

Abstract
The molecular chaperone, Heat Shock Protein 70 (Hsp70), is an emerging drug target for neurodegenerative diseases, because of its ability to promote degradation of microtubule-associated protein tau (MAPT/tau). Recently, we reported YM-08 as a brain penetrant, allosteric Hsp70 inhibitor, which reduced tau levels. However, the benzothiazole moiety of YM-08 is vulnerable to metabolism by CYP3A4, limiting its further application as a chemical probe. In this manuscript, we designed and synthesized seventeen YM-08 derivatives by systematically introducing halogen atoms to the benzothiazole ring and shifting the position of the heteroatom in a distal pyridine. In microsome assays, we found that compound JG-23 has 12-fold better metabolic stability and it retained the ability to reduce tau levels in two cell-based models. These chemical probes of Hsp70 are expected to be useful tools for studying tau homeostasis.
1. Introduction
Tau is an intrinsically disordered, microtubule-associated protein that is abundantly expressed in neurons.1, 2 When tau’s microtubule binding is disrupted, by either mutation3, 4 or post-translational modifications, such as phosphorylation5, 6 and acetylation7, 8, it is aberrantly localized to the cytosol. This dissociated tau is then prone to aggregate, where it can form neurofibrillary tangles (NFTs) that are associated with neuronal cell death9 and neurodegenerative disorders, including Alzheimer’s disease (AD) and frontotemporal dementia (FTD). These observations have driven interest in therapeutic approaches that modulate tau homeostasis,10, 11 including stabilizing microtubule12, reducing tau post-transcriptional modification8, 13, and enhancing tau degradation through inhibition of the molecular chaperones Hsp90 and Hsp7014–16.
Heat Shock Protein 70 (Hsp70) is a molecular chaperone that binds to tau soon after its release from microtubules.17 There are two major, cytoplasmic orthologs of Hsp70 that are thought to perform this function: Hsc70 (HSPA8) and Hsp72 (HSPA1A). These isoforms are highly conserved and, after they bind tau, they either stabilize its re-binding to microtubules or promote its degradation.18 Despite the similarities between the orthologs, Hsp72 is more tightly linked to tau degradation.19 Pharmacological studies have also supported this idea. Specifically, a brain penetrant, irreversible Hsp72 inhibitor, methylene blue (MB), reduces levels of insoluble tau and improves learning and memory in tauopathy mouse models.20 Likewise, other classes of Hsp70 inhibitors, including dihydropyridines and rhodacyanines, which likely bind to both Hsp72 and Hsc70,21 reduce tau levels in cellular and tissue models.22 23, 24 These molecules appear to work by stabilizing the conformation of both Hsp70s, which seems to promote tau degradation.25 Unlike MB, which has promiscuous activity, the rhodacyanines are relatively selective for Hsp70 family members in cells,26 so there is interest in further developing them as chemical probes.
One rhodacyanine, MKT-077, was first identified as an inhibitor of Hsp70s in cancer models, but it was later shown to decrease both phosphorylated and total tau levels in HeLaC3 cells.23 We found that MKT-077 binds to an allosteric site that is conserved among Hsp70 isoforms21 and it is considered to be a pan-Hsp70 inhibitor. Here, the term “inhibitor” is somewhat incomplete, because, while binding to this allosteric site inhibits ATP turnover, it also stabilizes the conformation of Hsc70 and Hsp72 that promotes degradation of bound proteins, such as tau. Thus, molecules in the chemical series could be useful probes in neurodegenerative disease models because they accelerate tau degradation. Towards that goal, recent medicinal chemistry efforts have started to improve the potency of MKT-077. For example, a close MKT-077 derivative, YM-01, was found to have improved membrane permeability and it promotes tau degradation at concentrations ~ 1 μM. Importantly, treatment with YM-01 leads to preferential degradation of pathogenic tau, while normal, microtubule-associated tau is resistant, in a mouse brain slice model.24. While these findings are promising, the quaternary amine in YM-01 leads to its accumulation in mitochondria and limits its blood-brain barrier (BBB) permeability. To solve this issue, a neutral analog, YM-08 (Figure 1), was synthesized by replacing the pyridinium with a pyridine. YM-08 retained binding to Hsp70s and it still had the ability to decrease tau levels in HeLaC3 cells, while it also had improved brain exposure.16 However, YM-08 is rapidly metabolized and its half-life is less than three minutes in mouse liver microsome assays, significantly limiting its applications in vivo.
Figure 1.
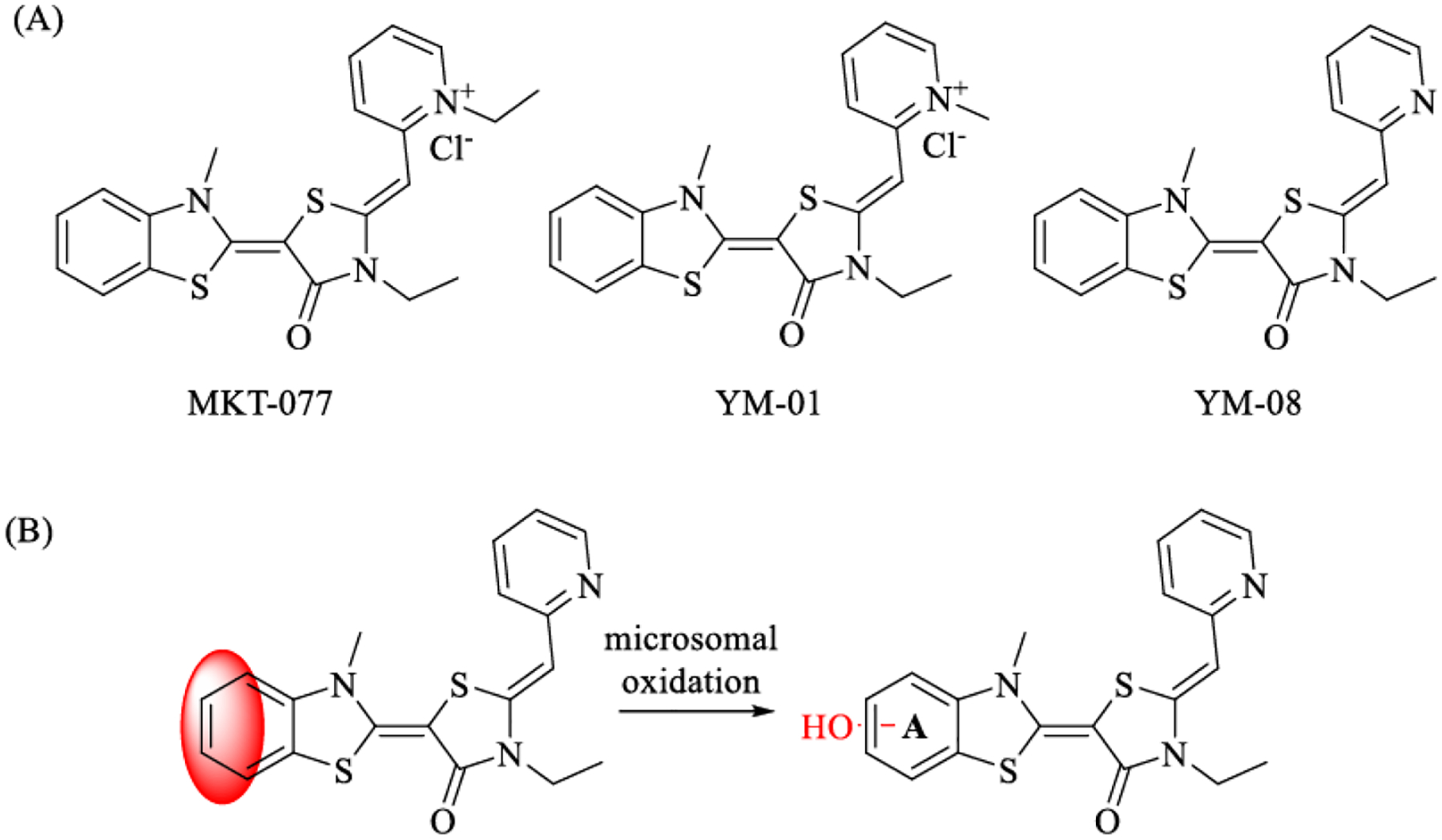
Previously reported allosteric Hsp70 inhibitors and its metabolism. (A). Chemical structures of MKT-077, YM-01 and YM-08; (B). Benzothiazole is the metabolic liable site.
In a metabolite identification study, we had previously identified the benzothiazole in YM-08 as the major site for Phase I oxidation (Figure 1).16 However, it was not clear whether oxidation occurred at the 3-, 4-, 5- or 6- positions. Here, we explored whether introducing halogens to specific sites on the benzothiazole ring may block this oxidation and increase the lifetime of YM-08 analogs. This approach seemed feasible because docking of YM-08 to Hsc70 suggested that the benzothiazole is located in a hydrophobic pocket that would accept modification by halogens. In this study, we reported the synthesis of seventeen YM-08 analogs and the selection of JG-23 as a molecule with improved metabolic stability and tau-reducing activity.
2. Results and Discussion
To understand which sites might enhance metabolic stability, we sought to systematically install halogens at the 3-, 4– 5- and 6-ring positions in the benzothiazole. In addition, we envisioned swapping the position of the pyridine nitrogen to reveal any distal, electronic effects. Accordingly, seventeen YM-08 analogs were designed and synthesized using a previously reported method as shown in Scheme 1.26, 27 Briefly, the synthesis started from the cyclization of substituted anilines with potassium ethyl xanthate, followed by methylation with methyl iodide under mild basic condition. The resulting substituted 2-(methylthio)benzothiazole was reacted with methyl p-toluenesulfonate to afford its methylthioiminium salt, which was subsequently condensed with 3-ethylrhodanine. The resulting compounds were activated by methyl p-toluenesulfonate, followed by the condensation of either 1-((1,3-dioxoisoindolin-2-yl)methyl)-2-methylpyridin-1-ium bromide or 1-((1,3-dioxoisoindolin-2-yl)methyl)-4-methylpyridin-1-ium bromide. The protecting group on the pyridine nitrogen was then removed in the presence of catalytic amount of aqueous ammonium hydroxide to yield the final products 1-17 in overall yields between 20 to 30% and purity greater than 97%. The compounds were purified by flash chromatography and characterized by LC-MS and 1H-NMR.
Scheme 1:
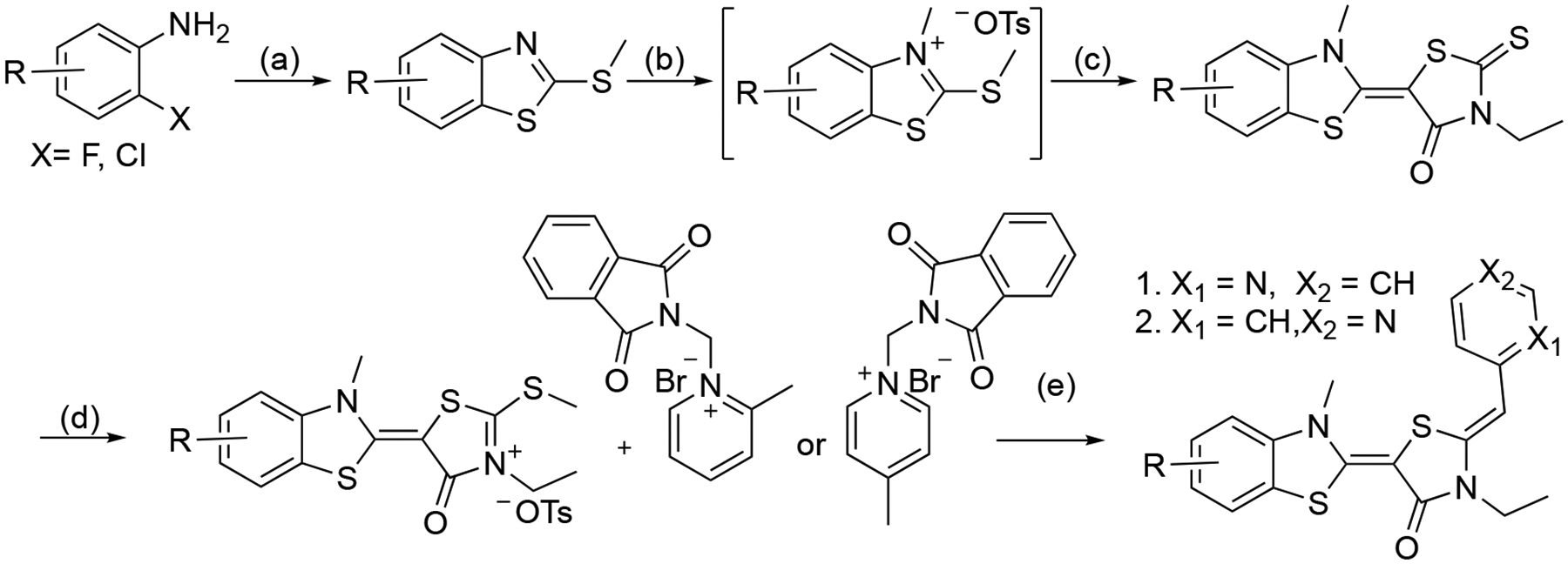
Reaction conditions: (a) 1) potassium ethylxanthate, DMF, 140 °C, 4 h; 2) MeI, NEt3, EtOH, 80 °C, 1 h; (b) p-TsOMe, anisole, 125 °C, 4 h; (c) 3-ethyl rhodanine, NEt3, MeCN, 25 °C, 4 h; (d) p-TsOMe, DMF, 135 °C, 3 h; (e) 1) NEt3, MeCN, 70 °C, 3 h; 2) DCM/MeOH, aq. NH3, r.t., 1 h.
To probe which benzothiazole position might be most important for microsome stability, we first measured the amount of unmodified compound remaining after incubation with mouse liver microsomes for 30 minutes at 37 °C, using dextromethorphan as a positive control28. First, we confirmed that YM-08 is rapidly metabolized in this experiment, such that its levels were below the limit of detection at 30 minutes (Table 1). Then, we found that introduction of fluorine at any of the 3-, 4-, 5- or 6-positions on YM-08 (compounds 1-4) modestly increased this stability (~5 to 20% remaining after 30 min). The analogs containing chlorine at the corresponding positions (compounds 5-8) were generally more stable. For example, the 5 and 6 were approximately 2- to 3-fold more stable than 1 and 2 (37 to 38%). In addition, across this series, the compounds with substituents at the 3- and 4-positions were generally better than those with halogens at the 5- and 6-positions.
Table 1:
Chemical structures and liver microsome stabilities of YM-08 analogs
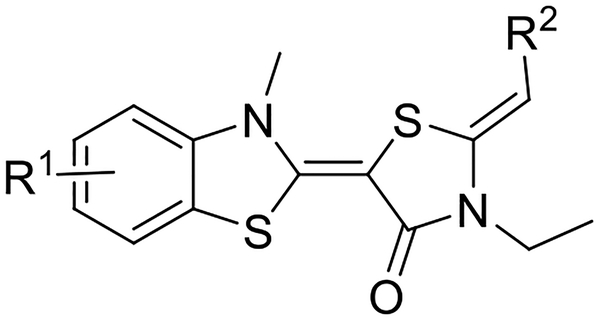
| |||
|---|---|---|---|
| Compound | R1 | R2 | Percentage at T30min |
| YM-08 | H | 2-pyridinyl | - |
| 1 | 3-F | 2-pyridinyl | 12 |
| 2 | 4-F | 2-pyridinyl | 19 |
| 3 | 5-F | 2-pyridinyl | 5 |
| 4 | 6-F | 2-pyridinyl | 12 |
| 5 | 3-Cl | 2-pyridinyl | 38 |
| 6 | 4-Cl | 2-pyridinyl | 37 |
| 7 | 5-Cl | 2-pyridinyl | 13 |
| 8 | 6-Cl | 2-pyridinyl | 17 |
| 9 | H | 4-pyridinyl | 6 |
| 10 | 3-F | 4-pyridinyl | 33 |
| 11 | 4-F | 4-pyridinyl | 23 |
| 12 | 5-F | 4-pyridinyl | 13 |
| 13 | 6-F | 4-pyridinyl | 30 |
| 14 | 3-Cl | 4-pyridinyl | n/t |
| 15 (JG-23) | 4-Cl | 4-pyridinyl | 52 |
| 16 | 5-Cl | 4-pyridinyl | 39 |
| 17 | 6-Cl | 4-pyridinyl | n/t |
Next, we examined the effects of switching the pyridine nitrogen from the ortho (YM-08) to para position (compound 9–17). In early experiments, we found that compound 9 was modestly more stable than YM-08 (Table 1), so it seemed worth exploring this series in more detail. Introduction of a fluorine at any of the four positions (compounds 10-13) improved microsome stability (~13 to 33% remaining). As in the previous series, replacement of fluorine with chlorine further improved metabolic stability. Specifically, 52% and 39% of compounds 15 (JG-23) and 16 were remaining, respectively. Compounds 14 and 17 had poor solubility and were thus excluded from further study (n/t; Table 1). Based on these results, we chose the top 4 compounds and measured their T1/2 in mouse liver microsome assays (Figure 2). The calculated T1/2 values from these experiments correlated with the single time point measurements: the half-lives of compounds 5, 6 and 16 are around 20 minutes, and JG-23 (15) has the longest T1/2 value (36 minutes).
Figure 2.
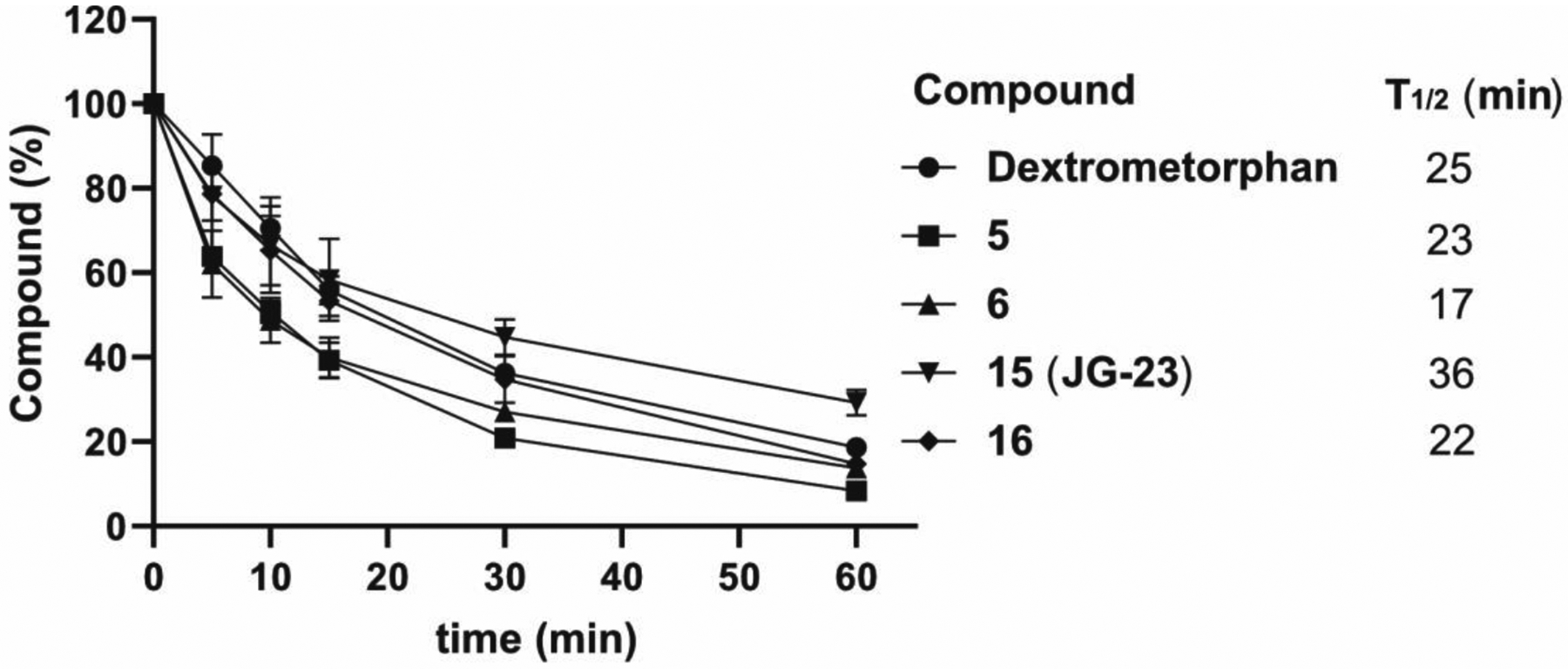
Microsomal stability of selected compounds. Dextrometorphan is used as positive control.
Finally, we tested the ability of JG-23 to decrease total tau (t-tau) levels in the HeLaC3 cell model. Cells were treated with JG-23 for 24 h at the indicated concentrations, and the t-tau levels were analyzed by Western blot. We found that JG-23 significantly decreased t-tau levels at concentrations above 10 μM (Figure 3a). Also, JG-23 did not activate cellular stress, as determined by the constant levels of Hsp72 and Hsp90 in the treated cells. Next, we asked whether JG-23 might also reduce t-tau levels in SH-SY5Y cells. This experiment was important because the HeLaC3 model over-expresses 0N4R tau, while it is expressed at physiological levels in the SH-SY5Y cells. Satisfyingly, we found that JG-23 and similar effects on t-tau and Hsp72 and Hsp90 in this model (Figure 3b), reducing t-tau by ~ 80%.
Figure 3.
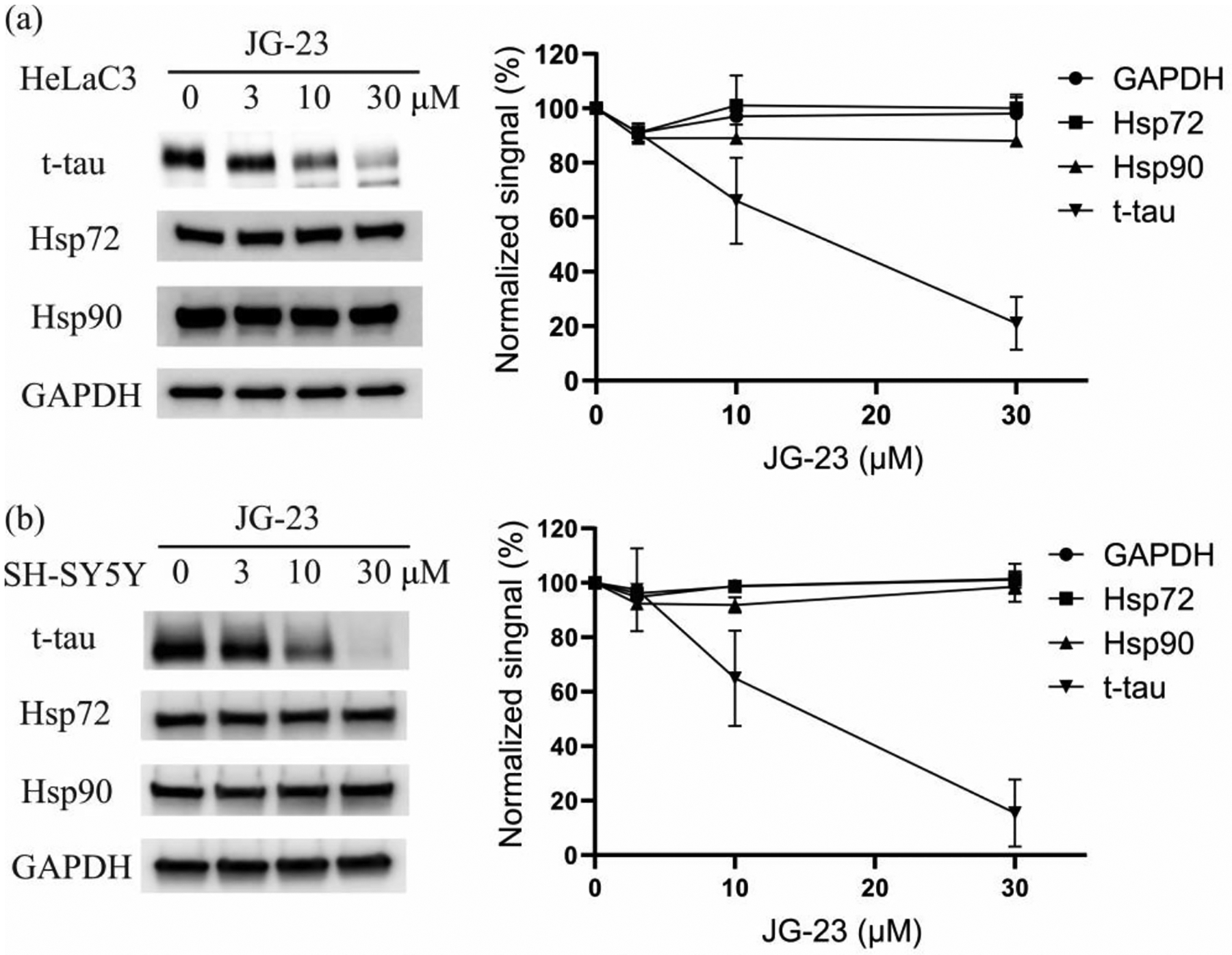
Compound JG-23 led to degradation of t-Tau without eliciting heat shock response in HeLaC3 (a) and SH-SY5Y (b) cells. HeLaC3 or SH-SY5Y cells were treated with JG-23 for 24 h at indicated concentration, lysed and western blots performed. Results are representative of at least two independent experiments.
3. Conclusions
Allosteric inhibitors of Hsp70, which favor a pro-degradation conformation of the chaperone, are potentially promising chemical probes for understanding tau homeostasis. However, the poor metabolic stability of earlier molecules, such as YM-08, had limited such studies. Here, guided by knowledge that P450-mediated metabolism occurs in the benzothiazole ring, we focused on introducing halogens at the 3-, 4-, 5- and 6-positions. Two series were designed and synthesized, revealing that a 4-chloro modified analog, JG-23, is 12-fold more stable than YM-08. One of the interesting aspects of these findings was that shifting the heteroatom in the distal pyridine generally improves stability, despite the fact that metabolism does not occur in this ring. It seems possible that the pyridine impacts either P450 binding or oxidation activity, perhaps mediated by electronic effects through the conjugated pi system. Importantly, these modifications did not seem to target binding, as compound JG-23 retained the ability to promote t-tau degradation in two cellular models. Future work will focus on improving potency and brain exposure, with the goal of creating a next-generation Hsp70 probe for studying tau turnover in the brain.
Supplementary Material
Acknowledgements
The authors thank Chad Dickey for providing the HeLaC3 cells. This work was supported by grants from the NIH (NS059690) and DoD (PC180716) to J.E.G.
References
- 1.Miyata Y, Koren J, Kiray J, Dickey CA, Gestwicki JE. Molecular chaperones and regulation of tau quality control: strategies for drug discovery in tauopathies. Future medicinal chemistry. 2011;3(12): 1523–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Narayanan RL, Durr UH, Bibow S, Biernat J, Mandelkow E, Zweckstetter M. Automatic assignment of the intrinsically disordered protein Tau with 441-residues. J Am Chem Soc. 2010;132(34): 11906–11907. [DOI] [PubMed] [Google Scholar]
- 3.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282(5395): 1914–1917. [DOI] [PubMed] [Google Scholar]
- 4.Coppola G, Chinnathambi S, Lee JJ, et al. Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Hum Mol Genet. 2012;21(15): 3500–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends in molecular medicine. 2009;15(3): 112–119. [DOI] [PubMed] [Google Scholar]
- 6.Morris M, Knudsen GM, Maeda S, et al. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nature neuroscience. 2015;18(8): 1183–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cook C, Carlomagno Y, Gendron TF, et al. Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Human Molecular Genetics. 2014;23(1): 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Min SW, Chen X, Tracy TE, et al. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nature medicine. 2015;21(10): 1154–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feinstein SC, Wilson L. Inability of tau to properly regulate neuronal microtubule dynamics: a loss-of-function mechanism by which tau might mediate neuronal cell death. Biochimica et biophysica acta. 2005;1739(2–3): 268–279. [DOI] [PubMed] [Google Scholar]
- 10.Brunden KR, Trojanowski JQ, Lee VM. Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nature reviews Drug discovery. 2009;8(10): 783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Young ZT, Mok SA, Gestwicki JE. Therapeutic Strategies for Restoring Tau Homeostasis. Csh Perspect Med. 2018;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makani V, Zhang B, Han H, et al. Evaluation of the brain-penetrant microtubule-stabilizing agent, dictyostatin, in the PS19 tau transgenic mouse model of tauopathy. Acta neuropathologica communications. 2016;4(1): 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melchior B, Mittapalli GK, Lai C, et al. Tau pathology reduction with SM07883, a novel, potent, and selective oral DYRK1A inhibitor: A potential therapeutic for Alzheimer’s disease. Aging cell. 2019;18(5): e13000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu Y, Ansar S, Michaelis ML, Blagg BS. Neuroprotective activity and evaluation of Hsp90 inhibitors in an immortalized neuronal cell line. Bioorganic & medicinal chemistry. 2009;17(4): 1709–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao H, Michaelis ML, Blagg BS. Hsp90 modulation for the treatment of Alzheimer’s disease. Advances in pharmacology. 2012;64: 1–25. [DOI] [PubMed] [Google Scholar]
- 16.Miyata Y, Li XK, Lee HF, et al. Synthesis and Initial Evaluation of YM-08, a Blood-Brain Barrier Permeable Derivative of the Heat Shock Protein 70 (Hsp70) Inhibitor MKT-077, Which Reduces Tau Levels. ACS chemical neuroscience. 2013;4(6): 930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jinwal UK, O’Leary JC, Borysov SI, et al. Hsc70 Rapidly Engages Tau after Microtubule Destabilization. Journal of Biological Chemistry. 2010;285(22): 16798–16805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimura H, Schwartz D, Gygi SP, Kosik KS. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. The Journal of biological chemistry. 2004;279(6): 4869–4876. [DOI] [PubMed] [Google Scholar]
- 19.Fontaine SN, Martin MD, Akoury E, et al. The active Hsc70/tau complex can be exploited to enhance tau turnover without damaging microtubule dynamics. Human Molecular Genetics. 2015;24(14): 3971–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Congdon EE, Wu JW, Myeku N, et al. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy. 2012;8(4): 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rousaki A, Miyata Y, Jinwal UK, Dickey CA, Gestwicki JE, Zuiderweg ER. Allosteric drugs: the interaction of antitumor compound MKT-077 with human Hsp70 chaperones. J Mol Biol. 2011;411(3): 614–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans CG, Jinwal UK, Makley LN, Dickey CA, Gestwicki JE. Identification of dihydropyridines that reduce cellular tau levels. Chemical communications. 2011;47(1): 529–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jinwal UK, Miyata Y, Koren J 3rd, et al. Chemical manipulation of hsp70 ATPase activity regulates tau stability. J Neurosci. 2009;29(39): 12079–12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abisambra J, Jinwal UK, Miyata Y, et al. Allosteric heat shock protein 70 inhibitors rapidly rescue synaptic plasticity deficits by reducing aberrant tau. Biol Psychiatry. 2013;74(5): 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Young ZT, Rauch JN, Assimon VA, et al. Stabilizing the Hsp70-Tau Complex Promotes Turnover in Models of Tauopathy. Cell Chem Biol. 2016;23(8): 992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shao H, Li XK, Moses MA, et al. Exploration of Benzothiazole Rhodacyanines as Allosteric Inhibitors of Protein-Protein Interactions with Heat Shock Protein 70 (Hsp70). J Med Chem. 2018;61(14): 6163–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shao H, Gestwicki JE. Neutral analogs of the heat shock protein 70 (Hsp70) inhibitor, JG-98. Bioorganic & medicinal chemistry letters. 2020;30(5): 126954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.C S, S I, Y Y, et al. Species Differences in the Pharmacokinetic Parameters of Cytochrome P450 Probe Substrates between Experimental Animals, such as Mice, Rats, Dogs, Monkeys, and Microminipigs, and Humans. Journal of drug metabolism and toxicology. 2014;5(6). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.