

Abstract
In the current pandemic, finding an effective drug to prevent or treat the infection is the highest priority. A rapid and safe approach to counteract COVID‐19 is in silico drug repurposing. The SARS‐CoV‐2 PLpro promotes viral replication and modulates the host immune system, resulting in inhibition of the host antiviral innate immune response, and therefore is an attractive drug target. In this study, we used a combined in silico virtual screening for candidates for SARS‐CoV‐2 PLpro protease inhibitors. We used the Informational spectrum method applied for Small Molecules for searching the Drugbank database followed by molecular docking. After in silico screening of drug space, we identified 44 drugs as potential SARS‐CoV‐2 PLpro inhibitors that we propose for further experimental testing.
Keywords: drug repurposing, ISM, molecular docking, Papain-like protease, SARS-CoV-2
Combined in silico machinery, consisting of long‐ and short‐range molecular interaction methods is capable of filtering large compound databases, yielding a good candidate for a receptor. The advantage of this approach is a fast assessment of a candidate, starting from basic information on both protein and small organic molecule – FASTA sequence and SMILES notation.
Introduction
Long time frames, high costs, and high failure rates are associated with the traditional drug discovery process. In the current pandemic finding an effective drug is the highest priority, and therefore drug repurposing is of great help in the fight against the SARS‐CoV‐2. FDA‐approved drugs with known safety and clinical profile allow for the reposition of drugs in the treatment of COVID‐19. Despite the challenges that accompany drug repurposing, the ability to identify new uses of old drugs in a relatively short time is a significant incentive to focus on drug reposition for COVID‐19. [1] In the search for an‐ti‐SARS‐CoV‐2 molecules, various computational methods were applied combined with experimental approaches, and thousands of articles were published. [2]
SARS‐CoV‐2 has a single‐strand, positive‐sense RNA genome 1 with at least ten open reading frames (ORFs). [3] The largest ORF1ab encompassing around two‐thirds of the virus genome encodes two large overlapping polyproteins, the pp1a and pp1ab, essential for viral replication and transcription, which go through proteolytic cleavage, generating 16 non‐structural proteins (NSP).[ 3 , 4 , 5 , 6 ]
The processing of two large viral polyproteins is autocatalytic proteolysis processed by virally encoded cysteine proteases. Papain‐like protease (PLpro), encoded by NSP3, recognizes the LXGG tetrapeptide motif found in‐between viral proteins nsp1 and nsp2, nsp2 and nsp3, and processes the replicase polyprotein 1a (pp1a) and replicase polyprotein 1ab (pp1ab) on the N‐termini into nsp1, nsp2, and nsp3, essential for viral replication.[ 7 , 8 ] PLpro is a monomer with an active site that comprises a Cys111/His272/Asp286 canonical catalytic triad. The SARS‐CoV‐2 PLpro modulates the host immune system via deubiquitination and deISGylation from the host cell proteins resulting in inhibition of the host antiviral innate immune response.[ 9 , 10 ]
Many different virtual screening procedures, using different approaches to identify PLpro inhibitors, were published in the last months. A recent study that reports screening of deubiquitinase inhibitors and cysteine protease inhibitors reports findings of potent PLpro inhibitors with IC50 values below ten μM. One of these molecules has an exceptional IC50 value of 0.56 μM but shows only partial inhibition of a PLpro, so it is considered to produce its inhibitory effect with binding to the allosteric site of PLpro. [11] In this study, 24 known drugs were selected as promising noncovalent and covalent inhibitors of the SARS‐CoV‐2 PLpro by combining protein‐ligand interaction fingerprint similarities, conventional docking scores, and MM‐GBSA‐binding free energies selected. In addition, the study was extended to investigate compounds that have reached phase III clinical trials with different pharmacological classes. [11] Finally, in a study by Xu and co‐workers, [12] two tests were established to screen PLpro inhibitors for protease and anti‐ISGylation activities. Screening the library of clinically approved drugs led to the discovery of sodium tanshinon IIA sulfonate and chloroxine with IC50 values lower than ten μM that directly interact with PLpro, and simulations of coupling and molecular dynamics show their molecular binding mechanisms.
A very recent paper reported potential antiviral effects of hypericin relative to the well‐known noncovalent PLpro inhibitor GRL‐0617. The molecules were investigated by molecular dynamics and PELE Monte Carlo simulations highlighting favourable binding of hypericin and GRL‐0617 to the naphthalene binding pocket of PLpro. Further results from in vitro fluorogenic enzymatic assays with hypericin showed concentration‐dependent inhibition of both PLpro protease and deubiquitinating activities. [13] A recent review reported a detailed overview of the targets (Mpro and PLpro) from a structural and medicinal chemistry point of view, with recently reported protease inhibitors and the challenges in developing effective protease inhibitors. In addition, one paragraph overviews the studies with molecular modelling and in silico virtual screening against SARS‐CoV‐2. [14] Another review summarized potential inhibitors targeting non‐structural proteins 3 (PLpro and Mac1) and 5 (3CLpro/Mpro) of SARS‐CoV‐2 in the recent review. [15]
In silico strategy of repurposing approved drugs has been employed to fight COVID‐19 in the current study. We used a virtual screening protocol with combined sequential filters based on long‐range and short‐range interactions to select candidates for PLpro inhibitors. By Informational spectrum method (ISM), a virtual spectroscopy method for the study of the long‐range protein‐protein interaction (ISM), applied to small molecules (ISM‐SM), we analysed small molecule‐protein interaction. The advantage of this virtual approach is its ability to rapidly scan huge molecular libraries demanding low data preparation, requiring only protein sequence and SMILES molecules notation of drug candidate. First, the ISM‐SM was used to search the Drugbank database, followed by molecular docking. By using this combined protocol, 44 compounds were selected for further experimental testing
Results and Discussion
Informational Spectrum Method Analysis
In the present study, we have used the Informational spectrum method (ISM) for the structure/function analysis of SARS‐CoV‐2 protein PLpro. The informational spectrum (IS) of PLpro contains two characteristic peaks, at the dominant frequencies F(0.383), and F(0.279), shown in Figure 1. To find the domains of a protein crucial for the information related to the three frequencies, PLpro was computationally scanned. As a result of scanning with the ISM algorithm, we identified regions with the highest amplitudes at these frequencies and shown that the regions, including residues 248–312 and 60–124, are essential for the information represented by the frequency F(0.383) and F(0.279), respectively. This finding is in accordance with the co‐crystalized covalently bound peptide‐like inhibitor VIR250 [16] (Figure 2). Namely, those two dominant frequencies correspond to the inhibitor domain of the enzyme, mapping the regions of key amino acids and the binding site of the reported co‐crystallized inhibitors. We further searched CS of Drugbank [17] candidates with PLpro at the F(0.383) and F(0.279) to find potential inhibitor candidates. With this search, we selected 44 candidate drugs (Table 1).
Figure 1.
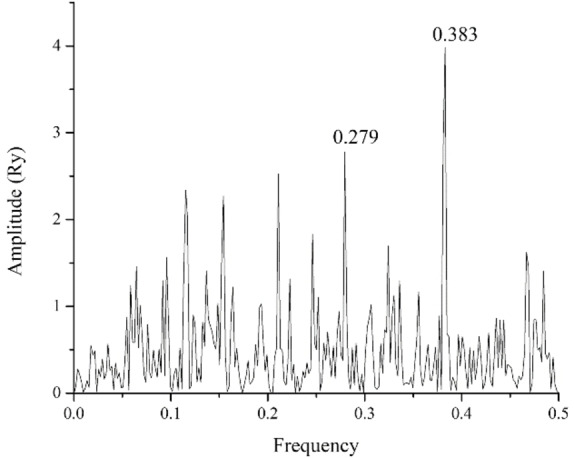
ISM spectrum of SARS‐CoV‐2 Papain like protease.
Figure 2.
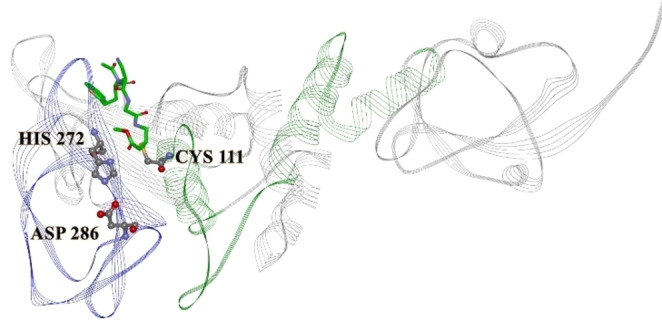
Crystal structure of PLpro, with a marked catalytic triad (PDBID 6WUU). Marked regions correspond F(0.383) 248–312 – blue, and F(0.279) 60–124 – green. The bound compound is the co‐crystallized covalent peptide inhibitor VIR250.
Table 1.
Docking scores of the compounds binding to the PLpro inhibitor binding site.
|
Compound name |
Drugbank ID |
Frequency |
VINA binding energy (kcal/mol) |
Similarity distance |
Negative binding energy/distance similarity |
|---|---|---|---|---|---|
|
VIR250 |
– |
0.279 |
– |
– |
– |
|
GRL0167 |
– |
0.382 |
−10.4 |
– |
– |
|
XR8‐89 |
– |
0.279 |
−10.2 |
– |
– |
|
VBY501 |
– |
0.382 |
−10.2 |
– |
– |
|
Ergometrine |
DB01253 |
0.382 |
−8.0 |
1.0983 |
7.2840 |
|
Indacaterol |
DB05039 |
0.279 |
−7.6 |
1.2657 |
6.0046 |
|
Phentolamine |
DB00692 |
0.279 |
−8.3 |
1.7025 |
4.8752 |
|
Metergoline |
DB13520 |
0.382 |
−9.4 |
2.0399 |
4.6081 |
|
Vandetanib |
DB05294 |
0.382 |
−7.0 |
1.65 |
4.2424 |
|
Epicriptine |
DB11275 |
0.382 |
−10.3 |
2.5965 |
3.9669 |
|
Osimertinib |
DB09330 |
0.279 |
−7.6 |
2.1215 |
3.5824 |
|
Oxamniquine |
DB01096 |
0.279 |
−6.7 |
1.8842 |
3.5559 |
|
Dihydro‐alpha‐ergocryptine |
DB11274 |
0.382 |
−8.9 |
2.5484 |
3.4924 |
|
Tubocurarine |
DB01199 |
0.279 |
−7.4 |
2.3355 |
3.1685 |
|
Clidinium |
DB00771 |
0.279 |
−7.0 |
2.2219 |
3.1505 |
|
Methylergometrine |
DB00353 |
0.382 |
−7.6 |
2.4437 |
3.1100 |
|
Stiripentol |
DB09118 |
0.382 |
−6.8 |
2.2338 |
3.0441 |
|
Orciprenaline |
DB00816 |
0.279 |
−6.6 |
2.1833 |
3.0229 |
|
Clonidine |
DB00575 |
0.382 |
−5.9 |
2.0595 |
2.8648 |
|
Bosutinib |
DB06616 |
0.279 |
−7.4 |
2.8336 |
2.6115 |
|
Nabumetone |
DB00461 |
0.382 |
−7.0 |
2.7469 |
2.5483 |
|
Lacosamide |
DB06218 |
0.382 |
−6.4 |
2.559 |
2.5010 |
|
Almitrine |
DB01430 |
0.382 |
−7.7 |
3.1207 |
2.4674 |
|
Fidaxomicin |
DB08874 |
0.382 |
−8.1 |
3.3195 |
2.4401 |
|
Eugenol |
DB09086 |
0.382 |
−5.9 |
2.4456 |
2.4125 |
|
Mephenesin |
DB13583 |
0.382 |
−6.2 |
2.631 |
2.3565 |
|
Sacubitril |
DB09292 |
0.279 |
−7.8 |
3.3286 |
2.3433 |
|
Medifoxamine |
DB13219 |
0.382 |
−6.9 |
3.0265 |
2.2799 |
|
Methdilazine |
DB00902 |
0.382 |
−6.9 |
3.0404 |
2.2694 |
|
Methscopolamine bromide |
DB00462 |
0.382 |
−6.6 |
3.0323 |
2.1766 |
|
Benzyl alcohol |
DB06770 |
0.382 |
−5.1 |
2.4 |
2.1250 |
|
Digoxin |
DB00390 |
0.279 |
−8.8 |
4.1656 |
2.1125 |
|
Phenoxyethanol |
DB11304 |
0.382 |
−5.4 |
2.6076 |
2.0709 |
|
Diacetyl benzoyl lathyrol |
DB11260 |
0.279 |
−7.5 |
3.746 |
2.0021 |
|
Undecoylium chloride iodine complex |
DB09377 |
0.279 |
−4.9 |
2.5165 |
1.9471 |
|
Cisatracurium |
DB00565 |
0.382 |
−5.9 |
3.037 |
1.9427 |
|
Quinine |
DB00468 |
0.382 |
−7.0 |
3.6113 |
1.9384 |
|
Dichlorobenzyl alcohol |
DB13269 |
0.279 |
−5.1 |
2.7908 |
1.8274 |
|
Bepotastine |
DB04890 |
0.382 |
−6.7 |
3.7253 |
1.7985 |
|
Terconazole |
DB00251 |
0.382 |
−7.3 |
4.0701 |
1.7936 |
|
Citalopram |
DB00215 |
0.382 |
−7.0 |
3.9308 |
1.7808 |
|
Ivabradine |
DB09083 |
0.382 |
−7.0 |
4.3764 |
1.5995 |
|
Escitalopram |
DB01175 |
0.382 |
−6.3 |
4.3764 |
1.4395 |
|
Cinoxate |
DB15467 |
0.382 |
−5.7 |
4.3016 |
1.3251 |
|
Troleandomycin |
DB13179 |
0.279 |
−6.3 |
4.9497 |
1.2728 |
|
Guanidine |
DB00536 |
0.382 |
−4.0 |
3.5501 |
1.1267 |
|
Meglumine |
DB09415 |
0.382 |
−4.4 |
4.6809 |
0.9400 |
|
Dimercaprol |
DB06782 |
0.382 |
−2.9 |
3.3001 |
0.8788 |
Ligand‐based Screening
To assess the pharmacological activity of our candidates, similarly as in the recently reported study on COVID‐19 drug repurposing, [11] we carried ligand‐based screening, calculating the molecular similarity using Principal Component Analysis (PCA) based on Molecular Interaction Fields (MIF) descriptors. [18] All 44 selected drugs from the previous step (ISM‐SM) were imported in Pentacle software, [19] protonated at pH 7.4, and aligned towards the principal moment of inertia. In ligand‐based virtual screening, we used the centroid distance method as criteria for the similarity between co‐crystallized inhibitors GRL0167, XR8‐89, VBY501, and candidate compounds. Similarity distance, compound ranks and PCA plots are reported in Supporting Information (Tables S1–S2, Figure S1). The best candidate, according to molecular similarity is Ergometrine, followed by Methylergometrine and Clidinium. However, the ligand‐based pharmacological similarity is not enough to assess the compound's potency towards a certain target, and therefore, structure‐based approaches are the next steps in virtual screening.
Molecular Docking
To further filter the selected compounds, we carried molecular docking into the site of reported co‐crystallized PLpro inhibitors, using PLpro‐GRL 0617 complex structure (PDB ID 7CJM). [20] The binding energies values were compared to the docked co‐crystallized ligands from different PLpro structures. From the initial docking, as top candidates were found Epicriptine and Metergoline, targeting the inhibitor binding site (Supporting Information, Table S3). Epicriptine is the top candidate, with more favourable docking energy (−10.3 kcal/mol) than literature inhibitors XR8‐89 [21] and VBY501, [22] suggesting it could be a potentially promising inhibitor of SARS‐CoV‐2 PLpro. Metergoline is the second‐best candidate, binding to the PLpro slightly weaker than literature inhibitor VBY501 (−9.4 kcal/mol). Types of intermolecular interactions that candidates form with amino acid residues are hydrogen bonds, aromatic π‐π, alkyl‐π, cation‐π interactions. Comparing the binding patterns of our top candidates to the literature inhibitors, it is noticeable that they are conserved. Thus, both Epicriptine (Figure 3) and Metergoline (Figure 4) form a salt bridge with Asp164 (Figure 5), analogously to GRL 0617 (Figure 6). Metergoline, in common with GRL 0617, forms a hydrogen bond with Gln269 via carboxyl oxygen. In all three cases, the aromatic moieties are oriented towards Tyr 268. In addition, Epicriptine forms cation‐π interaction with Tyr 268.
Figure 3.
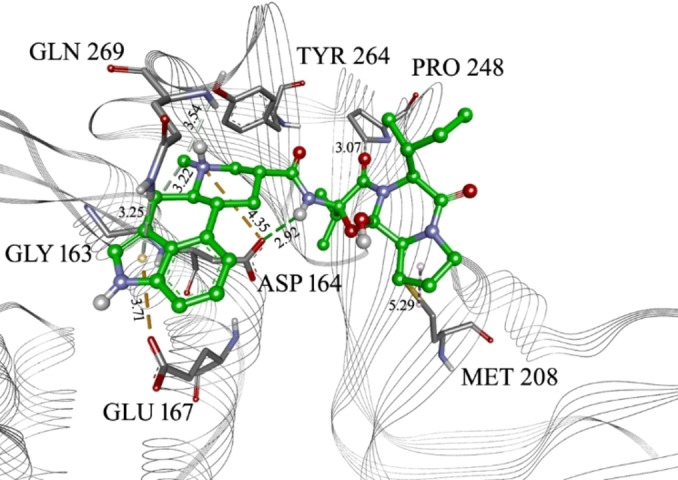
Epicriptine in the PLpro inhibitor binding site, with marked interacting amino acid residues and corresponding distances. Green lines: hydrogen bonds; light green: N−H‐π interactions or carbon hydrogen bond; orange: electrostatic or anion‐π interactions; purple: alkyl‐π interactions. The distance of each interaction is presented in Å.
Figure 4.
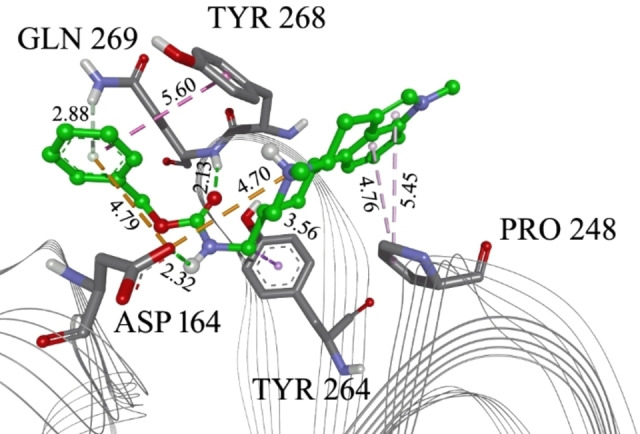
Metergoline in the PLPro inhibitor binding site. with marked interacting amino acid residues and corresponding distances. Green lines: hydrogen bonds; light green: N−H‐π interactions; orange: electrostatic interactions; purple: alkyl‐π interactions, magenta: π‐π interactions. The distance of each interaction is presented in Å.
Figure 5.
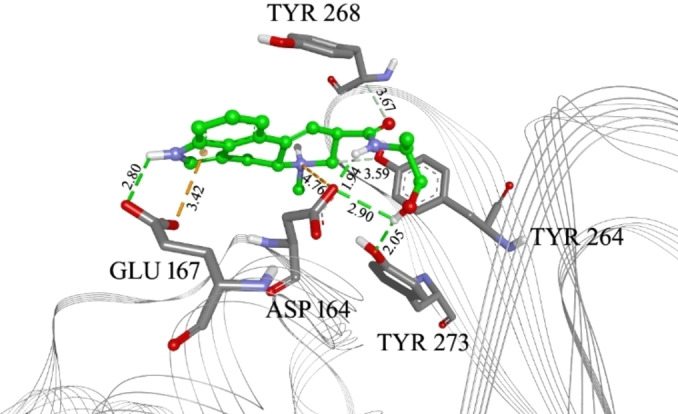
Ergometrine in the PLpro inhibitor binding site, with marked interacting amino acid residues and corresponding distances. Green lines: hydrogen bonds; light green: N−H‐π interactions or carbon hydrogen bonds; orange: electrostatic interactions or anion‐π interactions; purple: alkyl‐π interactions, magenta: π‐π interactions. The distance of each interaction is given in Å.
Figure 6.
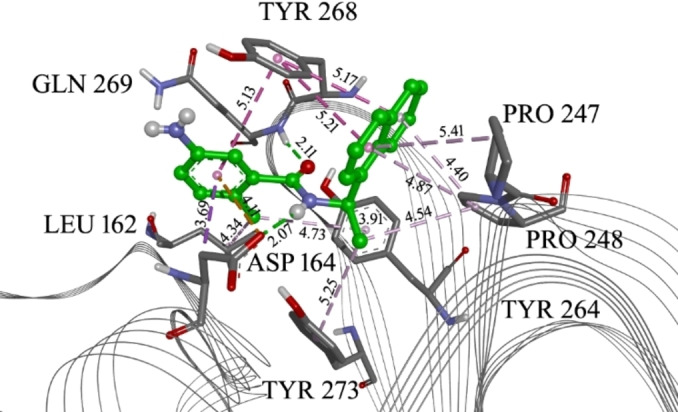
Co crystalized ligand GRL 0617 in the PLpro allosteric site with marked interacting amino acid residues and corresponding distances. Green lines: hydrogen bonds; purple: alkyl‐π/hydrophobic interactions. The distance of each interaction is given in Å.
However, to include both molecular similarity and binding energy from molecular docking as a consensus, we decided to assign their negative quotient values to the compounds as a basis for a ranking. As a more favourable candidate should contain lower values of binding energy and similarity distance closer to 0, this was an appropriate consideration. In that manner, the best candidate through the best balance between docking energy and molecular similarity is Ergometrine (Figure 5). This does not significantly affect the ranking of Epicriptine and Metergoline, as they are still among the top candidates (Table 1).
The summary of protein‐ligand intermolecular interactions is presented in Table 2.
Table 2.
Interaction scheme between PLpro inhibitor binding site amino acid residues and ligands (X depicts the presence of interaction)
|
Aminoacid residue |
GRL 0617 |
Epicriptine |
Methergoline |
Ergometrine |
|---|---|---|---|---|
|
ASP164 |
X |
X |
X |
X |
|
GLN269 |
X |
X |
X |
|
|
GLU167 |
|
X |
|
X |
|
GLY163 |
|
X |
|
|
|
LEU162 |
X |
|
|
|
|
MET208 |
|
X |
|
|
|
PRO247 |
X |
X |
|
|
|
PRO248 |
X |
X |
X |
|
|
TYR264 |
X |
X |
X |
X |
|
TYR268 |
X |
|
X |
X |
|
TYR273 |
X |
|
|
X |
Molecular Dynamics Simulations
For further confirmation our selection, we decided to check the stability of receptor‐ligand complexes using MD simulations. Prepared complexes of PLpro and Metergoline were subjected to MD simulation. During the 80 ns of the production phase, the complex remained stable and intermolecular interactions persisted, with eventual re‐arranging of ligand's flexible aromatic residue. In the RMSD plots, the receptor showed convergence of the system (Figure S2). For further insight into complex stabilities, we calculated binding free energy. For that purpose, we chose metadynamics.[ 23 , 24 ]
We performed well‐tempered metadynamics simulations of the Plpro‐Metergoline complex after the MD production phase to estimate the binding free energy. For the centroid of PLpro catalytic site, we chose the backbone carbon atoms (residue list 165, 166, 207, 208, 243, 244). We chose all atoms of a candidate. The initial distance between the two centroids in the docked conformation was 9.77 Å, and we altered this distance by up to 35 Å.
Projection of the distance between the two centroids on the x‐axis of the simulation cell is presented in Figure 7. Regarding the PMF energy profile, the PMF energy initially increased to 10 kcal/mol until the distance reached approximately 12 Å. This happens around 2.9 ns of simulation and corresponds to the breaking of the salt bridge between positive N atom in metergoline and Asp 164 in Plpro. Further on, metergoline conserves aromatic interactions with Tyr 268 and Tyr 264, eventually breaking them when reaching a distance around 25 Å (3.8 ns) and leaving the binding site, yielding an additional 2 kcal/mol to PMF. Later on, metergoline reaches the maximum distance of 35 Å, in an unbound state. In a simulation time scale, this takes place around 4.1 ns. In further metadynamics simulation, the force drives it back to the binding site, but metergoline occupies a different domain, and from that point, the PMF is no any more reliable. Therefore, the range we consider is between 4.11 and 4.16 ns. This is presented in Figure S3 and metadynamics movie in Supporting Information.
Figure 7.
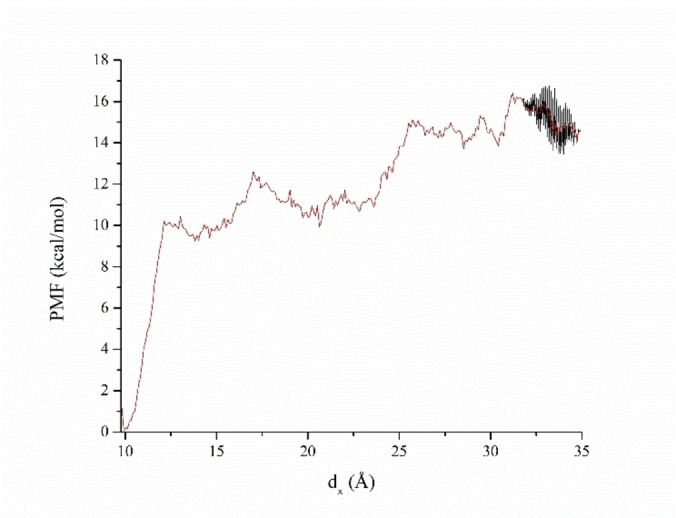
PMF during the metadynamics simulation of the Plpro‐metergoline complex. The Red line represents averaged multiple PMF curves in the range from 4.11 and 4.16 ns of the metadynamics simulation.
The binding free energy value is the negative difference between the initial and final states of PMF. We averaged the stable PMF area between 25.5 and 35 Å to calculate the binding free energy. The corresponding PMF difference, according to descriptive statistical analysis, was −14.9 kcal/mol; that is, we estimated the binding free energy of the Plpro candidate complex as ΔG=−14.9 kcal/mol. The standard error of the calculation was 0.6 kcal/mol, so the final result was written as ΔG=(−14.9±0.6) kcal/mol. Similarly, in the case of Epicriptine, we estimated ΔG=(−12.0±0.6) kcal/mol. Thus, the metadynamics binding free energy calculations confirm strong binding of our candidates to PLpro. However, longer simulations are required for the convergence of PMF through multiple binding/unbinding events. In this respect, we consider those binding energy values as estimated or semi quantitative.
Discussion
The continuous increase of drug‐resistant pathogens is a challenge for the treatment of infectious diseases, and drug repurposing represents an alternative approach for the fast identification of effective therapeutics. [25] Computational drug repurposing was an essential step in successfully affording agents against other pathogenic diseases, [26] inflammation, [27] cancer. [28]
The fundamental problem in a search for preventive and therapeutic options to respond to threats of a pandemic is a costly, time‐consuming, and risky process of drug development.
A promising therapeutic strategy for many viral diseases and the most rational in the current pandemic is drug repurposing. Given that the in silico approach allows rapid screening of large collections of compounds, computational drug repurposing offers a promising route when time is a critical factor. Various computational predictions approaches have been developed to identify drug repositioning opportunities against SARS‐CoV‐2.[ 29 , 30 ]
However, since it is challenging to simulate complex biological structures computationally, using state‐of‐the‐art methods has advantages and limitations. It has been shown that the use of combined in silico approaches provide solid grounds for repurposed hypothesis worthy of experimental investigation. [31] Therefore we used the VS protocol based on a combined in silico approach, which implies both short‐ and long‐range interactions between interacting molecules. The concept that we applied in selecting candidate molecules for the treatment of SARS‐CoV‐2 infection includes molecular characteristics responsible for long‐term recognition between biological molecules. The ISM was used in this work for the structure/function analysis of the SARS‐CoV‐2 protein PLpro and identification of the main informational characteristic of the protein, which corresponds to the protein key biological function. At the beginning of the pandemic, by using the ISM for the first time was suggested a potential SARS‐CoV‐2 receptor, therapeutic / vaccine target, and proposed SARS‐CoV‐2 cell to cell transmission. [32] In another recent study, ISM was used for analysing SARS‐CoV‐2 Orf3b, suggesting that this protein acts as a modulator of the interferon signalling network. [33]
Recently by using the same combined VS protocol as in this study, we have selected candidate SARS‐CoV‐2 Mpro inhibitors and proposed 57 compounds for further experimental testing. [34] Also, the same approach has been successfully applied in analysing GPCR drugs from the Golden dataset. [35]
The molecular descriptors EIIP/AQVN on which the ISM method is based were previously validated as a valuable parameters for in silico drug repositioning of small molecule inhibitors against HIV, Ebola virus, and influenza infection.[ 36 , 37 , 38 , 39 ] By this approach, ibuprofen was selected as an inhibitor of the Ebola virus infection, and later this in silico prediction was confirmed in vitro.[ 40 , 41 ]
Anti SARS‐CoV‐2 activity was previously shown in other studies for some of the candidates PLpro inhibitors from the current study, however, against a different target or without a known mechanism of action. The potential multitarget activity of drugs proposed as PLpro inhibitors may help overcome drug resistance in COVID‐19.
For the high‐ranked drug in our study‐ergometrine, it was reported by Mostafa et al. that among the tested FDA‐approved drugs revealed high docking scores for the five protease proteins. [42]
Epicriptine, an antiparkinsonian agent, high ranked from our study was reported in another in silico study as a drug that forms complexes with SARS‐CoV‐2′s spike protein with molecular mechanics interaction energy lower than −50.00 kcal/mol. [43]
Using bioinformatics analysis and experimentally, it was shown that Metergoline, the top‐ranked candidate for PLpro inhibitor according to our study, prevents SARS‐CoV‐2 infection primarily by interfering with viral entry. [44]
One of the best ranked PLpro inhibitors from our computational study is digoxin. In an in vitro study, digoxin leads to SARS‐CoV‐2 inhibition at the post‐entry stage of the viral life cycle. [45] Digoxin is a cardiovascular drug with antiviral activity against several coronaviruses and is proposed as a potential COVID‐19 therapeutic, with possible additional therapeutic effects for patients with cardiovascular disease. [45]
Osimertinib, a promising PLpro catalytic site inhibitor candidate according to our study, was identified as an inhibitor for spike‐mediated entry that showed greater than 50 % rescue of the SARS‐CoV‐2 cytopathic effect. [46] Recently in a high throughput screening assay for SARS‐CoV‐2, bosutinib was identified as a specific SARS2‐S pseudovirus entry inhibitor that significantly inhibited SARS2 replication. [47] In our analysis, several clinically used antidepressants, including citalopram, proposed as PLpro inhibitor, showed antiviral action against SARS‐CoV‐2. [48]
Quinine, proposed as PLpro inhibitor in this work, inhibited SARS‐CoV‐2 infection and the toxicological and pharmacological profile seems more favourable when compared to its progeny drugs hydroxychloroquine or chloroquine. [49] It is also suggested that quinine with a reputation record as medication against feverish illnesses might be able to mitigate the cytokine storm associated with severe COVID‐19. [49] Methdilazine was reported for activity against SARS‐CoV‐2 infection and was among the top confirmed anti‐SARS‐CoV‐2 compounds and was suggested as a PLpro inhibitor in this study. [50]
For some of the compounds like fidaxomicin, proposed in our study as PLpro inhibitor, it has been previously proposed antiviral activity from virtual screens against other SARS‐CoV‐2 targets, but without supporting biological data. [51]
Conclusion
The ISM‐SM approach has the advantage compared to other in silico approaches for its capability to determine long‐distance molecular recognition and targeting between protein and ligand (the distance of 5 to 1000 Å). This approach determines with immense precision the location of the protein domains with the possible binding site and enables the selection of small molecules which have great specificity for proposed domains. Moreover, rapid scanning of large compound libraries is undemanding since it is needed only protein sequence and SMILES molecules notation for data preparation. PLpro inhibitor candidates proposed in our computational study should be further experimentally tested in searching for safe, effective new treatments against SARS‐CoV‐2.
Experimental Section
Informational Spectrum Method
In this work, we analyse SARS‐CoV‐2 protein Papain like protease protein using the informational spectrum method (ISM). A comprehensive explanation of the sequence analysis based on ISM is available elsewhere. [52] According to this approach, a sequence (protein or DNA) is transformed into a signal by assignment of numerical values of each element (amino acid or nucleotide). These values correspond to electron‐ion interaction potential (EIIP), [53] determining the electronic properties of amino acid/nucleotides, which are essential for their long‐distance molecular interactions (the distance of 5 to 1000 Å). The EIIP descriptors are easily calculated using the following [Eq (1 and 2:
| (1) |
| (2) |
Where i is a type of the chemical element, Z is the valence of the i‐th chemical element, n is the number of the i‐th chemical element atoms in the compound, m is the number of types of chemical elements in the compound and N is the total number of atoms.
The EIIP signal is then transformed using Fast Fourier Transform (FFT) into information spectrum (IS) as a representation of a sequence in the form of a series of frequencies and amplitudes [Eq. 3]:
| (3) |
Where m is the summation index, x(m) is the m‐th member of a given numerical “signal” series (from a transformed, encoded primary protein sequence in our case), N is the total number of points in this series), n is the number of a discrete frequency (ranging from 1 on up to N/2) in the DFT, X(n) are the discrete Fourier transformation amplitude coefficients corresponding to each discrete frequency n, and 2π*(n/N) is the phase angle at each given m in the amino‐acid series of the protein in question.
However, in the case of protein analysis, the relevant information is primarily presented in energy density spectrum, which is defined as follows [Eq. 4]:
| (4) |
By this, the virtual spectroscopy method is feasible to analyse protein sequences without any previous experimental data functionally. Its extension for small molecules, ISM‐SM was developed and published recently. [35] A small molecule is imported in smiles notation and decoded by atomic groups into an array of corresponding EIIP values. Using FFT, the corresponding IS of a small molecule is computed. This spectrum is further multiplied by IS of the protein receptor to obtain a Cross‐spectrum (CS). Cross‐spectral function is the function which determines common frequency characteristics of two signals. For discrete series it is defined as follows [Eq. 5]:
| (5) |
Where X(n) and DFT coefficients of the series x(m), and Y(n)* are complex conjugated DFT coefficients of the series Y(m).
From common frequencies in CS, one can determine whether protein interacts with a small molecule and determine the corresponding binding region in the protein.
Data Preparation
FASTA SARS‐CoV‐2 PLpro sequence was downloaded from UNIPROT and corresponding IS was calculated. A set of 1490 approved Drugbank [17] drugs with corresponding SMILES was subjected to IS and CS calculation with PLpro. All calculations were carried using our in‐house software. PDB structures of PLpro in complexes with inhibitors (in brackets), encoded 6WUU [16] (VIR250), 7CJM [20] (GRL0167), 7JIW [22] (VBY501) and 7LBR [21] (XR8‐89) were downloaded from RCSB Protein Bank Database.
Ligand‐based Screening
To screen selected compounds from Drugbank, both learning set compounds and candidates from the previous step were converted to 3D SDF format from SMILES. GRIND descriptors of the molecules were calculated, based on molecular interaction field (MIF) probes. [19] The computation method for descriptor generation was GRID with step 0.5. Applied probes (mapped regions of molecule surface) were DRY (hydrophobic interactions) O (hydrogen bond acceptor) N1 (hydrogen bond donor) and TIP (molecular shape descriptor). The discretization Method was AMANDA [18] with a scale factor of 0.55. The encoding Method was MACC2 and weights were the following: DRY: −0.5, O: −2.6, N1: −4.2, TIP: −0.75. The number of PCA components was set to five. Explained variance of such obtained model was 70.69 %. Then, co‐crystallized compounds were imported and served for screening the candidate compound database. All calculations were carried in Pentacle software version 1.06 for Linux. [54]
Molecular Docking
Molecular docking of selected candidates into the crystal structure of PLpro was carried. Receptor three‐dimensional structure was downloaded from RCSB, PDB ID 7CJM. [20] All ligands, waters and ions were removed from PDB file. Two grid boxes with dimensions 24×24×24 Å were set to span all amino acid residues interacting with co‐crystallised inhibitor GRL 0617. The (x,y,z) centres of the grid boxes was (26.0, 70.0, −1.0). Selected drugs from the previous step were converted from SMILES to 3D SDF and further to PDB files and protonated at physiological pH. Geometry optimization was carried in MOPAC 2016 [55] at PM7 [56] level of theory. Default software settings for hydrophobic and hydrophilic terms in docking search function were used. Exhaustiveness was set to 50. Molecular docking was carried in Autodock Vina 1.1.2. [57] To validate the proper approach, the co‐crystalized ligands were re‐docked into their original receptor structures and RMSD values were measured (Table S4).
Figures were made in BIOVIA Discovery Studio 2017, Schrodinger Maestro 11.1 and Origin 9.0 software.
Molecular Dynamics Simulations
To further examine and confirm the stability of our docked complexes, we carried molecular dynamics (MD) simulations, followed by metadynamics[ 23 , 24 ] binding free energy calculation. The protein‐ligand complexes, obtained from docking poses were prepared for MD simulation by adding 20 Å water layers from both positive and negative sides of all three x, y and z‐axis, resulting in water box dimension 83×131×96 Å. NaCl ions were added to meet the physiological conditions of 0.15 M ionic strength, resulting in a system of ∼94 000 atoms. The system was subject to a 10 000 step energy minimization, 250 NVE ps equilibration, and 100 ns NPT MD production. Pressure and temperature were set to 1 bar and 310 K, respectively, using a Berendsen thermostat, and the applied integration step was 1 fs. In all simulations, periodic boundary conditions with particle‐mesh Ewald calculations were implemented. The cut‐off was set to 12 Å. A CHARMM36 force field [58] was used for protein, water and ions, and CGenFF [59] was used for ligand. For the metadynamics simulation, we chose one variable, the distance between centroids of protein amino acid (aa) and all atoms of a ligand. The lower boundary (minimal distance value) was set to the initial distance between centroids of receptor‐ligand atom groups, which was obtained from the coordinates of the docked structure optimized by MD production. The upper boundary (maximal value) was set to the distance at which the ligand was located at the edge of the PBC cell. The resulting change in free energy between initial and equilibrated states of the peptide in the water layer was designated the binding free energy of the complex. Collective variable trajectory frequency (frequency of generating free energy files) was set to 10 000 ps. The lower wall constant (lowest value of applied force, units in kcal/(mol*A)) was set to 1.0, the upper wall constant (highest value of applied force) was set to 20.0, and width (the force resolution) was set to 0.1. For the main atoms, we selected backbone carbon atoms from the receptor, and for reference, we selected backbone carbon atoms from the peptide. The hill weight (amount of PMF energy that is gradually added to a system during simulation) was set to 0.1 kcal/mol, the hill width was set to 1.0 Å, and the new hill frequency was set to 100 ps. Bias temperature was set to 1550 K. All free energy files generated during simulation were collected. The total metadynamics simulation time was 10 ns, and the integration step was 1 fs. The PMF output was averaged, and appropriate binding free energy was calculated. MD calculations were carried in NAMD 2.14. [60] The trajectories were analysed in VMD 1.9.3. [61]
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
This work was supported by the Science Fund of the Republic of Serbia, as part of the Special Research Program on COVID 19 – the project COVIDTARGET – Repurposing of drugs for prevention and treatment of COVID‐19.
M. Sencanski, V. Perovic, J. Milicevic, T. Todorovic, R. Prodanovic, V. Veljkovic, S. Paessler, S. Glisic, ChemistryOpen 2022, 11, e202100248.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.33774/chemrxiv‐2021‐39cd6‐v2).
Data Availability Statement
The data that support the findings of this study are openly available in Drugbank Online at https://go.drugbank.com/releases/latest, reference number 518. These data were derived from the following resources available in the public domain:, https://www.[resource1]; [Resource 2], https://www.[resource2]; …
References
- 1. Ng Y. L., Salim C. K., Chu J. J. H., Pharmacol. Ther. 2021, 228, 107930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Singh N., Villoutreix B. O., Comput. Struct. Biotechnol. J. 2021, 19, 2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Malik Y. A., Malays. J. Pathol. 2020, 42, 3. [PubMed] [Google Scholar]
- 4. Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., Song Z.-G., Hu Y., Tao Z.-W., Tian J.-H., Pei Y.-Y., Yuan M.-L., Zhang Y.-L., Dai F.-H., Liu Y., Wang Q.-M., Zheng J.-J., Xu L., Holmes E. C., Zhang Y.-Z., Nature 2020, 579, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mousavizadeh L., Ghasemi S., J. Microbiol., Immunol. Infect. 2021, 54, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Qiu Y., Xu K., STEMedicine 2020, 1, e39. [Google Scholar]
- 7. McClain C. B., Vabret N., Signal Transduction Targeted Ther. 2020, 5, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sun G., Xue L., He Q., Zhao Y., Xu W., Wang Z., Stem Cell Res. 2021, 52, 102219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lindner H. A., Fotouhi-Ardakani N., Lytvyn V., Lachance P., Sulea T., Ménard R., J. Virol. 2005, 79, 15199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Devaraj S. G., Wang N., Chen Z., Chen Z., Tseng M., Barretto N., Lin R., Peters C. J., Tseng C.-T. K., Baker S. C., Li K., J. Biol. Chem. 2007, 282, 32208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Delre P., Caporuscio F., Saviano M., Mangiatordi G. F., Front. Chem. 2020, 8, 1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu Y., Chen K., Pan J., Lei Y., Zhang D., Fang L., Tang J., Chen X., Ma Y., Zheng Y., Zhang B., Zhou Y., Zhan J., Xu W., Int. J. Biol. Macromol. 2021, 188, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liang J. J., Pitsillou E., Ververis K., Guallar V., Hung A., Karagiannis T. C., Chem. Phys. Lett. 2022, 788, 139294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Amin Sk. A., Banerjee S., Ghosh K., Gayen S., Jha T., Bioorg. Med. Chem. 2021, 29, 115860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yan F., Gao F., Comput. Struct. Biotechnol. J. 2021, 19, 4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rut W., Lv Z., Zmudzinski M., Patchett S., Nayak D., Snipas S. J., el Oualid F., Huang T. T., Bekes M., Drag M., Olsen S. K., Sci. Adv. 2020, 6, DOI: 10.1126/sciadv.abd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wishart D. S., Feunang Y. D., Guo A. C., Lo E. J., Marcu A., Grant J. R., Sajed T., Johnson D., Li C., Sayeeda Z., Assempour N., Iynkkaran I., Liu Y., Maciejewski A., Gale N., Wilson A., Chin L., Cummings R., Le D., Pon A., Knox C., Wilson M., Nucleic Acids Res. 2018, 46, D1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Durán A., Martínez G. C., Pastor M., J. Chem. Inf. Model. 2008, 48, 1813. [DOI] [PubMed] [Google Scholar]
- 19. Durán Á., Zamora I., Pastor M., J. Chem. Inf. Model. 2009, 49, 2129. [DOI] [PubMed] [Google Scholar]
- 20. Fu Z., Huang B., Tang J., Liu S., Liu M., Ye Y., Liu Z., Xiong Y., Zhu W., Cao D., Li J., Niu X., Zhou H., Zhao Y. J., Zhang G., Huang H., Nat. Commun. 2021, 12, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shen Z., Ratia K., Cooper L., Kong D., Lee H., Kwon Y., Li Y., Alqarni S., Huang F., Dubrovskyi O., Rong L., Thatcher G. R., Xiong R., bioRxiv 2021, preprint DOI: 10.1101/2021.02.13.431008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Osipiuk J., Azizi S.-A., Dvorkin S., Endres M., Jedrzejczak R., Jones K. A., Kang S., Kathayat R. S., Kim Y., Lisnyak V. G., Maki S. L., Nicolaescu V., Taylor C. A., Tesar C., Zhang Y.-A., Zhou Z., Randall G., Michalska K., Snyder S. A., Dickinson B. C., Joachimiak A., Nat. Commun. 2021, 12, 743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Laio A., Parrinello M., Proc. Natl. Acad. Sci. USA 2002, 99, 12562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barducci A., Bussi G., Parrinello M., Phys. Rev. Lett. 2008, 100, 20603. [DOI] [PubMed] [Google Scholar]
- 25. Zheng W., Sun W., Simeonov A., Br. J. Pharmacol. 2018, 175, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hassan A. H. E., Phan T.-N., Yoon S., Lee C. J., Jeon H. R., Kim S.-H., No J. H., Lee Y. S., J. Enzyme Inhib. Med. Chem. 2021, 36, 1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hassan A. H. E., Park H. R., Yoon Y. M., Kim H. I., Yoo S. Y., Lee K. W., Lee Y. S., Bioorg. Chem. 2019, 84, 444. [DOI] [PubMed] [Google Scholar]
- 28. Farag A. K., Hassan A. H. E., Ahn B. S., Park K. D., Roh E. J., J. Enzyme Inhib. Med. Chem. 2020, 35, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dotolo S., Marabotti A., Facchiano A., Tagliaferri R., Briefings Bioinf. 2020, 22, 726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Olubiyi O. O., Olagunju M., Keutmann M., Loschwitz J., Strodel B., Molecules 2020, 25, 3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baig M. H., Ahmad K., Roy S., Ashraf J. M., Adil M., Siddiqui M. H., Khan S., Kamal M. A., Provazník I., Choi I., Curr. Pharm. Des. 2016, 22, 572. [DOI] [PubMed] [Google Scholar]
- 32. Veljkovic V., Vergara-Alert J., Segalés J., Paessler S., F1000Research 2021, 9, DOI: 10.12688/f1000research.22149.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Veljkovic V., Paessler S., ResearchSquare 2020, preprint DOI: 10.21203/rs.2.24483/v1. [Google Scholar]
- 34. Sencanski M., Perovic V., Pajovic S. B., Adzic M., Paessler S., Glisic S., Molecules 2020, 25, 3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sencanski M., Sumonja N., Perovic V., Glisic S., Veljkovic N., Veljkovic V., arXiv 2019, preprint: arXiv:1907.02713 [q-bio.BM]. [Google Scholar]
- 36. Tintori C., Manetti F., Veljkovic N., Perovic V., Vercammen J., Hayes S., Massa S., Witvrouw M., Debyser Z., Veljkovic V., Botta M., J. Chem. Inf. Model. 2007, 47, 1536. [DOI] [PubMed] [Google Scholar]
- 37. Veljkovic V., Goeijenbier M., Glisic S., Veljkovic N., Perovic V. R., Sencanski M., Branch D. R., Paessler S., F1000Research 2015, 4, 104, DOI: 10.12688/f1000research.6436.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Radosevic D., Sencanski M., Perovic V., Veljkovic N., Prljic J., Veljkovic V., Mantlo E., Bukreyeva N., Paessler S., Glisic S., Front. Cell. Infect. Microbiol. 2019, 9, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Matejin S., Bukreyeva N., Radosevic D., Sencanski M., Mantlo E., Veljkovic V., Glisic S., Paessler S., Antiviral Ther. 2020, 24, 589. [DOI] [PubMed] [Google Scholar]
- 40. Zhao Y., Ren J., Harlos K., Jones D. M., Zeltina A., Bowden T. A., Padilla-Parra S., Fry E. E., Stuart D. I., Nature 2016, 535, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Paessler S., Huang C., Sencanski M., Veljkovic N., Perovic V., Glisic S., Veljkovic V., Front. Biosci. (Landmark edition) 2018, 23, 947. [DOI] [PubMed] [Google Scholar]
- 42. Mostafa E. M., Gamal M., Ghoneim M. M., Hussein S., El-Ghorab A. H., Abdelgawad M. A., Musa A., Pharmacogn. J. 2021, 13, 110. [Google Scholar]
- 43. Marion I. U., Marion A., ChemRxiv. 2020, preprint DOI: 10.26434/chemrxiv.13292768.v2. [Google Scholar]
- 44. Ginex T., Garaigorta U., Ramírez D., Castro V., Nozal V., Maestro I., García-Cárceles J., Campillo N. E., Martinez A., Gastaminza P., Gil C., Pharmaceuticals 2021, 14, DOI: 10.3390/ph14040332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cho J., Lee Y. J., Kim J. H., il Kim S., Kim S. S., Choi B.-S., Choi J.-H., Sci. Rep. 2020, 10, 16200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen C. Z., Xu M., Pradhan M., Gorshkov K., Petersen J. D., Straus M. R., Zhu W., Shinn P., Guo H., Shen M., Klumpp-Thomas C., Michael S. G., Zimmerberg J., Zheng W., Whittaker G. R., ACS Pharmacol. Transl. Sci. 2020, 3, 1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang L., Pei R.-J., Li H., Ma X.-N., Zhou Y., Zhu F.-H., He P.-L., Tang W., Zhang Y.-C., Xiong J., Xiao S.-Q., Tong X.-K., Zhang B., Zuo J.-P., Acta Pharmacol. Sin. 2021, 42, 1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fred S. M., Kuivanen S., Ugurlu H., Casarotto P. C., Levanov L., Saksela K., Vapalahti O., Castrén E., bioRxiv 2021, preprint DOI: 10.1101/2021.03.22.436379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Große M., Ruetalo N., Layer M., Hu D., Businger R., Rheber S., Setz C., Rauch P., Auth J., Fröba M., Brysch E., Schindler M., Schubert U., Viruses 2021, 13, 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen C. Z., Shinn P., Itkin Z., Eastman R. T., Bostwick R., Rasmussen L., Huang R., Shen M., Hu X., Wilson K. M., Brooks B. M., Guo H., Zhao T., Klump-Thomas C., Simeonov A., Michael S. G., Lo D. C., Hall M. D., Zheng W., Front. Pharmacol. 2021, 11, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Parvez Md. S. A., Karim Md. A., Hasan M., Jaman J., Karim Z., Tahsin T., Hasan Md. N., Hosen M. J., Int. J. Biol. Macromol. 2020, 163, 1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Veljković V., Cosić I., Dimitrijević B., Lalović D., IEEE Trans. Biomed. Eng. 1985, 32, 337. [DOI] [PubMed] [Google Scholar]
- 53. Veljković V., Slavić I., Phys. Rev. Lett. 1972, 29, 105. [Google Scholar]
- 54. Pastor M., Cruciani G., McLay I., Pickett S., Clementi S., J. Med. Chem. 2000, 43, 3233. [DOI] [PubMed] [Google Scholar]
- 55.MOPAC 2016, U. James J. P. Stewart, Stewart Computational Chemistry, Colorado Springs, CO, 2016.
- 56. Stewart J. J. P., J. Mol. Model. 2013, 19, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Trott O., Olson A. J., J. Comput. Chem. 2010, 31, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Huang J., MacKerell A. D. J., J. Comput. Chem. 2013, 34, 2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., Mackerell A. D. Jr, J. Comput. Chem. 2010, 31, 671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Phillips J. C., Hardy D. J., Maia J. D. C., Stone J. E., v Ribeiro J., Bernardi R. C., Buch R., Fiorin G., Hénin J., Jiang W., McGreevy R., Melo M. C. R., Radak B. K., Skeel R. D., Singharoy A., Wang Y., Roux B., Aksimentiev A., Luthey-Schulten Z., v Kalé L., Schulten K., Chipot C., Tajkhorshid E., J. Chem. Phys. 2020, 153, 44130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stone J., Gullingsrud J., Grayson P., Schulten K., in 2001 ACM Symposium on Interactive 3D Graphics (Eds.: Hughes J. F., Séquin C. H.), ACM SIGGRAPH, New York, 2001, pp. 191. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are openly available in Drugbank Online at https://go.drugbank.com/releases/latest, reference number 518. These data were derived from the following resources available in the public domain:, https://www.[resource1]; [Resource 2], https://www.[resource2]; …