

Abstract
A rapid reversed-phase ultra-high-performance liquid chromatography-high resolution mass spectrometry based mycobacterial lipidomics approach is described. This method enables the separation of various lipid classes including lipids specific to mycobacterial, such as methoxy mycolic acid and α-mycolic acid. Lipid separation occurs during a relatively short runtime of 14 minutes on a charged surface hybrid C18 column. A high-resolution quadrupole-time of flight mass spectrometer and a data independent acquisition mode allowed for the simultaneous acquisition of the full scan and collision induced dissociation fragmentation. The proposed method provides lipid detection results equivalent to or better than existing methods, but with a faster throughput and an overall higher sensitivity. The reversed-phase ultra-high-performance liquid chromatography-high resolution mass spectrometry method was shown to obtain structural information for lipids extracted from Mycobacterium smegmatis, but the method is applicable to the analysis of lipids from various bacterial and mammalian cell lines.
Keywords: Lipidomics, Mycolic acid, Mycobacteria, Liquid Chromatography, Mass Spectrometry
1. Introduction
Mass spectrometry-based approaches are now widely used to profile the global “regulome” (i.e., genes, mRNAs, proteins, and metabolites) [1, 2]. The metabolome of an organism, in general, comprises amino acids, sugars, organic acids, and lipids, among other chemical classes. Advancements in lipid extraction procedures and analysis has provided a rich repertoire of novel lipid targets that has become an important area for clinical and pharmaceutical research [3–7]. Thus, the “lipidome” has emerged as a relatively nascent subset of the larger “metabolome” analysis due to the complexity and diversity of lipid chemical structures [2]. According to LIPIDMAPS (https://www.lipidmaps.org/), lipids are broadly classified into eight categories: fatty acids, glycerolipids, glycerophospholipids, sterol lipids, prenol lipids, sphingolipids, saccharolipids and polyketides [8]. Of these eight, sterol and sphingolipids are absent in the mycobacterial lipidome [9]. Given the diversity of lipid molecules [1], it has been widely accepted that existing metabolomics protocols, sample processing procedures, extraction solvents and chromatographic separation techniques are insufficient to obtain a complete characterization of the lipidome [10, 11].
Both MS and NMR techniques has made it possible to obtain accurate qualitative and quantitative characterization of lipids [1], but MS has, by far, been the dominating approach for lipid analysis [12]. GC-MS is a well-established and standard approach for fatty acid analyses [13, 14]. GC-MS typically relies on electron ionization (EI) and chemical derivatization (i.e., fatty acid methyl esterification) prior to analysis [4, 13]. As a result, the elucidation of the unsaturation level or the location of double bonds in the fatty acyl chain is difficult by GC-MS due to extensive ion fragmentation. Therefore, high-throughput (HT) quantification of lipids by GC-MS is generally laborious and time-consuming. In contrast, liquid chromatography coupled to high-resolution mass spectrometry avoids the need for derivatization and is thus more amenable for high-throughput analysis [15]. Recently, ultra-high-performance liquid chromatography (UHPLC)-MS has become a preferred technique for HT-lipid analysis over direct infusion (DI) or GC-MS due to its ability to achieve isomer/isobar separations as well as being able to minimize the effects of ion suppression [16, 17]. In addition, LC-MS analysis provides a retention time (Rt) that, when combined with exact mass, enhances the confidence in lipid identification, especially when compared to DI methods. Nevertheless, LC-MS protocols are still time-consuming and resource intensive and require further optimization for efficient application to lipidomics [17, 18].
Currently, most lipidomics studies and method development efforts have emphasized eukaryotic cells (mammalian cells, Saccharomyces cerevisiae, Arabidopsis thaliana, etc.) or common prokaryotic model organisms like Escherichia coli [19]. In contrast, lipid information from medically important pathogens is often scarce and limited. Moreover, pathogenic organisms often have unique lipid profiles that are distinct from non-pathogenic model organisms. For example, the mycobacterium genus includes some of the most important clinically relevant human pathogens (e.g., Mycobacterium tuberculosis, M. leprae, M. ulcerans, etc.) that are responsible for over 1.5 million deaths each year [20–22]. This genus comprises acid-fast species, which is characterized by a lipid-rich cell wall that is thicker than most bacteria and produces a complex and unique lipidome [23]. In fact, lipids comprise about 30 to 60% of the dry-cell weight of mycobacteria. The presence of such a unique and extensive lipid profile is thought to play a major role in conferring pathogenicity and drug-resistance in mycobacteria [23]. In fact, several specialized MS databases, such as the M. tuberculosis lipid database (Mtb LipidDB), MycoMap and MycoMass (specific for mycobacterium genus) have emerged as valuable resources for the analysis of the mycobacteria lipidome [9, 24].
The mycobacteria lipidome consist of six lipid categories: fatty acyls (FA), glycerolipids (GL), glycerophospholipids (GP), polyketides (PK), prenol lipids (PR) and saccharolipids (SL) [21, 24]. Among these 6 lipid categories there are 15 main lipid classes: acyltrehaloses (Ac-T), diacylglycerols (DAG), fatty acids and conjugates (FA-conjs), fatty esters (FE), glycerophosphoethanolamines (PE), glycerophosphoglycerols (PG), glycerophosphoinositols (PI), glycerophosphoglycerophosphoglycerols (CL), glycerophosphoinositolglycans (PI-G), linear polyketides (L-PK), monoacylglycerols (MAG), polyprenols (Po-PR), polyketide hybrids (PKH), quinones and hydroquinones (Q-PR), triacylglycerols (TAG) [24]. Major structural lipids in the plasma membrane include phospholipids, glycosylated phospholipids (phosphatidylinositol mannosides (PIM)), lipomannans (LM), and lipoarabinomannans (LAM)) while cell wall components include peptidoglycan, arabinogalactan, glycolipids, and mycolic acids (MA) [25]. Of these, MAs are a hallmark of the mycobacteria cell envelope and are critical for survival [20, 21]. MAs are highly non-polar long chain fatty acid lipids. Notably, a primary mode of action for first-line anti-tuberculosis drugs, such as isoniazid, is the inhibition of MA biosynthesis [21, 22, 26]. Accordingly, significant effort has been devoted to deciphering the cellular biosynthesis of MAs as a therapeutic target for controlling tuberculosis. Critical to this effort is characterizing the mycobacteria lipidome. Herein, we describe a rapid and simple reversed-phase high-performance liquid chromatography-high resolution mass spectrometry (RP-UHPLC-HRMSE) strategy to detect and identify multiple classes of lipids from the Mycobacterium genus (Figure 1). The general applicability of our strategy is demonstrated by characterizing the Mycobacterium smegmatis lipidome, a non-pathogenic mycobacterium commonly used as a surrogate for M. tuberculosis [27].
Figure 1.
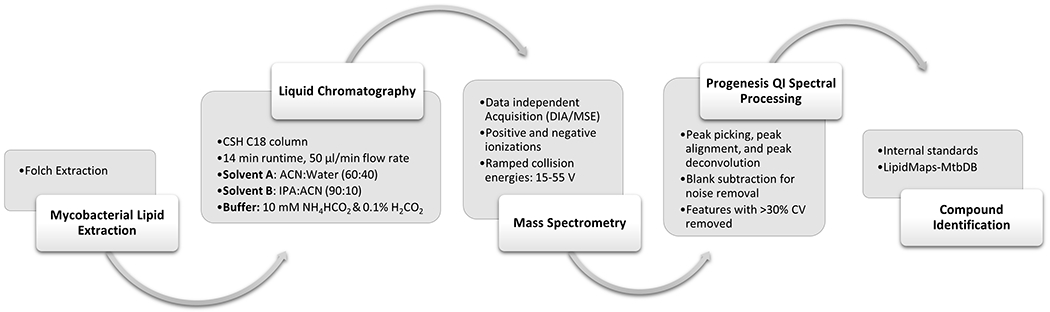
Flow diagram of a reversed-phase high-performance liquid chromatography-high resolution mass spectrometry strategy to detect and identify multiple classes of lipids from mycobacteria.
2. Material and Methods
2.1. Chemicals and Standards
EquiSPLASH™ LIPIDOMIX® Quantitative Mass Spec Internal Standard, α-mycolic acid (C80) and methoxy cis α-mycolic acid were purchased from Avanti Polar Lipids (Birmingham, AL). Optima LC-MS grade acetonitrile plus 0.1% formic acid, water plus 0.1% formic acid, methanol and 2-propanol were purchased from Fisher Chemical (Waltham, MA). LC grade chloroform was purchased from Sigma (St. Louis, MO). All other chemicals were purchased from Sigma (St. Louis, MO) and were of analytical grade quality. A 10 mM stock solution of ammonium formate at pH 6.2 was used as a standard buffer.
2.2. Preparation of Standard Samples
The EquiSPLASH™ LIPIDOMIX® stock mixture of 13 lipids was purchased as a 100 ¼g/mL solution in methanol (Table S1). A stock solution of α-mycolic acid (α-MA, 0.5 mg/mL) and a stock solution of α-methoxy mycolic acid (α-MMA, 0.75 mg mL−1) (Figure S1) were prepared in 50% chloroform in methanol (v/v). All stock solutions were stored at −80°C in amber vials until analysis. Prior to analysis, aliquots from each of the three stock solutions were combined and then diluted in 2-propanol to a volume of 100 μL for a final concentration of 1 ¼g/mL for each of the 15 lipids in the standard sample. 2 μL of the standard lipid sample was injected for each LC-MS experiment.
A second standard sample was prepared by extracting lipids from the non-pathogenic M. smegmatis wild-type MC2 155 strain. Briefly, M. smegmatis cells were cultured in 250 mL flasks in 50 ml of Middlebrook 7H9 broth supplemented with Tween-20, glycerol, bovine serum albumin (BSA), sodium chloride and D-glucose; and then grown overnight at 37°C in an orbital shaker at 200 rpm. Cells were harvested when the optical density (OD600) of the media reached 1.6 and then pelleted by centrifugation at 4000 rpm for 15 min at 4°C, and then flash frozen in liquid N2. Prior to extraction, the bacterial cells were washed with Nanopure water and pulse sonicated for 1 minute at 70% amplitude. Lipids were extracted as described by Folch et al. [28] by the addition of MeOH:H2O:CHCl3 (1:1:2 v/v). The upper aqueous phase was preserved for metabolomics analysis and the lipid enriched organic layer (lower phase) was dried using a CentriVap benchtop vacuum concentrator (Labconco). Just prior to MS analysis, the dried samples were reconstituted in 600 μL of a 2-propanol/acetonitrile (90/10, v/v) mixture containing 0.1% formic acid and 10 mM ammonium formate. 1 μL of the M. smegmatis lipid sample was injected for each LC-MS experiment.
2.3. RP-UHPLC-HRMSE Analysis of Lipids
Two commonly used Waters Acquity ultra-high performance liquid chromatography columns, a high strength silica (HSS-T3, 100 Å pore size, 1.8 um particle size, 1.0 mm x 50 mm column) and a charged surface hybrid (CSH C18, 130 Å pore size, 1.7 um particle size, 1.0 mm x 50 mm column), were selected for the development of the proposed RP-UHPLC-HRMSE method. The columns were individually evaluated for their ability to effectively separate mycobacterial lipids. For analysis, 2 μL (1 μg/mL) of the composite lipid mixture (EquiSplash + mycolic acids) was injected onto each of the LC columns using a Waters Acquity M class UHPLC class system coupled to a Waters Xevo G2-XS QTOF high resolution mass spectrometer. Mobile phase A was composed of an acetonitrile/water (60/40, v/v) mixture containing ammonium formate (10 mM, pH 6.2) and formic acid (0.1%). Mobile phase B was composed of a 2-propanol/acetonitrile (90/10, v/v) mixture containing ammonium formate (10 mM, pH 6.2) and formic acid (0.1%). The mobile phases were introduced into the MS at a constant flow rate of 50 μL/min (low flow rate, LFR). The gradient parameters for the 22-minute protocol on the HSS-T3 and CSH-C18 columns and the 14-minute protocol on the CSH-C18 column are listed in Tables 1A and 1B, respectively. The CS22-LFR method (Table 1A) used a 1 mm inner diameter CSH-C18 column with a 1.7 μm particle size and a 22-minute gradient at a flow rate of 50 ¼l/min. The HS22-LFR method (Table 1A) used an HSS-T3 column with a 1.8 μm particle size and a 22-minute gradient at a flow rate of 50 μl/min. The CS14-LFR method (Table 1B) simply reduced the CS22-LFR method to a 14-minute runtime.
Table 1.
Ultra-high performance liquid chromatography gradient parameters for our proposed method
| A) Parameters for the 22-minute runtime | B) Parameters for the 14-minute runtime | ||||||
|---|---|---|---|---|---|---|---|
| CS22-LFR and HS22-LFR | CS14-LFR | ||||||
| Time (min) | Flow (¼L/min)a | Solvent A (%)b | Solvent B (%)c | Time (min) | Flow (¼L/min)a | Solvent A (%)b | Solvent B (%)c |
| Initial | 50 | 60 | 40 | Initial | 50 | 60 | 40 |
| 2 | 57 | 43 | 1 | 57 | 43 | ||
| 12 | 46 | 54 | 6 | 46 | 54 | ||
| 12.10 | 30 | 70 | 6.10 | 30 | 70 | ||
| 18 | 1 | 99 | 9 | 1 | 99 | ||
| 20 | 1 | 99 | 12.00 | 1 | 99 | ||
| 20.10 | 60 | 40 | 12.10 | 60 | 40 | ||
| 22 | 60 | 40 | 14 | 60 | 40 | ||
Flow rate of the mobile phase.
Solvent composition: acetonitrile/water (60/40, v/v) mixture containing ammonium formate (10 mM, pH 6.2) and formic acid (0.1%).
Solvent composition: 2-propanol/acetonitrile (90/10, v/v) mixture containing ammonium formate (10 mM, pH 6.2) and formic acid (0.1%).
Lipids were analyzed in both positive and negative ionization (electrospray ionization, ESI) modes to obtain a comprehensive lipidome coverage. The mass spectrometer was operated under the following conditions: capillary voltage, 3 kV (for positive) and 2 kV (for negative); cone voltage, 30 V; source temperature, 120°C; desolvation temperature, 550°C; desolvation gas flow, 600 L/h, cone gas 50 L/h and acquisition in MSE continuum mode with either positive or negative ionization. The mass spectra were acquired over a mass range of m/z 50 to 2,000 with a scan time of 0.5 seconds. The MSE mode was operated with a low collision energy of 4 V. The high collision energy was ramped from 15 to 55 V. To ensure mass accuracy, the LockSpray interface (LockMass™) was set to Leucine Enkepahlin ([M+H]+/[M-H]− = 556.2771/554.2624 m/z). All experiments were conducted in triplicate (n=3).
2.4. Literature LC Lipidomics Methods
For validating the applicability of the proposed RP-UHPLC-HRMSE method, our CS14-LFR gradient scheme was compared against previously published LC methods (Tables 1B and 2) that were used for analyzing the lipidomes from M. tuberculosis [29] and Corynebacterium glutamicum [30]. The purpose of these comparisons was intended to place the performance of our proposed protocol in the context of acceptable parameters. It is important to note that our LC-MS method comparisons primarily focused on the differences in the LC gradients and the composition of the mobile phases. Conversely, column type, column dimensions, flow rate, and the mass spectrometer parameters (see above) were all kept constant in our study to minimize the number of variables in the comparisons. Specifically, the HS30-HFR method (Table 2A) used a 1 mm inner diameter HSS-T3 column with a 1.8 μm particle size and a 30-minute gradient with a 50 ¼l/min flow rate. The HS30-HFR method used the same mobile phases as the CS22-LFR, HS22-LFR and CS14-LFR methods, and the same column as the HS22-LFR method (Table 1). The HS22-HFR method (Table 2B) was reduced to a runtime of 22 minutes and used 100% water as mobile phase A and 100% acetonitrile as mobile phase B. The flow rate was also increased to 100 ¼l/min. The HS25-HFR method (Table 2C) increased the runtime to 25 minutes and returned the flow rate to 50 ¼l/min while using the same mobile phases as HS22-HFR. Overall, there were a few technical differences between the study reported herein and the prior literature studies, which are briefly summarized below.
Table 2.
Ultra-high performance liquid chromatography gradient parameters for literature methods.
| A) | B) | C) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HS30-LFRa | HS22-HFRb | HS25-LFRc | |||||||||
| Time (min) | Flow (¼L/min)d | Solvent A (%)e | Solvent B (%)f | Time (min) | Flow (¼L/min)d | Solvent A (%)g | Solvent B (%)h | Time (min) | Flow (¼L/min)d | Solvent A (%)g | Solvent B (%)h |
| Initial | 50 | 68 | 32 | Initial | 100 | 99 | 1 | Initial | 50 | 99 | 1 |
| 1.5 | 55 | 45 | 2 | 90 | 10 | 2 | 90 | 10 | |||
| 4 | 48 | 52 | 6 | 70 | 30 | 6 | 70 | 30 | |||
| 5 | 42 | 58 | 8 | 50 | 50 | 8 | 50 | 50 | |||
| 8 | 34 | 66 | 12 | 25 | 75 | 12 | 25 | 75 | |||
| 11 | 30 | 70 | 15 | 1 | 99 | 15 | 1 | 99 | |||
| 14 | 25 | 75 | 20 | 0 | 100 | 20 | 0 | 100 | |||
| 18 | 3 | 97 | 21 | 0 | 100 | 21 | 0 | 100 | |||
| 21 | 3 | 97 | 21.50 | 99 | 1 | 21.50 | 99 | 1 | |||
| 25 | 3 | 97 | 22 | 99 | 1 | 25 | 99 | 1 | |||
| 25.10 | 68 | 32 | |||||||||
| 30 | 68 | 32 | |||||||||
Method adapted from Klatt et al. (2018).
Method adapted from Raghunandan et al. (2019).
Method modified from Raghunandan et al. (2019).
Flow rate of solvents (mobile phase).
Solvent composition: Acetonitrile/water (60/40, v/v) mixture containing ammonium formate (10 mM, pH 6.2) and formic acid (0.1%).
Solvent composition: 2-propanol/acetonitrile (90/10, v/v) mixture containing ammonium formate (10 mM, pH 6.2) and formic acid (0.1%).
Solvent composition: 100% water containing 0.1% formic acid.
Solvent composition: 100% acetonitrile containing 0.1% formic acid.
The M. tuberculosis lipidomics study [29] utilized an HSS-T3 column with different dimensions (2.1 × 100 mm, 1.8 ¼m) to detect lipids using a 20-minute gradient runtime with a flow rate of 400 μL/min. This LC method was adapted in this study as HS22-HFR. HS25-LFR was a modification of HS22-HFR with a longer 25-min gradient runtime and a lower flow rate. Importantly, acetonitrile and water were used in the mobile phases for both HS22-HFR and HS25-LFR. The C. glutamicum lipidomics study utilized a C18 column (Phenomenex Kinetex, 2.6 μm EVO C18 100Å) to detect lipids using a 30-minute gradient runtime with a flow rate of 260 μL/min. This LC method was adapted in this study as HS30-LFR. The mobile phases used by HS30-LFR were identical to our CS14-LFR method.
Importantly, height equivalent to a theoretical plate (HETP) versus flow rate plots (Figure S2) have previously demonstrated that an optimal flow rate for a 2.1 mm inner diameter column is 450 ¼l/min, which reduces to 100 ¼l/min for a 1 mm inner diameter column [31–34]. Since the inner diameter of all the columns used in this study was 1 mm, the literature flow rates of 400- to 260 μL/min were reduced to 100- 50 μL/min. Our UHPLC system has a back pressure upper limit of 10,000 PSI which restricted the flow rate to 50 μL/min when 2-propanol was used as the mobile phase in a 1 mm column. The flow rate could only be increased to 100 μL/min when 2-propanol was not used. Simply, 2-propanol (2.27 cP) has a significantly higher viscosity than acetonitrile (0.37 cP). Thus, a constant flow rate of 50 μL/min was used for all experiments except for HS22-HFR where the flow rate was increased to 100 μL/min. This allowed for us to determine if an increase in flow rate yielded an overall improvement in the performance of an LC-MS method.
2.5. Data Processing
All mass spectra were processed using MassLynx (version 4.2) and MSE Data Viewer (Waters, Billerica, MA). Preliminary fragment information was identified using DI-MS spectra and validated based on theoretical fragmentation obtained from ChemDraw (PerkinElmer, Waltham, MA) and the scientific literature [35–37]. Lipid nomenclature followed the Mtb-LIPIDMAPS classification system that is based on the classification system by Sartain et al. [38]. Non-common lipid abbreviations are indicated in parenthesis. Mass spectra of M. smegmatis lipid samples were further processed using Progenesis® QI for metabolomics (v2.4, Nonlinear Dynamics, Newcastle, UK). Progenesis® QI offers an automated workflow pipeline for data analysis including peak picking, peak alignment, peak deconvolution, and feature identification associated with retention time and m/z value. For positive ionization, adducts were assigned as [M+H]+, [M+Na]+, [M+NH4]+, [M–H2O+H]+ or [M+K]+. For negative ionization, adducts were assigned as [M-H]−, [M-H2O-H]−, or [M+FA-H]− (FA, formic acid). Blank spectra were subtracted from experimental spectra to remove noise and artifact peaks. Similarly, spectral features with a coefficient of variation higher than 30% were removed. For tentative lipid assignments at MS1-level, mass spectral features were searched against LipidMaps-MtbDB (https://www.lipidmaps.org/tools/ms/Mtb_batch_bulk.html) [24, 39, 40]. Search parameters were defined as follows: accurate mass tolerance of 10 ppm for the parent ion, any lipid class from the mycobacterial database, and [M+H]+, [M+Na]+, [M+NH4]+ and [M-H]− adducts. Please note that MS2-level fragment data were not available for searching in the LipidMaps-MtbDB database.
3. Results
A modified RP-UHPLC-HRMSE method that can rapidly detect microbial lipids, including mycolic acids, is presented. The major lipid components of the mycobacterial cell wall α-MA and α-MMA were used as representative mycolic acid lipids (Figure S1). α-MA and α-MMA were combined with the thirteen deuterium-labeled lipids from the EquiSPLASH™ LIPIDOMIX® to produce a standard lipid mixture for evaluating and optimizing the RP-UHPLC-HRMSE method. In this regard, the standard mixture contained representatives from the following lipid categories: fatty acids, glycerolipids, glycerophospholipids, sterol lipids, and sphingolipids. The proposed RP-UHPLC-HRMSE strategy was optimized in phases by evaluating MS operating parameters, analytical columns, and finally the LC run time.
3.1. Optimization of MS Parameters to Detect the Lipids in the Standard Mixture
The first phase of the method development strategy focused on optimizing the precursor mass detection parameters and collision energy (CE) voltages for fragmentation. Precursor masses of the individual lipids in the composite mixture were qualitatively analyzed by performing an MS1-level scan of the lipids introduced via DI in both positive and negative modes. All 15 lipids, including the two MAs, were detected in either the negative or positive mode. Ceramide (Cer), cholesteryl ester (Chol Ester), diacylglycerol (DAG), lyso-phosphocholine (LPC), monoacylglycerol (MAG), glycerophosphocholine (PC), glycerophosphoethanolamine (PE), glycerophosphoserine (PS), sphingomyelin (SM), and triacylglycerol (TAG) preferentially ionized with positive polarity while lyso-phosphoethanolamine (LPE), glycerophosphoethanolamine (PE), glycerophosphoglycerol (PG), glycerophosphoinositol (PI) and both MAs (α-MA and α-MMA) showed better ionization in negative polarity (Table S1). SM, Cer, LPC and PC were also ionized in negative polarity as formate adducts [M+HCOO]−, while the MAs ionized in positive polarity as Na adducts [M+Na]+, albeit at a lower response. Ionization of MAs in positive polarity as [M+Na]+ adducts has been previously reported [23]. In contrast, Chol Ester and the glycerolipids (TAG, DAG, and MAG) only ionized with positive polarity (Table S1).
The efficiency of different CE voltages to fragment individual lipid species was surveyed by applying voltages ranging from 15 to 35 V in both the positive and negative ionization mode. As the optimal CE energy required for fragmentation varied widely across the lipid classes, a CE ramp from 15 to 35 V was deemed necessary to observe qualitative fragmentation of all lipids. Lipid assignments were verified (Table S1) based on calculated m/z values and MS2 theoretical fragmentation information [41, 42]. MAs were observed to ionize better with negative ionization [20]. Therefore, α-MA and α-MMA were assessed in negative polarity with a 15 to 35 V CE ramp. However, the CE voltage ramp did not induce the expected side-chain fragmentation as previously observed with DI-MS/MS at a CE voltage of 55 V (Figure S3). Consequently, α-MA and α-MMA were individually subjected to a range of ramp CE voltages from 15-35 V to 15-55 V (Figures S4–S5). Ramping up the CE voltage to 55 V provided adequate energy to fragment the MA side chains. Thus, our RP-UHPLC-HRMSE strategy used a CE voltage ramp of 15 to 35 V in the positive mode and a CE ramp voltage of 15 to 55 V in the negative mode to derive the expected lipid fragments from a single LC-MSE analysis (Figure 2).
Figure 2.
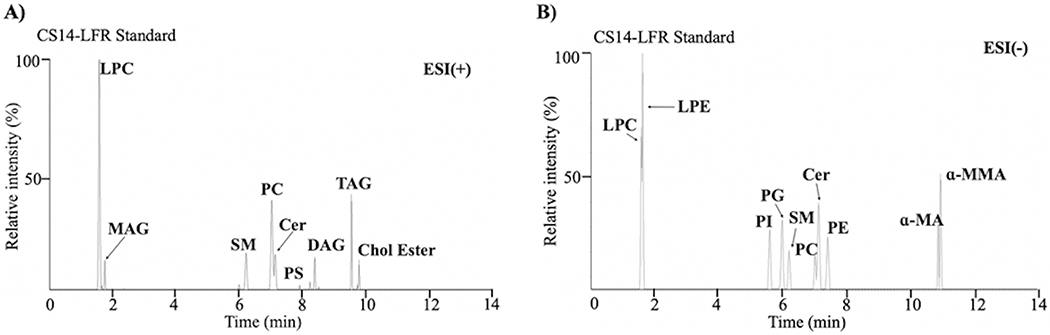
(A) Chromatogram of the standard lipid mixture in the positive ionization mode. The analytes were separated on an Acquity ultra-high performance liquid chromatography charged surface hybrid column with an acetonitrile/water (60/40, v/v) mixture containing ammonium formate (10 mM, pH 6.2) and formic acid (0.1%) (Solvent A), and a 2-propanol/acetonitrile (90/10, v/v) mixture containing ammonium formate (10 mM, pH 6.2) and formic acid (0.1%). (Solvent B). (B) Chromatogram of the standard lipid mixture in the negative ionization mode. The analytes were separated in 14 minutes on an Acquity ultra-high performance liquid chromatography charged surface hybrid column with an acetonitrile/water (60/40, v/v) mixture containing ammonium formate (10 mM, pH 6.2) and formic acid (0.1%) (Solvent A), and a 2-propanol/acetonitrile (90/10, v/v) mixture containing ammonium formate (10 mM, pH 6.2) and formic acid (0.1%). (Solvent B). Abbreviations of the lipids correspond to: Cer: ceramide, Chol Ester: cholesteryl ester, DAG: diacylglycerol, LPC: lyso-phosphocholine, LPE: lyso-phosphoethanolamine, α-MA: α-mycolic acid, MAG: monoacylglycerol, MMA: methoxy mycolic acid, PC: glycerophosphocholine, PE: glycerophosphoethanolamine, PG: glycerophosphoglycerol, PI: glycerophosphoinositol, PS: glycerophosphoserine, SM: sphingomyelin, and TAG: triacylglycerol.
3.2. Optimization of UHPLC Conditions to Detect the Lipids in the Standard Mixture
In the next phase, the performance of the two analytical columns were compared to identify a system that provided the best lipid separation. Charged surface enhanced (CSH) and high strength silica (HSS) T3 columns were selected for evaluation since these C18 columns are widely used in lipidomics research [43]. CSH particles were designed to offer a low level surface charge that promotes peak symmetry in low-ionic-strength mobile phases [44]. Previous studies have reported the suitability of the CSH column for the separation of different lipid molecular species and lipid isomers [43, 45]. Similarly, the HSS column has been extensively used for both metabolomics and lipidomics studies, which suggests a potential suitability for a coupled omics experiment with improved high-throughput capabilities [16, 17, 43]. Our preliminary results showed that the CSH column performed comparatively better than the HSS column. The CSH column demonstrated improved lipid separations, while maintaining sharp and symmetric peak shapes. For example, 12 out of 14 peaks exhibited smaller peak widths on the CSH column (Table 1). Specifically, the α-MA and α-MMA peak shapes were considerably improved on the CSH column compared to the HSS column (Figures S6–S7). Therefore, subsequent method development and optimization utilized the better performing CSH column.
The final stage of the method development strategy involved optimizing the LC gradient runtime. A preliminary gradient runtime was developed based on generic lipidomics methods having runtimes ranging from 20 to 25 minutes. Accordingly, the initial version of the method design had a total gradient runtime of 22 minutes, which included end-run column equilibration (Table 1). The rational for this elution gradient was based on three main principals: (1) a protocol complimentary to untargeted metabolomics – emphasize non-polar lipids, (2) an elution gradient that contains three primary phases that first target polar lipids, moderately non-polar lipids, and then non-polar lipids to optimize resolution within each group, and (3) maximize the detection and separation of mycolic acids, which are important lipids to mycobacteria. This was achieved by using a higher percentage of non-polar solvents (i.e., 2-propanol), by using relatively slow gradients separated by rapid, step transitions, and by using mycolic acid standards to experimentally validate gradient performance.
Though initial method provided a separation of all the lipids in the standard mixture, it was still not sufficient for high throughput lipidomics experiments with a high number of samples. Based on the results from our initial method, the runtime was revised to a shorter 14 minutes (i.e., CS14-LFR, Table 1), which included an approximate 2-minute end-run column equilibration time. Notably, the CS14-LFR protocol did not compromise on the analytical separation of the lipids. In this revised method, all the lipids in the standard mixture, including the MAs, were detected with baseline or near baseline separation. Quantitative analysis of the LC spectra showed that the average minimal peak resolution for two mycolic acids on the CSH column was 0.99 ± 0.06 for the 14-minute runtime while the resolution on the CSH and HSS columns at a 22-minute runtime were 0.8 ± 0.2 and 0.97 ± 0.01, respectively (Figure 3 and Table S3). Therefore, our optimized CS14-LFR method with the shortest 14-minute runtime was able to elute all the 15 lipids from the standard mixture with equal to or better resolution than the longer runtimes (Figure 4). Please note, all LC-MS methods described in this study relied on a combination of positive and negative ionization modes for lipid detection.
Figure 3.
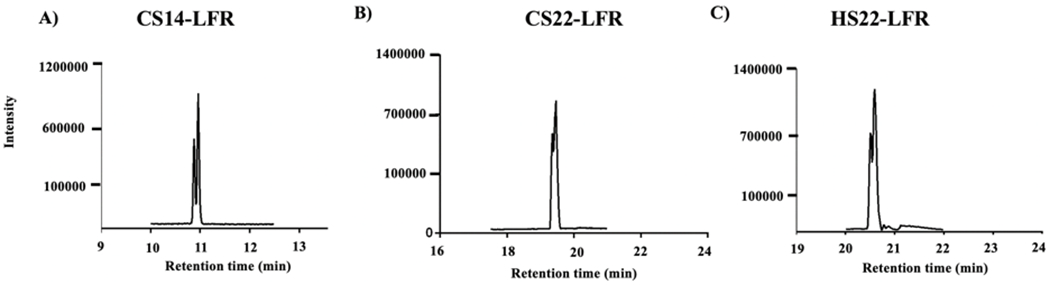
Chromatograms illustrating the resolution between two mycolic acids on a (A) charged surface hybrid column with a 14-minute run time, (B) a charged surface hybrid column with a 22-minute run time, and (C) a high strength silica column with a 22-minute run time. The details of the chromatographic gradients are given in Table 1.
Figure 4.
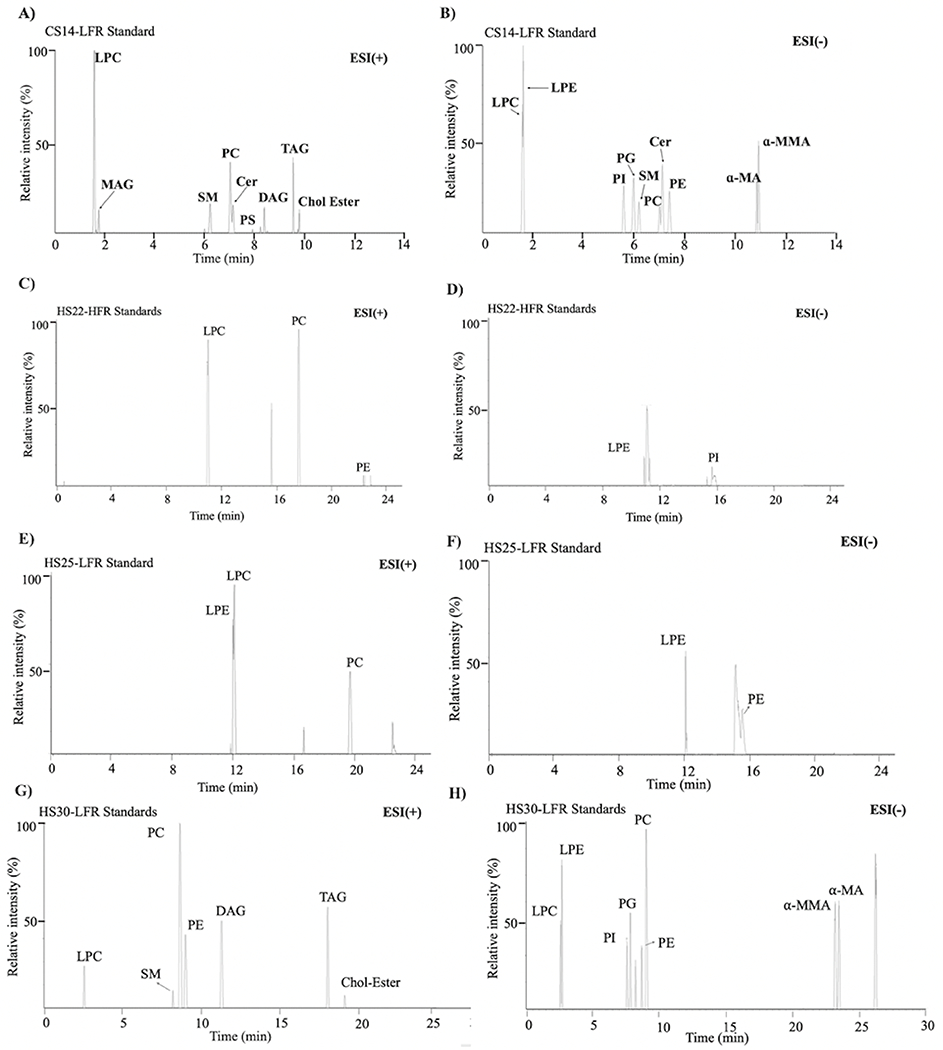
Illustrative examples of chromatograms obtained for the standard lipid mixture using the (A, B) CS14-LFR, (C, D) HS22-HFR, (E, F) HS25-LFR, and (G, H) HS30-LFR methods with (left) positive and (right) negative ionization modes.
3.2. Application of the RP-UHPLC-HRMSE Strategy to Detect Lipids in M. smegmatis Cell Lysate
Lipids were also extracted from lysed M. smegmatis bacterial cells to illustrate the applicability of the proposed CS14-LFR method to real-world analyses. A representative chromatogram is shown in Figure 5. The raw LC-MS spectra were processed using Progenesis QI for Metabolomics and yielded a total of 5433 spectral features, 2574 features in positive mode and 2859 features in negative mode. The raw and median intensity of these features was 5.7x104 ± 1x103 and 2.9 x103 ± 1 x102, respectively (Table S4). Feature identification was then carried out using LipidMaps-MtbDB [24, 39, 40], which resulted in identifying a total of 308 lipids (MS1-level) consisting of 111 and 197 lipids being detected in the positive and negative ionization modes, respectively. Overall, 16 FA-Conj, 15 FA-Esters, 12 PKH, 12 Ac-T, 2 Po-PR, and 1 LPK were identified, which corresponds to mycobacterial specific lipid classes. Similarly, 166 PI-G, 3 CL, 9 PI, 12 PE, 20 TAG, 8 PG, 20 DAG, and 13 MAG lipids were identified, which corresponds to general lipid classes. The median peak intensities for these identified lipids were 2.70 x103 ± 8 x105 (Table S5). These results are summarized in Figures 6 to 8.
Figure 5.
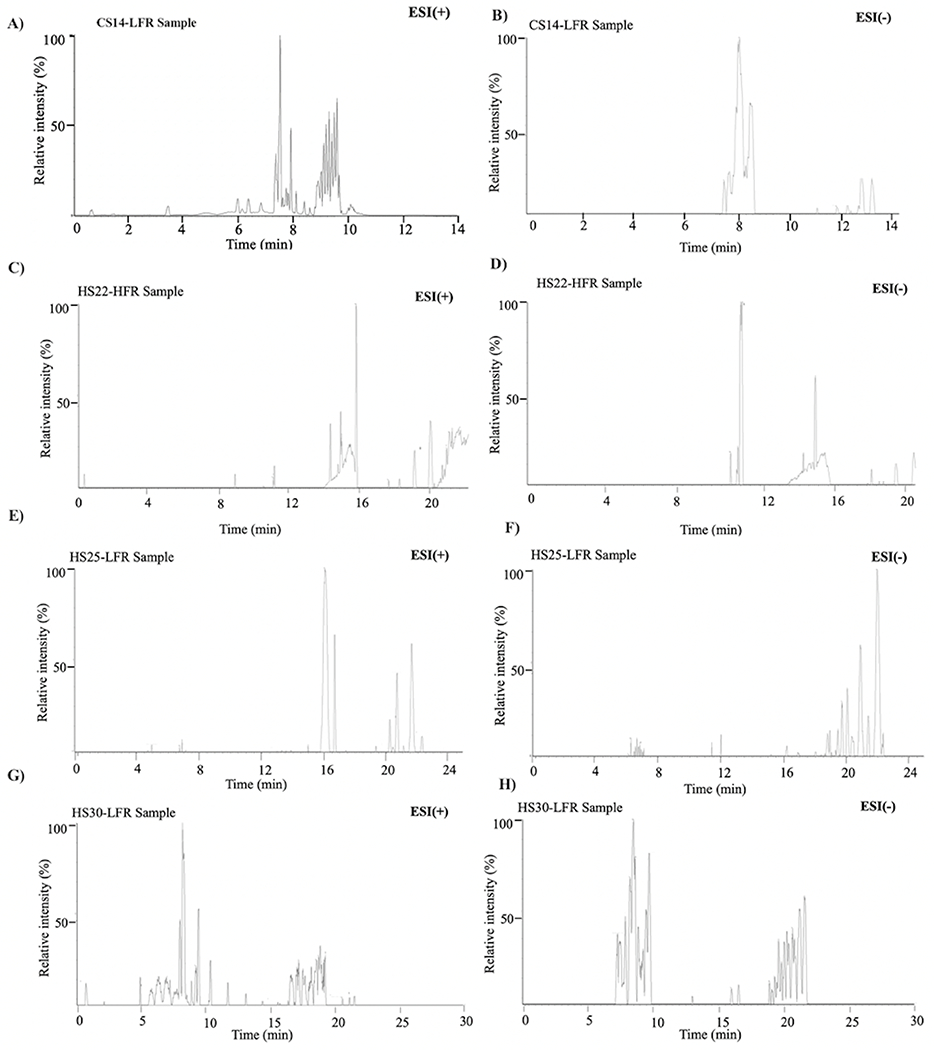
Illustrative examples of chromatograms obtained for the lipid mixture extracted from M. smegmatis cell lysates using the (A, B) CS14-LFR, (C, D) HS22-HFR, (E, F) HS25-LFR, and (G, H) HS30-LFR methods with (left) positive and (right) negative ionization modes.
Figure 6.
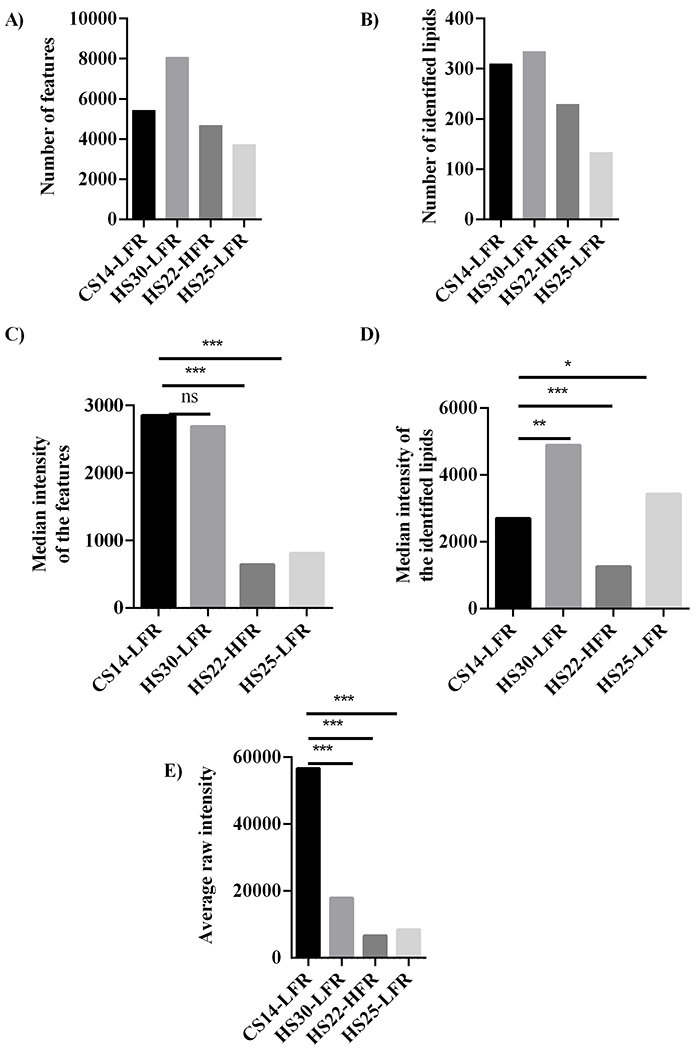
Bar plots summarizing the (A) total number of features from both the positive and negative ionization mode, (B) the total number of identified lipids from both the positive and negative ionization mode, (C) the median intensity of all spectral features, (D) median intensity of spectral features only for the identified lipids, and (E) the average value of the raw intensities of all features for methods CS14-LFR, HS30-LFR, HS22-HFR, and HS25-LFR. Statistical significance is indicated as: no statistical difference, NS, p < 0.05, *, p < 0.01, **, and p < 0.001, ***.
Figure 8.
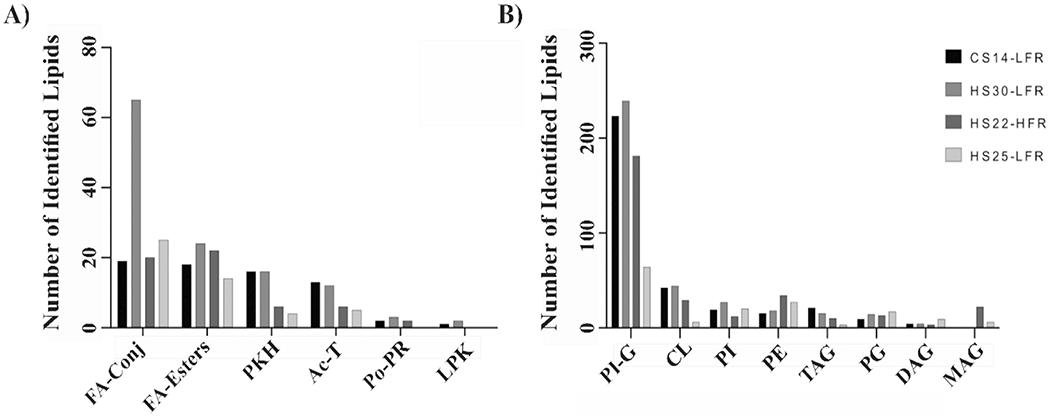
Bar plots summarizing the number of (A) mycobacterial specific or (B) general lipid classes identified by the CS14-LFR, HS30-LFR, HS22-HFR, and HS25-LFR methods. Abbreviations of the lipid classes correspond to: acyltrehaloses (Ac-T), diacylglycerols (DAG), fatty acids and conjugates (FA-conjs), fatty esters (FE), glycerophosphoethanolamines (PE), glycerophosphoglycerols (PG), glycerophosphoglycerophosphoglycerols (CL), glycerophosphoinositols (PI), glycerophosphoinositolglycans (PI-G), linear polyketides (L-PK), monoacylglycerols (MAG), polyprenols (Po-PR), polyketide hybrids (PKH), quinones and hydroquinones (Q-PR), triacylglycerols (TAG).
3.4. Performance Comparison of LC Methods
The overall potential of our CS14-LFR method for improving lipidomics efficiency was evaluated by comparing its performance against three mycobacterial lipidomics methods (i.e., HS30-LFR, HS22-HFR, HS25-LFR) that were previously described in the literature (Table 2). Importantly, these literature methods were primarily used to benchmark the performance of our CS14-LFR method and to assess the coverage of the lipidome. These three literature methods had significantly longer total runtimes, which ranged from 22 to 30 minutes, compared to our 14-minute runtime. Accordingly, our CS14-LFR method is 1.6 to 2.1 times faster than these standard lipidomics approaches. This is a very significant and impactful improvement considering that a given lipidomics study may have a 100 or more samples. Of course, increasing throughput is irrelevant if it sacrifices overall sensitivity and/or the number of features/lipids detected.
3.4.1. Impact of Mobile Phase Polarity on Lipid Detection
The HS25-HFR and HS25-LFR methods used water for mobile phase A and acetonitrile for mobile phase B. These mobile phases were notably different from both the CS14-LFR and HS30-LFR methods, which used an acetonitrile/water mixture and a 2-propanol/acetonitrile mixture for mobile phases A and B, respectively (Tables 1 and 2). Given the polarity index (PI) for these solvents, 2-propanol (PI 3.92), acetonitrile (PI 5.8), and water (PI 10.2), the mobile phases for the HS25-HFR and HS25-LFR methods were significantly more polar than both CS14-LFR and HS30-LFR. Not surprisingly, the polarity of the mobile phases is critical for the optimal separation of lipids and for maximizing the diversity of lipids detected and extracted from a biological sample. Simply, non-polar, hydrophobic lipids will have a higher affinity for non-polar solvents. In general, decreasing the polarity of the mobile phase will result in a decrease in retention times, but it will also impact which lipids are captured or eluted during the relatively higher polarity of mobile phase A. Importantly, the relative solubility of the lipids in the mobile phase will also impact what lipids make it onto the column. In this regard, non-polar solvents were expected to yield a better outcome for untargeted lipidomics.
Indeed, a comparison of the results with the standard mixture showed that HS25-LFR and HS25-HFR did not capture non-polar lipids such as glycerolipids (MAG, DAG, TAG) or Chol-Ester (Figure 4 and Table S6). An increase in the flow rate from 50 to 100 μL/min did not improve the lipid detection. CS14-LFR and HS30-LFR performed better than HS25-LFR and HS22-HFR and captured all the lipids in the standard mixture with near baseline separation.
Similarly, CS14-LFR and HS30-LFR performed better than HS25-LFR and HS22-HFR in detecting lipids from the M. smegmatis cellular extract (Figures 6–8). CS14-LFR and HS30-LFR detected a considerably larger number of lipids and features with an overall higher peak intensity. Our CS14-LFR method detected 5433 features from the M. smegmatis bacterial cell extract, which compares well to the 8063 features detected by the HS30-LFR method. Our CS14-LFR method out-performed both the HS22-HFR and HS25-LFR methods, which only detected 3727 and 4679 features, respectively (Figure 6A). CS14-LFR did equally well in the number of lipids identified. CS14-LFR detected 308 lipids compared to 334 lipids identified by HS30-LFR, 229 lipids identified by HS22-HFR, and 133 lipids detected by HS25-LFR (Figure 6B and Table S7). Clearly, a decrease in the polarity of the mobile phase solvents significantly improved the overall performance of the LC-MS lipidomics method.
A longer runtime would also be expected to lead to the identification of more features and lipids, but our CS14-LFR method did substantially better than the two methods with longer run-times. Of course, the differences in the mobile phase polarities may have also contribute to these observed differences in lipid retention times and detection. For example, consider non-polar lipids such as TAGs, which are in the glycerolipid class of lipids. A total of 20 TAGs were detected with the CS14-LFR method, whereas; only 3 or 2 TAGs were captured by the HS22-HFR or HS25-LFR method, respectively. CS14-LFR compared well to the HS30-LFR method, which had the longest runtime of 30 minutes. Again, CS14-LFR and HS30-LFR shared the same mobile phases, but used different elution gradients (Table 1B, Table 2A). Interestingly, only 12 TAGs were detected with the HS30-LFR method, which may be a result of differences in the elution gradient. Specifically, the CS14-LFR gradient utilized a higher concentration of a non-polar eluent (90% 2-propanol) during the remaining minutes of the LC program. Thus, CS14-LFR provides a reasonable trade-off, a 2.1 times faster runtime that still detects about 92% of the observable lipids.
3.4.2. Differences in Sensitivity and Coverage
Enhancing signal sensitivity is equally as important as maximizing the coverage of the lipidome. Simply, a higher signal-to-noise ratio will likely improve reproducibility and reduce between group and within group variance. A comparison of feature intensities showed that our CS14-LFR method produced the highest median peak intensities of all the methods evaluated. In particular, CS14-LFR yielded peak intensities over 3-times the intensities produced by methods HS22-HFR and HS25-LFR (Figure 6C). Feature intensities for identified lipids were also compared between the four methods. Again, our CS14-LFR method yielded statistically significant higher median peak intensities compared to HS22-HFR (Figure 6D). Conversely, median peak intensities were statistically lower for CS14-LFR compared to HS25-LFR and HS30-LFR. A comparison of the combined average raw intensities of the features detected in both positive and negative ionization mode for our CS14-LFR method with those of the other three methods indicated that the overall sensitivity of CS14-LFR was significantly higher (p<0.001) (Figure 6E and Tables S4–S5).
The detected features were further processed to assign lipids at the MS1-level, which were then compared across the four LC-MS methods. Of the 308 lipids identified by the CS14-LFR method, only 105, 61 and 34 of these lipids were detected by HS30-LFR, HS22-HFR and HS25-LFR, respectively. This is a particularly worrisome observation that gradient runtimes can drastically change the complete set of lipids that are detected despite using identical samples and instrumentation.
A comparison of the median normalized abundances of the lipids common to CS14-LFR and each of the other methods yielded a Pearson correlation of 0.85 for HS30-LFR, 0.74 for HS22-HFR, and 0.54 for HS25-LFR. In addition, normalized intensities of the lipids common to CS14-LFR and to either of the other three methods showed no statistically significant difference (Figures 5C–7A). Conversely, a comparison of the median peak intensities for the common lipids showed our CS14-LFR method yielded statistically significant higher peak intensities than HS22-HFR and HS25-LFR (Figures 7D, E). CS14-LFR yielded statistically equivalent peak intensities to HS30-LFR (Figure 7F). Overall, our shorter run time CS14-LFR method performed comparable to or better than the longer run-time methods regarding lipidome coverage.
Figure 7.
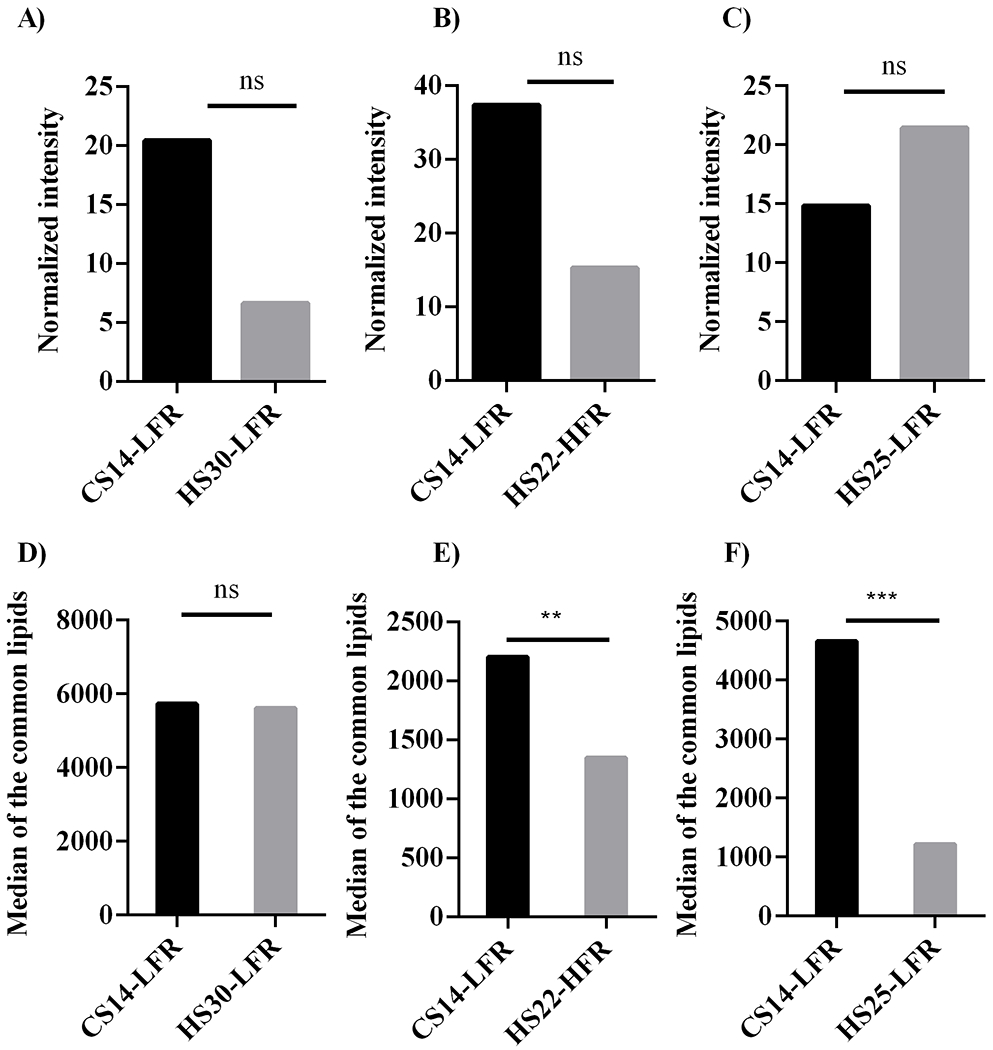
Bar plots comparing the normalized lipid peak intensities from CS14-LFR to (A) HS30-LFR, (B) HS22-HFR, and (C) HS25-LFR. Bar plots comparing the median lipid peak intensities from CS14-LFR to (D) HS30-LFR, (E) HS22-HFR, and (F) HS25-LFR. Statistical significance is indicated as: no statistical difference, NS, p < 0.05, *, p < 0.01, **, and p < 0.001, ***.
We also examined the coverage of the 15 main lipid classes following the Mtb. LipidMaps data base classification system created by Sartain et. al. [24]. Both our CS14-LFR method and the HS30-LFR method identified lipids from 13 of the 15 main lipid classes, which included 6 of the 7 mycobacterial specific lipid main classes (Figure 8). Thus, our CS14-LFR method captured the same number of lipid main classes within half the time of the HS30-LFR method. HS22-HFR and HS25-LFR identified 13 or 12 of the 15 main lipid classes, respectively. This included either 5 or 4 of the 7 mycobacterial specific lipid main classes, respectively. In general, our CS14-LFR method performed better than either the HS22-HFR or HS25-LFR methods. Therefore, it is prudent to conclude that CS14-LFR provides for a high-throughput lipid analysis without compromising on sensitivity or lipidome coverage. Overall, our proposed method yields a comparable characterization of the M. smegmatis lipidome while achieving a 1.6 to 2.1 times faster throughput.
4. Discussion
Sample throughput and molecular coverage are two important considerations for any omics analysis. A major bottleneck in LC-MS based lipidomics is lipid identification. Thus, untargeted lipidomic workflows usually rely on lipid separation with a reversed-phase C18 column and a solvent gradient to facilitate lipid identification [24]. Although more complex than direct-injection ionization, chromatographic separation of the lipids from a complex biological mixture prior to ionization offers several advantages. For example, chromatography separates molecules of similar mass, which enables individualized detection while reducing ion suppression.25 Furthermore, column retention predicts the polarity of unknown molecules, which facilitates their identification when combined with exact mass. Thus, the acquisition of accurate masses and retention times along with detailed fragmentation patterns is key to obtaining reliable lipid assignments. Accordingly, the MSE acquisition mode provides a unique advantage and a preferred choice for our RP-UHPLC-HRMSE strategy. The acquisition of a mass spectrum in a data independent mode allows for all the ions observed in the MS1-level scan to be fragmented simultaneously. Thus, the MSE acquisition mode delivers a comprehensive tandem MS spectrum in a single analytical run [46]. Altogether, LC-MS creates a two-dimensional accurate mass retention time (AMRT) area to resolve individual components from mixtures containing thousands of ions [9]. CSH and HSS columns have been routinely used in LC-MS lipidomic studies [43]. Our analysis of these columns as part of our RP-UHPLC-HRMSE method development identified CSH as a preferred choice due to the better lipid separation and peak shape (Figures 2, S6).
Most LC-MS run-times for eukaryotic lipid analyses are over 20 minutes and are greater than 35 minutes for mycobacterial lipidomics [9, 24, 38, 47]. A keyword query of the scientific literature published since 2011 found only 25 papers that analyzed mycolic acids using LC-MS (Figures S8). Of these, three studies reported LC run times between 15 and 20 minutes while the remaining studies reported run times of >35 minutes. Notably, the relatively short run times relied on a mycolic acid specific extraction protocol or used a mycolic acid derivatization (mycolic acid methyl ester, MAME) schema. However, it is important to note that the additional extraction protocol or the added derivatization step significantly lengthened the overall experimental time beyond the short LC run proposed in the present study [48, 49]. Other published studies have analyzed microbial lipids using GC instead of LC [23]. However, GC lipidomic methods have considerably longer run times, which can vary from 16 minutes to over 60 minutes depending on the carbon chain-length of the mycolic acids [50–53]. Moreover, GC analysis required derivatization of the lipids that can further add to the total sample analysis time. To the best of the authors knowledge, there are no published protocols that report analytical runtimes for bacterial lipid analysis lower than 15 minutes.
A long LC or GC run time makes high throughput lipidomic analyses challenging, if not impractical, for large cohorts of hundreds to thousands of samples. Moreover, not all lipids ionize with only one polarity, making it necessary for LC-MS spectra to be acquired in both positive and negative modes for complete coverage of the lipidome. In effect, doubling the overall experiment time, which further emphasizes the importance of reducing the gradient run time to the shortest possible timeframe. Recently, Xuan et al. (2020) developed a rapid lipid profiling protocol that detected 481 lipids covering 20 common lipid subclasses from 40 μL of human serum within 13 minutes [16]. In effect, Xuan et al. (2020) demonstrates the potential of a shorter chromatographic time for enhancing the coverage of the lipidome. In this regard, the proposed method (Table 1B) provides a rapid, 14 minutes analytical method for analyzing mycobacterial lipids compared to other mycobacterial lipidomic methods.
The annotation of lipids by comparing only precursor accurate mass with existing databases will lead to numerous potential matches and ambiguous identifications. This problem can be rectified by basing lipid identification on a MS/MS analysis. GC-MS has been widely used for the analysis of mycolic acids, but suffers from complex EI fragmentation patterns that may be difficult to interpret [23]. In contrast, LC-MS may facilitate multistage fragmentation (MSn), which produces distinct and identifiable fragmentation patterns. For example, mycolic acids are a unique class of mycobacterial lipids that are not found in eukaryotes. Hong et al. (2012) determined the chemical composition of 65 homologous mycolic acids from Segniliparus mycolates [54]. They also identified three different α-mycolic acid subclasses: α+-mycolates, α-mycolates and short α’-mycolates, further illustrating the diversity of mycolic acids. Finer structures of the R group (Figure S3) of the mycolic acid can be determined by MS when coupled to a collision-induced dissociation (CID) process [23]. Indeed, Song et al. (2009) described a rapid and informative ESI-MS/MS protocol for mycolic acid profiling that was able to identify individual R groups [37]. This led to an enhanced characterization of the structural diversity within this lipid class. Plumb et al. (2006) described a new data independent acquisition mode (MSE), which has been introduced as a rapid approach for generating molecular fragments in LC-MS studies [46]. The MSE acquisition mode allows for the structures of numerous lipids to be confirmed in a single analytical run by the simultaneous acquisition of an exact mass precursor and fragment ion data. The spectral data obtained by MSE is comparable to conventional LC-MS/MS [46]. To include the MSE acquisition mode in our LC-MS scheme, an optimal CE voltage to uniformly fragment mycobacterial lipids needed to be identified. To this end, we explored a range of CE voltages and determined that a ramp voltage from 15 to 55 V was needed to effectively fragment the diversity of lipids, including mycolic acids, that are found in mycobacteria. Figure 2 demonstrates the complete identification of the 15 lipids from a standard mixture comprising 5 distinct lipid categories. Thus, the MSE acquisition mode combined with a rapid LC run time and a CSH column enabled us to efficiently identify the type of lipids commonly found in mycobacterium species.
The high-throughput analysis of eukaryotic lipids has gained in popularity over the years, but bacterial lipidomics, specifically mycobacterial lipidomics, is still an emerging field. As a result, there is a need to develop and optimize LC-MS lipidomics techniques to enhance the understanding of the mycobacterial lipidome. Furthermore, the identification of unknown lipids at either an MS1 or MS2 level presents a similar challenge. In this regard, databases specific to mycobacterial lipids are limited in their structure prediction capabilities. For example, the LipidMaps-MtbDB prediction tool consolidated approximately 2,500 precursor ions from LC-MS lipid profiling, but it does not contain fragment ion information [24, 39, 40]. Accordingly, labor-intensive, and impractical manual data curation is necessary to achieve lipid assignments at the MS2 level. Achieving an MS3 level for lipid identification by using deuterated standards is even more difficult because of the large size of the lipidome (over 46,000 lipids in the Lipid Maps database) and the limited number of commercially available lipids. Hence, improving the lipidomics analysis of mycobacterial requires efficient data acquisition protocols, improvements in data analysis, and enhancements to reference data.
Mycobacterium tuberculosis displays one of nature’s most complex lipid envelopes, containing an inner phospholipid-bilayer and an outer layer of mycolic acids and other conjugated lipids (i.e. glycolipids) [55]. Lipidomics would also be essential for detecting the role of lipids in host-pathogen relations, which in turn, can contribute to therapeutic advances and biomarker development [47]. To this end, we described herein a RP-UHPLC-HRMSE method for the rapid, efficient, and comprehensive coverage of the mycobacterial lipidome. The described method incorporates a relatively short 14-minutes LC run time (per ionization mode) to enable the high-throughput analysis of mycobacterial samples. The acquisition of samples in both positive and negative modes with varying CEs is essential to cover the entire diversity of lipid alterations and modifications in mycobacterial lipidome. This is highlighted by the fact that some phospholipids (e.g., PI, PG) were only ionizable in the negative ionization mode while the identification of the acyl chain length of mycolic acids required a CE higher than 35 V.
5. Conclusion
A simple and robust method for the high-throughput identification of various mycobacteria lipids was presented. Our modified LC-MS method separates various lipids within 14 minutes using a CSH column, which facilitates a short sample analysis turn-over rate and a high-throughput. The RP-UHPLC-HRMSE method specifically provides for the identification of mycolic acids, which are unique to mycobacteria. Mycobacteria have a complex and lipid-rich cell wall, which makes lipid analysis challenging and time consuming, requiring our specialized and optimized LC-MS protocol. The applicability of the RP-UHPLC-HRMSE method to mycobacterial lipidomics research was demonstrated by characterizing the extracted lipidome from M. smegmatis. A total of 308 lipids were detected, which consisted of 12 acyltrehaloses, 3 diacylglycerols, 16 fatty acids and conjugates, 15 fatty esters, 166 glycerophosphoinositolglycans, 32 glycerophosoglycerophosphoglycerols, 12 glycerophosphoethanolamines, 9 glycerophosphoinositols, 8 glycerophosphoglycerols, 1 linear polyketide, 15 polyketide hybrids, 2 polyprenols, and 17 triacylglycerols. Importantly, our rapid RP-UHPLC-HRMSE method yielded comparable or better results to standard protocols with significantly longer (1.6x to 2.1x) LC run times. Our RP-UHPLC-HRMSE method exhibited a higher sensitivity and a comparable or higher amount of detected lipids or lipid main classes. An unexpected and concerning outcome of our investigation was the observation that a change in the LC gradient resulted in an essentially unique set of detected lipids despite using the same sample and instrumentation. Only 11 to 34% of the identified lipids were common between the four compared methods. Overall, our LC-MS method may facilitate the discovery of novel and unusual mycobacterial lipids, uncover important clinical and pathogenic differences between bacterial strains, or enhance our understanding of lipid metabolism in infection and drug resistance in high-throughput mycobacterial lipidomics area.
Supplementary Material
6. Acknowledgments
This material is based upon work supported by the National Science Foundation under Grant Number 1660921 and was supported in part by funding from the Nebraska Center for Integrated Biomolecular Communication (P20GM113126, NIGMS). The research was performed in facilities renovated with support from the National Institutes of Health (RR015468-01).
Footnotes
Supplementary material.
Supplementary materials include figures of the chemical structures and predicted properties for α-mycolic acid and methoxy cis mycolic acid; height equivalent to a theoretical plate versus flow rate plots for different sized ultra-high-performance liquid chromatography columns; direct-injection mass spectrometry-mass spectrometry spectrum of α-mycolic acid; mass spectrometry elevated collision energy mass spectrum of methoxy cis mycolic acid; mass spectrometry elevated collision energy mass spectrum of α-mycolic acid; ultra-high-performance liquid chromatography-quadrupole time of flight-mass spectrometry chromatogram of the mixture of the EquiSPLASH™ standard and mycolic acids; extracted ion chromatogram of methoxy cis mycolic acid and α-mycolic acid; bar plots of the number of PubMed papers published per year containing the keywords: mycolic acid, liquid chromatography, and mass spectrometry, and the runtimes of the published liquid-chromatography-mass spectrometry methods for mycobacterial lipidomics studies. Tables include a list of peak widths and retention times for lipids from the standard mixture; summary of column resolution; summary of spectral features for the four liquid-chromatography-mass spectrometry methods; summary of the statistical analysis of the four liquid-chromatography-mass spectrometry methods; summary of liquid-chromatography-mass spectrometry data for standard lipids; retention times obtained for lipids from the standard mixture for each liquid-chromatography-mass spectrometry method; lipid retention times identified by each of the four liquid-chromatography-mass spectrometry methods
Declaration of Competing Interest
The authors declare no competing financial interest. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of National Institutes of Health, National Science Foundation, or their constituents.
7. References
- [1].Shevchenko A, Simons K, Lipidomics: coming to grips with lipid diversity, Nat. Rev. Mol. Cell Biol 11(8) (2010) 593–598. 10.1038/nrm2934. [DOI] [PubMed] [Google Scholar]
- [2].Wenk MR, Lipidomics: New Tools and Applications, Cell (Cambridge, MA, U. S.) 143(6) (2010) 888–895. 10.1016/j.cell.2010.11.033. [DOI] [PubMed] [Google Scholar]
- [3].Meikle PJ, Wong G, Barlow CK, Kingwell BA, Lipidomics: Potential role in risk prediction and therapeutic monitoring for diabetes and cardiovascular disease, Pharmacol. Ther 143(1) (2014) 12–23. 10.1016/j.pharmthera.2014.02.001. [DOI] [PubMed] [Google Scholar]
- [4].Wheelock CE, Goss VM, Balgoma D, Nicholas B, Brandsma J, Skipp PJ, Snowden S, Burg D, D’Amico A, Horvath I, Chaiboonchoe A, Ahmed H, Ballereau S, Rossios C, Chung KF, Montuschi P, Fowler SJ, Adcock IM, Postle AD, Dahlen S-E, Rowe A, Sterk PJ, Auffray C, Djukanovic R, Application of ‘omics technologies to biomarker discovery in inflammatory lung diseases, Eur. Respir. J 42(3) (2013) 802–825. 10.1183/09031936.00078812. [DOI] [PubMed] [Google Scholar]
- [5].Yang L, Li M, Shan Y, Shen S, Bai Y, Liu H, Recent advances in lipidomics for disease research, J. Sep. Sci 39(1) (2016) 38–50. 10.1002/jssc.201500899. [DOI] [PubMed] [Google Scholar]
- [6].Zhao Y-Y, Cheng X-L, Lin R-C, Lipidomics applications for discovering biomarkers of diseases in clinical chemistry, Int. Rev. Cell Mol. Biol 313 (2014) 1–26. 10.1016/B978-0-12-800177-6.00001-3. [DOI] [PubMed] [Google Scholar]
- [7].Perrotti F, Rosa C, Genovesi D, Perrotti F, Rosa C, Genovesi D, Cicalini I, Del BP, Cicalini I, Sacchetta P, Del BP, Pieragostino D, Cicalini I, Sacchetta P, Del BP, Pieragostino D, Advances in Lipidomics for Cancer Biomarkers Discovery, Int J Mol Sci 17(12) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fahy E, Subramaniam S, Brown HA, Glass CK, Merrill AH, Murphy RC, Raetz CRH, Russell DW, Seyama Y, Shaw W, Shimizu T, Spener F, van Meer G, VanNieuwenhze MS, White SH, Witztum JL, Dennis EA, A comprehensive classification system for lipids, Journal of Lipid Research 46(5) (2005) 839–862. 10.1194/jlr.e400004-jlr200. [DOI] [PubMed] [Google Scholar]
- [9].Layre E, Sweet L, Hong S, Madigan CA, Desjardins D, Young DC, Cheng T-Y, Annand JW, Kim K, Shamputa IC, McConnell MJ, Debono CA, Behar SM, Minnaard AJ, Murray M, Barry CE 3rd, Matsunaga I, Moody DB, A comparative lipidomics platform for chemotaxonomic analysis of Mycobacterium tuberculosis, Chem Biol 18(12) (2011) 1537–1549. 10.1016/j.chembiol.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang R, Li B, Lam SM, Shui G, Integration of lipidomics and metabolomics for in-depth understanding of cellular mechanism and disease progression, Journal of Genetics and Genomics 47(2) (2020) 69–83. 10.1016/j.jgg.2019.11.009. [DOI] [PubMed] [Google Scholar]
- [11].Subramaniam S, Fahy E, Gupta S, Sud M, Byrnes RW, Cotter D, Dinasarapu AR, Maurya MR, Bioinformatics and systems biology of the lipidome, Chem Rev 111(10) (2011) 6452–6490. 10.1021/cr200295k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li M, Zhou Z, Nie H, Bai Y, Liu H, Recent advances of chromatography and mass spectrometry in lipidomics, Anal Bioanal Chem 399(1) (2011) 243–9. 10.1007/s00216-010-4327-y. [DOI] [PubMed] [Google Scholar]
- [13].Teramoto K, Suga M, Sato T, Wada T, Yamamoto A, Fujiwara N, Characterization of Mycolic Acids in Total Fatty Acid Methyl Ester Fractions from Mycobacterium Species by High Resolution MALDI-TOFMS, Mass Spectrom (Tokyo) 4(1) (2015) A0035. 10.5702/massspectrometry.A0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Murphy RC, Axelsen PH, Mass spectrometric analysis of long-chain lipids, Mass Spectrom Rev 30(4) (2011) 579–99. 10.1002/mas.20284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wu Z, Shon JC, Liu KH, Mass Spectrometry-based Lipidomics and Its Application to Biomedical Research, J Lifestyle Med 4(1) (2014) 17–33. 10.15280/jlm.2014.4.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xuan Q, Zheng F, Yu D, Ouyang Y, Zhao X, Hu C, Xu G, Rapid lipidomic profiling based on ultra-high performance liquid chromatography–mass spectrometry and its application in diabetic retinopathy, Analytical and Bioanalytical Chemistry 412(15) (2020) 3585–3594. 10.1007/s00216-020-02632-6. [DOI] [PubMed] [Google Scholar]
- [17].Cajka T, Fiehn O, Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry, Trends Analyt Chem 61 (2014) 192–206. 10.1016/j.trac.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fenaille F, Barbier Saint-Hilaire P, Rousseau K, Junot C, Data acquisition workflows in liquid chromatography coupled to high resolution mass spectrometry-based metabolomics: Where do we stand?, J. Chromatogr. A 1526 (2017) 1–12. 10.1016/j.chroma.2017.10.043. [DOI] [PubMed] [Google Scholar]
- [19].Oursel D, Loutelier-Bourhis C, Orange N, Chevalier S, Norris V, Lange CM, Lipid composition of membranes of Escherichia coli by liquid chromatography/tandem mass spectrometry using negative electrospray ionization, Rapid Commun Mass Spectrom 21(11) (2007) 1721–8. 10.1002/rcm.3013. [DOI] [PubMed] [Google Scholar]
- [20].Daffé M, Draper P, The Envelope Layers of Mycobacteria with Reference to their Pathogenicity, Advances in Microbial Physiology, Elsevier, 1997, pp. 131–203. [DOI] [PubMed] [Google Scholar]
- [21].Marrakchi H, Lanéelle M-A, Daffé M, Mycolic Acids: Structures, Biosynthesis, and Beyond, Chem Biol 21(1) (2014) 67–85. 10.1016/j.chembiol.2013.11.011. [DOI] [PubMed] [Google Scholar]
- [22].Layre E, Lee HJ, Young DC, Martinot AJ, Buter J, Minnaard AJ, Annand JW, Fortune SM, Snider BB, Matsunaga I, Rubin EJ, Alber T, Moody DB, Molecular profiling of Mycobacterium tuberculosis identifies tuberculosinyl nucleoside products of the virulence-associated enzyme Rv3378c, Proc Natl Acad Sci U S A 111(8) (2014) 2978–2983. 10.1073/pnas.1315883111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Crick PJ, Guan XL, Lipid metabolism in mycobacteria—Insights using mass spectrometry-based lipidomics, Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 1861(1) (2016) 60–67. 10.1016/j.bbalip.2015.10.007. [DOI] [PubMed] [Google Scholar]
- [24].Sartain MJ, Dick DL, Rithner CD, Crick DC, Belisle JT, Lipidomic analyses of Mycobacterium tuberculosis based on accurate mass measurements and the novel “Mtb LipidDB”, Journal of lipid research 52(5) (2011) 861–872. 10.1194/jlr.M010363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Crellin PK, Luo C-Y, Morita YS, Metabolism of Plasma Membrane Lipids in Mycobacteria and Corynebacteria, Lipid Metabolism, InTech, 2013. [Google Scholar]
- [26].Nataraj V, Varela C, Javid A, Singh A, Besra GS, Bhatt A, Mycolic acids: deciphering and targeting the Achilles’ heel of the tubercle bacillus, Mol Microbiol 98(1) (2015) 7–16. 10.1111/mmi.13101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sundarsingh T JA, Jambulingam R, Rajan A, Shankar V, Features of the biochemistry of Mycobacterium smegmatis, as a possible model for Mycobacterium tuberculosis, J Infect Public Health (2020). 10.1016/j.jiph.2020.06.023. [DOI] [PubMed] [Google Scholar]
- [28].Folch J, Lees M, Sloane Stanley GH, A simple method for the isolation and purification of total lipides from animal tissues, J Biol Chem 226(1) (1957) 497–509. [PubMed] [Google Scholar]
- [29].Raghunandanan S, Jose L, Gopinath V, Kumar RA, Comparative label-free lipidomic analysis of Mycobacterium tuberculosis during dormancy and reactivation, Sci Rep 9(1) (2019) 3660. 10.1038/s41598-019-40051-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Klatt S, Brammananth R, O’Callaghan S, Kouremenos KA, Tull D, Crellin PK, Coppel RL, McConville MJ, Identification of novel lipid modifications and intermembrane dynamics in Corynebacterium glutamicum using high-resolution mass spectrometry, J Lipid Res 59(7) (2018) 1190–1204. 10.1194/jlr.M082784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Huang Y, A Quality by Design (QbD) Framework for Reversed-Phase Liquid Chromatography Method Development, 2016. [Google Scholar]
- [32].Narwate BM, Ghule PJ, Ghule AV, Darandale AS, Wagh JG, Ultra Performance Liquid Chromatography: a new revolutionn in liquid chromatography, International Journal of Pharmaceutics and Drug Analysis 2(1) (2014) 25–34. [Google Scholar]
- [33].McCabe DR, An introduction to UPLC technology improve productivity and data quality, Waters seminars (2007). [Google Scholar]
- [34].McCabe DR, Iraneta P, Walter TH, Reports of the Death of Fully Porous Particles are Greatly Exaggerated, Chromatography today Waters Corporation (2012). [Google Scholar]
- [35].Sanchez CL, Souders CL 2nd, Pena-Delgado CJ, Nguyen KT, Kroyter N, Ahmadie NE, Aristizabal-Henao JJ, Bowden JA, Martyniuk CJ, Neurotoxicity assessment of triazole fungicides on mitochondrial oxidative respiration and lipids in differentiated human SH-SY5Y neuroblastoma cells, Neurotoxicology 80 (2020) 76–86. 10.1016/j.neuro.2020.06.009. [DOI] [PubMed] [Google Scholar]
- [36].Utsunomiya A, Chino T, Utsunomiya N, Luong VH, Tokuriki A, Naganuma T, Arita M, Higashi K, Saito K, Suzuki N, Ohara A, Sugai M, Sugawara K, Tsuruta D, Oyama N, Hasegawa M, Homeostatic Function of Dermokine in the Skin Barrier and Inflammation, J Invest Dermatol 140(4) (2020) 838–849 e9. 10.1016/j.jid.2019.09.011. [DOI] [PubMed] [Google Scholar]
- [37].Song SH, Park KU, Lee JH, Kim EC, Kim JQ, Song J, Electrospray ionization-tandem mass spectrometry analysis of the mycolic acid profiles for the identification of common clinical isolates of mycobacterial species, J Microbiol Methods 77(2) (2009) 165–77. 10.1016/j.mimet.2009.01.023. [DOI] [PubMed] [Google Scholar]
- [38].Subramaniam S, Fahy E, LipidMaps Core Update, Nature Precedings (2007). 10.1038/npre.2007.23.1. [DOI] [Google Scholar]
- [39].Schrimpe-Rutledge AC, Codreanu SG, Sherrod SD, McLean JA, Untargeted Metabolomics Strategies-Challenges and Emerging Directions, J Am Soc Mass Spectrom 27(12) (2016) 1897–1905. 10.1007/s13361-016-1469-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sud M, Fahy E, Cotter D, Brown A, Dennis EA, Glass CK, Merrill AH Jr., Murphy RC, Raetz CRH, Russell DW, Subramaniam S, LMSD: LIPID MAPS structure database, Nucleic Acids Res. 35(Database Iss) (2007) D527–D532. 10.1093/nar/gkl838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Murphy RC, Tandem Mass Spectrometry of Lipids: Molecular Analysis of Complex Lipids. New Developments in Mass Spectrometry, Royal Society of Chemistry (2015) 10–220. [Google Scholar]
- [42].C. Lipidomics Standards Initiative, Lipidomics needs more standardization, Nat Metab 1(8) (2019) 745–747. 10.1038/s42255-019-0094-z. [DOI] [PubMed] [Google Scholar]
- [43].Damen CW, Isaac G, Langridge J, Hankemeier T, Vreeken RJ, Enhanced lipid isomer separation in human plasma using reversed-phase UPLC with ion-mobility/high-resolution MS detection, J Lipid Res 55(8) (2014) 1772–83. 10.1194/jlr.D047795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].M.S.a.K.J. Fountain, Rapid Method Development through Proper Column Selection, 2012. [Google Scholar]
- [45].Giorgis Isaac NM, Gethings Lee A., Mullin Lauren, Plumb Robert S., A Robust and Reproducible Reversed-Phase Lipid Profiling Method for Large Sample Sets, 2020. [Google Scholar]
- [46].Plumb RS, Johnson KA, Rainville P, Smith BW, Wilson ID, Castro-Perez JM, Nicholson JK, UPLC/MSE; a new approach for generating molecular fragment information for biomarker structure elucidation, Rapid Communications in Mass Spectrometry 20(14) (2006) 2234–2234. 10.1002/rcm.2602. [DOI] [PubMed] [Google Scholar]
- [47].Appala K, Bimpeh K, Freeman C, Hines KM, Recent applications of mass spectrometry in bacterial lipidomics, Analytical and Bioanalytical Chemistry (2020). 10.1007/s00216-020-02541-8. [DOI] [PubMed] [Google Scholar]
- [48].Perumal J, Dinish US, Bendt AK, Kazakeviciute A, Fu CY, Ong ILH, Olivo M, Identification of mycolic acid forms using surface-enhanced Raman scattering as a fast detection method for tuberculosis, Int J Nanomedicine 13 (2018) 6029–6038. 10.2147/IJN.S171400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tsai YT, Salzman V, Cabruja M, Gago G, Gramajo H, Role of long-chain acyl-CoAs in the regulation of mycolic acid biosynthesis in mycobacteria, Open Biol 7(7) (2017). 10.1098/rsob.170087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Nishiuchi Y, Baba T, Yano I, Mycolic acids from Rhodococcus, Gordonia, and Dietzia, J Microbiol Methods 40(1) (2000) 1–9. 10.1016/s0167-7012(99)00116-5. [DOI] [PubMed] [Google Scholar]
- [51].Rivera-Betancourt OE, Karls R, Grosse-Siestrup B, Helms S, Quinn F, Dluhy RA, Identification of mycobacteria based on spectroscopic analyses of mycolic acid profiles, Analyst 138(22) (2013) 6774–85. 10.1039/c3an01157g. [DOI] [PubMed] [Google Scholar]
- [52].Singh A, Varela C, Bhatt K, Veerapen N, Lee OY, Wu HH, Besra GS, Minnikin DE, Fujiwara N, Teramoto K, Bhatt A, Identification of a Desaturase Involved in Mycolic Acid Biosynthesis in Mycobacterium smegmatis, PLoS One 11(10) (2016) e0164253. 10.1371/journal.pone.0164253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].de Carvalho C, Fischer MA, Kirsten S, Wurz B, Wick LY, Heipieper HJ, Adaptive response of Rhodococcus opacus PWD4 to salt and phenolic stress on the level of mycolic acids, AMB Express 6(1) (2016) 66. 10.1186/s13568-016-0241-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hong S, Cheng T-Y, Layre E, Sweet L, Young DC, Posey JE, Butler WR, Moody DB, Ultralong C100 mycolic acids support the assignment of Segniliparus as a new bacterial genus, PloS one 7(6) (2012) e39017–e39017. 10.1371/journal.pone.0039017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jackson M, The mycobacterial cell envelope-lipids, Cold Spring Harb Perspect Med 4(10) (2014) a021105. 10.1101/cshperspect.a021105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.