

Abstract
SARS-CoV-2 encodes two viral cysteine proteases, the main protease (Mpro) and the papain-like protease (PLpro), both of which are validated antiviral drug targets. PLpro is involved in the cleavage of viral polyproteins as well as immune modulation by removing ubiquitin and interferon-stimulated gene product 15 (ISG15) from host proteins. Therefore, targeting PLpro might be a two-pronged approach. Several compounds including YM155, cryptotanshinone, tanshinone I, dihydrotanshinone I, tanshinone IIA, SJB2-043, 6-thioguanine, and 6-mercaptopurine were recently identified as SARS-CoV-2 PLpro inhibitors through high-throughput screenings. In this study, we aim to validate/invalidate the reported PLpro inhibitors using a combination of PLpro target-specific assays including enzymatic FRET assay, thermal shift binding assay (TSA), and cell-based FlipGFP assay. Collectively, our results showed that all compounds tested either did not show binding or led to denaturation of PLpro in the TSA binding assay, which might explain their weak enzymatic inhibition in the FRET assay. In addition, none of the compounds showed cellular PLpro inhibition as revealed by the FlipGFP assay. Therefore, more efforts are needed to search for potent and specific SARS-CoV-2 PLpro inhibitors.
Keywords: SARS-CoV-2, papain-like protease, GRL0617, FlipGFP, antiviral
The COVID-19 pandemic is a timely reminder for the urgent need of antivirals, especially broad-spectrum antivirals that could be used as the first-line defense against not only the current pandemic but also future pandemics.1 SARS-CoV-2, SARS-CoV, and MERS-CoV are the three coronaviruses in the β-coronavirus family that caused pandemics/epidemics in humans.2 SARS-CoV and MERS-CoV had higher mortality rates than SARS-CoV-2.3 However, SARS-CoV-2 has a much higher transmission rate than SARS-CoV and MERS-CoV, which leads to far greater infection cases and death tolls.
SARS-CoV-2 shares 86% sequence identity with SARS-CoV, which renders the rapid understanding of its viral pathogenicity feasible. SARS-CoV-2 expresses two viral proteases during the viral replication, the main protease (Mpro; nsp5) and the papain-like protease (PLpro; nsp3). Both are cysteine proteases and have been validated as antiviral drug targets.4−6 PLpro and Mpro cleave the viral polyproteins pp1a and pp1ab at 3 and more than 11 sites, respectively, resulting in individual functional viral proteins for the assembly of a viral replication complex. Compared to PLpro, Mpro is a more amenable drug target and has been the central focus of COVID-19 antiviral discovery. Structurally disparate compounds have been reported as Mpro inhibitors from either drug repurposing screening or rational design.7,8 Although a large number of reported Mpro inhibitors were later proven to be promiscuous cysteine modifiers,9−13 several Mpro inhibitors have been validated as specific inhibitors and have shown in vivo antiviral efficacy in animal model studies.14−18 Significantly, two Pfizer Mpro inhibitors PF-07304814 and PF-07321332 are advanced to human clinical trials.17,18
PLpro is the second viral cysteine protease that cleaves the viral polypeptide at three different sites during viral replication. In addition, PLpro modulates host immune response by cleaving ubiquitin and ISG15 (interferon-induced gene 15) from host proteins.19−21 In contrast to Mpro inhibitors, the development of PLpro inhibitors is still at its infancy.22 The most promising PLpro inhibitors are the naphthalene-based GRL0617 series of compounds (Figure 1A).6,21,23−25 However, their antiviral potency and pharmacokinetic properties need to be further optimized for the in vivo animal model study. PLpro specifically recognizes the P4-P1 sequence Leu-X-Gly-Gly that is conserved among the nsp1/2, nsp2/3, and nsp3/4 cleavage sites at the viral polyprotein.26 The featureless P1 and P2 binding pockets present a grand challenge in developing potent PLpro inhibitors.26 As the drug-binding site is far away from the catalytic cysteine 111 (>10 Å), the majority of the reported PLpro inhibitors are noncovalent inhibitors.7 Although peptidomimetic covalent PLpro inhibitors have been reported, no antiviral activity is shown.26 To identify additional novel PLpro inhibitors, several high-throughput screenings have been performed and structurally disparate compounds are found to inhibit PLpro (Figure 1B).27−30 It is intriguing that structurally diverse compounds can act on the same protein; we therefore are interested in validating/invalidating the reported PLpro inhibitors using a combination of orthogonal assays including the activity-based FRET enzymatic assay, the thermal shift binding (TSA) assay, and the cell-based FlipGFP assay. The purpose is to prioritize well-characterized PLpro inhibitors for medicinal chemistry optimization. The FRET assay is the gold standard assay for protease, which measures the activity of the purified enzyme in the presence of a compound. However, the FRET assay results might be complicated by the fluorescence interference compound, alkylating reagent, or redox compound. The TSA assay is a binding assay that measures the direct binding between the small molecule and the protein. High-affinity binders generally stabilize the protein, leading to the increase in the melting temperature. The cell-based FlipGFP assay measures the cellular protease activity and can rule out compounds that are cytotoxic, membrane-impermeable, or lack the cellular target engagement due to off-target effects. Collectively, in contrast to the literature-reported results, our data showed that the examined non-GRL0617-based PLpro inhibitors (Figure 1B) had weak enzymatic inhibition and no cellular PLpro inhibition; therefore, they should not be referenced as specific PLpro inhibitors.
Figure 1.
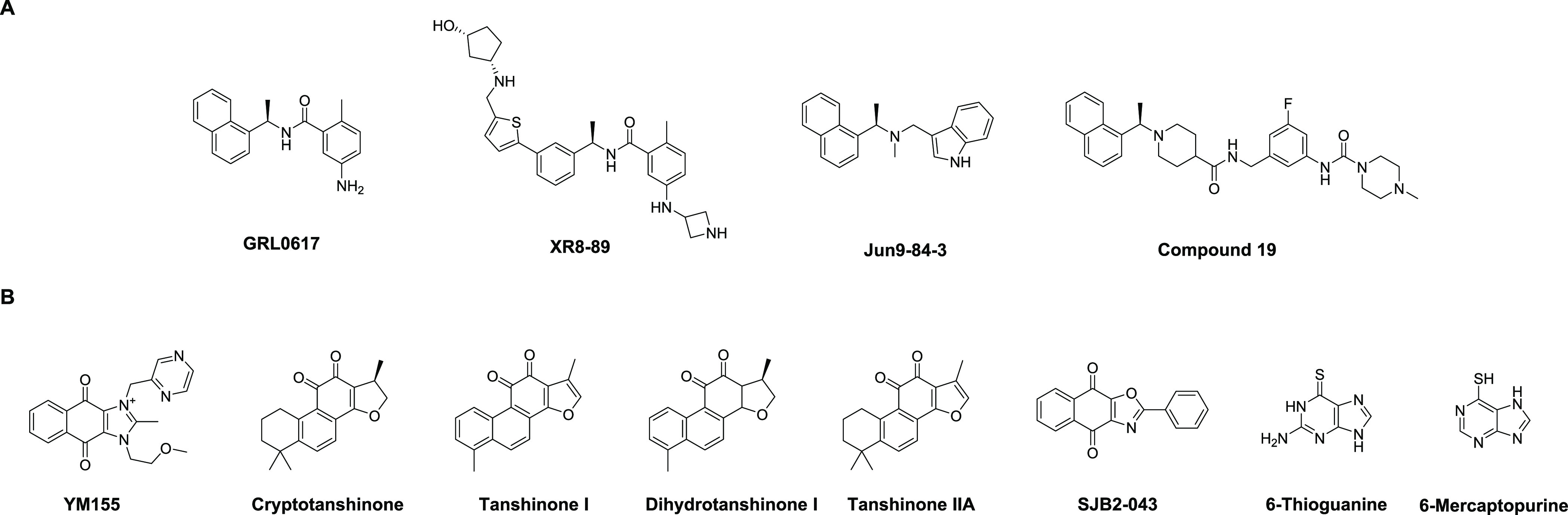
SARS-CoV-2 PLpro inhibitors. (A) GRL0617 analogs. (B) Structurally disparate PLpro inhibitors identified from high-throughput screening or drug repurposing screening of bioactive compounds.
Results
Through a high-throughput screening of over 6,000 bioactive compounds using a quenched fluorescent substrate RLRGG-AMC, Zhao et al. identified four compounds, YM155, cryptotanshinone, tanshinone I, and GRL0617, as potent PLpro inhibitors with IC50 values from 1.39 to 5.63 μM.29 In the plaque reduction assay, YM155, cryptotanshinone, and tanshinone l inhibited SARS-CoV-2 replication in Vero E6 cells with EC50 values of 0.17, 0.70, and 2.26 μM, respectively. The X-ray crystal structure of SARS-CoV-2 PLpro with YM155 was solved (PDB: 7D7L), revealing three different binding sites that are located in the thumb domain, the zinc-finger motif, and the substrate-binding pocket. In the substrate binding pocket, YM155 induces a conformational change of Y268 on the BL2 loop and forms a π-stacking interaction, which is similar to the binding mode of GRL0617. Binding at the thumb domain likely impedes the interaction between PLpro and ISG15. The third binding site at the zinc-finger domain might perturb its stability; however, this mechanism remains to be validated.
In our validation study, the positive control GRL0617 showed similar enzymatic inhibition with an IC50 of 1.67 μM (Figure 2A). However, we found that the enzymatic inhibition potency for YM155, cryptotanshinone, and tanshinone I against PLpro was ∼10-fold less active than previously reported and the IC50 values from our study were 20.16, 52.24, and 18.58 μM, respectively (Figure 2A and Table 1). The discrepancy might be caused by the different substrates used. In Zhao et al.’s study, RLRGG-AMC was used, while we used a FRET substrate Dabcyl-FTLRGG/APTKV-Edans, which spans both the P and P′ sites. Nevertheless, in the thermal shift binding assay, YM155 had no effect on the stability of PLpro, while cryptotanshinone and tanshinone I caused destabilization of the protein (Figure 2B and Table 1). In the cell-based FlipGFP assay,6 the result for YM155 was not conclusive as it was cytotoxic to the 293T cells (Figure 2C). Both cryptotanshinone and tanshinone I were not active (EC50 > 60 μM) (Figure 2C). Overall, our results indicate that cryptotanshinone and tanshinone I were not specific PLpro inhibitors, while YM155 might be a specific PLpro inhibitor. However, the large discrepancy between the enzymatic inhibition (IC50 = 20.16 μM) and the reported cellular antiviral activity of YM155 (EC50 = 0.17 μM) suggests that the other mechanisms might contribute to its potent antiviral activity against SARS-CoV-2.
Figure 2.

Pharmacological characterization of SARS-CoV-2 PLpro inhibitors. (A) Enzymatic inhibitory activity against SARS-CoV-2 PLpro in the FRET-based assay. (B) Thermal shift assay of the SARS-CoV-2 PLpro inhibitors in stabilizing SARS-CoV-2 PLpro. The asterisk (*) means that the melting peak was not observed in the presence of the inhibitor. The dashed red line indicates the mean of SARS-CoV-2 PLproTm in the absence of testing inhibitors. (C) Cell-based FlipGFP assay for the quantification of the cellular activity of SARS-CoV-2 PLpro inhibitors. Representative images of the FlipGFP-PLpro assay with the positive control GRL0617 and the negative control GC-376. Dose–response curves of the ratio of GFP/mCherry fluorescence with SARS-CoV-2 PLpro inhibitors were plotted at right side column; mCherry signal alone was used to normalize the protein expression level or calculate compound cytotoxicity.
Table 1. Validation/Invalidation of SARS-CoV-2 PLpro Inhibitorsa.

N.T., not tested.
In another study, cryptotanshinone and two analogs dihydrotanshinone I and tanshinone IIA were identified as SARS-CoV-2 PLpro inhibitors through a high-throughput screening (HTS).28 Cryptotanshinone, dihydrotanshinone I, and tanshinone IIA inhibited SARS-CoV-2 PLpro with IC50 values of 1.336, 0.5861, and 1.571 μM, respectively. It is noted that no complete inhibition was observed for cryptotanshinone; therefore, the IC50 value might not be trustworthy. To rule out the possibility of fluorescence interference, these compounds were also tested in the gel-based PLpro assay using the GST-nsp2/3-MBP substrate. All these compounds inhibited the digestion of the protein substrate with tanshinone IIA and cryptotanshinone showing higher potency than dihydrotanshinone I. Dihydrotanshinone I also inhibited the deubiquitinase and deISGlase activities of PLpro in the gel-based digestion assay. In the SARS-CoV-2 antiviral assay in Vero E6 cells, dihydrotanshinone I had the highest potency with an EC50 of 8.148 μM, while cryptotanshinone and tanshinone I were not active (EC50 > 200 μM). The lack of correlation between the enzymatic inhibition and the cellular antiviral activity might be due to cell membrane permeability or off-target effects.
In our validation study, both dihydrotanshinone I and tanshinone IIA were greater than 10-fold less potent in the enzymatic assay with IC50 values of 33.01 and 15.30 μM, respectively (Figure 2A and Table 1). Dihydrotanshinone I did not bind to PLpro as shown by the results from the TSA assay (Figure 2B). Tanshinone IIA led to denaturation of the protein (no melting peak). Dihydrotanshinone I and tanshinone IIA were not active in the FlipGFP assay (IC50 > 20 μM) (Figure 2C). Overall, it appears that dihydrotanshinone I and tanshinone IIA were not specific SARS-CoV-2 PLpro inhibitors, and the antiviral activity of dihydrotanshinone I might involve other mechanisms.
Cho et al. reported SJB2-043 as a SARS-CoV-2 PLpro inhibitor through a focused screening of a library of deubiquitinase inhibitors.27 SJB2-043 did not achieve complete inhibition and had an apparent IC50 of 0.56 μM. When Ub-AMC was used as a substrate, the IC50 of SJB2-043 was 0.091 μM. Similarly, no complete inhibition was achieved. In comparison, the positive control GRL0617 showed complete inhibition when both Z-LRGG-AMC and Ub-AMC were used as substrates, and the IC50 values were 1.37 and 1.80 μM, respectively. Molecular docking suggests that SJB2-043 binds to an allosteric site in PLpro.
When repeated in our assay, SJB2-043 did not inhibit SARS-CoV-2 PLpro (IC50 > 60 μM) (Figure 2A). The discrepancy might be caused by different methods used for data fitting. In our assay, SJB2-043 did not achieve more than 50% inhibition at the highest concentration tested, which is 60 μM; therefore, we deemed the IC50 greater than 60 μM. In the TSA binding assay, SJB2-043 led to the destabilization of PLpro (ΔTm = −8.72 °C) (Figure 2B). The result from the FlipGFP assay was not conclusive as SJB2-043 was cytotoxic (Figure 2C). Overall, SJB2-043 might not be a specific SARS-CoV-2 PLpro inhibitor and only shows partial inhibition.
Swaim et al. reported that 6-thioguanine inhibited SARS-CoV-2 replication in Vero E6 cells with an EC50 of 2.13 μM.30 Mechanistic studies showed that 6-thioguanine had potent inhibition against PLpro in cells and in vitro using the TAP-nsp123, TAP-nsp23, and Pro-ISG15-HA substrates. Another study from Fu et al. showed that 6-thioguanine inhibited SARS-CoV-2 PLpro in the enzymatic assay with an IC50 of 72 μM.31 In our study, 6-thioguanine was not active in the FRET-based enzymatic assay (IC50 > 60 μM) (Figure 2A). 6-Thioguanine also did not show binding in the TSA assay nor inhibition in the FlipGFP assay (Figure 2B,C). As such, the antiviral activity of 6-thioguanine might involve other mechanisms. 6-Mercaptopurine, which is an analog of 6-thioguanine, was similarly not active in all three assays (Figure 2 and Table 1). Therefore, it can be concluded that 6-thioguanine and 6-mercaptopurine were not SARS-CoV-2 PLpro inhibitors.
Discussion
Drug repurposing is often viewed as a fast-track drug discovery approach since it can potentially bypass the length safety tests before entering clinical trials.32 However, we should remain cautiously optimistic about this approach.33 Since the existing bioactive compounds were not specifically designed and optimized for the screening drug target, the identified hits need to be vigorously characterized to validate the target specificity. SARS-CoV-2 Mpro and PLpro are cysteine proteases that are prone to nonspecific inhibition by alkylating agents or redox cycling compounds. Indeed, our recent studies, together with others, have collectively shown that a number of reported Mpro inhibitors including ebselen, carmofur, disulfiram, and shikonin are promiscuous nonspecific cysteine protease inhibitors.9−13 With our continuous interest in the validation/invalidation of literature-reported Mpro and PLpro inhibitors, in this study, we characterized eight PLpro inhibitors using a combination of FRET-based enzymatic assay, TSA binding assay, and cell-based FlipGFP-PLpro assay. It is expected that specific inhibitors will show consistent results from all three assays. Among the compounds tested, cryptotanshinone, tanshinone I, dihydrotanshinone I, and tanshinone IIA were also previously reported as potent SARS-CoV PLpro inhibitors with IC50 values ranging from 0.8 to 8.8 μM.34 Interestingly, they also inhibited SARS-CoV Mpro with IC50 values ranging from 14.4 to 226.7 μM. Since Mpro and PLpro do not share structural and sequence similarities, the dual inhibitory activities of these tanshinones, coupled with the weak PLpro enzymatic inhibition from our study, suggest that they might have a promiscuous mechanism of action.
Similarly, 6-thioguanine and 6-mercaptopurine were previously reported as SARS-CoV PLpro inhibitors with IC50 values of 5.0 and 21.6 μM, respectively.35,36 In addition, Cheng et al. reported that 6-thioguanine and 6-mercaptopurine are competitive inhibitors of MERS-CoV PLpro with IC50 values of 24.4 and 26.9 μM, respectively.37 In contrast, YM155 and SJB2-043 were not previously shown to inhibit SARS-CoV PLpro. Given the 82.6% sequence similarity between SARS-CoV-2 PLpro and SARS-CoV PLpro, it is not a surprising finding that cryptotanshinone, tanshinone I, dihydrotanshinone I, tanshinone IIA, and 6-thioguanine were also reported as SARS-CoV-2 PLpro inhibitors from recent studies. Nevertheless, our independent study has shown that all eight compounds had drastically reduced enzymatic inhibition against SARS-CoV-2 PLpro compared to reported values (Table 1). In addition, all compounds either had no effect or destabilize PLpro as shown by the TSA binding assay (Table 1). Furthermore, cryptotanshinone, tanshinone I, dihydrotanshinone I, tanshinone IIA, 6-thioguanine, and 6-mercaptopurine were not active in the FlipGFP-PLpro assay, suggesting that there is a lack of cellular PLpro target engagement. YM155 and SJB2-043 were cytotoxic; therefore, it remains unknown whether they can selectively bind to PLpro inside the cell. From the chemical structure perspective, YM155, cryptotanshinone, tanshinone, dihydrotanshinone I, tanshinone IIA, and SJB2-043 are quinones, which are known as cysteine modifiers.38 Overall, our study calls for more stringent validation of the reported SARS-CoV-2 PLpro inhibitors to avoid the failure in the follow-up lead optimization and translational development.
In addition to the compounds examined (Table 1), ebselen39 and its analogs,40 as well as disulfiram,39,41,42 were also reported as SARS-CoV-2 PLpro inhibitors. Our previous results have shown that both ebselen and disulfiram are promiscuous PLpro inhibitors and their inhibition against PLpro is abolished in the presence of reducing reagent DTT.10 In addition, ebselen and disulfiram also inhibit several other unrelated cysteine proteases in the absence of DTT, suggesting that both compounds are nonspecific cysteine modifiers.
It is important to highlight that the focus of this study is the pharmacological characterization of reported SARS-CoV-2 PLpro inhibitors. Although our results have shown that cryptotanshinone, tanshinone I, dihydrotanshinone I, tanshinone IIA, and 6-thioguanine are nonspecific PLpro inhibitors, they have been reported to inhibit SARS-CoV-2 replication in cell culture (Table 1). Therefore, our results should not be interpreted as a discouragement to further pursue them as potential SARS-CoV-2 antivirals. If their antiviral activity is validated, follow-up studies are needed to delineate their mechanism of action.
Materials and Methods
Chemicals and Peptide Substrate
The SARS-CoV-2 PLpro substrate Dabcyl-FTLRGG/APTKV-Edans was synthesized by the solid-phase synthesis, and the detailed procedure was described in our previous publication.6 The testing compounds were ordered from the following sources: tanshinone I (Sigma T5330), dihydrotanshinone I (Sigma D0947), tanshinone IIA (Sigma SML2517), cryptotanshinone (Sigma C5624), YM-155 (APExBIO A4221), and SJB2-043 (APExBIO A3823).
SARS-CoV-2 PLpro Expression and Purification
SARS-CoV-2 papain-like protease (PLpro) gene (ORF 1ab 1564–1876) from strain BetaCoV/Wuhan/WIV04/2019 with Escherichia coli codon optimization was ordered from GenScript in the pET28b(+) vector. The detailed expression and purification procedures were previously described.6
FRET-Based Enzymatic Assay
For the IC50 measurement with the FRET-based assay, the reaction was carried out in 96-well format with 100 μL of 200 nM PLpro protein in a PLpro reaction buffer (50 mM HEPES (pH 7.5), 5 mM DTT, and 0.01% Triton X-100); 1 μL of testing compounds at various concentrations was added to each well and was incubated at 30 °C for 30 min. The reaction was initiated by adding 1 μL of 1 mM FRET substrate and was monitored in a Cytation 5 image reader with filters for excitation at 360/40 nm and emission at 460/40 nm at 30 °C for 1 h. The initial velocity of the enzymatic reaction was calculated from the initial 10 min enzymatic reaction. The IC50 was calculated by plotting the initial velocity against various concentrations of testing compounds by the use of a four-parameter variable slope dose–response curve in Prism 8 software.
Differential Scanning Fluorimetry (DSF)
The thermal shift binding assay (TSA) was carried out using a Thermo Fisher QuantStudio 5 Real-Time PCR system as described previously.6
Cell-Based FlipGFP-PLpro Assay
The cell-based FlipGFP-PLpro assay was established as previously described. Briefly, 293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% heat-inactivated FBS and 1% penicillin–streptomycin antibiotics in a 37 °C incubator with 5% CO2. A 96-well Greiner plate (catalog no. 655090) was seeded with 293T cells to overnight 70–90% confluency. Fifty nanograms of pcDNA3-flipGFP-PLpro-T2A-mCherry plasmid and 50 ng of pcDNA3.1-SARS2-PLpro were used for each well in the presence of transfection reagent TransIT-293 (Mirus) per the manufacturer’s protocol. Three hours after transfection, 1 μL of testing compound was added to each well at 100-fold dilution. Two days after transfection, images were obtained with a Cytation 5 imaging reader (Biotek) with GFP and mCherry channels. SARS-CoV-2 PLpro protease activity was evaluated by the ratio of GFP signal intensity over the mCherry signal intensity. The compound FlipGFP-PLP assay IC50 value was calculated by plotting the GFP/mCherry signal to the testing compound concentration with a four-parameter variable slope dose–response function in Prism 8. The mCherry signal alone was utilized to determine the compound cytotoxicity.
Acknowledgments
This research was supported by the National Institutes of Health (NIH) (grants AI147325, AI157046, and AI158775) and the Arizona Biomedical Research Centre Young Investigator grant (ADHS18-198859) to J.W.
Author Contributions
C.M. performed the enzymatic assay, thermal shift binding assay, and FlipGFP assay. J.W. designed and supervised this study. J.W. wrote the manuscript with contribution from C.M.
The authors declare no competing financial interest.
This article is made available via the ACS COVID-19 subset for unrestricted RESEARCH re-use and analyses in any form or by any means with acknowledgement of the original source. These permissions are granted for the duration of the World Health Organization (WHO) declaration of COVID-19 as a global pandemic.
References
- Lu L.; Su S.; Yang H.; Jiang S. Antivirals with common targets against highly pathogenic viruses. Cell 2021, 184, 2021. 10.1016/j.cell.2021.02.013. [DOI] [PubMed] [Google Scholar]
- Cui J.; Li F.; Shi Z. L. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 2019, 17, 181–192. 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-On Y. M.; Flamholz A.; Phillips R.; Milo R. SARS-CoV-2 (COVID-19) by the numbers. eLife 2020, 9, e57309 10.7554/eLife.57309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Sacco M. D.; Hurst B.; Townsend J. A.; Hu Y.; Szeto T.; Zhang X.; Tarbet B.; Marty M. T.; Chen Y.; Wang J. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res 2020, 30, 678–692. 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco M. D.; Ma C.; Lagarias P.; Gao A.; Townsend J. A.; Meng X.; Dube P.; Zhang X.; Hu Y.; Kitamura N.; Hurst B.; Tarbet B.; Marty M. T.; Kolocouris A.; Xiang Y.; Chen Y.; Wang J. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against M(pro) and cathepsin L. Sci. Adv. 2020, 6, eabe0751 10.1126/sciadv.abe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Sacco M. D.; Xia Z.; Lambrinidis G.; Townsend J. A.; Hu Y.; Meng X.; Szeto T.; Ba M.; Zhang X.; Gongora M.; Zhang F.; Marty M. T.; Xiang Y.; Kolocouris A.; Chen Y.; Wang J. Discovery of SARS-CoV-2 Papain-like Protease Inhibitors through a Combination of High-Throughput Screening and a FlipGFP-Based Reporter Assay. ACS Cent. Sci. 2021, 7, 1245–1260. 10.1021/acscentsci.1c00519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Brindisi M.; Shahabi D.; Chapman M. E.; Mesecar A. D. Drug Development and Medicinal Chemistry Efforts toward SARS-Coronavirus and Covid-19 Therapeutics. ChemMedChem 2020, 15, 907–932. 10.1002/cmdc.202000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong M.; Su H.; Zhao W.; Xie H.; Shao Q.; Xu Y. What coronavirus 3C-like protease tells us: From structure, substrate selectivity, to inhibitor design. Med. Res. Rev. 2021, 41, 41. 10.1002/med.21783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Tan H.; Choza J.; Wang Y.; Wang J. Validation and invalidation of SARS-CoV-2 main protease inhibitors using the Flip-GFP and Protease-Glo luciferase assays. Acta. Pharm. Sin. B 2021, 10.1016/j.apsb.2021.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Hu Y.; Townsend J. A.; Lagarias P. I.; Marty M. T.; Kolocouris A.; Wang J. Ebselen, Disulfiram, Carmofur, PX-12, Tideglusib, and Shikonin Are Nonspecific Promiscuous SARS-CoV-2 Main Protease Inhibitors. ACS Pharmacol Transl Sci 2020, 3, 1265–1277. 10.1021/acsptsci.0c00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurard-Levin Z. A.; Liu C.; Jekle A.; Jaisinghani R.; Ren S.; Vandyck K.; Jochmans D.; Leyssen P.; Neyts J.; Blatt L. M.; Beigelman L.; Symons J. A.; Raboisson P.; Scholle M. D.; Deval J. Evaluation of SARS-CoV-2 3C-like protease inhibitors using self-assembled monolayer desorption ionization mass spectrometry. Antiviral Res. 2020, 182, 104924. 10.1016/j.antiviral.2020.104924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W.; Cho C.-C. D.; Geng Z. Z.; Ma X. R.; Allen R.; Shaabani N.; Vatansever E. C.; Alugubelli Y. R.; Ma Y.; Ellenburg W. H.; Yang K. S.; Qiao Y.; Ji H.; Xu S.; Liu W. R. Cellular Activities of SARS-CoV-2 Main Protease Inhibitors Reveal Their Unique Characteristics. bioRxiv 2021, 10.1101/2021.06.08.447613. [DOI] [Google Scholar]
- Ma C.; Wang J. Dipyridamole, chloroquine, montelukast sodium, candesartan, oxytetracycline, and atazanavir are not SARS-CoV-2 main protease inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2024420118 10.1073/pnas.2024420118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao J.; Li Y. S.; Zeng R.; Liu F. L.; Luo R. H.; Huang C.; Wang Y. F.; Zhang J.; Quan B.; Shen C.; Mao X.; Liu X.; Sun W.; Yang W.; Ni X.; Wang K.; Xu L.; Duan Z. L.; Zou Q. C.; Zhang H. L.; Qu W.; Long Y. H.; Li M. H.; Yang R. C.; Liu X.; You J.; Zhou Y.; Yao R.; Li W. P.; Liu J. M.; Chen P.; Liu Y.; Lin G. F.; Yang X.; Zou J.; Li L.; Hu Y.; Lu G. W.; Li W. M.; Wei Y. Q.; Zheng Y. T.; Lei J.; Yang S. SARS-CoV-2 M(pro) inhibitors with antiviral activity in a transgenic mouse model. Science 2021, 371, 1374–1378. 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dampalla C. S.; Zheng J.; Perera K. D.; Wong L. R.; Meyerholz D. K.; Nguyen H. N.; Kashipathy M. M.; Battaile K. P.; Lovell S.; Kim Y.; Perlman S.; Groutas W. C.; Chang K. O. Postinfection treatment with a protease inhibitor increases survival of mice with a fatal SARS-CoV-2 infection. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2101555118 10.1073/pnas.2101555118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cáceres C. J.; Cardenas-Garcia S.; Carnaccini S.; Seibert B.; Rajao D. S.; Wang J.; Perez D. R. Efficacy of GC-376 against SARS-CoV-2 virus infection in the K18 hACE2 transgenic mouse model. Sci. Rep. 2021, 11, 9609. 10.1038/s41598-021-89013-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boras B.; Jones R. M.; Anson B. J.; Arenson D.; Aschenbrenner L.; Bakowski M. A.; Beutler N.; Binder J.; Chen E.; Eng H.; Hammond H.; Hammond J.; Haupt R. E.; Hoffman R.; Kadar E. P.; Kania R.; Kimoto E.; Kirkpatrick M. G.; Lanyon L.; Lendy E. K.; Lillis J. R.; Logue J.; Luthra S. A.; Ma C.; Mason S. W.; McGrath M. E.; Noell S.; Obach R. S.; MN O. B.; O’Connor R.; Ogilvie K.; Owen D.; Pettersson M.; Reese M. R.; Rogers T. F.; Rosales R.; Rossulek M. I.; Sathish J. G.; Shirai N.; Steppan C.; Ticehurst M.; Updyke L. W.; Weston S.; Zhu Y.; White K. M.; Garcia-Sastre A.; Wang J.; Chatterjee A. K.; Mesecar A. D.; Frieman M. B.; Anderson A. S.; Allerton C. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat. Commun. 2021, 12, 6055. 10.1038/s41467-021-26239-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D. R.; Allerton C. M. N.; Anderson A. S.; Aschenbrenner L.; Avery M.; Berritt S.; Boras B.; Cardin R. D.; Carlo A.; Coffman K. J.; Dantonio A.; Di L.; Eng H.; Ferre R.; Gajiwala K. S.; Gibson S. A.; Greasley S. E.; Hurst B. L.; Kadar E. P.; Kalgutkar A. S.; Lee J. C.; Lee J.; Liu W.; Mason S. W.; Noell S.; Novak J. J.; Obach R. S.; Ogilvie K.; Patel N. C.; Pettersson M.; Rai D. K.; Reese M. R.; Sammons M. F.; Sathish J. G.; Singh R. S. P.; Steppan C. M.; Stewart A. E.; Tuttle J. B.; Updyke L.; Verhoest P. R.; Wei L.; Yang Q.; Zhu Y. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- Freitas B. T.; Durie I. A.; Murray J.; Longo J. E.; Miller H. C.; Crich D.; Hogan R. J.; Tripp R. A.; Pegan S. D. Characterization and Noncovalent Inhibition of the Deubiquitinase and deISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect Dis 2020, 6, 2099–2109. 10.1021/acsinfecdis.0c00168. [DOI] [PubMed] [Google Scholar]
- Shin D.; Mukherjee R.; Grewe D.; Bojkova D.; Baek K.; Bhattacharya A.; Schulz L.; Widera M.; Mehdipour A. R.; Tascher G.; Geurink P. P.; Wilhelm A.; van der Heden van Noort G. J.; Ovaa H.; Müller S.; Knobeloch K.-P.; Rajalingam K.; Schulman B. A.; Cinatl J.; Hummer G.; Ciesek S.; Dikic I. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm T.; Ebert G.; Calleja D. J.; Allison C. C.; Richardson L. W.; Bernardini J. P.; Lu B. G.; Kuchel N. W.; Grohmann C.; Shibata Y.; Gan Z. Y.; Cooney J. P.; Doerflinger M.; Au A. E.; Blackmore T. R.; van der Heden van G. J.; Geurink P. P.; Ovaa H.; Newman J.; Riboldi-Tunnicliffe A.; Czabotar P. E.; Mitchell J. P.; Feltham R.; Lechtenberg B. C.; Lowes K. N.; Dewson G.; Pellegrini M.; Lessene G.; Komander D. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J 2020, 39, e106275 10.15252/embj.2020106275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baez-Santos Y. M.; St John S. E.; Mesecar A. D. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res. 2015, 115, 21–38. 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z.; Ratia K.; Cooper L.; Kong D.; Lee H.; Kwon Y.; Li Y.; Alqarni S.; Huang F.; Dubrovskyi O.; Rong L.; Thatcher G. R. J.; Xiong R. Design of SARS-CoV-2 PLpro Inhibitors for COVID-19 Antiviral Therapy Leveraging Binding Cooperativity. J. Med. Chem. 2021, 10.1021/acs.jmedchem.1c01307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan H.; Liu J.; Shen J.; Dai J.; Xu G.; Lu K.; Han C.; Wang Y.; Xu X.; Tong Y.; Xiang H.; Ai Z.; Zhuang G.; Hu J.; Zhang Z.; Li Y.; Pan L.; Tan L. Development of potent and selective inhibitors targeting the papain-like protease of SARS-CoV-2. Cell Chem. Biol. 2021, 28, 855–865.e9. 10.1016/j.chembiol.2021.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipiuk J.; Azizi S. A.; Dvorkin S.; Endres M.; Jedrzejczak R.; Jones K. A.; Kang S.; Kathayat R. S.; Kim Y.; Lisnyak V. G.; Maki S. L.; Nicolaescu V.; Taylor C. A.; Tesar C.; Zhang Y. A.; Zhou Z.; Randall G.; Michalska K.; Snyder S. A.; Dickinson B. C.; Joachimiak A. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021, 12, 743. 10.1038/s41467-021-21060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rut W.; Lv Z.; Zmudzinski M.; Patchett S.; Nayak D.; Snipas S. J.; El Oualid F.; Huang T. T.; Bekes M.; Drag M.; Olsen S. K. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti-COVID-19 drug design. Sci. Adv. 2020, 6, eabd4596 10.1126/sciadv.abd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho C. C.; Li S. G.; Lalonde T. J.; Yang K. S.; Yu G.; Qiao Y.; Xu S.; Ray Liu W. Drug Repurposing for the SARS-CoV-2 Papain-Like Protease. ChemMedChem 2022, 17, e202100455 10.1002/cmdc.202100455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C. T.; Tan K. W.; Wu M.; Ulferts R.; Armstrong L. A.; Ozono E.; Drury L. S.; Milligan J. C.; Zeisner T. U.; Zeng J.; Weissmann F.; Canal B.; Bineva-Todd G.; Howell M.; O’Reilly N.; Beale R.; Kulathu Y.; Labib K.; Diffley J. F. X. Identifying SARS-CoV-2 antiviral compounds by screening for small molecule inhibitors of Nsp3 papain-like protease. Biochem. J. 2021, 478, 2517–2531. 10.1042/BCJ20210244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Du X.; Duan Y.; Pan X.; Sun Y.; You T.; Han L.; Jin Z.; Shang W.; Yu J.; Guo H.; Liu Q.; Wu Y.; Peng C.; Wang J.; Zhu C.; Yang X.; Yang K.; Lei Y.; Guddat L. W.; Xu W.; Xiao G.; Sun L.; Zhang L.; Rao Z.; Yang H. High-throughput screening identifies established drugs as SARS-CoV-2 PLpro inhibitors. Protein Cell 2021, 12, 877–888. 10.1007/s13238-021-00836-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaim C. D.; Dwivedi V.; Perng Y.-C.; Zhao X.; Canadeo L. A.; Harastani H. H.; Darling T. L.; Boon A. C. M.; Lenschow D. J.; Kulkarni V.; Huibregtse J. M. 6-Thioguanine blocks SARS-CoV-2 replication by inhibition of PLpro. iScience 2021, 24, 103213. 10.1016/j.isci.2021.103213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z.; Huang B.; Tang J.; Liu S.; Liu M.; Ye Y.; Liu Z.; Xiong Y.; Zhu W.; Cao D.; Li J.; Niu X.; Zhou H.; Zhao Y. J.; Zhang G.; Huang H. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 2021, 12, 488. 10.1038/s41467-020-20718-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushpakom S.; Iorio F.; Eyers P. A.; Escott K. J.; Hopper S.; Wells A.; Doig A.; Guilliams T.; Latimer J.; McNamee C.; Norris A.; Sanseau P.; Cavalla D.; Pirmohamed M. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov 2019, 18, 41–58. 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- Mercorelli B.; Palù G.; Loregian A. Drug Repurposing for Viral Infectious Diseases: How Far Are We?. Trends Microbiol. 2018, 26, 865–876. 10.1016/j.tim.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. Y.; Kim J. H.; Kim Y. M.; Jeong H. J.; Kim D. W.; Park K. H.; Kwon H. J.; Park S. J.; Lee W. S.; Ryu Y. B. Tanshinones as selective and slow-binding inhibitors for SARS-CoV cysteine proteases. Bioorg. Med. Chem. 2012, 20, 5928–5935. 10.1016/j.bmc.2012.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Chou C. Y.; Chang G. G. Thiopurine analogue inhibitors of severe acute respiratory syndrome-coronavirus papain-like protease, a deubiquitinating and deISGylating enzyme. Antiviral Chem. Chemother. 2009, 19, 151–156. 10.1177/095632020901900402. [DOI] [PubMed] [Google Scholar]
- Chou C. Y.; Chien C. H.; Han Y. S.; Prebanda M. T.; Hsieh H. P.; Turk B.; Chang G. G.; Chen X. Thiopurine analogues inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biochem. Pharmacol. 2008, 75, 1601–1609. 10.1016/j.bcp.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K. W.; Cheng S. C.; Chen W. Y.; Lin M. H.; Chuang S. J.; Cheng I. H.; Sun C. Y.; Chou C. Y. Thiopurine analogs and mycophenolic acid synergistically inhibit the papain-like protease of Middle East respiratory syndrome coronavirus. Antiviral Res. 2015, 115, 9–16. 10.1016/j.antiviral.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. W.; Heinze J.; Haehnel W. Site-specific binding of quinones to proteins through thiol addition and addition-elimination reactions. J. Am. Chem. Soc. 2005, 127, 6140–6141. 10.1021/ja050974x. [DOI] [PubMed] [Google Scholar]
- Sargsyan K.; Lin C.-C.; Chen T.; Grauffel C.; Chen Y.-P.; Yang W.-Z.; Yuan H. S.; Lim C. Multi-targeting of functional cysteines in multiple conserved SARS-CoV-2 domains by clinically safe Zn-ejectors. Chem. Sci. 2020, 11, 9904–9909. 10.1039/D0SC02646H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weglarz-Tomczak E.; Tomczak J. M.; Talma M.; Burda-Grabowska M.; Giurg M.; Brul S. Identification of ebselen and its analogues as potent covalent inhibitors of papain-like protease from SARS-CoV-2. Sci. Rep. 2021, 11, 3640. 10.1038/s41598-021-83229-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M. H.; Moses D. C.; Hsieh C. H.; Cheng S. C.; Chen Y. H.; Sun C. Y.; Chou C. Y. Disulfiram can inhibit MERS and SARS coronavirus papain-like proteases via different modes. Antiviral Res. 2018, 150, 155–163. 10.1016/j.antiviral.2017.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T.; Fei C. Y.; Chen Y. P.; Sargsyan K.; Chang C. P.; Yuan H. S.; Lim C. Synergistic Inhibition of SARS-CoV-2 Replication Using Disulfiram/Ebselen and Remdesivir. ACS Pharmacol Transl Sci 2021, 4, 898–907. 10.1021/acsptsci.1c00022. [DOI] [PMC free article] [PubMed] [Google Scholar]