the SEQUEST‐HT engine against the UniProt Human Reference Proteome (2019), supplemented with in‐house‐curated sequences of common contaminant proteins. The following parameters were used: a tolerance level of 10 ppm for MS1 and 0.03 Da for MS2 and the false discovery rate of the Percolator decoy database search was set to 1%. As a Trypsin/Lys‐C combination was used as the digestion enzyme, two missed cleavages were allowed and the minimal peptide length was set to six amino acids. The carbamidomethylation (+57.021 Da) of cysteine was set as a fixed modification, and the oxidation (+15.995 Da) of methionine and GlyGly (+114.043 Da) modification of lysine were allowed as a variable modification, as well as protein N terminus acetylation (+42.011 Da). Scoring and localization of the GlyGly site was done using the ptmRS node (default settings). Protein and peptide were quantified based on the precursor abundance area, and only peptides quantified in all three replicates were considered for further analysis. Protein or peptide quantification values were exported for further analysis in Microsoft Excel and GraphPad Prism v.8.
Multiple sequence alignments and consensus sequences
To acquire a list of homologous sequences, we conducted a BLAST search using each human E2 enzyme gene query sequence, extracted from UNIPROT (Apweiler et al, 2004). We took the top 10,000 hits and inspected each family set manually and established a cutoff that included all hits corresponding to the E2 family member. Some E2 enzymes have several isoforms. In this case, all corresponding sequences were grouped together in one dataset. To limit potential bias in the alignment, redundant sequences were removed from the dataset with use of the MEME Suite (v5.1.1, build 2020‐02‐01) Purge tool (Bailey et al, 2009). A score threshold of 900 was used. The resulting sequences in the dataset were then aligned using the multiple sequence alignment tool Mafft (v7.471, build 2020‐07‐03; Katoh & Standley, 2013). To enhance the quality of the multiple sequence alignment (MSA) and remove poorly aligned regions and leftover spurious sequences, we employed the trimming tool triMAl (v1.4.rev.22, build 2015‐05‐21; Capella‐Gutierrez et al, 2009). The alignment was trimmed using a gap threshold of 0.75 resulting in the removal of positions with gaps that occurred in more than 25% of the sequences. A consensus sequence for each E2 was then created from the trimmed MSA using the EMBOSS package Cons (v6.6.0.0, build 2013‐07‐15; Rice et al, 2000). Consensus residues at each position were determined through Cons using a plurality value of 0.1. All steps described up to this point were repeated for all members of the E2 enzyme family, resulting in 23 E2 enzyme consensus sequences. Finally, we utilized the PROMALS3D server to create an MSA of these consensus sequences (Pei et al, 2008). PROMALS3D was used since it takes both structural and sequence information into account, resulting in a higher quality MSA.
Phylogenetic tree, Weblogos, and Shannon entropy
A maximum likelihood phylogenetic tree was constructed using Protest3, which used the Blosum62 + G + F substitution matrix. We then plotted the tree as an unrooted radial phylogram (Fig EV2A). To illustrate the amino acid charge conservation variation at each position in the protein sequences for each E2 enzyme family member, we constructed weblogos using Weblogo3 (Figs 2I and EV2C; Crooks et al, 2004). We also conducted a Shannon entropy calculation on this alignment (Fig EV2B). To calculate the amino acid (AA) variation at all positions in all 23 E2 consensus sequences, we utilized the Calculate_AA_Variation function in the R Package BALCONY (v0.2.10, build 2019‐02‐28) and applied it to each E2 family member MSA (Pluciennik et al, 2018). Based on the AA variability analysis, we were able to calculate the average charge for each position in all E2 family alignments using the formula (K% + R% + H%)−(D% + E%). We then colored each E2HTH according to this score.
Bio‐layer interferometry
Bio‐layer interferometry (BLI) was used to quantitatively analyze Ub binding to UBE2R2 (Figs 4A and EV3C and D). Using a standard buffer of 0.22 µM filtered 20 mM HEPES pH 8.0, 200 mM NaCl, 25 μg/ml of GST‐UBE2R2 was flowed onto anti‐GST biosensors (Sartorius Bio, Göttingen, Germany) on an Octet Red96 machine (FortéBio, Fremont, Ca). A titration of Ub was added (125 µM, 250 µM, 375 µM, 500 µM, 750 µM, 1250 µM, 2000 µM, and 4,000 µM—for D143K and R149E only) and then allowed to dissociate. Baseline was subtracted from signals and individual experiments were aligned to dissociation. Global fit analysis for each replicate was performed using Octet Analysis 10.0 for n ≥ 3 experiments. Binding values were used to plot and fit K d values with GraphPad Prism v.9.
Mechanically transduced immunosorbent assay (METRIS)
To form the isopeptide‐linked UBE2R2~Ub, 10 µM of UBE2R2 containing a C93K substitution was mixed with 2 µM E1, 80 µM Ub containing an N‐terminal strep‐tag (WSHPQFEKGENLYFGSC appended to the N terminus of wild‐type Ub), and 10 mM Mg‐ATP in a buffer solution of 50 mM Tris pH 9.5, 50 mM NaCl, and 2 mM 2‐mercaptoethanol. The reaction was allowed to proceed overnight at 35°C before being purified by Strep‐tactin Sepharose resin and SEC into 25 mM Tris pH 8, 50 mM NaCl, and 1 mM DTT.
UBE2R2 wild‐type and its variants were labeled at cysteines using Poly(ethylene glycol) [N‐(2‐maleimidoethyl)carbamoyl]methyl ether 2‐(biotinylamino)ethane (Sigma 757748) (Biotin‐maleimide) as described previously (Petell et al, 2021). Ub was biotinylated via an N‐terminal cysteine that is commonly used fluorescent labeling. Biotinylation reactions were carried out with ~5 equivalent excess of Biotin‐maleimide and allowed to shake gently at 4°C for 5 h. Excess Biotin‐maleimide was removed using overnight dialysis and the proteins were stored frozen for future use.
Biotinylated Ub was then inserted into a tube with streptavidin‐coated ferromagnetic particles, provided by Spherotech. The amount of Ub added was such to coat each bead 50x the theoretical limit to ensure all binding sites on the beads were covered. The bead and peptide solution was left to react at room temperature for at least 2 h.
The substrates are avidin‐coated glass slides, provided by Arrayit or Nanocs, with a ligand density of 1.1 × 1010 ligands per mm2. Microfluidic channels were created on this substrate using two pieces of double‐sided tape, provided by 3 M. The pieces of double‐sided tape were cut to a width of several mms and a length of at least 25 mm. The pieces of tape were placed parallel to each other and at a distance of approximately 5 mm apart. Then, a glass coverslip, provided by VWR, was placed on top of the tape to create channels approximately 22 × 5 mm.
A solution of biotinylated E2 proteins was then inserted into the channel. The amount of proteins inserted was again enough to coat the channel surface 50× the theoretical limit to ensure that all of the sites on the substrate were coated. The substrate and solution were left in a sealed container for two hours to allow the proteins time to bind to the substrate. After two hours, the solution was washed from the channel to remove any excess protein that was not attached to the substrate. Then, the solution of Ub‐coated ferromagnetic beads was diluted approximately 2,000×. 10 µl of this solution was then extracted and inserted into the channel. The channel was sealed with epoxy and magnetized by an external permanent neodymium magnet. The substrate was placed in the slide holder at the center of the Helmholtz Coil Inspired Experimental Apparatus.
Three pairs of coils were secured in the Helmholtz Coil Inspired Experimental Apparatus, made of aluminum T‐slots, and attached to an optical breadboard. The coils have an inner diameter of 7 cm and an outer diameter of 13 cm. The coils consist of approximately 150 turns of gauge 19 copper wire. Two sinusoidal signals, phase shifted by 90 degrees, were generated in Matlab. Those signals were sent to a National Instruments USB X Series DAQ and then passed through a 300W amplifier (150W/channel) before being sent through each pair of coils. The magnetic field is large enough (approximately 10 mT) to ensure alignment of the rotational frequency of the particles with the frequency Ω of the magnetic field. The signal from the amplifier was routed to a Rigol Oscilloscope to measure the frequency and the voltage. The sample holder is made from 6,061 aluminum and attached to an OMAX binocular microscope which functions as our 3D optical stage. Data acquisition was accomplished via a CMOS camera mounted on a C‐mount DIN objective tube assembly. The camera was connected to a computer for visualization, video capture, and subsequent analysis.
To analyze particle motion, we converted the video captured from the CMOS camera and converted it into an .avi file using ArcSoft Media Converter 8. The .avi file was then converted into a sequence of .jpeg images. To analyze the motion of the active particles, we used Able Particle Tracker. The data were then imported into Mathematica for subsequent analysis. To record the distance each particle travels, we use a normalized term, the rolling parameter, which is defined by the following equation, where D is the particle diameter, ω is the frequency and τ is the actuation time.
Molecular dynamics simulations on UBE2R
The UBE2R E2 domain was taken from PDB: 6NYO, the isopeptide‐linked ubiquitin was removed and the active site cysteine was reinstalled using Rosetta fixedbb and we also used this protocol to make the R149E variant (Williams et al, 2019). MD simulations were run using the CHARMM36 force field and NAMD simulation software. The structure was solvated in a rectilinear water box composed of TIP3P water molecules (r = 10.0 Å). PSF generation and solvation were done using the autopsf and solvate plug‐ins of the VMD molecular visualization program. During simulation, the system was subjected to optimization stabilization, heating to 298 K, then an equilibration of 2 picoseconds of minimization using 2 femtosecond time steps. Runs were executed using Langevin dynamics, the Particle Mesh Ewald method, and constant pressure control. Both the WT and R149E variant were subjected to 2, 20 ns simulations (Fig 4E). All analysis was carried out using the MDanalysis python scripting language (Michaud‐Agrawal et al, 2011).
Molecular dynamics simulations of UBE2S~Ub
The 1.93 Å human UBE2S (PDB: 1ZDN) and the 2.80 Å human Ub (PDB: 4RF0) crystal structures were used to prepare wild‐type and variant structural models for molecular dynamics simulations of UBE2S in complex with Ub (Sheng et al, 2012; Bailey‐Elkin et al, 2014). The apo UBE2S structure consisted of residues 9‐156 of chain A from 1ZDN. Four replicate molecular dynamics simulations of 500 ns length were completed for apo UBE2S. Trajectories from UBE2S were visually inspected for an opening between Cα carbons of N97 and E126 of at least 11 Å, which was large enough to allow the formation of the thioester linkage of Ub to UBE2S without steric hindrance. One snapshot was selected from the wild‐type UBE2S trajectories for formation of the thioester conjugation to Ub. This conjugated structure provided the starting conformation for a new round of molecular dynamics simulations.
The Amber v18 software package was used to run all‐atom molecular dynamics simulations of the wild‐type apo and UBE2S~Ub complexes in explicit solvent (Salomon‐Ferrer et al, 2013). The LEaP program was used to neutralize the charge of the systems and to generate a truncated octahedron of TIP3P water molecules of at least 16 Å depth around the solute. The UBE2S~Ub complex systems required up to 20,500 water molecules. The ff14SB force field was used for parameterization of protein molecules (Maier et al, 2015). Parameters were calculated for the thioester bond using antechamber with the AM1‐BCC charge method and amber atom types. Simulations were run using the pmemd GPU‐accelerated code (Salomon‐Ferrer et al, 2013). Equilibration consisted of 5,000 cycles of minimization, followed by 500 ps of heating using an NVT ensemble, and 500 ps of density equilibration using an NPT ensemble. For heating, the target temperature was increased linearly from 0K to 300K over the course of the 500 ps using a Berendsen thermostat with a relaxation time of 0.5 ps. A Monte Carlo barostat was used for density equilibration. The production run was 500 ns in total length in the NPT ensemble with a Langevin Thermostat with a collision frequency of 1.0 ps−1, and a Berendsen barostat. All simulations were under periodic boundary conditions with a 2 fs time‐step and with hydrogen atoms constrained by SHAKE. Snapshots were saved every 10 ps. Four replicates of 500 ns each were run of each of the systems. Coordinate trajectories underwent initial processing using cpptraj.
NMR spectroscopy and data analysis
All NMR experiments were carried out on a Bruker Avance III 850 spectrometer equipped with 5mm triple‐resonance cryogenic probes at 25°C. 100 µM of 15N‐labeled UBE2SUBC wild‐type and variant samples were prepared with and without the addition of 1.2 mM un‐labeled Ub. 15N‐1H HSQC spectra were collected using standard Bruker hsqcfpf3gpphwg pulse program. Spectra were processed and analyzed using NMRPipe/NMRDraw and Sparky 3.110 (Delaglio et al, 1995). Resonance assignments were adapted from previous studies (Wickliffe et al, 2011; Brown et al, 2016). Chemical shift perturbations were calculated as performed in previous studies (Wickliffe et al, 2011). The lineshape analyses were carried out using the software TITAN, which was accessed via an NMRbox Virtual Machine environment (Waudby et al, 2016). TITAN’s “No Exchange” binding model and default HSQC pulse program were used to fit 1H and 15N chemical shifts as well as apparent transfer relaxation rates for all unambiguously assigned resonances. Errors in the fitted values were determined by Titan’s default standard error from the estimated covariance matrix.
Rosetta modeling
To generate the ensemble of conjugated E2 structures, we used the E2 thioester protocol in Rosetta (Saha et al, 2011). This protocol is a tethered docking protocol which we have previously used to construct and analyze the conformational ensemble of conjugated Ub (Yin et al, 2020). We have previously observed that this conjugated docking quickly settles in a local minimum and then remains in that relative orientation. Therefore, to increase sampling we ran shorter number of Monte Carlo steps, 500, and made 100,000 models. We analyzed the top 200 structures to look for electrostatic interactions with the HTH. To determine if APC2 might sterically clash with these structures, we generated a relaxed model of APC2‐UBE2S based on APC2 (PDB: 6TLJ) and UBE2S from (5L9T) and selected the highest scoring model that contained an interaction between UBE2S E132 and APC2 K562 which we previously determined with a charge‐swapped mutation (Brown et al, 2016; Alfieri et al, 2020).
Cell extract experiments
Cell extract experiments (Fig 6F) were performed similar to previous (Williamson et al, 2009a; Martinez‐Chacin et al, 2020). HelaS3 were grown in DMEM (Gibco) supplemented with 10% fetal bovine serum (FBS) (Sigma) and penicillin/streptomycin 1× (Gibco) at 37°C with 5% CO2. Confluent cells were seeded into 15 cm plates at a density of 3 million per plate and allowed to grow for 24 h, after which cells were transfected with 30 ng of two UBE2S siRNAs or Firefly control using Lipofectamine RNAiMAX reagent (Life Technologies) following manufacturer’s instructions. After 8 h, 2 mM thymidine was added to the medium. After 24 h, cells were washed once with PBS then twice with DMEM and released for 3 h before being arrested with 100 ng/ml of nocodazole in DMEM for 11 h. Cells were released for 2 h to obtain a G1 population then harvested for extract preparation. Cells were pelleted at 500 g for 5 min, washed with PBS, centrifuged, and washed once more with PBS before being placed on ice and being resuspended in 2× volume of the cell pellet cold PBS. Cells were transferred to Eppendorf tubes and centrifuged at 200 g and 4°C for 5 min. Supernatant was carefully aspirated and 1.5× cell pellet volume cold SB buffer (20 mM HEPES pH 7.5, 1.5 mM MgCl2, 1 mM DTT, 5 mM KCl, 2 µg/ml aprotinin, 10 µg/ml leupeptin, and 1 mM AEBSF) was added, along with 30 µl energy mix (375 mM creatine phosphate, 50 mM ATP, and 50 mM MgCl2, pH 8.0). Cells were incubated for 30 min on ice, being mixed well every 5 min before being frozen in liquid nitrogen, quick thawed in a 30°C water bath, frozen and thawed once more before being sheared with a 20 1/2G needle seven times and a 25 3/8G needle twice. The sheared solution was centrifuged at 2,500 g, 4°C for 5 min before the supernatant was taken and cleared by centrifuging again at 20,000 g, 4°C for 30 min. The supernatant was taken, aliquoted, and frozen in liquid nitrogen before being stored at −80°C until use.
For degradation assays, prepared extracts were used at 1:2 ratio, diluted with SB buffer, 1 mg/ml ubiquitin (final), and energy mix. UBE2S wild‐type or ER/ER was added at 1 µM final before reactions were allowed to proceed at 30°C. At the indicated time points, aliquots were taken and quenched in 4x SDS loading buffer before being boiled for 5 min and separated on SDS–PAGE gels (Bio‐Rad). Proteins were transferred to nitrocellulose membranes (Bio‐Rad) and blocked with PBST with 5% non‐fat dry milk overnight (UBE2S and Cyclin B), or for one hour (Vinculin) before being washed three times with PBST and being incubated overnight with indicated antibodies at concentrations below in PBST with 5% BSA. UBE2S and Cyclin B primary antibodies were incubated for one hour at RT, and then, all membranes were washed with PBS‐T and incubated with appropriate secondary antibodies for one hour at RT. After being washed three more times with PBST, membranes were incubated with ECL (ClarityTM, Bio‐Rad) for 5 min and visualized by film. Antibody concentrations: Vinculin (Santa Cruz Biotechnology, Cat. #sc‐25336, 1:1,000 dilution), UBE2S (Cell Signaling Technologies, Cat. #9630, 1:5,000 dilution), CyclinB (Abcam, Cat. #ab32053, 1:10,000 dilution).
Author contributions
KAW, DLB, AEN, JB, BRST, TB, LG, AO designed research supervised by JWH, JPS, QZ, MJE, JSH, and NGB; KAW, DLB, AEN, JB, BRST, TB, LG, AO, JPS, and JSH performed research and/or contributed new reagents; KAW, DLB, AEN, JB, AO, JWH, JPS, QZ, MJE, JSH, and NGB analyzed data; and KAW, DLB, JSH, and NGB wrote the paper.
Conflict of interest
J.W.H. is a consultant and founder of Caraway Therapeutics and is a founding advisory board member of Interline Therapeutics.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgments
Our work is supported by NIH T32GM135095 (KAW); NIH T32GM008570 (DLB); NIH R01AG011085 (JWH); start‐up funds from UNC (QZ); start‐up funds from UoP (JSH); UNC University Cancer Research Fund (UCRF), NIH R01GM120309, and the American Cancer Society RSG‐18‐220‐01‐TBG (MJE); and NIH R35GM128855 and UCRF (NGB).
The EMBO Journal (2022) 41: e108823
Contributor Information
Joseph S Harrison, Email: jharrison@pacific.edu.
Nicholas G Brown, Email: nbrown1@med.unc.edu.
Data availability
The raw MS data have been deposited to the MassIVE repository (https://massive.ucsd.edu/) with the dataset identifier MSV000088403 (https://massive.ucsd.edu/ProteoSAFe/FindDatasets?query=MSV000088403).
References
- Alfieri C, Tischer T, Barford D (2020) A unique binding mode of Nek2A to the APC/C allows its ubiquitination during prometaphase. EMBO Rep 21: e49831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M et al (2004) UniProt: the Universal Protein knowledgebase. Nucleic Acids Res 32: D115–D119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aristarkhov A, Eytan E, Moghe A, Admon A, Hershko A, Ruderman JV (1996) E2‐C, a cyclin‐selective ubiquitin carrier protein required for the destruction of mitotic cyclins. Proc Natl Acad Sci USA 93: 4294–4299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek K, Krist DT, Prabu JR, Hill S, Klugel M, Neumaier LM, von Gronau S, Kleiger G, Schulman BA (2020a) NEDD8 nucleates a multivalent cullin‐RING‐UBE2D ubiquitin ligation assembly. Nature 578: 461–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek K, Scott DC, Schulman BA (2020b) NEDD8 and ubiquitin ligation by cullin‐RING E3 ligases. Curr Opin Struct Biol 67: 101–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey‐Elkin BA, Knaap RC, Johnson GG, Dalebout TJ, Ninaber DK, van Kasteren PB, Bredenbeek PJ, Snijder EJ, Kikkert M, Mark BL (2014) Crystal structure of the Middle East respiratory syndrome coronavirus (MERS‐CoV) papain‐like protease bound to ubiquitin facilitates targeted disruption of deubiquitinating activity to demonstrate its role in innate immune suppression. J Biol Chem 289: 34667–34682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodrug T, Welsh KA, Hinkle M, Emanuele MJ, Brown NG (2021) Intricate regulatory mechanisms of the anaphase‐promoting complex/cyclosome and its role in chromatin regulation. Front Cell Dev Biol 9: 687515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremm A, Freund SM, Komander D (2010) Lys11‐linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nat Struct Mol Biol 17: 939–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NG, VanderLinden R, Watson ER, Qiao R, Grace CRR, Yamaguchi M, Weissmann F, Frye JJ, Dube P, Ei Cho S et al (2015) RING E3 mechanism for ubiquitin ligation to a disordered substrate visualized for human anaphase‐promoting complex. Proc Natl Acad Sci USA 112: 5272–5279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NG, VanderLinden R, Watson ER, Weissmann F, Ordureau A, Wu K‐P, Zhang W, Yu S, Mercredi PY, Harrison JS et al (2016) Dual RING E3 architectures regulate multiubiquitination and ubiquitin chain elongation by APC/C. Cell 165: 1440–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown N, Watson E, Weissmann F, Jarvis M, VanderLinden R, Grace C, Frye J, Qiao R, Dube P, Petzold G et al (2014) Mechanism of polyubiquitination by human anaphase‐promoting complex: RING repurposing for ubiquitin chain assembly. Mol Cell 56: 246–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buetow L, Huang DT (2016) Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat Rev Mol Cell Biol 17: 626–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella‐Gutierrez S, Silla‐Martinez JM, Gabaldon T (2009) trimAl: a tool for automated alignment trimming in large‐scale phylogenetic analyses. Bioinformatics 25: 1972–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Zhang Z, Yang J, McLaughlin SH, Barford D (2015) Atomic structure of the APC/C and its mechanism of protein ubiquitination. Nature 522: 450–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaugule VK, Arkinson C, Rennie ML, Kamarainen O, Toth R, Walden H (2020) Allosteric mechanism for site‐specific ubiquitination of FANCD2. Nat Chem Biol 16: 291–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clague MJ, Urbe S, Komander D (2019) Breaking the chains: deubiquitylating enzyme specificity begets function. Nat Rev Mol Cell Biol 20: 338–352 [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14: 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Mariano J, Tsai YC, Kalathur RC, Kostova Z, Li J, Tarasov SG, McFeeters RL, Altieri AS, Ji X et al (2009) Allosteric activation of E2‐RING finger‐mediated ubiquitylation by a structurally defined specific E2‐binding region of gp78. Mol Cell 34: 674–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6: 277–293 [DOI] [PubMed] [Google Scholar]
- Deshaies RJ, Joazeiro CA (2009) RING domain E3 ubiquitin ligases. Annu Rev Biochem 78: 399–434 [DOI] [PubMed] [Google Scholar]
- Dou H, Buetow L, Sibbet GJ, Cameron K, Huang DT (2012) BIRC7‐E2 ubiquitin conjugate structure reveals the mechanism of ubiquitin transfer by a RING dimer. Nat Struct Mol Biol 19: 876–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddins MJ, Carlile CM, Gomez KM, Pickart CM, Wolberger C (2006) Mms2‐Ubc13 covalently bound to ubiquitin reveals the structural basis of linkage‐specific polyubiquitin chain formation. Nat Struct Mol Biol 13: 915–920 [DOI] [PubMed] [Google Scholar]
- Garnett MJ, Mansfeld J, Godwin C, Matsusaka T, Wu J, Russell P, Pines J, Venkitaraman AR (2009) UBE2S elongates ubiquitin chains on APC/C substrates to promote mitotic exit. Nat Cell Biol 11: 1363–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdoiu S, Yamoah K, Wu K, Pan ZQ (2007) Human Cdc34 employs distinct sites to coordinate attachment of ubiquitin to a substrate and assembly of polyubiquitin chains. Mol Cell Biol 27: 7041–7052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundogdu M, Walden H (2019) Structural basis of generic versus specific E2‐RING E3 interactions in protein ubiquitination. Protein Sci 28: 1758–1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haakonsen DL, Rape M (2019) Branching out: improved signaling by heterotypic ubiquitin chains. Trends Cell Biol 29: 704–716 [DOI] [PubMed] [Google Scholar]
- Hill S, Harrison JS, Lewis SM, Kuhlman B, Kleiger G (2016) Mechanism of lysine 48 selectivity during polyubiquitin chain formation by the Ube2R1/2 ubiquitin‐conjugating enzyme. Mol Cell Biol 36: 1720–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis MA, Brown NG, Watson ER, VanderLinden R, Schulman BA, Peters JM (2016) Measuring APC/C‐dependent ubiquitylation in vitro. Methods Mol Biol 1342: 287–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Vargyas M, Vasko‐Szedlar J, Roux B, Im W (2008) PBEQ‐Solver for online visualization of electrostatic potential of biomolecules. Nucleic Acids Res 36: W270–W275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones WM, Davis AG, Wilson RH, Elliott KL, Sumner I (2019) A conserved asparagine in a ubiquitin‐conjugating enzyme positions the substrate for nucleophilic attack. J Comput Chem 40: 1969–1977 [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly A, Wickliffe KE, Song L, Fedrigo I, Rape M (2014) Ubiquitin chain elongation requires E3‐dependent tracking of the emerging conjugate. Mol Cell 56: 232–245 [DOI] [PubMed] [Google Scholar]
- Khago D, Fucci IJ, Byrd RA (2020) The role of conformational dynamics in the recognition and regulation of ubiquitination. Molecules 25: 5933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RW, Deshaies RJ, Peters JM, Kirschner MW (1996) How proteolysis drives the cell cycle. Science 274: 1652–1659 [DOI] [PubMed] [Google Scholar]
- Kirkpatrick DS, Hathaway NA, Hanna J, Elsasser S, Rush J, Finley D, King RW, Gygi SP (2006) Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat Cell Biol 8: 700–710 [DOI] [PubMed] [Google Scholar]
- Kleiger G, Saha A, Lewis S, Kuhlman B, Deshaies RJ (2009) Rapid E2–E3 assembly and disassembly enable processive ubiquitylation of cullin‐RING ubiquitin ligase substrates. Cell 139: 957–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D, Rape M (2012) The ubiquitin code. Annu Rev Biochem 81: 203–229 [DOI] [PubMed] [Google Scholar]
- Kumar P, Magala P, Geiger‐Schuller KR, Majumdar A, Tolman JR, Wolberger C (2015) Role of a non‐canonical surface of Rad6 in ubiquitin conjugating activity. Nucleic Acids Res 43: 9039–9050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liess AKL, Kucerova A, Schweimer K, Yu LU, Roumeliotis TI, Diebold M, Dybkov O, Sotriffer C, Urlaub H, Choudhary JS et al (2019) Autoinhibition mechanism of the ubiquitin‐conjugating enzyme UBE2S by autoubiquitination. Structure 27: 1195–1210 [DOI] [PubMed] [Google Scholar]
- Lin Y, Hwang WC, Basavappa R (2002) Structural and functional analysis of the human mitotic‐specific ubiquitin‐conjugating enzyme, UbcH10. J Biol Chem 277: 21913–21921 [DOI] [PubMed] [Google Scholar]
- Liwocha J, Krist DT, van der Heden van Noort GJ, Hansen FM, Truong VH, Karayel O, Purser N, Houston D, Burton N, Bostock MJ et al (2021) Linkage‐specific ubiquitin chain formation depends on a lysine hydrocarbon ruler. Nat Chem Biol 17: 272–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz S, Bhattacharyya M, Feiler C, Rape M, Kuriyan J (2016) Crystal Structure of a Ube2S‐Ubiquitin Conjugate. PLoS One 11: e0147550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C (2015) ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J Chem Theory Comput 11: 3696–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Chacin RC, Bodrug T, Bolhuis DL, Kedziora KM, Bonacci T, Ordureau A, Gibbs ME, Weissmann F, Qiao R, Grant GD et al (2020) Ubiquitin chain‐elongating enzyme UBE2S activates the RING E3 ligase APC/C for substrate priming. Nat Struct Mol Biol 27: 550–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty RK, Henrici RC, Tan S (2014) Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature 514: 591–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger MB, Pruneda JN, Klevit RE, Weissman AM (2014) RING‐type E3 ligases: master manipulators of E2 ubiquitin‐conjugating enzymes and ubiquitination. Biochim Biophys Acta 1843: 47–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer HJ, Rape M (2014) Enhanced protein degradation by branched ubiquitin chains. Cell 157: 910–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud‐Agrawal N, Denning EJ, Woolf TB, Beckstein O (2011) MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J Comput Chem 32: 2319–2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder MPC, Witting K, Berlin I, Pruneda JN, Wu K‐P, Chang J‐G, Merkx R, Bialas J, Groettrup M, Vertegaal ACO et al (2016) A cascading activity‐based probe sequentially targets E1‐E2‐E3 ubiquitin enzymes. Nat Chem Biol 12: 523–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne HC, Irving E, Forment JV, Schmidt CK (2021) E2 enzymes in genome stability: pulling the strings behind the scenes. Trends Cell Biol 31: 628–643 [DOI] [PubMed] [Google Scholar]
- Ozkan E, Yu H, Deisenhofer J (2005) Mechanistic insight into the allosteric activation of a ubiquitin‐conjugating enzyme by RING‐type ubiquitin ligases. Proc Natl Acad Sci USA 102: 18890–18895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page RC, Pruneda JN, Amick J, Klevit RE, Misra S (2012) Structural insights into the conformation and oligomerization of E2~ubiquitin conjugates. Biochemistry 51: 4175–4187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei J, Kim BH, Grishin NV (2008) PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res 36: 2295–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petell CJ, Randene K, Pappas M, Sandoval D, Strahl BD, Harrison JS, Steimel JP (2021) Mechanically transduced immunosorbent assay to measure protein‐protein interactions. Elife 10: e67525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce NW, Kleiger G, Shan SO, Deshaies RJ (2009) Detection of sequential polyubiquitylation on a millisecond timescale. Nature 462: 615–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plechanovova A, Jaffray EG, Tatham MH, Naismith JH, Hay RT (2012) Structure of a RING E3 ligase and ubiquitin‐loaded E2 primed for catalysis. Nature 489: 115–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluciennik A, Stolarczyk M, Bzowka M, Raczynska A, Magdziarz T, Gora A (2018) BALCONY: an R package for MSA and functional compartments of protein variability analysis. BMC Bioinformatics 19: 300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluska L, Jarosch E, Zauber H, Kniss A, Waltho A, Bagola K, Delbrück M, Löhr F, Schulman BA, Selbach M et al (2021) The UBA domain of conjugating enzyme Ubc1/Ube2K facilitates assembly of K48/K63‐branched ubiquitin chains. EMBO J 40: e106094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruneda JN, Littlefield PJ, Soss SE, Nordquist KA, Chazin WJ, Brzovic PS, Klevit RE (2012) Structure of an E3:E2~Ub complex reveals an allosteric mechanism shared among RING/U‐box ligases. Mol Cell 47: 933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruneda JN, Stoll KE, Bolton LJ, Brzovic PS, Klevit RE (2011) Ubiquitin in motion: structural studies of the ubiquitin‐conjugating enzyme approximately ubiquitin conjugate. Biochemistry 50: 1624–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao R, Weissmann F, Yamaguchi M, Brown NG, VanderLinden R, Imre R, Jarvis MA, Brunner MR, Davidson IF, Litos G et al (2016) Mechanism of APC/CCDC20 activation by mitotic phosphorylation. Proc Natl Acad Sci USA 113: E2570–E2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reverter D, Lima CD (2005) Insights into E3 ligase activity revealed by a SUMO‐RanGAP1‐Ubc9‐Nup358 complex. Nature 435: 687–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A (2000) EMBOSS: the European molecular biology open software suite. Trends Genet 16: 276–277 [DOI] [PubMed] [Google Scholar]
- Rout MK, Hodge CD, Markin CJ, Xu X, Glover JN, Xiao W, Spyracopoulos L (2014) Stochastic gate dynamics regulate the catalytic activity of ubiquitination enzymes. J Am Chem Soc 136: 17446–17458 [DOI] [PubMed] [Google Scholar]
- Sadowski M, Suryadinata R, Lai X, Heierhorst J, Sarcevic B (2010) Molecular basis for lysine specificity in the yeast ubiquitin‐conjugating enzyme Cdc34. Mol Cell Biol 30: 2316–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A, Lewis S, Kleiger G, Kuhlman B, Deshaies RJ (2011) Essential role for ubiquitin‐ubiquitin‐conjugating enzyme interaction in ubiquitin discharge from Cdc34 to substrate. Mol Cell 42: 75–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon‐Ferrer R, Gotz AW, Poole D, Le Grand S, Walker RC (2013) Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J Chem Theory Comput 9: 3878–3888 [DOI] [PubMed] [Google Scholar]
- Saluja A, Kalonia DS (2008) Nature and consequences of protein‐protein interactions in high protein concentration solutions. Int J Pharm 358: 1–15 [DOI] [PubMed] [Google Scholar]
- Schelpe J, Monte D, Dewitte F, Sixma TK, Rucktooa P (2016) Structure of UBE2Z enzyme provides functional insight into specificity in the FAT10 protein conjugation machinery. J Biol Chem 291: 630–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serniwka SA, Shaw GS (2009) The structure of the UbcH8‐ubiquitin complex shows a unique ubiquitin interaction site. Biochemistry 48: 12169–12179 [DOI] [PubMed] [Google Scholar]
- Sheng YI, Hong JH, Doherty R, Srikumar T, Shloush J, Avvakumov GV, Walker JR, Xue S, Neculai D, Wan JW et al (2012) A human ubiquitin conjugating enzyme (E2)‐HECT E3 ligase structure‐function screen. Mol Cell Proteomics 11: 329–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart MD, Ritterhoff T, Klevit RE, Brzovic PS (2016) E2 enzymes: more than just middle men. Cell Res 26: 423–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Li B, Bharadwaj R, Zhu H, Ozkan E, Hakala K, Deisenhofer J, Yu H (2001) APC2 Cullin protein and APC11 RING protein comprise the minimal ubiquitin ligase module of the anaphase‐promoting complex. Mol Biol Cell 12: 3839–3851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vittal V, Shi L, Wenzel DM, Scaglione KM, Duncan ED, Basrur V, Elenitoba‐Johnson KSJ, Baker D, Paulson HL, Brzovic PS et al (2015) Intrinsic disorder drives N‐terminal ubiquitination by Ube2w. Nat Chem Biol 11: 83–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waudby CA, Ramos A, Cabrita LD, Christodoulou J (2016) Two‐dimensional NMR lineshape analysis. Sci Rep 6: 24826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmann F, Petzold G, VanderLinden R, Huis in 't Veld PJ, Brown NG, Lampert F, Westermann S, Stark H, Schulman BA, Peters J‐M (2016) biGBac enables rapid gene assembly for the expression of large multisubunit protein complexes. Proc Natl Acad Sci USA 113: E2564–E2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DA, Buell AK, Knowles TP, Welland ME, Dobson CM (2010) Protein aggregation in crowded environments. J Am Chem Soc 132: 5170–5175 [DOI] [PubMed] [Google Scholar]
- Wickliffe KE, Lorenz S, Wemmer DE, Kuriyan J, Rape M (2011) The mechanism of linkage‐specific ubiquitin chain elongation by a single‐subunit E2. Cell 144: 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KM, Qie S, Atkison JH, Salazar‐Arango S, Alan Diehl J, Olsen SK (2019) Structural insights into E1 recognition and the ubiquitin‐conjugating activity of the E2 enzyme Cdc34. Nat Commun 10: 3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson A, Jin L, Rape M (2009a) Preparation of synchronized human cell extracts to study ubiquitination and degradation. Methods Mol Biol 545: 301–312 [DOI] [PubMed] [Google Scholar]
- Williamson A, Wickliffe KE, Mellone BG, Song L, Karpen GH, Rape M (2009b) Identification of a physiological E2 module for the human anaphase‐promoting complex. Proc Natl Acad Sci USA 106: 18213–18218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn PJ, Religa TL, Battey JN, Banerjee A, Wade RC (2004) Determinants of functionality in the ubiquitin conjugating enzyme family. Structure 12: 1563–1574 [DOI] [PubMed] [Google Scholar]
- Wu K, Kovacev J, Pan ZQ (2010a) Priming and extending: a UbcH5/Cdc34 E2 handoff mechanism for polyubiquitination on a SCF substrate. Mol Cell 37: 784–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Merbl Y, Huo Y, Gallop JL, Tzur A, Kirschner MW (2010b) UBE2S drives elongation of K11‐linked ubiquitin chains by the anaphase‐promoting complex. Proc Natl Acad Sci USA 107: 1355–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, VanderLinden R, Weissmann F, Qiao R, Dube P, Brown N, Haselbach D, Zhang W, Sidhu S, Peters J‐M et al (2016) Cryo‐EM of mitotic checkpoint complex‐bound APC/C reveals reciprocal and conformational regulation of ubiquitin ligation. Mol Cell 63: 593–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Klenerman D, Finley D (2020) N‐terminal ubiquitination of amyloidogenic proteins triggers removal of their oligomers by the proteasome holoenzyme. J Mol Biol 432: 585–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin G, Zhang J, Nair V, Truong V, Chaia A, Petela J, Harrison J, Gorfe AA, Campbell SL (2020) KRAS ubiquitination at lysine 104 retains exchange factor regulation by dynamically modulating the conformation of the interface. iScience 23: 101448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, King RW, Peters JM, Kirschner MW (1996) Identification of a novel ubiquitin‐conjugating enzyme involved in mitotic cyclin degradation. Curr Biol 6: 455–466 [DOI] [PubMed] [Google Scholar]
- Ziemba A, Hill S, Sandoval D, Webb K, Bennett EJ, Kleiger G (2013) Multimodal mechanism of action for the Cdc34 acidic loop: a case study for why ubiquitin‐conjugating enzymes have loops and tails. J Biol Chem 288: 34882–34896 [DOI] [PMC free article] [PubMed] [Google Scholar]