

Abstract
Spontaneous chemical modifications play an important role in human disease and aging at the molecular level. Deamidation and isomerization are known to be among the most prevalent chemical modifications in long-lived human proteins and are implicated in a growing list of human pathologies, but the relatively minor chemical change associated with these processes has presented a longstanding analytical challenge. Although the adoption of high-resolution mass spectrometry has greatly aided the identification of deamidation sites in proteomic studies, isomerization (and the isomeric products of deamidation) remain exceptionally challenging to characterize. Herein, we present a liquid chromatography/mass spectrometry-based approach for rapidly characterizing the isomeric products of Gln deamidation using diagnostic fragments that are abundantly produced and capable of unambiguously identifying both Glu and isoGlu. Importantly, the informative fragment ions are produced through orthogonal fragmentation pathways, thereby enabling the simultaneous detection of both isomeric forms while retaining compatibility with shotgun proteomics. Furthermore, the diagnostic fragments associated with isoGlu pinpoint the location of the modified residue. The utility of this technique is demonstrated by characterizing the isomeric products generated during in vitro aging of a series of glutamine-containing peptides. Sequence-dependent product profiles are obtained, and the abundance of deamidation-linked racemization is examined. Finally, comparisons are made between Gln deamidation which is relatively poorly understood, and asparagine deamidation which has been more thoroughly studied.
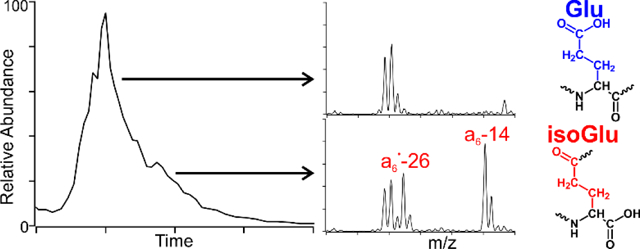
Graphical Abstract
Introduction
Asparagine and Gln are uniquely unstable amino acids that can spontaneously deamidate under physiological conditions.1,2 Deamidation can modulate protein function,3 and in some cases may be beneficial.4 Nonetheless, it is more commonly perceived as an unwanted chemical modification that is disruptive to normal cellular function,5 and is potentially a critical quality attribute in biological therapeutics.6 Regardless of the origin, deamidation represents an important spontaneous degradation that can alter protein behavior and is associated with numerous age-related diseases,7 including Alzheimer’s,8,9,10,11 multiple sclerosis,12 prion-related encephalopathies,13 and cataracts.14,15
The mechanism of asparagine deamidation has been studied thoroughly, but Gln deamidation has been investigated less, though it is likely to follow an analogous chemical trajectory.1,16–17,18 Both amino acids can deamidate through direct hydrolysis of the side chain, yielding the corresponding acidic amino acid (Asp or Glu). However, asparagine deamidation more commonly proceeds through a succinimide intermediate which forms as the backbone nitrogen of the C-terminal neighboring residue attacks the amide side chain. The resulting cyclic intermediate is susceptible to hydrolysis at two locations, yielding the isomeric products aspartic acid and isoaspartic acid (Asp and isoAsp). Additionally, the succinimide ring is racemization prone,19 therefore four potential products are possible: L-Asp, L-isoAsp, D-Asp, and D-isoAsp. The corresponding mechanism for Gln deamidation could yield a glutarimide intermediate and, in theory, could produce the corresponding four isomers of Glu (L-Glu, D-Glu, L-isoGlu, and D-isoGlu) as depicted in Scheme 1. However, experimental evidence for all four Glu isomers is lacking, leaving open the question as to whether D-isomers form during Gln deamidation.
Scheme 1.

Deamidation of Gln and isomerization of Glu can both proceed through the glutarimide intermediate, potentially yielding four isomers of Glu.
Deamidation causes a mass shift of 0.984 Dalton that is readily detected with high-resolution mass spectrometry,20,21 but identification of the four products of deamidation requires deeper analysis than a simple MS1 scan.22,23 Fragmentation techniques such as electron-transfer dissociation (ETD) and electron-capture dissociation (ECD) have been utilized to identify isoAsp24 and isoGlu25 residues by producing peptide fragments with characteristic mass-shifts. Unfortunately unique mass fragments do not exist for L- versus D- epimers, meaning that such fragments cannot be used to distinguish them. Not only is isomerization a difficult modification to detect, the heterogeneous nature and chemical similarity of deamidation products has contributed to the difficulty in identifying the specific structural perturbations responsible for protein dysfunction.26,27 One of the most illustrative examples of the impact of deamidation arises in RNase U2, which has long been known to lose activity as it deamidates.28 Recently, crystal structures have revealed that the native α-helix is partially unfolded when Asn32 deamidates to produce L-isoAsp.29 Top-down experiments have suggested that deamidation induces protein unfolding.30 An even more pronounced loss of function has been described in a therapeutic monoclonal antibody wherein a single isoAsp deactivates the entire antigen binding region.31 Recently, in vitro aging has proven valuable for predicting in vivo degradation.32 These observations suggest that deamidation and isomerization are important modifications in biological aging and also merit consideration in the design of biological therapeutics.33,34
Despite the growing interest in spontaneous degradation and protein aging, Gln deamidation studies remain sparse. Although the first report of glutarimide formation during in vitro aging was reported nearly 3 decades ago,15 many of the subsequent aging studies have focused on asparagine deamidation and/or aspartic acid isomerization. For instance, studies on asparagine have elucidated the role of pH, temperature, and secondary structure, while Gln remains relatively unexplored.35,36 This is likely due to the timescale associated with Gln deamidation1 and the difficulty associated with detecting isomers via conventional proteomics. While the most rapid asparagine deamidation reactions have half-lives of ~1 day, the corresponding Gln deamidation has a half-life of nearly 2 years.37 Still, Gln warrants further attention because it is known to be biologically significant, especially in extremely long-lived proteins such as the crystallins found in the eye lens,38,39 and in brain proteins where isoGlu has recently been detected.40 Interestingly, within the lens crystallin proteins, deamidation at specific Gln sites has been reported to be more abundant than nearby asparagine deamidation sites,41,42 although the overall extent of asparagine deamidation remains higher.38 Unlike asparagine, some portion of the total deamidation observed at Gln may be due to the enzymatic activity of tissue transglutaminase (TTG), which has been associated with deamidation and crosslinking in ß-crystallins in vitro.43 TTG likely plays a role in lens aging,44 and is associated with cataractogenesis but a detailed understanding of this enzyme is still emerging. Nonetheless, Gln deamidation in the lens increases with age, and it is associated with aggregation, loss of function, and insolubility.25
We have previously utilized radical-directed dissociation (RDD) in conjunction with reverse-phase liquid chromatography to identify and characterize isomerization and deamidation.45,46,47 In a recent study on cataractous human lens samples, we detected Gln deamidation and were intrigued by the observation of tandem MS peptide fragments arising from the Glu sidechain that appeared to diminish across an LC peak, hinting at the possibility of Glu/isoGlu coelution.48 In this study, we develop a liquid-chromatography/mass spectrometry approach to rapidly identify and characterize the products of Gln deamidation and Glu isomerization. In particular, we improve on existing techniques by providing a means for positive identification of canonical Glu as well as isoGlu, while interfacing with a common reverse phase liquid chromatography setup. After validating the approach using synthetic Glu isomers, we apply the technique to a series of model peptides subjected to in vitro aging. Similarities with asparagine deamidation and aspartic acid isomerization are discussed in addition to several important differences.
Experimental Section
Materials.
Organic solvents and reagents were purchased from Fisher Scientific, Sigma-Aldrich, or Acros Organics and used without further purification. FMOC-protected amino acids and Wang resins were purchased from Anaspec, Inc or Chem-Impex International. Glu isomers were purchased from CarboSynth.
Peptide Synthesis.
Peptides were synthesized manually following an accelerated FMOC-protected solid-phase peptide synthesis protocol.49 Para-iodobenzoic acid (4IB) was attached to the N-terminus during synthesis. Following synthesis, peptides were purified using a Phenomenex Jupiter Proteo C12 4 μm 90 Å 250 mm x 4.6 mm column. Purified peptides were stored frozen in 50/50 acetonitrile/water (v/v).
Aging Studies.
Crystallin proteins were extracted from human lens tissue and modified with 4IB as previously described.47 Briefly, human lens cortices and nuclei were separated using a trephine before homogenization. The water soluble and water insoluble proteins were subsequently fractionated by centrifugation, digested with trypsin, and analyzed by LCMS. Aliquots of each digest were modified with 4IB to facilitate RDD. Synthetic peptides were deamidated at 37 °C in microcentrifuge tubes submerged in a sand-bath style incubator. The initial deamidation studies were performed in 0.1M Tris buffer at pH 7.7. Accelerated studies were conducted in 0.2M Tris with the addition of 300mM NH3 at pH 8.7. The isomerization study was carried out in 0.2M ammonium acetate at pH 5. Aliquots were collected at various time-points and stored frozen as lyophilized powder until analysis.
Analysis.
Following aging studies, peptides were analyzed via LCMS. An Agilent 1100 binary pump was used with a Thermo BetaBasic-18 3 μm C18 150 mm x 2.1 mm column interfaced to a Thermo Fisher Scientific LTQ mass spectrometer with a standard electrospray ionization source. Samples were eluted using water with 0.1% formic acid as mobile phase A, and acetonitrile with 0.1% formic acid as mobile phase B. Synthetic standards were prepared as ~10 μM samples in 49.5/49.5/1 methanol/water/acetic acid (v/v) and infused into a modified LTQ linear ion trap using the standard electrospray ionization source. The LTQ was modified with a quartz window to allow fourth harmonic (266 nm) laser pulses from an Nd:YAG laser to irradiate the trapped ion cloud which allows for photoinitiated RDD.50 Following photodissociation, the radical peptide population was isolated and collisionally activated to promote radical migration and fragmentation.
Risomer scores.
To distinguish isomers based on fragmentation patterns, we utilize an approach first developed by Tao et al.51 Briefly, Risomer scores were calculated using equation 1 where R1 and R2 refer to the ratios of a pair of fragment ions that change the most between two spectra. Identical fragmentation patterns would yield an Risomer score of 1, indicating no discrimination, while the magnitude of the Risomer score increases with larger differences between the spectra.
| (1) |
R values >2.4 for RDD are statistically significant and are indicative of isomers that can be distinguished based on fragmentation patterns.44
Results and Discussion
We previously identified numerous sites of isomerization and deamidation within the crystallin proteins of the human lens.45 Our isomer proteomics approach employs LC/MSn and RDD to access unique and structurally sensitive fragmentation pathways which are capable of discerning peptide isomers. By evaluating the peptide fragmentation patterns, isomers can often be identified even when complete chromatographic separation is not possible. For instance, during the analysis of a fraction from a 72-year-old water-insoluble cortex, the 4IB modified peptide 85AVHLPSGGQYK95 from γS-crystallin was found to be heavily deamidated at Q93, but the deamidated peak is only partially resolved chromatographically (Figure 1a). Examination of the RDD data reveals that the fragmentation pattern at the beginning of the LC peak differs from the latter scans within the peak, indicating the presence of two species that are not chromatographically resolved. In particular, the −59E and −72E Glu side chain losses are notably absent in the leading shoulder of the peak but are observed in the trailing edge of the peak (Figure 1b). These side chain losses are commonly observed for Glu but cannot be generated when the side chain is modified to the isoGlu form (see supporting information figure S1 and reference 52 for additional mechanistic details.) We hypothesized that systematic differences in the radical fragmentation behavior for Glu and isoGlu could explain these observations.
Figure 1.

(a) The γS-crystallin AVHLPSGGQYK peptide is found to be deamidated in 72-year-old human lens tissue, yielding a broad peak at ~54 minutes. (b) The full MS are identical, however the RDD fragmentation patterns differ from the leading and trailing edge of the peak, indicating coeluting isomers. Notably, the −59E and −72E Glu side chain losses are absent in the leading edge of the peak but appear in the trailing edge.
To evaluate the fragmentation patterns of Glu isomers, we synthesized all four potential isomers of 4IB-VHLGGEGYK, which is derived from the γS-crystallin peptide sequence with modifications to facilitate in vitro aging (discussed below).1 RDD was performed on the [M+2H]2+ ions of each peptide. Figure 2 highlights the key differences and similarities between the fragmentation patterns. Fragments common to all four isomers are labelled in black, while fragments that positively identify Glu are labelled in blue, and those that are unique to isoGlu are labelled in red. Relative intensities in all four spectra are normalized to the base peak, which is common to all four isomers.
Figure 2.

RDD fragmentation enables unambiguous identification of Glu and isoGlu containing peptides. Fragments common to all isomers are labelled in black. Fragments that positively identify Glu are labelled in blue, while fragments unique to isoGlu are labelled in red. Peaks in outer segments are normalized to the intensity of −56L peak.
Several diagnostic fragments are apparent in Figure 2. For instance, L- and D-Glu can be identified by the abundant Glu side chain losses (labelled −59E2+ and −72E2+) and a6 ion that are absent in the isoGlu-containing peptides. Conversely, both isoGlu peptides produce mass-shifted backbone fragments which are characteristic of the unnatural ethylene group in the peptide backbone. Notably, these backbone mass-shifts are abundantly observed in peptides derived from both N-terminal (‘a6• - 26’and ‘a6 - 14’) as well as the C-terminal (‘x3• + 14’ and ‘x3 + 26’) fragments. This observation is useful because fragment ions may be favored for one terminus in a typical LCMS run (i.e. C-terminal fragments are more commonly observed for tryptic peptides). In addition, loss of COOH is much more abundant for the isoGlu peptides. Full spectra are provided in the supporting information.
The fragments observed in Fig. 2 arise from radical migration pathways localized at the Glu and isoGlu residues and are therefore expected to be largely sequence independent. RDD pathways that would yield the observed fragments are illustrated in Scheme 2. Migration of the initially formed radical to the β-carbon position of canonical Glu (depicted in red in Scheme 2a) leads to formation of normal a/x ions following β-scission. This pathway yields the a6 ion, which cannot form in isoGlu. The analogous pathway in isoGlu leads to loss of the entire unnatural sidechain COOH group as shown in Scheme 2b. Other pathways can also lead to loss of −45 Da, so while the loss is characteristic for this peptide, it may not always be so. However, fragmentation at isoGlu mediated by radical migration to the α-carbon and γ-carbon positions produces signature fragmentation due to the unnatural backbone. Abstraction of the α-hydrogen leads to cleavage between the two ethylene carbons, yielding ‘a - 14’ and ‘x• + 14’ backbone fragments (Scheme 2c). Alternatively, γ-hydrogen abstraction facilitates cleavage between the backbone ethylene and the α-carbon, producing ‘x + 26’ and ‘a• - 26’ fragment ions as depicted in Scheme 2d. In both cases, the fragments are unique to isoGlu containing peptides and pinpoint the location of the unnatural backbone modification.
Scheme 2.

(a) Radical fragmentation pathways depict formation of an a-type and x•-ion pair at the canonical Glu residue following hydrogen abstraction from the β-carbon (original site labelled in red in all four mechanisms). b) For isoGlu, the analogous pathway leads to loss of COOH. (b,c) IsoGlu residues also yield mass-shifted a- and x-type ions due to the additional backbone carbon, following α- or γ- hydrogen abstraction.
Having established a method for isomer discrimination, we tested the utility of the approach by aging in vitro the same peptide with a Glu→Gln substitution. 4IB-VHLGGQGYK (referred to as GQG) was aged in 100 mM Tris buffer at pH 7.7, 37 °C and monitored by LCMS. The chromatogram in Figure 3a depicts the 176-day (~6 month) aging time-point where the Gln-containing precursor peak at ~44 minutes is followed by two deamidated product peaks. After 176 days, the peptide is 19.6% deamidated, corresponding to a half-life of 559 days, which is comparable to aging studies under similar conditions.1 Of the deamidation products, the Glu peak represents 5.5% of the total peptide signal, while the isoGlu peak represents 14.1%, yielding a ratio of 2.6 which parallels previous observations for asparagine deamidation where ring cleavage favors the formation of the unnatural isomer in unstructured systems (i.e. isoAsp or isoGlu).15 The aging experiment was repeated after increasing the pH to 8.9 with ammonia, which is known to accelerate asparagine deamidation in general53 without significantly altering the product profile.46 The results are shown in Figure 3b, and the deamidation reaction is found to be greatly accelerated for Gln as well. After just two weeks, the peptide is 9.4% deamidated, representing a 12-fold increase in rate relative to Tris alone. Interestingly, the isoGlu:Glu ratio is slightly elevated to 3.3 in the accelerated sample, suggesting that direct hydrolysis (which cannot form isoGlu and is not known to be accelerated by ammonia) may contribute more to deamidation over the longer 176-day period. To confirm the deamidation product identities, RDD was employed continuously throughout the LCMS run. The characteristic fragmentation pattern of the first deamidated peak is shown in Figure 3c where the −59E and −72E fragments confirm the presence of the canonical Glu residue. Conversely, the second deamidated peak generated the diagnostic isoGlu mass-shifted backbone fragments, as shown in Figure 3d. Full spectra are available in the SI.
Figure 3.

(a) Incubation of the 4IB-VHLGGQGYK peptide yields 19.6% deamidated products after 176 days. (b) Addition of ammonia greatly accelerates the deamidation process, yielding 9.4% deamidation within 14 days. (c,d) RDD confirms that the leading deamidation peak contains the canonical Glu residue, while the latter peak contains the isoGlu product.
The amino acid residue immediately C-terminal to Gln or Asn is known to strongly influence the ring formation rate.1 To expose the impact of ring formation kinetics on the deamidation products, we incubated 4IB VHLGGQAYK and 4IB VHLGGQSYK (referred to as GQA and GQS respectively) in tris buffer with ammonia accelerant. In the case of the GQS peptide, only one product peak was observed by LCMS. However, the RDD spectra contained both -59E and −72E side chain losses and mass-shifted a-type and x-type backbone fragments, confirming the presence of both Glu and isoGlu in one coeluting peak. The individual product ratios were therefore extracted using calibration curves derived from spectra obtained from synthetic standards of both isomers. Additional details and data are available in the SI. The isomer product distributions for GQG, GQA, and GQS are shown in Figure 4 for easy comparison.
Figure 4.

Relative deamidation product ratios following ammonia accelerated deamidation for 4IB-VHLGGQGYK (t=14d), 4IB-VHLGGQAYK (t=127d), 4IB-VHLGGQSYK (t=127d).
Interestingly, the GQG peptide isomer distribution parallels that of asparagine, yielding more isoGlu than Glu by ~3:1. However, both GQA and GQS yield inverted isomer distributions, where there is roughly 3x as much canonical Glu. Although ammonia was used to accelerate deamidation for all three peptides, the slow rates for GQA and GQS required 127 days incubation compared with only 14 days for GQG. It appears that this extended timeframe is sufficient for direct hydrolysis to be competitive and increase the amount of Glu produced. Given that alanine and serine are among the least bulky side chains (which typically influence rate),1 these results suggest that direct hydrolysis may outcompete glutarimide ring formation for all sequences except QG motifs. This finding may explain why isoGlu has been difficult to identify in biological samples, despite the comparatively common observation of Gln deamidation.
As a final comparison between Gln and Asn deamidation, the extent of D- isomer production during glutarimide formation was examined. The succinimide ring formed during asparagine deamidation is known to be racemization prone,18 yielding D-isomers with ~10% relative abundance independent of peptide sequence and deamidation rate.46 Clarke and Houk previously rationalized the formation of D- isomers by comparing the proton affinity of the succinimide α-carbon using ab initio quantum mechanics calculations (RHF/6–31+G*) and density-functional theory (B3LYP/6–31+G*)18 where they found the succinimide to be 18 kcal/mol more acidic than its amide counterpart, corresponding to a pKa shift of 10.8 pH units. A comparable effect is expected for the glutarimide ring which is chemically similar. To evaluate this possibility, 4IB-VHLGGQGYK was allowed to deamidate for 64 days under ammonia acceleration. Unfortunately, neither pair of L- and D-isomers for 4IB-VHLGGEGYK separate by chromatography, but L-isoGlu and D-isoGlu synthetic standards were used to construct a calibration curve that could quantitate the amount of D-isoGlu. The results indicate ~4% of the product corresponds to D-isoGlu (see Figure S5). This is lower than the amount of D-isoAsp typically observed during deamidation of Asn,46 suggesting that the glutarimide ring may be less prone to racemization.
Given the parallels noted so far between Asn/Gln, the potential for Glu to mirror the isomerization behavior of Asp was explored. Drawing from the known pH dependence of asparagine deamidation (which is favored under neutral to basic conditions)52 and aspartic acid isomerization (which is favored under acidic conditions),54 we incubated the all-L isomer form of 4IB-VHLGGEGYK in ammonium acetate, at pH 5. The aging data following 2 months of incubation (61 days) is shown in Figure 5. Chromatographically, only one peak is observed (Figure 5a), suggesting that no detectable amount of isoGlu formed within 61 days. During the majority of the peak, only one species is present in the full mass spectrum at 595.2 m/z, matching the doubly protonated L-Glu precursor (Figure 5b left panel), however a minor second peak appears at 586.2 m/z (−18Da) toward the trailing end of the peak (Figure 5b right panel), consistent with the observation of the glutarimide ring. To verify the identity of the −18 Da peak, we performed RDD (Figure 5c). The −18 Da loss is found to be localized on peptide fragments which contain the Glu residue, as expected following formation of a glutarimide ring. More specifically, the b3 fragment is canonical while the c7 is shifted by −18 Da. Aside from the Glu residue, these two fragments differ only by the presence of glycine residues which cannot easily account for the loss of −18 Da. Notably, glutarimide ring formation for Glu proceeds ~37x slower than the corresponding reaction for Gln at pH 7.7. Nevertheless, Glu can form the glutarimide ring in an analogous manner to Gln, establishing another parallel between the Glu/Gln aging processes. It is expected that the same four isomers generated by Gln deamidation would be produced following the re-opening of the glutarimide ring detected in Fig. 5 because it is identical to that produced during Gln deamidation.
Figure 5.

Aging results for 4IB-VHLGGEGYK peptide under acidic conditions. (a) Only one peak is observed by LCMS, suggesting that no isoGlu has formed within 61 days. (b) Trace amounts of a coeluting species at −18 Dalton, consistent with the glutarimide, is observed during the trailing edge of the main peak. (c) RDD fragmentation confirms the presence of the glutarimide intermediate.
Conclusion
Asn and Gln deamidation share many similarities and have been grouped together as nearly equivalent aging processes, but our in vitro aging studies have revealed important differences. As previously reported with asparagine, ammonia is effective at accelerating Gln deamidation. Yet, Gln peptides are found to be more variable in terms of the distributions of isomers that are formed, which seem to be more strongly influenced by primary sequence. Even under accelerating conditions, direct hydrolysis was found to be a competitive process for all sequences except QG motifs. Direct hydrolysis is therefore likely to contribute a larger percentage of the overall deamidation at Gln in the majority of cases. Nonetheless, it is clear that isoGlu can form for certain sequences (i.e. QG motifs) or perhaps in highly flexible regions in extremely long-lived proteins. In these situations, RDD excels at discriminating Glu from isoGlu, with positive identifications enabled by the −59E and −72E fragments which are unique to the canonical Glu side chain, and mass shifted backbone fragment pairs ‘x• + 14’ and ‘x + 26’, and ‘a• - 26’ and ‘a - 14’ which can be used to pinpoint the location of isoGlu. Additionally, we have demonstrated that the level of isoGlu in an aged sample can be quantitatively assessed post hoc by use of a calibration curve. While the combination of ammonia acceleration with RDD-based isomer characterization is poised to enable more comprehensive in vitro aging studies, the technique may prove most useful in the analysis of complex aged human tissue such as the eye lens and brain where long-lived proteins are abundant and often highly deamidated. RDD benefits greatly by enabling positive identification of both Glu and isoGlu through orthogonal fragmentation channels while maintaining compatibility with shotgun proteomics.
Supplementary Material
Acknowledgements
The authors are grateful for funding from the NIH (NIGMS grant R01GM107099).
Footnotes
Supporting Information
Full mass spectra, detailed mechanisms, and calibration curves are provided.
References
- 1.Robinson NE; Robinson ZW; Robinson BR; Robinson AL; Robinson JA; Robinson ML; Robinson AB J. Pept. Res. 2004, 63 (5), 426–436. [DOI] [PubMed] [Google Scholar]
- 2.Robinson NE; Robinson AB Proc. Natl. Acad. Sci. 2001, 98 (3), 944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curnis F; Longhi R; Crippa L; Cattaneo A; Dondossola E; Bachi A; Corti AJ Biol. Chem. 2006, 281 (47), 36466–36476. [DOI] [PubMed] [Google Scholar]
- 4.Reissner KJ; Aswad DW Cell. Mol. Life Sci. 2003, 60 (7), 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarke S Ageing Res. Rev. 2003, 2 (3), 263–285. [DOI] [PubMed] [Google Scholar]
- 6.Yan Q, Huang M, Lewis MJ, and Hu P MAbs 2018, 10, 901–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindner H; Helliger W Exp. Gerontol. 2001, 36 (9), 1551–1563. [DOI] [PubMed] [Google Scholar]
- 8.Lam YPY; Wootton CA; Hands-Portman I; Wei J; Chiu CKC; Romero-Canelon I; Lermyte F; Barrow MP; O’Connor PB Chem. Commun. 2018, 54 (98), 13853–13856. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen PT; Zottig X; Sebastiao M; Bourgault S Biochemistry 2017, 56 (29), 3808–3817. [DOI] [PubMed] [Google Scholar]
- 10.Roher AE; Lowenson JD; Clarke S; Wolcow C; Wang R; Cotter RJ; Reardon IM; Zurcher-Neely HA; Heinrikson RL; Ball MJ; Greenberg BD J. Biol. Chem. 1993, 268 (5), 3072–3083. [PubMed] [Google Scholar]
- 11.Lambeth TR; Riggs DL; Talbert LE; Tang J; Coburn E; Kang AS; Noll J; Augello C; Ford BD; Julian RR ACS Cent. Sci. 2019, 5 (8), 1387–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedrich MG; Hancock SE; Raftery MJ; Truscott RJW Acta Neuropathol. Commun. 2016, 4 (1), 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandmeier E; Hunziker P; Kunz B; Sack R; Christen P Biochem. Biophys. Res. Commun. 1999, 261 (3), 578–583. [DOI] [PubMed] [Google Scholar]
- 14.Truscott RJW; Schey KL; Friedrich MG Trends Biochem. Sci. 2016, 41 (8), 654–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takata T; Oxford JT; Demeler B; Lampi KJ Protein Sci. 2008, 17 (9), 1565–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geiger T; Clarke S J. Biol. Chem. 1987, 262 (2), 785–794. [PubMed] [Google Scholar]
- 17.Rivers J; McDonald L; Edwards IJ; Beynon RJ J. Proteome Res. 2008, 7 (3), 921–927. [DOI] [PubMed] [Google Scholar]
- 18.Capasso S; Mazzarella L; Sica F; Zagari A J. Chem. Soc., Chem. Commun. 1991, 23, 1667–1668. [Google Scholar]
- 19.Radkiewicz JL; Zipse H; Clarke S; Houk KN J. Am. Chem. Soc. 1996, 9148–9155. [DOI] [PubMed] [Google Scholar]
- 20.Yang H; Fung EYM; Zubarev AR; Zubarev RA J. Proteome Res. 2009, 8 (10), 4615–4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang H; Fung EYM; Zubarev AR; Zubarev RA J. Proteome Res. 2009, 8 (10), 4615–4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang S; Kaltashov IA An 18 O-Labeling Assisted LC/MS Method for Assignment of Aspartyl/Isoaspartyl Products from Asn Deamidation and Asp Isomerization in Proteins. Anal. Chem. 2013, 85 (13), 6446–6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maeda H; Takata T; Fujii N; Sakaue H; Nirasawa S; Takahashi S; Sasaki H; Fujii N Rapid Survey of Four Asp Isomers in Disease-Related Proteins by LC-MS Combined with Commercial Enzymes. Anal. Chem. 2015, 87 (1), 561–568. [DOI] [PubMed] [Google Scholar]
- 24.Cournoyer JJ; Lin C; O’Connor PB Anal. Chem. 2006, 78 (4), 1264–1271. [DOI] [PubMed] [Google Scholar]
- 25.Li X; Lin C; O’connor PB Anal. Chem. 2010, 82 (9), 3606–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lampi KJ; Wilmarth PA; Murray MR; David LL Prog. Biophys. Mol. Biol. 2014, 115 (1), 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lyon YA; Collier MP; Riggs DL; Degiacomi MT; Benesch JLP; Julian RR J. Biol. Chem. 2019, 294 (19), 7546–7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uchida T; Shibata Y J. Biochem. 1981, 90 (2), 463–471. [DOI] [PubMed] [Google Scholar]
- 29.Noguchi S Biopolymers 2010, 93 (11), 1003–1010. [DOI] [PubMed] [Google Scholar]
- 30.Soulby AJ; Heal JW; Barrow MP; Roemer RA; O’Connor PB Protein Sci. 2015, 24 (5), 850–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rehder DS; Chelius D; Mcauley A; Dillon TM; Xiao G; Crouse-zeineddini J; Vardanyan L; Perico N; Mukku V; Brems DN; Matsumura M; Bondarenko PV Biochemistry 2008, 2518–2530. [DOI] [PubMed] [Google Scholar]
- 32.Tran JC; Tran D; Hilderbrand A; Andersen N; Huang T; Reif K; Hotzel I; Stefanich EG; Liu Y; Wang J Anal. Chem. 2016, 88 (23), 11521–11526. [DOI] [PubMed] [Google Scholar]
- 33.Wakankar AA; Borchardt RT J. Pharm. Sci. 2006, 95 (11), 2321–2336. [DOI] [PubMed] [Google Scholar]
- 34.Xu A; Kim HS; Estee S; Viajar S; Galush WJ; Gill A; Hötzel I; Lazar GA; McDonald P; Andersen N; Spiess C Mol. Pharm. 2018, 15 (10), 4529–4537. [DOI] [PubMed] [Google Scholar]
- 35.Patel K; Borchardt RT Pharm. Res. 1990, 7 (8), 787–793. [DOI] [PubMed] [Google Scholar]
- 36.Yüksel KÜ; Gracy RW Arch. Biochem. Biophys. 1986, 248 (2), 452–459. [DOI] [PubMed] [Google Scholar]
- 37.Robinson NE; Robinson ZW; Robinson BR; Robinson AL; Robinson JA; Robinson ML; Robinson AB J. Pept. Res. 2004, 63 (5), 426–436. [DOI] [PubMed] [Google Scholar]
- 38.Hanson SR; Smith DL; Smith JB Exp. Eye Res. 1998, 67 (3), 301–312. [DOI] [PubMed] [Google Scholar]
- 39.Hains PG; Truscott RJW Investig. Ophthalmol. Vis. Sci. 2010, 51 (6), 3107–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Serra A; Gallart-Palau X; Wei J; Sze SK Anal. Chem. 2016, 88 (21), 10573–10582. [DOI] [PubMed] [Google Scholar]
- 41.Hains PG; Truscott RJW J. Proteome Res. 2007, 6 (10), 3935–3943. [DOI] [PubMed] [Google Scholar]
- 42.Asomugha CO; Gupta R; Srivastava OP Mol.Vis. 2010, 16, 476–494. [PMC free article] [PubMed] [Google Scholar]
- 43.Boros S; Wilmarth PA; Kamps B; de Jong WW; Bloemendal H; Lampi K; Boelens WC Exp. Eye Res. 2008, 383–393. [DOI] [PubMed] [Google Scholar]
- 44.Lorand L; Hsu LK; Siefring GE; Rafferty NS Proc. Natl. Acad. Sci. 2006, 78 (3), 1356–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tao Y; Julian RR Anal. Chem. 2014, 86 (19), 9733–9741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lyon YA; Sabbah GM; Julian RR J. Proteome Res. 2017, 16 (4), 1797–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Riggs DL; Gomez SV; Julian RR ACS Chem. Biol. 2017, 12 (11), 2875–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lyon YA; Sabbah GM; Julian RR Exp. Eye Res. 2018, 171, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hood CA, Fuentes G, Patel H, Page K, Menakuru M, Park JH J. Pept. Sci. 2008, 14, 97–101. [DOI] [PubMed] [Google Scholar]
- 50.Ly T, Julian RR J. Am. Chem. Soc. 2008, 130, 351–358. [DOI] [PubMed] [Google Scholar]
- 51.Tao WA; Zhang D; Nikolaev EN; Cooks RG J. Am. Chem. Soc. 2000, 122 (43), 10598–10609. [Google Scholar]
- 52.Sun Q; Nelson H; Ly T; Stoltz BM; Julian RR J. Proteome Res. 2009, 8 (2), 958–966. [DOI] [PubMed] [Google Scholar]
- 53.Tyler-Cross R; Schirch V J. Biol. Chem. 1991, 266 (33), 22549–22556. [PubMed] [Google Scholar]
- 54.Aswad DW; Paranandi MV; Schurter BT J. Pharm. Biomed. Anal. 2000, 21 (6), 1129–1136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.