

ABSTRACT
Explant culture is a more suitable method than enzyme digestion for the isolation of keloid fibroblasts (KFs), but it has a longer isolation period. In this study, we propose a long-term explant culture method. Unlike in the conventional explant culture method, we continued culturing explants to isolate KFs rather than discarding them during passage. We demonstrated that keloid explants could be cultured for more than 4 months to continuously yield enriched KFs, and the KFs from the repeatedly cultured explants had shorter isolation times. The biological behavior and fibrotic phenotypic characteristics of the KFs from the explants cultured long term were investigated, and no statistical differences were found compared with the KFs from the original explants. In conclusion, the long-term explant culture method was shown to be efficient for harvesting a large, homogeneous population of KFs. The high efficiency as well as ease of operation and sample saving make this method convenient for researchers working with KFs.
KEYWORDS: Explant culture, keloid fibroblast, primary cell culture, cell separation
Introduction
Keloids, benign fibro-proliferative cicatricial neoplasia with unknown physiopathogenesis, are characterized by local fibroblast proliferation and collagen overproduction [1,2]. Although multiple medical and surgical therapies have been used for the treatment of keloids, none of these treatments has been adequately evaluated in high-quality studies, and there is no universally accepted treatment approach. Therefore, further studies are needed to explicate the pathogenesis of keloids and identify the best treatment method.
Cell culture is the most basic, core biotechnology used in the study of keloids, especially in the absence of a keloid animal model [3]. It allows the identification of the mechanism of action of cellular regulators at gene expression and cell signaling levels [4]. Keloid fibroblasts (KFs) – similar to normal dermal fibroblasts in growth characteristics, cell volume, karyotype, and other aspects [5] – are hyperproliferative in keloids and are the major contributors to the abnormal proliferation of the keloid extracellular matrix (ECM) [6]; therefore, they are highly valued in research on the occurrence and development of keloids.
Currently, the primary KFs are isolated mainly through enzyme digestion and explant culture [7–10]. To isolate KFs by enzyme digestion, the specimens are dissected into pieces and digested with collagenase (with or without trypsin) in a constant-temperature shaker at 37°C for several hours. The resulting suspension is then filtered and centrifuged to obtain the KFs. Despite the short time required to obtain many single KFs, enzyme digestion is complicated, expensive, and often requires the use of specialized instruments, therefore limiting its use in the generation of large volumes of tissues, with the probability of cell contamination also increasing [11]. Histologically, keloids are rich in thickened, hyalinized bundles of collagen fibers [12], which places a heavy burden on collagenase during separation of cells by enzyme digestion. In addition, the enzyme digestion method also has the problem of reduced viability of the separated cells, due to the lysing activity [13]. It has been shown that mechanical cell separation produces less damage to DNA than collagenase separation [14–16]. Compared with enzyme digestion, the simple explant culture method seems to be more suitable for the isolation of keloid fibroblasts. Specimens are minced and attached to culture flasks containing medium, and the KFs are then isolated from the edges of the pieces. However, the time required to harvest KFs by this method is usually 7–14 days [17]. To some extent, this lengthens the duration of the experiment.
Previously, dental pulp stem cells have been isolated from human dental pulp explants sub-cultured seven times without displacement [18]. These cells have been shown to be homogeneous, express mesenchymal stem cell surface markers, and be capable of multidirectional differentiation. However, no studies on long-term explant culture for the isolation of KFs have been conducted. Therefore, in this study, we propose an innovative long-term keloid explant culture method, based on the hypothesis that keloid explants can be cultured for a long time to consistently separate homogeneous KFs. We aim to optimize the method for keloid fibroblast isolation and to provide a useful reference for researchers.
Materials and methods
Patients and keloid samples
Keloid samples grown for over a year without infection were collected from patients without any previous history of treatment. Keloid specimens were acquired through surgical excision, after informed consent was obtained. The procedures for processing human tissues and cells were approved by the Ethics Committee of Shandong University Provincial Hospital.
Isolation and culture of keloid fibroblasts
Harvesting of the first batch of KFs was carried out using the conventional explant culture method described previously by Tucci-Viegas et al. [17], with adaptations. Keloid tissues were placed in 50 mL conical tubes with phosphate-buffered saline (PBS, Basalmedia, Shanghai, China) containing penicillin (1000 U/mL, Basalmedia, Shanghai, China) and streptomycin (1000 μg/mL, Basalmedia, Shanghai, China) for 40 min, after the keloid tissues were washed with PBS several times. Then, the keloid tissues, after epidermal removal, were minced into 1.5 mm × 1.5 mm × 1.5 mm fragments. The dermal fragments – the original explants – were equally treated, seeded in 25 cm2 culture flasks, and incubated in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Grand Island, NY, USA) containing 20% fetal bovine serum (FBS, Gibco, Grand Island, NY, USA), penicillin (100 U/mL, Basalmedia, Shanghai, China), and streptomycin (100 μg/mL, Basalmedia, Shanghai, China), for a week. The medium was then replaced with fresh medium containing 10% FBS every 2 days, with the height of the medium not exceeding the thickness of the keloid explants.
Subculturing (passage) was performed when cellular confluence reached approximately 80%. The culture medium was aspirated, and the culture flasks containing the explants and the KFs were washed with PBS. After 0.25% trypsin (Gibco, Grand Island, NY, USA) was added, the flasks were kept in an incubator for approximately 2 min. Trypsin was neutralized with DMEM plus 10% FBS, and the cellular suspension was sucked into conical tubes. The culture flasks containing the explants were then washed with PBS several times until the KFs were almost invisible under the microscope. All the cellular suspensions were centrifuged, and KFs were seeded in 25 cm2 culture flasks containing DMEM with 10% FBS and 1% antibiotics. The explants were equally treated again and incubated in DMEM plus 20% FBS for 2 days, and the medium was replaced with fresh medium containing 10% FBS. The same procedure was repeated to culture the explants for a third time, a fourth time, and so on (Figure 1). KFs isolated from the first culture of the explants (KFs-E1), second culture of the explants (KFs-E2), tenth culture of the explants (KFs-E10), and twentieth culture of the explants (KFs-E20) were chosen as the sample groups for this study. KFs from the fourth passage (P4) were used in subsequent experiments.
Figure 1.
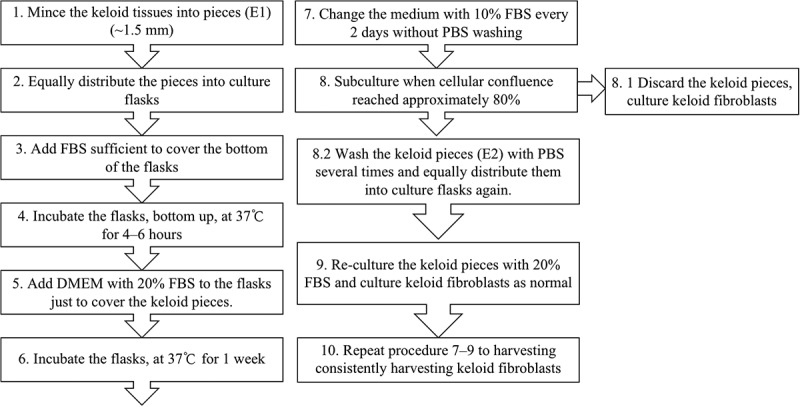
Flow chart showing general steps of traditional explant culture method (1–8.1) and long-term explant culture method (1–10).
Cell counting kit-8 assay
Cell proliferation was analyzed using the cell counting kit-8 assay (CCK-8, Dojindo Laboratories, Mashiki, Japan). Briefly, 4 × 103 cells from each sample were seeded into each well of a 96-well plate and starved in an incubator for 24 h. At days 1, 2, 3, and 4, the CCK-8 solution was added to the KFs (10 μL per well), and the cells were incubated at 37°C for 2 h. The absorbance was measured at 450 nm using a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA); the optical density value from each well represented the proliferation of KFs.
Cell cycle analysis
As previously described by Zhou et al. [7], 1 × 106 P4 KFs from each sample were fixed in 70% pre-cooled ethanol at 4°C overnight. Subsequent steps were carried out according to the instructions provided in the Cell Cycle Kit (7sea Biotech, Shanghai, China). Flow cytometric analysis was performed using a flow cytometer (Beckman Coulter, Fullerton, CA, USA) equipped with ModiFit LT v2.0.
Cell migration assay
The P4 KFs from each group were separately inoculated into 6-well plates. A 200 μL pipette tip was used to scratch the cell monolayer when the cells reached more than 90% confluence, and the medium was then replaced with serum-free medium for 24 h. Images were acquired at 0 and 24 h after scratching. The results were derived from measured areas in six randomly selected fields in a well, and ImageJ software (NIH, Washington, DC, USA) was used to perform the cell migration assay.
RNA isolation and real-time quantitative polymerase chain reaction
Each group of KFs was harvested to analyze the mRNA expression levels of genes associated with fibrosis by quantitative polymerase chain reaction (qPCR). A SteadyPure Universal RNA Extraction Kit II (ACCURATE BIOLOGY, Hunan, China) was used for total RNA extraction, according to the manufacturer’s instructions. Reverse transcription was performed to obtain the complementary DNA (cDNA), after which qPCR was performed with SYBR Green Pro Taq HS qPCR Kit (ACCURATE BIOLOGY, Hunan, China). Relative expression levels were evaluated by the comparative CT method (ΔΔCT) and normalized to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The human primer sequences were as follows: alpha smooth muscle actin (α-SMA): forward CATCATGCGTCTGGATCTGG, reverse GGACAATCTCACGCTCAGCA; collagen I (COL-1): forward GGCGGCCAGGGCTCCGACCC, reverse AATTCCTGGTCTGGGGCACC; collagen III (COL-3): forward TGGTGTTGGAGCCGCTGCCA, reverse CTCAGCACTAGAATCTGTCC; fibronectin (FN): forward GCCACTGGAGTCTTTACCACA, reverse CCTCGGTGTTGTAAGGTGGA; GAPDH: forward TCACCATCTTCCAGGAGCG, reverse CTGCTTCACCACCTTCTTGA.
Immunofluorescence assay
Briefly, each group of P4 KFs was seeded onto 6-well plates with sheet glass and incubated for 48 h in regular culture medium. Primary antibodies, for α-SMA (14,395-1-AP, 1:200, Servicebio), COL-1 (14,695-1-AP, 1:500, Proteintech), COL-3 (22,734-1-AP, 1:500, Proteintech), and FN (A12932, 1:200, Abclonal) and the secondary antibody (A23220, 1:200, Abbkine) were diluted according to the manufacturer’s instructions. DAPI (C0065, Solarbio) was used for nuclear counterstaining. Images for α-SMA–positive, COL-1–positive, COL-3–positive, or FN–positive (green) and DAPI nuclear-stained (blue) cells were obtained under a fluorescence microscope (Olympus).
Western blot analysis
As previously described by Liang et al. [19], each flask of cells was lysed in 500 mL of lysis buffer and subjected to Western blot analysis. Approximately 20 mg of total protein was separated by SDS-PAGE, transferred to a PVDF membrane, blocked by 5% nonfat milk, and incubated overnight in diluted primary antibodies according to the manufacturer’s instructions, followed by incubation with horseradish peroxidase-conjugated secondary antibodies for 1 h. The target proteins were visualized using an electrochemiluminescence reagent (Millipore Sigma). Images were captured using an Amersham Imager 680 (GE, Boston, MA, USA). Densitometric analysis of the bands was performed using ImageJ software (NIH, Washington, DC, USA).
Hematoxylin-eosin staining
To detect the changes in histology and pathology, hematoxylin-eosin (HE) staining was performed on fresh keloid tissues extracted by surgical excision, and the keloid explants were cultured for 4 months. Paraffin sections were prepared first; the process included tissue fixation, dehydration, wax leaching, embedding, and sectioning. Then, the tissues were stained using the HE dye solution set (Servicebio, Wuhan, China), as per the manufacturer’s instructions, after dewaxing. Finally, all tissue sections were dehydrated, sealed, and observed under a microscope.
Statistics and data analysis
Each experiment was repeated more than thrice. GraphPad Prism 8 software was used for all statistical analyses. Differences among groups were analyzed using one-way analysis of variance. All data were presented as the mean ± standard deviation (SD), and statistical significance was set at P < 0.05.
Results
In this study, we tested a long-term explant culture method to isolate primary KFs, aiming to provide a useful reference for researchers requiring an appropriate method that optimizes KF isolation. After this unconventional method was shown to consistently isolate KFs in a certain time period, the biological behavior and fibrotic phenotypic characteristics of the KFs from the long-term cultured explants were compared with those of the original explants to determine whether these cells were homogeneous with those obtained using the conventional explant culture method. In addition, histological analysis was performed on the explants cultured for four months, using HE staining.
Explant culture of keloid tissues
We observed that many small, round, and bright cells could be seen around the original keloid explants 48 h after adherence. Approximately 4 days later, a few spindle-shaped cells, considered KFs, began to migrate from the edges of the explants, after which they increased rapidly in number and became arranged radially around each explant. At approximately 10–14 days, the fusiform KFs attained more than 80% confluence. Compared with the original explants, the repeatedly attached explants were observed to produce KFs isolation more quickly, and satisfactory confluence of KFs occurred on the fifth day (Figure 2a). We also subsequently discovered that the explants could be cultured for more than 4 months despite an increase in staleness. All the cells were uniform in shape, size, and arrangement, although the number of cells migrating from the explants in the same amount of time decreased as time progressed (Figure 2b). In addition, we observed that KFs growing through adherence could not tolerate PBS. When PBS was added to the flasks, the KFs shrank and became round within a few minutes; they were then suspended in the PBS solution, as in the digestion of cells by trypsin. Therefore, in the proposed long-term explant method, we advocate repeated washing of explants with PBS after the first trypsin digestion of the cells around the explants, which can also induce shedding of the residual fibroblasts to a large extent.
Figure 2.
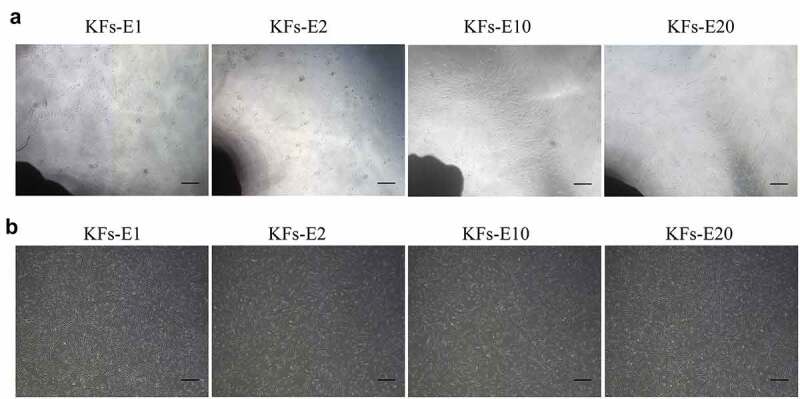
Homogeneous KFs isolated from long-term cultured keloid explants. a Representative images of keloid explants cultured for 12, 5, 5, and 5 days after the 1st, 2nd, 10th, and 20th cycle of explant culture, respectively (× 40, bar = 200 μm). b Representative images of P1 KFs derived from E1, E2, E10 and E20, respectively (× 40, bar = 200 μm).
Analysis of cell proliferation and cell cycle
As shown in Figure 3a, the CCK-8 assay showed no significant differences in growth characteristics among KFs-E1, KFs-E2, KFs-E10, and KFs-E20 during the four-day time period (P > 0.05 vs KFs-E1). That is, the KFs isolated from the long-term cultured explants of keloid did not exhibit an increase or decrease in their proliferative ability.
Figure 3.
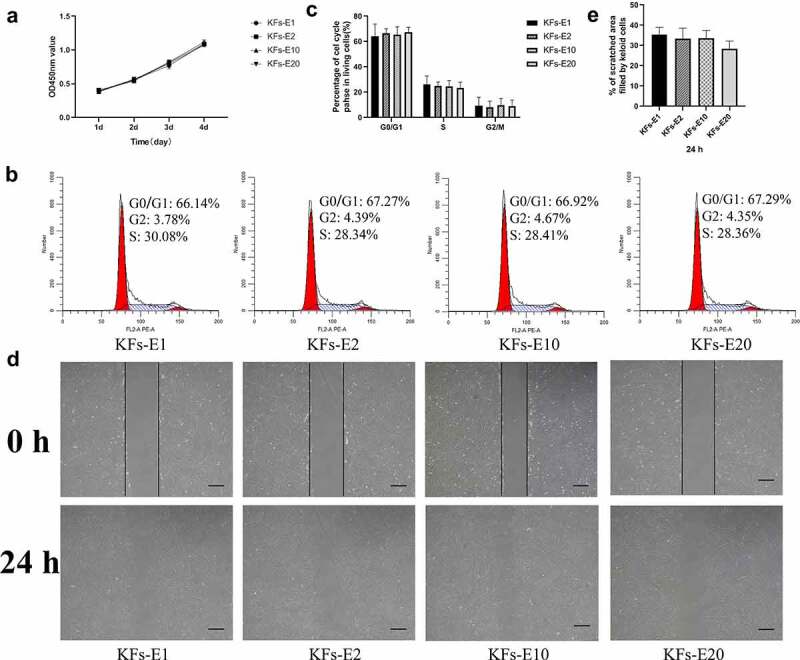
Comparison of biological behavior of KFs-E2, KFs-E10, and KFs-E20 with KFs-E1. a The CCK-8 assay was used to analyze the proliferation of KFs-E1, KFs-E2, KFs-E10, and KFs-E20 at days 1, 2, 3, and 4 (P > 0.05 vs KFs-E1). b Representative cell cycle data plotted for each group. c Flow cytometry analysis of cell cycle for each group (P > 0.5 vs KFs-E1). d Representative images obtained at 0 and 24 h after scratching (× 40, bar = 200 μm). e Semiquantitative analysis of scratch assay results (P > 0.1 vs KFs-E1).
Flow cytometry analysis revealed no statistical differences among the four groups (P > 0.5 vs KFs-E1, Figure 3b&C). However, we observed that KFs isolated from explants of different keloids displayed different cell cycles, which may be related to the explants being derived from different keloid regions [17].
Analysis of cell migratory capacity
After 24 h of culturing, the scratch assay revealed similar migratory capacities for KFs-E2, KFs-E10, and KFs-E20 compared with KFs-E1, when observed under a microscope (Figure 3d). At both time points, semiquantitative analysis by measuring areas further confirmed that there were no statistical differences between the KFs isolated from the long-term cultured explants and KFs-E1 (P > 0.1 vs KFs-E1, Figure 3e). Taken together, these experiments confirmed that there was no significant difference in the biological behavior of KFs isolated from long-term culture explants.
Analysis of fibrotic factors
As presented in Figure 4a, at the transcriptional level, the gene expression of COL-1, COL-3, FN, and α-SMA showed no statistical differences in KFs-E2, KFs-E10, and KFs-E20 compared with KFs-E1, although differences were sometimes noticeable among the four groups when a only single experiment was performed (P > 0.1 vs KFs-E1).
Figure 4.
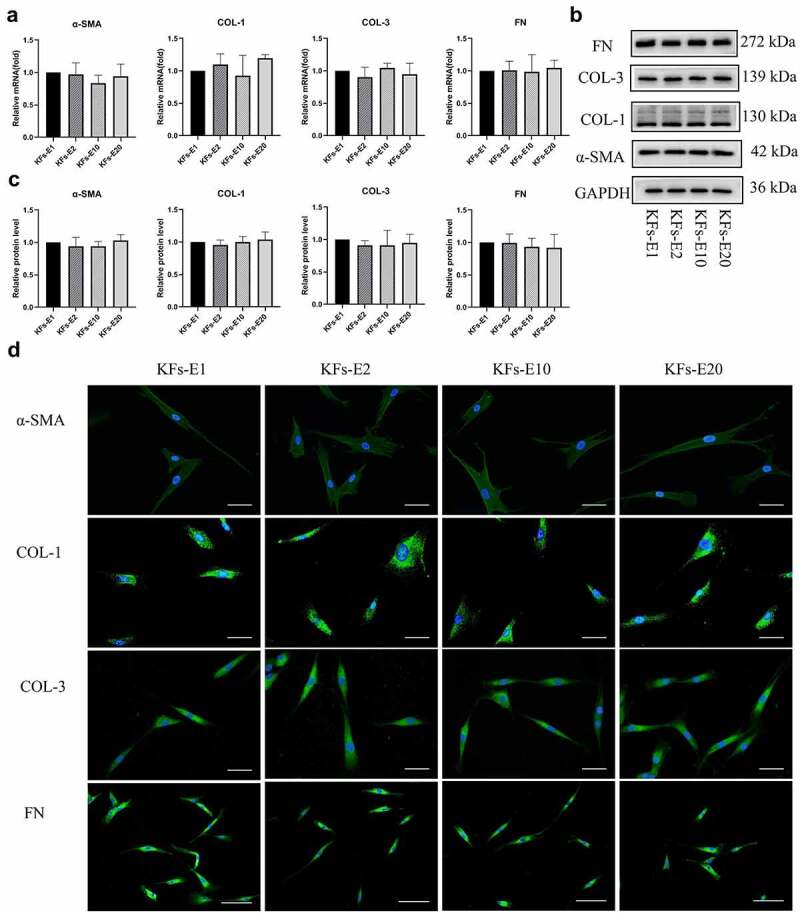
Comparison of ECM related gene expression of KFs-E2, KFs-E10, and KFs-E20 with that of KFs-E1. a qPCR analysis comparing α-SMA, COL-1, COL-3, and FN expression in KFs-E2, KFs-E10, and KFs-E20 with that in KFs-E1 (P > 0.1 vs KFs-E1). b Western blot analysis of α-SMA, COL-1, COL-3, and FN production. c Semi-quantification of the Western blot results (P > 0.5 vs KFs-E1). d Fluorescence microscopy image of intracellular α-SMA, COL-1, COL-3, and FN expression (α-SMA, COL-1 and COL-3, × 400, bar = 20 μm. FN, × 200, bar = 50 μm).
Consistent with the PCR results, Western blotting also showed that the production of COL-1, COL-3, FN, and α-SMA proteins in the four groups was without significant differences, as shown in Figure 4b, and semiquantitative analysis produced the same conclusion (P > 0.5 vs KFs-E1, Figure 4c). Similar production of COL-1, COL-3, FN, and α-SMA proteins was also confirmed by immunofluorescence staining of KFs-E1, KFs-E2, KFs-E10, and KFs-E20 (Figure 4d). In conclusion, we confirmed at different levels that KFs isolated from long-term cultured explants did not exhibit significant changes in their fibrotic phenotypic characteristics.
Histological analysis
HE staining revealed histopathological changes in long-term cultured explants. Compared with fresh keloid tissues, we found that the explants cultured for 4 months had lost the normal histological structure of keloid tissues, manifesting as more loosely arranged bundles of collagen fiber. No obvious vascular structure was observed in explants cultured for 4 months. The long spindle-shaped cells were no longer attached to the ECM and appeared to be activated (Figure 5).
Figure 5.
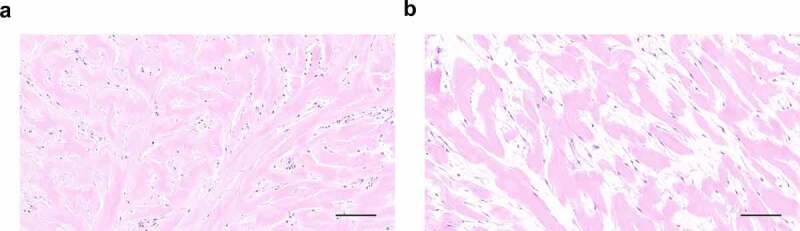
Histological analysis of original explants (a) with explants cultured for 4 months (b) via HE staining.
Discussion
Explant culture, an age-old technique, has been successfully used to isolate primary cells from different tissues [10,20,21]. Compared with enzyme digestion, the explant culture method seems to be more suitable for the isolation of KFs because it is relatively simpler, more economical, and less harmful to cells and their DNA [11–16]. However, because the absence of proteolytic enzymes impedes the dissociation of cells attached to the ECM [22], satisfactory proliferation of KFs requires approximately 7–14 days when using the explant culture method [17]; this is similar to the 10–14 days observed in our study. This factor may have contributed to the longer experimental period.
In the long-term explant culture method, we found that KFs from repeatedly cultured explants had shorter isolation times. The mechanisms behind this result were analyzed as follows: on the one hand, the time required for cell isolation is somewhat dependent on the adhesion period required in the explants, with original explants often requiring longer adhesion periods. Culture flasks containing explants were required to be stationary in the incubator for a week to provide a good adhesion environment. When the explants were re-cultured, stable adherence was observed 48 h later; this may be because the cells surrounding the explants were not completely separated. When the explants were cultured again, the cells surrounding the explants continued to grow while maintaining adherence, thus promoting the attachment of explants. Faster attachment of the explants, in turn, caused more fibroblasts to be isolated from the explants more quickly.
On the other hand, it seems that in the explant method, cells start to migrate out of the tissue in a process such as wound healing and gradually exit the tissue while maintaining their interaction with the tissue explant [22]. When the cells isolated from the original explant were digested, the explant and the fibroblasts in it were still activated for the wound response process. In addition, the fibroblasts in the re-cultured explants were no longer tightly attached to the ECM, which also contributed to the shorter separation time for re-cultured explants.
It has been shown that some keloid-derived fibroblast characteristics are lost when cells are removed from the in vivo microenvironment of the keloid lesion and passaged in vitro, which may be attributed to changes in the extracellular environment, such as those in the cytokine milieu [23,24]. In addition, some studies on isolating mesenchymal stem cells (MSCs) suggested that co-culture with the originating tissue rather than isolated monoculture supports an appropriate microenvironment that provides matrix compounds and survival factors for maintaining important properties of the primary cells [22,25]. The fact that continuous explant culture can establish an in vitro microenvironment that augments MSC acquisition while maintaining stemness and carrying identical properties over time was evidenced by some detailed research findings [25]. Consistent with the results of these studies, no statistical differences in the proliferation and migration ability of the KFs isolated from the long-term cultured keloid explants and no loss of fibrosis phenotype were detected in this study. This confirmed the ability of long-term cultured keloid explants – despite increasing staleness – to allow KFs to retain their biological characteristics and fibrotic phenotype, suggesting that each batch of KFs obtained from long-term culture explants should be considered as KFs in passage 1. For example, we should treat KFs isolated from E20 as KFs in passage 1 rather than as KFs in passage 20 isolated from E1. However, there are limitations to the present study. It is not clear how long the culture of keloid explants can be continued for KF isolation, or how this period of time is related to the size of the original explants. Further studies are needed to clarify these factors, which were not considered in this study.
Conclusion
This study suggested that long-term culture of keloid explant could consistently isolate homogeneous KFs, and the KFs from the repeatedly cultured explants had shorter isolation times. Therefore, we propose this unconventional explant culture process as an efficient, reproductive, and inexpensive technique with potential research utility. This method may be preferred over enzyme digestion or conventional explant culture methods for harvesting human keloid fibroblasts for scientific research.
Acknowledgements
We appreciate the technical support from the Central Laboratory of Shandong Provincial Hospital. We would also like to thank other research groups in the Central Laboratory for their technical guidance.
Funding Statement
This work was supported by the National Natural Science Foundation of China under Grant [number 81873938].
Research highlights
Keloid explants can be cultured long term to continuously isolate keloid fibroblasts
Keloid fibroblasts from repeatedly cultured explants have shorter isolation times
Keloid fibroblasts isolated from explants cultured long term are homogeneous
The long-term explant culture method can reduce the duration of the experiment
Disclosure statement
No potential conflict of interest was reported by the author(s).
Author contributions
Conception and design: J. Li, R. Huo.
Acquisition of specimens: Y. Zou, S. Guo, Z. Huang.
Performing experiments: J. Li, Y. Zou, S. Guo, Z. Huang.
Analysis and interpretation of data: R. Huo, S. Wang, J. Li.
Writing, and/or revision of the manuscript: R. Huo, S. Wang, and J. Li.
References
- [1].Berman B, Maderal A, Raphael B.. Keloids and hypertrophic scars: pathophysiology, classification, and treatment. Dermatol Surg. 2017;43(Suppl 1):S3–S18. [DOI] [PubMed] [Google Scholar]
- [2].Teofoli P, Barduagni S, Ribuffo M, et al. Expression of Bcl-2, p53, c-jun and c-fos protooncogenes in keloids and hypertrophic scars. J Dermatol Sci. 1999;22(1):31–37. [DOI] [PubMed] [Google Scholar]
- [3].Duong HS, Zhang Q, Kobi A, et al. Assessment of morphological and immunohistological alterations in long-term keloid skin explants. Cells Tissues Organs. 2005;181(2):89–102. [DOI] [PubMed] [Google Scholar]
- [4].Grimm S. The art and design of genetic screens: mammalian culture cells. Nat Rev Genet. 2004;5(3):179–189. [DOI] [PubMed] [Google Scholar]
- [5].Murray JC, Pollack SV, Pinnell SR. Keloids: a review. J Am Acad Dermatol. 1981;4(4):461–470. [DOI] [PubMed] [Google Scholar]
- [6].Xue M, Jackson CJ. Extracellular matrix reorganization during wound healing and its impact on abnormal scarring. Adv Wound Care (New Rochelle). 2015;4(3):119–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhou BY, Wang WB, Wu XL, et al. Nintedanib inhibits keloid fibroblast functions by blocking the phosphorylation of multiple kinases and enhancing receptor internalization. Acta Pharmacol Sin. 2020;41(9):1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang XM, Liu XM, Wang Y, et al. Activating transcription factor 3 (ATF3) regulates cell growth, apoptosis, invasion and collagen synthesis in keloid fibroblast through transforming growth factor beta (TGF-beta)/SMAD signaling pathway. Bioengineered. 2021;12(1):117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang Q, Wang P, Qin Z, et al. Altered glucose metabolism and cell function in keloid fibroblasts under hypoxia. Redox Biol. 2021;38:101815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sato C, Yamamoto Y, Funayama E, et al. Conditioned medium obtained from amnion-derived mesenchymal stem cell culture prevents activation of keloid fibroblasts. Plast Reconstr Surg. 2018;141(2):390–398. [DOI] [PubMed] [Google Scholar]
- [11].Baptista LS, Do Amaral RJ, Carias RB, et al. An alternative method for the isolation of mesenchymal stromal cells derived from lipoaspirate samples. Cytotherapy. 2009;11(6):706–715. [DOI] [PubMed] [Google Scholar]
- [12].Jumper N, Paus R, Bayat A. Functional histopathology of keloid disease. Histol Histopathol. 2015;30(9):1033–1057. [DOI] [PubMed] [Google Scholar]
- [13].Ishige I, Nagamura-Inoue T, Honda MJ, et al. Comparison of mesenchymal stem cells derived from arterial, venous, and Wharton’s jelly explants of human umbilical cord. Int J Hematol. 2009;90(2):261–269. [DOI] [PubMed] [Google Scholar]
- [14].Kosmehl T, Hallare AV, Reifferscheid G, et al. A novel contact assay for testing genotoxicity of chemicals and whole sediments in zebrafish embryos. Environ Toxicol Chem. 2006;25(8):2097–2106. [DOI] [PubMed] [Google Scholar]
- [15].Dietel M, Arps H, Gerding D, et al. Establishment of primary cell cultures: experiences with 155 cell strains. Klin Wochenschr. 1987;65(11):507–512. [DOI] [PubMed] [Google Scholar]
- [16].Rubin H. Cell aging in vivo and in vitro. Mech Ageing Dev. 1997;98(1):1–35. [DOI] [PubMed] [Google Scholar]
- [17].Tucci-Viegas VM, Hochman B, França JP, et al. Keloid explant culture: a model for keloid fibroblasts isolation and cultivation based on the biological differences of its specific regions. Int Wound J. 2010;7(5):339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Patil VR, Kharat AH, Kulkarni DG, et al. Long term explant culture for harvesting homogeneous population of human dental pulp stem cells. Cell Biol Int. 2018;42(12):1602–1610. [DOI] [PubMed] [Google Scholar]
- [19].Liang X, Zhang L, Wang S, et al. Exosomes secreted by mesenchymal stem cells promote endothelial cell angiogenesis by transferring miR-125a. J Cell Sci. 2016;129(11):2182–2189. [DOI] [PubMed] [Google Scholar]
- [20].Jing W, Xiao J, Xiong Z, et al. Explant culture: an efficient method to isolate adipose-derived stromal cells for tissue engineering. Artif Organs. 2011;35(2):105–112. [DOI] [PubMed] [Google Scholar]
- [21].Sanches BDA, Maldarine JDS, Tamarindo GH, et al. Explant culture: a relevant tool for the study of telocytes. Cell Biol Int. 2020;44(12):2395–2408. [DOI] [PubMed] [Google Scholar]
- [22].Hendijani F. Explant culture: an advantageous method for isolation of mesenchymal stem cells from human tissues. Cell Prolif. 2017;50(2):e12334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Moon JH, Kwak SS, Park G, et al. Isolation and characterization of multipotent human keloid-derived mesenchymal-like stem cells. Stem Cells Dev. 2008;17(4):713–724. [DOI] [PubMed] [Google Scholar]
- [24].Ehrlich HP, Desmoulière A, Diegelmann RF, et al. Morphological and immunochemical differences between keloid and hypertrophic scar. Am J Pathol. 1994;145(1):105–113. [PMC free article] [PubMed] [Google Scholar]
- [25].Otte A, Bucan V, Reimers K, et al. Mesenchymal stem cells maintain long-term in vitro stemness during explant culture. Tissue Eng Part C Methods. 2013;19(12):937–948. [DOI] [PubMed] [Google Scholar]