

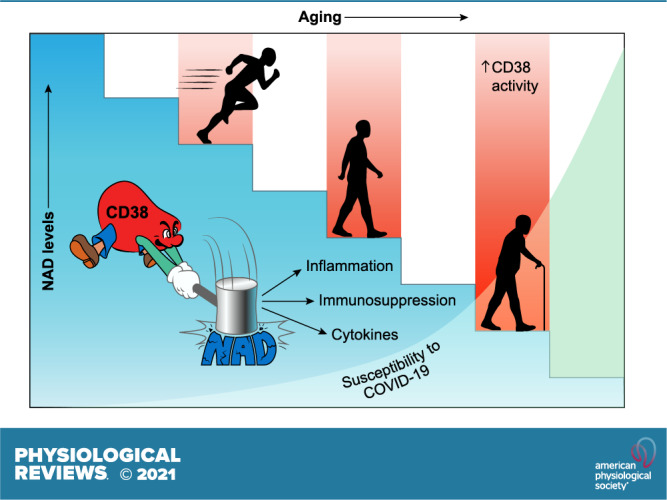
Keywords: CD38, COVID-19, inflammation, NAD, nicotinamide adenine dinucleotide
Abstract
During the COVID-19 pandemic, efforts have been made worldwide to develop effective therapies to address the devastating immune-mediated effects of SARS-CoV-2. With the exception of monoclonal antibody-mediated therapeutics and preventive approaches such as mass immunization, most experimental or repurposed drugs have failed in large randomized clinical trials (https://www.who.int/publications/i/item/therapeutics-and-covid-19-living-guideline). The worldwide spread of SARS-CoV-2 virus revealed specific susceptibilities to the virus among the elderly and individuals with age-related syndromes. These populations were more likely to experience a hyperimmune response characterized by a treatment-resistant acute lung pathology accompanied by multiple organ failure. These observations underscore the interplay between the virus, the biology of aging, and outcomes observed in the most severe cases of SARS-CoV-2 infection. The ectoenzyme CD38 has been implicated in the process of “inflammaging” in aged tissues. In a current publication, Horenstein et al. present evidence to support the hypothesis that CD38 plays a central role in altered immunometabolism resulting from COVID-19 infection. The authors discuss a critical but underappreciated trifecta of CD38-mediated NAD+ metabolism, aging, and COVID-19 immune response and speculate that the CD38/NAD+ axis is a promising therapeutic target for this disease.
CLINICAL HIGHLIGHTS
Cellular metabolic dysfunction is an important part of viral infections, including the COVID-19 pathogenesis. Understanding the underlying causes of this metabolic dysfunction could help with the development of effective complementary therapies for acute and postviral syndromes. The enzyme CD38, which is activated during inflammatory processes, may present a target in such conditions.
CD38 is a multifaceted ectoenzyme commonly recognized as both a marker of activated immune cells (1) and a nucleotidase linking the immune system and NAD+ metabolism through Ca2+ second messengers, adenosinergic signaling, epigenetic regulation, and leukocyte migration (1). As a pathogen reaches the site of infection, CD38 may 1) direct Ca2+ signaling important for viral endocytosis (1); 2) facilitate pathogen-fighting oxidative bursts from macrophages; 3) regulate interferon-stimulated genes (ISGs); 4) modulate ectoenzymatic adenosinergic networks; and ultimately 5) orchestrate a deadly hyperinflammatory response or cytokine storm (FIGURE 1). In the context of COVID-19, what may result is the accumulation of immune cells in the lungs and, in the most fulminate cases of SARS-CoV-2, a likely CD38-mediated thrombosis. Of additional concern in individuals with severe SARS-CoV-2 is the onset of bacterial secondary infections. CD38 is necessary to mediate NAD+-dependent bacterial engulfment, cytoskeleton rearrangements in phagocytes, and adenosine diphosphate ribose (ADPR)-dependent signaling required for migration of immune cells to the site of infection (2, 3). Taken together, CD38 may have a substantial role in both potentiating a SARS-CoV-2 infection and responding to a secondary bacterial infection.
FIGURE 1.
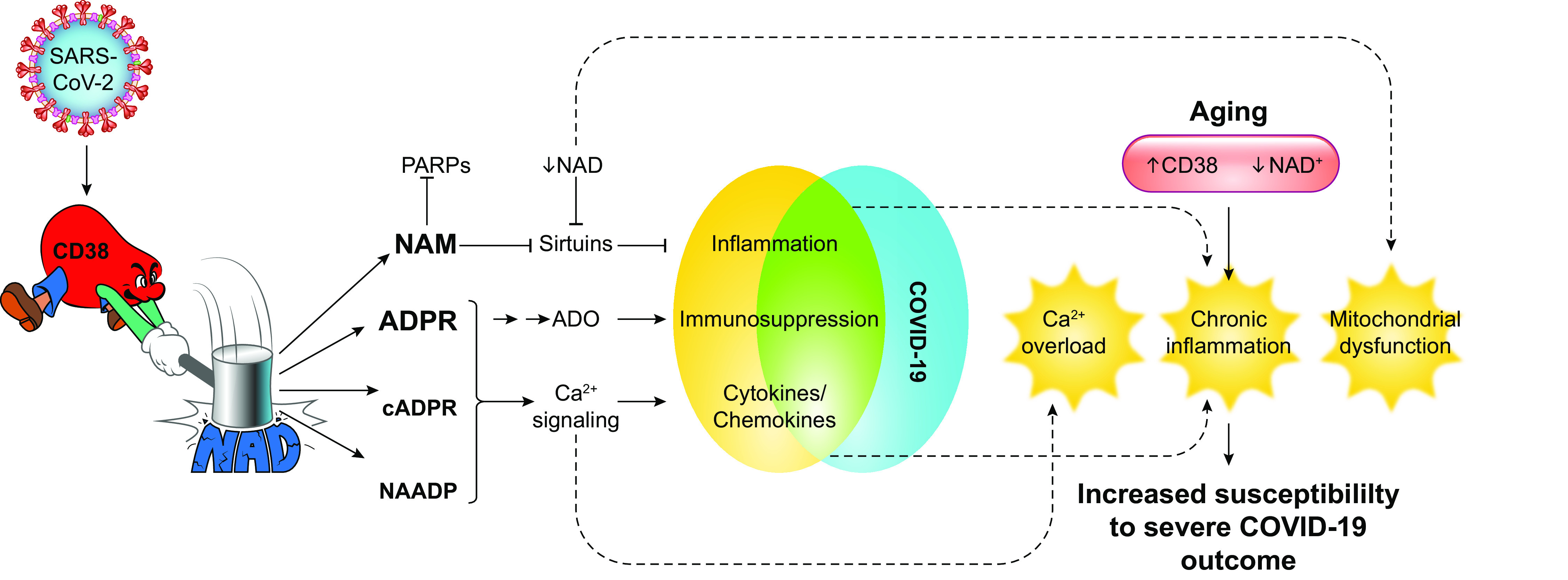
Possible interactions between CD38/NAD axis and COVID-19 pathophysiology. Horenstein et al. (1) propose that SARS-CoV-2 infection leads to increased CD38 expression. Metabolites generated by CD38 such as nicotinamide (NAM), adenosine diphosphate ribose (ADPR), cyclic adenosine diphosphate ribose (cADPR), and nicotinic acid adenine dinucleotide phosphate (NAADP) affect several pathways that ultimately may lead to an inflammatory profile typically identified in COVID-19 patients. Increased CD38 expression and decreased NAD+ levels are also common features of aging. The consequence of increased CD38 and reduced NAD+ levels may lead to Ca2+ overload, chronic inflammation, and impaired mitochondrial function and predispose elderly individuals to a severe COVID-19 infection. ADO, adenosine; PARP, poly(ADP-ribose) polymerase.
Horenstein et al. (1) highlight the relationship between CD38 activation and NAD+ decline as features of aging and discuss its potential relevance as modulator of COVID-19 disease in the elderly. Impaired or modified NAD+ metabolism has been described in mouse models of age-related disease and in certain human conditions (4). As reviewed by Wu and Zhang (5), CD38+ immune cells accumulate in tissues of old mice because of the presence of senescence-associated factors, causing a reduction in NAD+ levels in these tissues. In the context of COVID-19, it was observed that mice infected with a mouse coronavirus show increased accumulation of senescent cells and that senescent cells exposed to SARS-CoV-2 Spike protein-1 exhibit a hyperinflammatory response (6). Importantly, senolytic drugs have been shown to reduce senescence, inflammation, and mortality in old mice infected with coronavirus, suggesting that aged cells may have specific vulnerabilities to viral infections (1). As Horenstein et al. (1) speculate, hyperinflammation observed in COVID-19 infections may result in CD38 activation and NAD+ degradation, leading to a predisposition to severe COVID-19 outcomes that includes tissue fibrosis and tissue damage, particularly in the elderly (FIGURE 1).
Surprisingly, the CD38/NAD+ axis has not been thoroughly interrogated as a druggable target in the context of viral infection. To date, an increase in CD38+ immune cells is reported in patients infected with Epstein–Barr virus, in cytomegalovirus (CMV), and in human immunodeficiency virus (HIV) infections (7, 8). In SARS-CoV-2 disease, lymphocytopenia is a common feature; however, these cells, which are typically CD4+ and CD8+, are hyperactivated, as shown by the presence of a CD38/HLA-DR double-positive population (4). It is still unknown whether CD38 activity in these cells is sufficient to cause dysregulation of NAD metabolism (4). Presently, only a single in vitro study has investigated the relationship between CD38 and NAD metabolism in a respiratory syncytial virus (RSV) infection model (9). Taken together, there exists a gap in the evidence for dysregulated NAD+ metabolism due to increased CD38+ cells in viral infection.
Importantly, Horenstein et al. (1) highlight key therapeutic options targeting the CD38/NAD+ axis that could improve COVID-19 disease outcomes, including the use of inhibitors and monoclonal antibodies against CD38 to modulate NAD+ levels or administration of vitamin B3 precursors (e.g., nicotinamide, nicotinamide riboside, nicotinamide mononucleotide) to restore both NAD+ levels and normal immune function during viral infections (1). However, use of these approaches carries considerable caveats. Targeting CD38 to treat viral infections may predispose individuals to opportunistic bacterial infections, especially in primary viral respiratory tract infections. It has been observed that cancer patients treated with daratumumab, a monoclonal anti-CD38 antibody used to treat multiple myeloma, have a higher susceptibility not only to bacterial infections but also to viral infections (10). As the focus on CD38 as a therapeutic target in COVID-19 disease comes into clearer view, long-term or off-target effects of CD38 modulation will become clearer.
In conclusion, Horenstein et al. (1) provide a critical review of literature at the intersection of COVID-19 infection and NAD+ metabolism and hypothesize that the CD38/NAD+ axis plays a central role in COVID-19 pathology, particularly in the context of aging and senescence. The authors also highlight potential therapeutic approaches involving CD38/NAD+ modulation in COVID-19 disease (1). A major caveat is that direct evidence supporting the interaction of SARS-CoV-2 and CD38 is nascent and requires investigation at the bench and in the clinic. Beyond COVID-19 disease, modulators of the CD38/NAD+ axis have tremendous potential to address current and emerging viral illnesses.
GRANTS
The authors are supported by National Institutes of Health Grants CA-233790, AG-26094, and AG-58812.
DISCLOSURES
E.N.C. holds a patent on the use of CD38 inhibitors for metabolic diseases that is licensed by Elysium Health. E.N.C. is a consultant for TeneoBio, Calico, Mitobridge, and Cytokinetics. E.N.C. is on the advisory board of Eolo Pharma. E.N.C. owns stocks in TeneoBio. Research in E.N.C.’s laboratory has been conducted in compliance with Mayo Clinic Conflict of Interest policies. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
J.D.Z., S.K., and E.N.C. conceived and designed research; J.D.Z., S.K., and K.A.H. prepared figures; J.D.Z., S.K., K.A.H., and E.N.C. drafted manuscript; J.D.Z., S.K., K.A.H., and E.N.C. edited and revised manuscript; J.D.Z., S.K., K.A.H., and E.N.C. approved final version of manuscript.
REFERENCES
- 1.Horenstein AL, Faini AC, Malavasi F. CD38 in the age of COVID-19: a medical perspective. Physiol Rev 101: 1457–1486, 2021. [PMC doi: 10.1152/physrev.00046.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matalonga J, Glaria E, Bresque M, Escande C, Carbó JM, Kiefer K, Vicente R, León TE, Beceiro S, Pascual-García M, Serret J, Sanjurjo L, Morón-Ros S, Riera A, Paytubi S, Juarez A, Sotillo F, Lindbom L, Caelles C, Sarrias MR, Sancho J, Castrillo A, Chini EN, Valledor AF. The nuclear receptor LXR limits bacterial infection of host macrophages through a mechanism that impacts cellular NAD metabolism. Cell Rep 18: 1241–1255, 2017. doi: 10.1016/j.celrep.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 3.Partida-Sánchez S, Cockayne DA, Monard S, Jacobson EL, Oppenheimer N, Garvy B, Kusser K, Goodrich S, Howard M, Harmsen A, Randall TD, Lund FE. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat Med 7: 1209–1216, 2001. doi: 10.1038/nm1101-1209. [DOI] [PubMed] [Google Scholar]
- 4.Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, Liu S, Zhao P, Liu H, Zhu L, Tai Y, Bai C, Gao T, Song J, Xia P, Dong J, Zhao J, Wang FS. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med 8: 420–422, 2020. doi: 10.1016/S2213-2600(20)30076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu S, Zhang R. CD38-expressing macrophages drive age-related NAD+ decline. Nat Metab 2: 1186–1187, 2020. doi: 10.1038/s42255-020-00292-5. [DOI] [PubMed] [Google Scholar]
- 6.Camell CD, Yousefzadeh MJ, Zhu Y, Prata LG, Huggins MA, Pierson M, et al. Senolytics reduce coronavirus-related mortality in old mice. Science 373: eabe4832, 2021. doi: 10.1126/science.abe4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zidovec Lepej S, Vince A, Dakovic Rode O, Remenar A, Jeren T. Increased numbers of CD38 molecules on bright CD8+ T lymphocytes in infectious mononucleosis caused by Epstein-Barr virus infection. Clin Exp Immunol 133: 384–390, 2003. doi: 10.1046/j.1365-2249.2003.02219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Booiman T, Wit FW, Girigorie AF, Maurer I, De Francesco D, Sabin CA, Harskamp AM, Prins M, Franceschi C, Deeks SG, Winston A, Reiss P, Kootstra NA; Co-morBidity in Relation to Aids (COBRA) Collaboration. Terminal differentiation of T cells is strongly associated with CMV infection and increased in HIV-positive individuals on ART and lifestyle matched controls. PLoS One 12: e0183357, 2017. doi: 10.1371/journal.pone.0183357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schiavoni I, Scagnolari C, Horenstein AL, Leone P, Pierangeli A, Malavasi F, Ausiello CM, Fedele G. CD38 modulates respiratory syncytial virus-driven proinflammatory processes in human monocyte-derived dendritic cells. Immunology 154: 122–131, 2018. doi: 10.1111/imm.12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nahi H, Chrobok M, Gran C, Lund J, Gruber A, Gahrton G, Ljungman P, Wagner AK, Alici E. Infectious complications and NK cell depletion following daratumumab treatment of multiple myeloma. PLoS One 14: e0211927, 2019. doi: 10.1371/journal.pone.0211927. [DOI] [PMC free article] [PubMed] [Google Scholar]