

Abstract
The effects of healthy aging on the kidney, and how these effects intersect with superimposed diseases, are highly relevant in the context of the population’s increasing longevity. Age-associated changes to podocytes, which are terminally differentiated glomerular epithelial cells, adversely affect kidney health. This review discusses the molecular and cellular mechanisms underlying podocyte aging, how these mechanisms might be augmented by disease in the aged kidney, and approaches to mitigate progressive damage to podocytes. Furthermore, we address how biologic pathways such as those associated with cellular growth confound aging in humans and rodents.
Keywords: podocyte, aging, glomerulosclerosis, senescence, RNA sequencing, glomerulus
Scope of the Problem
The mechanisms underlying the aging process are diverse, and include general and cell type–specific aspects.1 Such mechanisms also apply to kidney aging, and several lines of evidence highlight the importance of studying aging and the kidney. The US population is growing older, and the US Census Bureau predicts that the number of Americans aged ≥65 years will more than double over the next 40 years. Similarly, Eurostat, the statistical office of the European Union, forecasts that 28% of Europeans will be aged >65 years by 2060. As life expectancy increases, the effect of advanced age on kidney health and function is becoming an increasingly important medical and socioeconomic factor. The GFR declines after age 40 years by 0.8%–1.0% per year,2,3 and kidneys from healthy donors aged 70–75 years have 48% fewer intact nephrons compared with young donors aged 19–29 years.4 This is consistent with an estimated annual loss of 6000–6500 nephrons after age 30 years.4,5 The progressive decrease in nephron number affects the kidney at multiple levels: (1) kidney function declines with age6–8; (2) glomerular diseases are disproportionately more common in people aged >65 years9,10; (3) the elderly have worse outcomes compared with those of younger individuals with the same disease11–14; (4) older age accelerates the transition from AKI to chronic kidney injury15; (5) new starts for chronic dialysis are now predominantly in the elderly population16–18; and (6) the kidney’s drug-metabolizing capacity declines in older people.
Clinical Relevance of Aged Podocytes
Characteristic physiologic, histologic, and molecular changes to the aged kidney have been well described and reviewed, and will not be summarized here.7,8,19–21 This review focuses on aging of the glomerular podocyte. The glomerular structure is a microcosm within the kidney, where its different cell types interact and communicate extensively. Aging results in age-dependent glomerulosclerosis and an accompanying decline in GFR, caused by changes in number, structure, and function to all four resident glomerular cell types.5,7,22,23,24–26 Compelling evidence points toward podocytes as the critical cell type in aging. Floege et al.27 were the first to describe age-related glomerulosclerosis as a podocyte disorder, subsequently confirmed by others.25,28 Clinically, the development of age-associated glomerulosclerosis is accompanied by a predictable decrease in podocyte number and density with advancing age.28–30 This contributes, for example, to a reduced life expectancy of transplanted kidneys,31 and is why glomerular diseases have a higher likelihood for a poorer outcome in older patients than in younger ones.12,32
But there are still many connections between age and podocyte function that have not been sufficiently studied. These include the direct pleiotropic cellular effects of therapeutics such as corticosteroids, or renin-angiotensin-aldosterone system inhibitors that alter biologic process/pathways in young podocytes; genetic predispositions such as the apolipoprotein L1 genotype; and infection with certain viruses, such as HIV and possibly severe acute respiratory syndrome coronavirus 2. Older age also negatively affects the normal function of podocytes in their role as important players in the health of other glomerular cell types, including making survival factors for glomerular endothelial cells,33 in regulating parietal epithelial cell differentiation,34 and in recruiting mesangial cells.
Finally, a critical clinical question that requires mechanistic understanding is how podocyte aging and disease intersect. Experimentally, disease-induced podocyte loss compounds the pre-existing podocyte depletion in the aged kidney, associated with increased glomerulosclerosis.32 As such, from a clinical treatment standpoint, it is important to know whether changes in gene expression during podocyte aging overlap or are distinct from gene expression changes in common podocyte diseases. One analysis comparing data from published studies showed the regulated pathways were largely distinct from those perturbed in diabetic kidney disease and focal segmental glomerulosclerosis.35 Yet, more studies are clearly needed to identify distinct and common pathways between aging and disease.
In this review, we do not discuss the normal structure or function of podocytes, but rather examine what is specifically known about podocyte aging. As expected, podocyte aging is a synthesis of pathways involved in the general aging process, with additional cell type–specific aspects.1 We limit our discussion to published data and do not apply information extrapolated from studies on general aging to podocyte aging.
Approaches to Study Podocyte Aging
Humans
Studying the biology of kidney aging in humans is the gold standard to assure clinical and therapeutic usability of the data. Yet, human studies face numerous barriers. First, longitudinal analyses are not possible because only a single time point of tissue collection is typically available from a given person. Second, proper sex- and race-matched controls are mostly limited. Third, comorbid conditions (such as obesity, diabetes, or hypertension) or environmental factors (such as exposure to cigarette smoke or air pollution) increase with age and confound changes directly attributed to aging alone,36 as do epigenetic changes.37 Fourth, although approaches have been optimized for small sample sizes, collecting sufficient kidney tissue for mRNA and protein studies is still challenging.
Over the years, several creative attempts have been made to overcome some of these barriers for human studies. Wiggins and Rule used implantation biopsies from donor kidneys for transplantation or radical nephrectomy for renal tumors.8,28,38,39 Bertram et al. used contrast-enhanced computed tomography and a renal biopsy to determine glomerular density and glomerular and tubular area in living people.21 Finally, the Kidney Precision Medicine Project is optimizing conditions to acquire material for cutting-edge technologies such as single-nucleus RNA-sequencing (RNA-seq).40,41 These workflows will become available to study aging in healthy people.
Animal Models
Because use of human kidneys for longitudinal and interventional studies of aging and comorbid conditions remains a challenge, animal models continue to be essential to advance understanding of podocyte aging. Yet, it is important to state that the interpretation of experimental data with respect to human aging always requires careful crossvalidation in human kidneys. Over the past few years, it has become increasingly clear there are significant species-dependent differences in kidney development and disease, and it is likely differences also exist for aging. Early important insights into podocytes and kidney aging were obtained from studying rats.30,42,43 Marmosets,44 canines,45 and minipigs46 have also been studied. However, largely because of the availability of transgenic animals, mice, and to a lesser extent, rats are now commonly used experimentally to study kidney and podocyte aging.
Importantly, several caveats need to be considered when using mice or rats for podocyte studies of aging (and for studies of other kidney cell types). First is the consideration of age. For example, mice aged for 12 months, 18 months, or 24 months correspond to human aged 42.5 years, 56 years, and 69 years, respectively.47,48 Thus, because a 12-month-old mouse is definitely not considered aged, and an 18-month-old mouse is really only middle-aged, we recommend only mice that are ≥24 months be considered a true model of “human” kidney aging. Rats that are aged 28 months are equivalent to a 70-year-old human.49 Second, species- and strain-dependent glomerular physiology affect age-associated glomerular changes. Similar to findings in aged humans reported by Rule et al.,50 aged mice typically do not develop proteinuria.51–53 They exhibit a 35% decrease their GFR between age 4 months and 24 months, at a rate of 6.3 ml/min per 1.73m2 per decade, but show only a very small increase in serum creatinine.7,54 In contrast, aged rats develop more overt changes, including proteinuria and lower GFR.55 Importantly, different mouse strains also are not equivalent with respect to aging. C57BL/6 mice are most susceptible to the development of glomerulosclerosis, spontaneous amyloidosis, and proteinuria as they age.56 Conversely, mice on a mixed genetic background have podocyte depletion by age 12 months,57,58 which is earlier in onset than in C57BL/6 mice. The reasons for these differences are unknown. Third, as mentioned above, an animal’s sex affects aging, with male sex typically associated with an earlier kidney aging phenotype, for reasons that are not well understood. One possibility lies in kidney morphology/homeostasis. Females have more cortical glomeruli per unit volume than males.59 Similarly, nitric oxide production, which affects renal hemodynamics, is better preserved in aged females than males.60,61 When males are housed together, they tend to fight more than females, often resulting in subclinical low-grade inflammation from wounds, which can affect aging. Fourth, housing conditions specific to a given vivarium can influence all strains, due to husbandry, diet, infectious and parasitic diseases, and differences in the microbiome. Fifth, the high costs for healthy aging of mice and rats for up to 24–27 months can be prohibitive and prevent exploratory experiments regularly performed in other scientific areas. Thus, considering models exhibiting accelerated podocyte aging are a more cost-effective possibility. These include genetically manipulated mice, such as those with reduced telomere lengths,62 with overexpression of p16 to induce senescence,63 or Inducible Changes in the Epigenome mice that have undergone induced DNA breaks to drive epigenetic changes that accelerate aging.64(preprint),65–67 A more physiologic alternative is inducing premature birth, which results in a low nephron mass that is more susceptible to aging effects.68 Finally, one might also consider replicating in models comorbid conditions that affect a large proportion of the human population, such as hypertension and/or obesity.
Cell Culture Models
Despite obvious limitations, cultured primary and immortalized human and mouse podocytes have advanced our understanding of podocyte biology and have allowed mechanistic dissections that require large number of pure cell populations.69,70 Unfortunately, with respect to aging, a standardized model to study this process in vitro is lacking. The most straightforward approach so far has been comparing primary podocytes harvested from aged and young mice. For example, this has provided information on the induction of replication-associated telomere shortening,71,72 genotoxic stress,73–76 and how low–molecular weight compounds77 or overexpression of senescent pathway regulators78 can induce senescence-like states. Finally, stem cell–derived kidney cells/organoids have offered a new model for study of kidney development and disease, although there are no published findings yet toward understanding podocyte aging.
Sex as a Biologic Variable
Over all, aging-related changes in the kidney typically occur earlier in males than females.60,79–82 Indeed, studies are increasingly identifying sex differences between male and female kidney cell types, such as proximal tubule cells or cells of the renin lineage.83–86 Unfortunately, in the current gold standard single cell/nuclear RNA-seq studies, glomerular cell types—and podocytes in particular—are numerically underrepresented unless they are specifically enriched.87–89 To circumvent this and obtain more detailed information on podocyte aging, we recently performed a study using an inducible podocyte-specific reporter mouse (NPHS2-rtTA|tetO-cre|tdTomato) to permanently label the podocyte population in juvenile animals and follow them through their lifetime.35 A comparison of labeled podocytes from young mice (aged 3 months, corresponding to approximately 20 years in humans)47,48 with podocytes from aged mice (22–24 months, corresponding to approximately 70+ years in humans)47,48 identified nearly 2500 genes with significant differential expression between young and aged podocytes.35 However, when analyzed with respect to sex differences, age differences explained most of the expression variance and overwhelmed the small expression variance of 7.2% associated with sex. Thus, this study suggests that in mouse podocytes, age-related changes may be largely independent of sex. However, further studies are needed to address this in more detail using human glomeruli, additional mouse strains, and a wider range of ages.
Podocyte Lifespan versus Healthspan
As we review the molecular and cellular changes to aged podocytes and their physiologic and pathologic consequences, consideration needs to be given to distinguishing a podocyte’s lifespan and healthspan (Figure 1). We view the lifespan of the individual podocyte as the length of time it lives, which translates to the total number of podocytes comprising the glomerular filtration barrier. Lifespan is binary (a podocyte is living or not). Healthspan is the part of the podocyte’s life that is spent in good health and able to perform normal cellular and molecular functions to maintain its physiology, structure, and function. Healthspan is not binary, and is best considered a continuous variable, changing dynamically throughout life.
Figure 1.
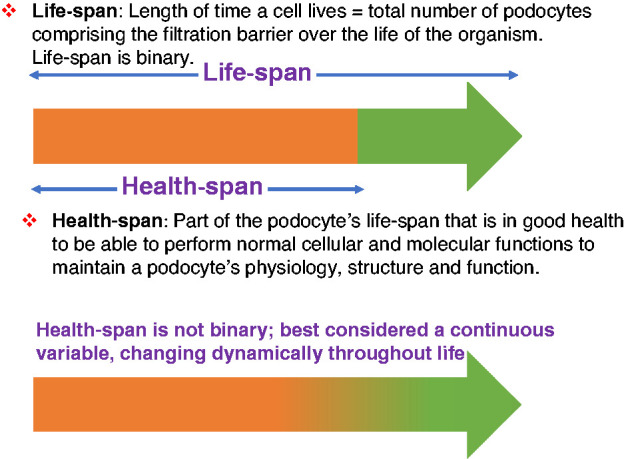
Podocyte lifespan and healthspan. The length of time that an individual podocyte lives is its lifespan (green arrow). Lifespan is binary, where cells are either alive or lost due to death and/or detachment. Conversely, healthspan (orange arrow) is the part of the podocyte’s lifespan spent in good health. It is not binary as it changes dynamically throughout life (gradient orange-green arrow).
Podocyte Depletion
It is generally accepted that terminally differentiated podocytes are unable to self-renew, yet some studies have shown postnatal nephron endowment with podocyte gain.90 Animal model studies have shown a direct correlation between podocyte depletion and glomerulosclerosis.91–93 Indeed, kidneys that exhibit glomerulosclerosis increasing with age50,94–96 also feature a decrease in absolute podocyte numbers (reduced lifespan) and density.23,30,32,43,57,58,97,98,99 It is important to note that this absolute loss of podocytes is accompanied by an increase in podocyte size (i.e., hypertrophy). Podocyte hypertrophy can partially compensate for the decreased podocyte numbers, yet the relative podocyte depletion is ultimately insufficient to match an overall reduction of podocyte density.29,100,101 In fact, podocyte density decreases in both aged rats43 and mice.23,32,58,97–99 Moreover, this effect is region specific, because the kidney’s cortical versus juxtamedullary compartments differ in age-related changes in podocyte numbers/density. Compared with young mice, aged mice have podocyte densities that are 45.6% and 39% lower in the cortex and juxtamedulla, respectively.32 This contrasts with the other glomerular cell types, where data lack consistency. Some studies have found the number of mesangial and glomerular endothelial cells increase on aging, so the density—the ratio between the glomerular volume and cell number—stays constant.30,102,103 Conversely, studies in aged Wistar/Lou rats have observed no change in mesangial hypercellularity,104 and research in Sprague-Dawley rats found a glomerular capillary loss with reduced proliferation.105
These findings from animal studies are supported by data in humans. Wiggins et al. reported 589±166 podocytes per glomerulus, with a density of 1166±310 per 106 µm3,106 which is similar to the 558 podocytes per glomerulus reported by Puelles et al.29 Moreover, aging is associated with both an absolute and relative depletion of podocytes.29 The podocyte reserve drops from more than 300 podocytes per 106 µm3 in young kidneys to less than 100 per 106 µm3 by age 70–80 years, resulting in an annual podocyte loss of approximately 0.9%.28 This age-dependent podocyte depletion has been shown to have high clinical relevance. A decrease in absolute podocyte number and density often result in glomerulosclerosis,102,107 and thus remains the best predictor for glomerulosclerosis risk in healthy aged kidneys.28,29,31,100 Moreover, podocyte number and integrity are also critical in transplanted kidneys,108 because kidney transplants with low podocyte endowment develop transplant glomerulopathy at a higher rate compared with those with normal podocyte density.109 Several mechanisms have been implicated with respect to the causes of podocyte depletion in aged kidneys.
Apoptosis
Although apoptosis occurs in aging podocytes (Figure 2),110,111 its overall contribution to podocyte depletion is technically difficult to quantify over a lifespan.112 Established assays (e.g., terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling) can capture only a short time span in the apoptotic program. Similarly, the underlying causes and inducers of age-dependent podocyte apoptosis are poorly understood. Transcriptomic data from aged podocytes showed both a decrease in survival factors and an increase in proapoptotic pathways (Table 1).35 Yet, their mechanistic contributions have not been experimentally tested. Other candidates can be extrapolated from published studies in nonaged podocytes.112 These include the age-dependent decrease of nephrin and podocin that likely reduces podocyte survival113; the age-dependent decrease of Wilms’ Tumor 1 that leads to increased WNT/β-catenin signaling, which in turn can induce podocyte death114; the age-dependent increase of TGF-β that directly induces apoptosis115; and the balance in autophagy, because age-dependent decrease in autophagy induces apoptosis and restoring autophagy therefore attenuates it.116
Figure 2.

Changes to podocytes in the aged human and rodent kidney. (A) Schema of the microcosm of cell types comprising a young glomerulus. (B) Glomerular size (hypertrophy) is increased in the aged kidney (ruler shown below). Podocytes detach, hypertrophy, efface, apoptose, and undergo pyroptosis. Parietal epithelial cells (PECs) become activated and decrease in number due to loss, mesangial cells are expanded, and glomerular endothelial cells become injured. Created with BioRender.com.
Table 1.
Differences in podocyte gene expression between young and old mice35
| Biologic Process | Genes Increased | Gene Decreased |
|---|---|---|
| Extracellular matrix proteins | Mmp13, Fn1 | Sema5a, Col4a1, Col4a5, Col4a4, Col18a1, Lamb2, Lamc1, Npnt, Dag1, Myh11 |
| Apoptosis | p53 pathway, TNF pathway, Faslg, Bim, Prf1erforin, Apaf1, Casp, Casp4. | Pou3f3, Fgfr2, Wt1, Erbb3, c-Met, Smad6 |
| Survival factors | Tgfb1, Tgfb3 | Pou3f3, Fgfr2, Wt1, Erbb3, c-Met, Egfr, Smo |
| Pyroptosis | Casp1, Nlrc4, Aim2, Gsdmd. | None |
| Cell cycle regulation | Tgfb1, Myc, Ccnd3, p21, Notch2, Schlafen-1Slfn1 | Fgfr2, Edn3, c-Met, Tbx3, Pkd2, Sox4, Meis2 |
| Hypertrophy | Akap13, Rgs2, P2rx4 | Lmcd1, Igfbp5 |
| Inflammasome and inflammation | Nlrp3LRP3, Casp1, Casp4, Nlrc4, Naip2, Naip5, and Naip6, Tnfa, multiple interferon genes, Il6, Il2, multiple complement component genes, Nfkb1, Nfkb2 | Nlrp6 |
| Senescence and SASPs | Irf5, Cebpb, and Jdp2, Ccl2, Ccl3, Ccl5, Cc8, Mmp12 Mmp13 | Igfbp5, Igfbp7 |
| Podocyte canonical and slit diaphragm genes | Nhsp1, Nhsp2, Lmx1B, Cdkn1c, Wt1, Podxl, Synpo,Vegfa, Cd2ap, Actn4, Ptpro, Trpc6 | |
| Tight junction genes | Vasp, Amica1 | Tjp1, Pard3, Pard6, Nedd4l, Pkci, multiple occludins, multiple claudins (including Cldn4, Cldn8), Cgn, Magi1, Magi2 |
| Transcription factors | Cebpa, Cebpb, Prdm1, Nr4a3, Tbx21, Bach1, Tcf7, Myc, Irf4, Runx3, Stat4, Nfkb2, Fosl2, Bcl6, Nfkb | Hnf1b, Grhl2, Ssim1, Lrx1, Creb3l1, Zbtbl6, Osr2, Foxo4, Pax8 and Tref1, Hsp27, Foxc2, Arf2, Itga3, Yap, Coro2b, Trpc6, Ptk2 |
| Actin and regulators, motility | Was, Wipf1, Flna, Evl, Myo9b, Myo5a | Kank1-Kank4, Dsp, Parva, Syne2, Myh10, Tpm1, Emp2, Myh14, Gja5, Vil1 |
| Crosstalk with other cells | Il1a/bIl1a, Il1b, C1qa, C1qb, Tgfb1, Mmp13, Cxcl16, Ccl4, Ccl5, Tnf, Vim, Ifng, Il18, Vcam1, Sema4d | Receptor ligand pairs: Vegf, Fgf, Wnt, Bmp, Egf, Erbb4, Slit2, Robo2 |
| Mitochondrial OXPHOS | Ncf1, Ncf2, Tlr2, Aif1, Nox2, P2rx7, Tnf, Prkcd Syk, Nrros | Complex I (Ndufa5, Ndufs2, Ndufs4 and Ndufb8), Complex II (Sdha, Sdhd, and Sdhc), Complex III (e.g., Uqcrb, Uqcrh, Uqcr11), Complex IV Cytochrome C oxidase components (e.g., Cox5b, Cox6c, Cox7b) |
| Glycolysis | Hk2, Hk3, Eno3 | Pfkm, Fbp1, Gpd1, Aldob |
| Lipids | Cd36, Apoe, Apobr, Soat1, Ptgs2, Fabp5, Ldlr, Cerk, Dgat1 | Elovl7, Elovl6, Scd2, Acadsb, Asah2, Ppargc1a, Mgll, Hadh, Acadm, Acad11, Acat1, Acsm, Acox1, Eci3, Ehhadh |
| Cell-cell junction | Cdh1, Chd2, Cdh3, Chd4, Rab25 along with Grhl2 | |
| Integrin-collagen interactions | Collagen chains (Col4a3, Col4a4, Col4a5) and Integrins (Itga3, Itgb6 Itbg8) | |
| DNA damage | Samhd1, Bach1, Eepd1, Hmga1, Uhrf1, Dna2, Dclre1c, Taok3, Topbp1, Uvrag, Rad9a | Spire2, Xpc, Tnks1bp1 |
SASP, senescence-associated secreted phenotype; OXPHOS, oxidative phosphorylation.
Pyroptosis
Our recent study showed that genes required for pyroptosis, a highly inflammatory form of lytic programmed cell death, are markedly increased in aged podocytes (Table 1, Figure 2). Pyroptosis is characterized by the activation of caspase-1, with cells undergoing lysis, swelling, pore formation, blebbing, and DNA fragmentation, and is typically initiated by the inflammasome.117 It generates an inflammatory response due to the activation and release of IL-1β and IL-18, followed by IFN-γ production.117
Podocyte Detachment
The detection of podocytes in the urine is considered a sensitive marker of ongoing glomerular damage (Figure 2).118 Viable podocytes can be detected in the urine.119 But attempts to determine their actual number is likely to result in an underestimation, because detached podocytes can undergo secondary apoptosis in the urine by the process called anoikis.120 Moreover, detachment of one podocyte can result in subsequent detachment of neighboring podocytes.121–123 Hodgin et al. showed that compared with younger people, humans aged >60 years had a 3.3-fold increase in podocytes in the urine, and many of podocytes were also stressed.28 Mechanistically, RNA-seq data identified several candidate mechanisms for podocyte detachment (Table 1). These include the significant downregulation of several cell-cell junction genes and the weakening of integrin-collagen interactions.
DNA Damage and Cell Cycle Control
Terminally differentiated podocytes cannot proliferate and self-renew, in part because of increased cell cycle inhibitors.124 During aging, several inhibitory cell cycle regulatory genes are further increased,35 further limiting any possibility for cell cycle re-entry and self-renewal. Moreover, those podocytes that do manage to enter the cell cycle are unable to assemble an efficient mitotic spindle, thereby triggering a mitotic catastrophe and causing DNA damage.125 As in injured nonaged podocytes,126 it is likely such DNA damage is an additional mechanism that limits proliferation and increases loss in aged podocytes due to changes in genes associated with cell cycle regulation (Table 1).
Podocyte Hypertrophy
Normal glomerular function requires podocytes to cover the underlying glomerular basement membrane (GBM). Glomerular volume increases up to 700% between childhood and adulthood, at a rate of 2.7% per year, although the mechanisms have not been well defined.29 This is accompanied by an increase in total podocyte cell mass, at a rate of 1.8% per year. However, as described above, podocyte numbers decrease with age and glomerular volume increases, resulting in reduced podocyte density.28 This creates a mismatch between the number of podocytes and the increasing glomerular volume, and, because podocytes cannot proliferate to increase their number, they cannot compensate for this mismatch.124 Thus, to continue to cover the GBM during aging, podocytes rely on compensatory hypertrophy, defined as an increase in size due to an increase in protein to DNA ratio (Figure 2).30 Although the average human podocyte volume is 335±136 µm3,106 the podocyte nuclear volume increases at a rate of 2% per year, and average podocyte cell volume increases by 3.2% per year.28
Wiggins et al. showed in rats that to cover the increased surface area, aging podocytes undergo five stages of hypertrophy.30 Stage 1 is the normal adult podocyte state; stage 2 is characterized by nonstressed hypertrophy with normal function; stage 3 features adaptive hypertrophy, in which podocyte function remains normal despite signs of podocyte stress; stage 4 involves decompensated hypertrophy, in which podocytes are stressed, have decreased machinery for normal function, and start losing function and exhibiting signs of cellular stress; and stage 5, in which failure of podocyte hypertrophy to meet the size demand leads to both relative and absolute podocyte loss. Over time, this compensatory hypertrophy becomes maladaptive, leading to glomerulosclerosis.25,28,30 In particular, GBM lacking podocytes has been critically linked to the scarring process.127 It is also noteworthy that unlike podocytes, two other glomerular cell types, endothelial and mesangial cells, proliferate in response to increased glomerular size.28
At the molecular level, the mechanisms of podocyte hypertrophy in the context of aging are less well understood. It is unclear to what extent they parallel the mechanisms of hypertrophy in nonaged podocytes. However, using cues from hypertrophy studies in nonaged podocytes, candidate pathways for aged podocytes include increased p21, p27, TGF-β, mammalian target of rapamycin (mTOR), and IL-6.35 We acknowledge that aging is inherently confounded with other biologic pathways. These include growth of the glomerulus as it adapts to changes in filtration needs. To further dissect these age-dependent adaptations, future aging studies would benefit from including several time points over the lifespan of the kidney.
Changes in Canonical Podocyte Gene Expression
Canonical genes are defined as proteins that are constitutively active in a particular cell type and are required for its unique shape and function. Podocytes contain numerous canonical genes, many of which are highly specific to this terminally differentiated epithelial cell type. The normal functioning of the slit diaphragm as a size and charge barrier requires constitutive expression of a set of specific proteins.128 Slit diaphragms are highly specialized adherens junctions that contain unique membrane proteins, such as nephrin and podocin; typical adherens junction proteins, such as cadherins and catenins; and scaffold proteins such as ZO-1, CD2AP, and MAGI-2.129 Decreased levels of these proteins lead to impaired signaling to the actin cytoskeleton, resulting in weakening of the foot processes, proteinuria, and at times, glomerulosclerosis.130 The expression of several critical proteins that constitute the slit diaphragm is significantly reduced in aged podocytes (Table 1). For example, Wiggins et al.30 showed that in 24-month-old rats (approximately age 60 years in humans), expression for Tcf21, Nphs1, Col4a5, Glepp1, and Wt1 was strongly reduced. Similarly, podocytes of aged mice have decreased mRNA and protein expression of many critical slit diaphragm, tight junction, and other canonical podocyte genes (Table 1).23,30,35,58 Considering the role of these proteins in nonaged podocytes, it is likely that expression changes will biologically affect the healthspan of podocytes as they progressively age.
Foot Process Effacement and Podocyte Stress
In effacement, a characteristic feature of podocyte injury, podocytes undergo a flattening in shape. It is a dynamic, ATP-consuming signaling-based process that involves the rearrangement of the actin cytoskeleton and integrin–focal adhesion dynamics. Effacement is accompanied by loss of the interdigitating pattern of the foot processes between neighboring podocytes, loss of adherence to the GBM, and loss of the subpodocyte space between the podocyte cell body and the GBM.128,131,132 Foot process effacement has been associated with reduced GFR due to changes in the ultrafiltration coefficient Kf.133 The same process is observed in aged human kidneys, in which the remaining podocytes in sclerosed glomeruli tend to be effaced, likely contributing to a decrease in kidney function (Figure 2).
The causes of age-associated podocyte effacement are not fully elucidated, but clearly involve remodeling of the cytoskeleton. In fact, many proteins involved in podocyte effacement in different clinical settings are also altered during aging.23,35,134,135 RNA-seq data35 identified actin and several other regulators involved in the regulation of actin polymerization and cell motility as altered in aged podocytes6 (Table 1). One major player appears to be desmin, a type III intermediate filament protein. Desmin connects the nuclear and cell membranes with actin filaments and microtubules and contributes to cell morphology and mechanics. Although desmin is present in healthy podocytes, it is increased on podocyte stress, and in aging.27,30 More importantly, increased desmin in aged podocytes in rats and mice precedes glomerulosclerosis, suggesting its dysregulation is a critical prerequisite for podocyte dysfunction.27,30 Together, these studies suggest the cytoskeleton is significantly affected during the aging process, and these effects likely contribute to marked disturbances in normal podocyte physiology in the aged kidney.
Reduced Synthetic Function and Reduced Crosstalk with Other Glomerular Cell Types
Resident glomerular cells communicate to maintain glomerular integrity and function. For example, a critical podocyte synthetic function is the production of angiopoietic factors required for the survival and health of neighboring glomerular endothelial cells.136 This crosstalk is best exemplified by a meta-analysis of recent RNA-seq data.35 We identified 55 ligand-receptor interactions that were specifically enriched in healthy podocytes. Although the individual contributions of these to podocyte health require evaluation, it is noteworthy that 26 of these receptor-ligand pairs were significantly reduced in aged podocytes (Table 1). These data suggest that aging podocytes are characterized by the lack or reduced activity of autocrine and perhaps paracrine signaling networks, such as the VEGF signaling pathway (Figure 3).35 This may explain why glomerular endothelial cell numbers are reduced in aged rats.105 Surprisingly, the opposite, an upregulation of ligand-receptor pairs in aged podocytes, was not observed, suggesting podocyte aging is rather characterized by the disappearance of homeostatic signaling events. Further studies are needed to assess the biologic effects of reduced podocyte ligand-receptor pairs on podocytes themselves and on neighboring glomerular cells in aging.
Figure 3.

Loss of autocrine and paracrine functions in aged podocytes. (A) In young human and rodent glomeruli, podocytes produce ligands (represented by circles, squares, and rectangles) that bind to receptors on podocytes for autocrine effects to maintain podocyte health. Likewise, podocyte ligands bind receptors on neighboring parietal epithelial cells and fenestrated glomerular endothelial cells for paracrine effects that maintain their health. (B) In the aged mouse kidney, data support a decrease in podocyte autocrine and paracrine functions, which adversely affects the health of podocytes, parietal epithelial cells, and glomerular endothelial cells. This is further compounded by an increased inflammatory and senescent-associated secretory phenotype (SASP), which in aged podocytes exert an injurious autocrine effect on podocytes and paracrine effect on neighboring parietal epithelial cells and glomerular endothelial cells. Created with BioRender.com.
Mechanisms Contributing to Podocyte Aging
In previous sections, we discussed multiple cellular and molecular manifestations of podocyte aging. However, when addressing the question of why podocytes age, at least four considerations that should be heeded:
The mechanisms underlying the podocyte aging process likely involve common aging-related factors shared with many other cell types (e.g., senescence, loss of autoph agy, p53, mTOR, NAD, AMPK, and NF-κB), and podocyte-specific factors (e.g., changes to specific transcription factors such as Grh2). Some of those are known (see below); others remain to be discovered.
Podocyte aging is a multifactorial process/pathway and not simply linear. Yet, the interactions between the pathways, their hierarchy, and chronology are mostly unexplored.
Although not firmly established, podocyte aging is almost certainly accelerated by age-related comorbid stressors on podocytes, such as obesity, diabetes, and hypertension, and environmental factors. These might act using pathways shared between aging and injury (e.g., mTOR, p16, AMPK signaling) or they could be distinct, culminating in the ultimate phenotype.
Crosstalk likely exists between the different glomerular cell types. For example, aged podocytes might elicit changes in the endothelial cells and vice versa (Figure 3).
Below we discuss published candidate mechanisms and their contribution to podocyte aging (Figure 4).
Figure 4.

Summary of mechanisms and subsequent consequences in aged podocytes. Multiple mechanisms (listed in the red box) contribute to podocyte aging in humans and rodents. These molecular and cellular changes decrease the podocyte’s lifespan and healthspan each leading to physiologic and pathologic consequences. Created with BioRender.com.
Senescence, Senescence-associated Secreted Phenotypes, and Inflammasome
Cellular senescence is a permanent cell cycle arrest, with telomere shortening and macromolecule accumulation, and is multifactorial in origin.137 Senescence develops acutely as a safeguard to prevent damaged cells from proliferating and replicating damaged DNA. Because senescent cells are resistant to apoptosis, they tend not to be cleared by macrophages, but instead accumulate within an organ. Chronic accumulation of senescent cells is damaging to an organ, largely because these cells release proteins comprising the senescence-associated secreted phenotype and other factors, which in turn directly or indirectly damage the surrounding cells.138 Although short-term exposure to these proteins is beneficial to cellular regeneration in certain tissues, chronic exposure can induce tissue damage,139 and also induce senescence of neighboring cells.140 The clinical relevance of the accumulation of senescent cells in aged mice was best illustrated in the INK-ATTAC mouse model that specifically ablated senescent (i.e., p16INK4-positive) cells.141 These aged mice exhibited improved kidney health and a reduction of age-associated glomerulosclerosis.
Although the classic senescence markers p16INK4 and SA-ßgal have been detected occasionally in podocytes from healthy nonaged human glomeruli,142 they are clearly increased in healthy aged kidneys and in nonaged kidneys with superimposed glomerular or diabetic kidney disease.39,142,143 Similarly, podocyte senescence has also been demonstrated in aged mice.23,144–146 Our RNA-seq study of aged podocytes identified many senescence-associated genes, including 11 SASP genes (Table 1).35 Surprisingly, classic inflammation-associated genes, and components of the inflammasome pathway, were significantly increased (Table 1).35 Both Nfkb1 and Nfkb2 are increased in aged podocytes and have been shown to be involved in glomerular aging.147 Thus, it is likely they are the transcriptional drivers for this inflammatory phenotype. However, because there is little recruitment of immune cells into aging glomeruli,148 aging podocytes are likely in a state of sterile inflammation (i.e., inflammation not caused by infection).149,150
Mitochondrial Dysfunction, Oxidative Stress, and Reduced Metabolism
Mitochondria generate most of the chemical energy needed for cellular homeostasis, and their dysfunction is often coupled to an increase in reactive oxygen species (ROS), a hallmark of aging in many cell types. We reported that in mice aged 24 months, podocytes had extensive mitochondrial damage, including disruption of the inner mitochondrial membranes, loss of cristae, changes to the mitochondrial matrix density, outer mitochondrial rupture, and increased expression of the ROS marker Nox4.23 These changes were progressive, because mitochondria of 26-month-old mice were even more severely affected. A transcriptomic study35 of aged mouse podocytes showed global downregulation of genes involved in mitochondrial oxidative phosphorylation (OXPHOS), including mitochondrial complexes I–IV, and the upregulation of several genes known to be activated by ROS, supporting a concomitant increase in ROS production (Table 1).
In addition, key metabolic pathways, including OXPHOS, the citric acid cycle, fatty acid oxidation (FAO), and amino acid catabolism, were reduced in aged podocytes (Table 1).35 Such changes in metabolism are well described for a majority of aged cells. Yet, in podocytes, the focus has largely been on OXPHOS.151 Indeed, aged podocytes show dramatically reduced expression of enzymes involved in energy production via OXPHOS (Table 1). In particular, the peroxisome proliferator-activated receptor γ coactivator 1-α, a central regulator of mitochondrial biogenesis and OXPHOS,152,153 was significantly downregulated in aged podocytes, and may thus be responsible for the overall decline in the mitochondrial energy production in aged podocytes.
It has recently been reported that anaerobic glycolysis—not OXPHOS—is the predominant energy source of podocytes.154 Surprisingly, we did not observe significant age-dependent changes in genes of the glycolysis pathway.23 We hypothesize that a reduction in OXPHOS might render aged podocytes even more reliant on glycolysis for energy production. This may be even further aggravated by the observed downregulation of Fatty Acid Oxidation (FAO) genes (Table 1). Podocytes primarily oxidize fatty acids instead of glutamine or pyruvate,154 and inhibition of FAO has been shown to increase the susceptibility of podocytes to palmitic acid–induced cell death in diabetic nephropathy.155 A similar process may be at play in podocyte aging. Taken together, decreased energy sources and metabolism accompanied by increased ROS likely cause progressive injury to aging podocytes.
Transcriptional Changes
Transcriptional regulation is critical for normal podocyte development and homeostasis.156 In line with the observed DNA damage in aged podocytes, the transcription factor p53 is upregulated, although this has not been functionally evaluated. In contrast, the majority of other transcription factors studied decrease in aged podocytes. A virtual inference of protein activity by enriched regulon (VIPER) analysis,157 which studies transcription factor gene expression and their regulatory networks, showed that many candidate transcription factors, their regulators, and their downstream targets were significantly downregulated in the aging podocyte (Table 1).35 One of the best examples, the transcription factor CAAT enhancer-binding protein alpha (Cebpα), a member of the basic-region leucine zipper family of proteins, is highly expressed in young podocytes158 but decreases in 20-month-old mice.144 Functionally, podocyte-specific deletion of Cebpα in aged mice induces podocyte senescence (increased p16 and SA-ßgal staining), reduced podocyte numbers, and reduced expression of several canonical podocyte markers.144 Moreover, in vitro studies show that adriamycin-induced aging of immortalized mouse podocytes reduces Cebpα levels and that Cebpα overexpression reversed these changes.144 Although the precise downstream mechanisms are still unclear, the authors hypothesize that Cebpα acts via the AMPK-mTOR pathway. Finally, the RE1-silencing transcription factor protects podocytes from injury in aging mice.159 Taken together, a robust decrease in transcription factors and their downstream targets is likely a major cause underlying the morphologic and physiologic changes in aged podocytes.
Sirtuins
The sirtuins (Sirt) comprise a family of nicotinamide adenine dinucleotide–dependent deacetylases that are involved in several cellular processes such as autophagy, and act largely through transcription repression by deacetylation.160 Sirt1 and Sirt6 have been implicated in normal podocyte function. A decrease in Sirt1 expression augments podocyte aging,145 and knockdown of Sirt1 in mouse podocytes resulted in an overall reduction in the number of podocytes, increased glomerulosclerosis and albuminuria, and reduced levels of key podocyte proteins.145 The changes were accompanied by elevated expression of the senescent markers p16INK4a and p19ARF. The underlying mechanism for Sirt1 is unknown, but the authors speculated that the effect on podocyte aging is dysregulation of proliferator-activated receptor γ coactivator 1-α, FOXO3, FOXO4, and NF-kB. Yet, Sirt1 knockdown mice also show elevated urinary levels of the cellular oxidative stress marker 8-OHdG, suggesting the overall effects of reduced Sirt1 levels are caused by increased oxidative stress and mitochondrial injury, as has been observed in other cell types.161 The deletion of Sirt6 from mouse podocytes were performed in 7-month-old mice (not considered aged), and studies need to be analyzed in older cohorts to solidify the authors’ claim that Sirt 6 is critical for the podocyte aging process per se.162
Finally, the missing aspect of these studies is that no study actually shows an age-dependent downregulation of Sirt mRNAs. In fact, our transcriptomic analysis showed that the only change in sirtuins between old and young podocytes was an age-dependent upregulation of Sirt7 (1.9-fold). Although counterintuitive, this may be a protective/adaptive response, as Sirt7-deficient mice show an accelerated aging phenotype in other tissues.163 Finally, sirtuins have been implicated in the beneficial effects of calorie restriction via the regulation of autophagy, which are described below.
Reduced Autophagy
Autophagy is a healthy, biologic “housekeeping” process that helps cells maintain homeostatic balance between the synthesis, degradation, and recycling of cellular proteins and organelles, with the ultimate purpose of supplying nutrients for survival.164 Podocytes, as other postmitotic cells, have a very high basal autophagocytic activity, which is essential for energy homeostasis.165 When podocytes are unable to perform autophagy, damage occurs, resembling an aging phenotype. Moreover, because decreased autophagy enhances the extent of injury in podocyte diseases such as FSGS,165 it is likely that decreased autophagy is augmenting podocyte injury in aged patients with podocyte disorders.
This correlation is also supported by the podocyte-specific deletion of autophagy-related 5 (Atg5), one of the key regulatory genes in the autophagy process. These mice exhibit reduced autophagy and characteristics of enhanced aging, namely, a decline in podocyte numbers and accumulation of damaged organelles and protein aggregates.165 Surprisingly, this phenotype appears to be independent of one of the major regulators of autophagy, the mTOR complex. This pathway has been shown to inhibit autophagy98 and, as expected, mTOR activity is higher in podocytes of aged mice.98 In aged podocytes, expression of Prkaa2, which encodes a protein that is part of the AMPK complex, and Pkhd1166—two known inhibitors of mTOR activity—is downregulated, whereas Card11, an activator of mTOR signaling,167 is upregulated 6.9-fold.35 Yet, administering the mTOR inhibitor rapamycin to 26.5-month-old mice (approximately age 70+ years in humans)47,48 reduced mTOR activity, but did not alter podocyte density.98 Thus, further studies are needed to understand the mTOR-independent regulation of autophagy in aged podocytes and whether this can be harnessed toward antiaging approaches.
Mitigating Podocyte Aging
There is a large effort to identify treatments and drugs that mitigate the effects of aging in humans. In our opinion, alleviating or reversing podocyte aging in the healthy aged kidney should not be the focus for therapy development. Instead, we believe the goal should be maintaining and improving podocyte lifespan and healthspan in states of stress. As previously reported,32 this would allow one to interfere with the acceleration of the aging process mediated by diseases. Such diseases include a wide array of disorders that either directly injure podocytes (e.g., FSGS and diabetic kidney disease), glomerular diseases in which podocytes are secondarily injured (e.g., IgA nephropathy, hypertension, obesity, and vasculitis), and conditions (and treatment of conditions) that result in a reduction of kidney mass or function (e.g., as a result of surgery, transplantation of older kidneys, and CKD). Thus, mitigating aging in podocytes will likely (as has been seen in other cell types168) require different drug combinations or personalized, disease-specific therapies. To date, several approaches or compounds have been reported to reduce podocyte aging.
Calorie Restriction
This approach is the oldest and most robust way to extend rodent life span systemically. In the kidney, Wiggins et al.30 showed that calorie restriction prevented age-associated podocyte hypertrophy in rats. Moreover, aged calorie-restricted rats exhibited lower mRNA levels of Wt1, Podxl, Nphs1, and Glepp1 compared with age-matched rats given an ad libidum diet, and they also displayed upregulation of the podocyte stress marker desmin.30 Similarly, we reported that calorie restriction of male F344 rats during aging limited their podocyte loss compared with their counterparts fed an ad libidum diet.43 The precise mechanisms underlying the effects of calorie restriction on podocyte biology is unknown, as is whether those mechanisms differ from those described for other aging cell types. One possible link between calorie restriction and its effects in podocytes is the sirtuins, which (as mentioned above) have been implicated as effectors of calorie restriction.169
SS-31
The mitochondrial antioxidant elamipretide (SS-31) is a small cell-permeable synthetic peptide that is in clinical trials for heart failure, primary mitochondrial myopathy, and other mitochondrial diseases.170 SS-31 improves mitochondrial function by enhancing electron transfer through cytochrome c, improving OXPHOS coupling, increasing ATP synthesis, and reducing ROS production.171,172 Interestingly, it is also linked to changes in Sirt1 levels.173 To explore SS-31’s role in podocyte aging, 24-month-old mice were treated for 8 weeks with either SS-31 or vehicle23; SS-31 preserved podocyte mitochondrial integrity, lowered the expression of the ROS-generating enzyme NOX4, and increased expression of genes encoding NduFA and Cox IV, enzymes in the mitochondria electron transport chain. At the cellular level, SS-31 reduced podocyte hypertrophy and foot process effacement, but did not affect podocyte density. It also reduced the injury marker desmin and preserved the expression of synaptopodin. Together, these results provide a strong rationale for the possibility of interfering with age-dependent decline in podocyte function. The most impressive aspect of this study was finding that despite administering the treatment to aged mice, it was still possible to preserve the remaining mitochondrial integrity and significantly improve podocyte health.
Rapamycin
The mTOR pathway regulates many essential cellular processes, including translation, transcription, and autophagy. Inhibiting mTOR induces autophagy, thereby sustaining cell survival under conditions of nutrient deprivation. It has been shown that increased mTOR signaling in podocytes causes disease and augments injury, whereas `reducing it specifically in podocytes limits injury.164 However, as mentioned above, rapamycin treatment had no effect on podocyte density in aged mice.98 However, it is noteworthy that, as in injury scenarios, inhibiting mTOR signaling can revert podocyte hypertrophy.174
Senolytics
The cancer drugs quercetin and dasatinib inhibit a broad spectrum of protein kinases and tyrosine kinases and have been shown to reduce markers of aging.175–177 This effect was also extended to the podocytes, in which administration of dasatinib and quercetin in a diabetes model of senescence improved kidney function, increased expression of WT1, and decreased p16 levels.178
Endothelin Receptor
Darusentan is a selective endothelin ETA receptor antagonist that is clinically evaluated as a treatment for congestive heart failure and hypertension. Interestingly, treating 23-month-old male Wistar rats with darusentan for 28 days resulted in substantial recovery of podocyte structure, accompanied by a decrease in expression of p21 and MMP9, but no changes in blood pressure or renin levels.179
Novel Targets
Our recent RNA-seq analysis comparing young with aged podocytes,35 using the Connectivity Map database,180 identified potential interventions to reverse the aged podocyte phenotype. This analysis yielded candidate genes that have been implicated in podocyte diseases. For example, Stat5 has been shown to be upregulated in FSGS.181 The analysis also identified therapeutics that have previously been used, such as the poly (ADP-ribose) polymerase inhibitor PJ-34, which blocks ROS species generation and protects podocytes in diabetic nephropathy.182
Because podocyte depletion is a critical pathophysiologic process in the aged kidney, one might ask if replacement can be augmented from the putative adult podocyte progenitors, glomerular parietal epithelial cells,34,58,183,184 and renin-producing cells.99 Both progenitors are detected in individual aged glomeruli in which podocytes are depleted, albeit in low numbers.34,58,99,183,184 It is noteworthy that these aged glomeruli have a normal morphology with no scarring. However, both types of podocyte progenitors decline in number with advancing age, and they acquire additional adverse changes, such as mesenchymal transformation, that limit their progenitor function.97,146 Thus, podocyte progenitor augmentation is not fully capable of countering the age-associated decrease in podocyte numbers. On the basis of this observation, boosting adult podocyte progenitors should be considered a target for future therapies to improve podocyte numbers.
Future Directions
Although certain pathways, such as senescence and genomic instability, are considered general hallmarks of aging, recent single-cell studies show changes occurring across multiple organs, and within each are cell type–specific changes.185 Yet, a unique podocyte aging signature is not currently available. Fortunately, the emergence of single-cell technologies and an associated ever-evolving analysis toolbox are providing the opportunity to study cell types such as the podocyte that are less abundant and to resolve the aging process at an unprecedented level of detail.
To understand the molecular and cellular mechanisms of podocyte aging, future studies should integrate longitudinal transcriptomic, proteomic, metabolomic, and epigenetic changes, and translate experimental studies to clinical settings. This would result in the identification of specific aging signatures for podocytes and their temporal profile. Such signatures will likely include pathways implicated in the general aging process (e.g., insulin/IGF, p53, mTOR, AMPK, and NF-κB147,186,187), and podocyte-specific ones. A holistic analysis of these pathways will allow researchers to determine if and how aging in podocytes differs from that of other cell types and whether developing therapeutics specifically targeting aged podocytes is possible. Are the pathways controlling podocyte lifespan similar to or different from those of podocyte healthspan?
A wide-open opportunity exists to fill an important clinical gap, namely our understanding of the mechanistic intersection between podocyte disease and aging. Definitive data remain scarce for determining if disease-related stressors might hasten natural podocyte aging,32 and/or if aging lowers the threshold to disease-induced podocyte injury. In practical terms, this means the efficacy of currently used clinical therapeutics for podocyte disorders should be thoroughly evaluated in elderly adults. This is particularly important if, as we suspect, pathways are common to podocyte injury and aging. Such knowledge will have substantial therapeutic implications in clinical practice for the elderly patient population with superimposed glomerular disease. Finally, new therapeutics limiting or even reversing the rate of podocyte aging might have beneficial effects in podocyte diseases that lack effective treatment options.
Disclosures
A. Shaw reports being empl oyed by Genentech; reports having an ownership interest in Roche; and reports other interests/relationships with NephCure. J. Pippin reports having patents and inventions with University of Washington TechTransfer Invention Licensing Unit. All remaining authors have nothing to disclose.
Funding
This work is supportedby National Institute of Diabetes and Digestive and Kidney Diseases grants 5 R01 DK 056799-10, 5 R01 DK 056799-12, and 1 R01 DK097598-01A1.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
References
- 1.da Costa JP, Vitorino R, Silva GM, Vogel C, Duarte AC, Rocha-Santos T: A synopsis on aging-Theories, mechanisms and future prospects. Ageing Res Rev 29: 90–112, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lang J, Katz R, Ix JH, Gutierrez OM, Peralta CA, Parikh CR, et al. : Association of serum albumin levels with kidney function decline and incident chronic kidney disease in elders. Nephrol Dial Transplant 33: 986–992, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pottel H, Hoste L, Dubourg L, Ebert N, Schaeffner E, Eriksen BO, et al. : An estimated glomerular filtration rate equation for the full age spectrum. Nephrol Dial Transplant 31: 798–806, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Denic A, Lieske JC, Chakkera HA, Poggio ED, Alexander MP, Singh P, et al. : The substantial loss of nephrons in healthy human kidneys with aging. J Am Soc Nephrol 28: 313–320, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoy WE, Douglas-Denton RN, Hughson MD, Cass A, Johnson K, Bertram JF: A stereological study of glomerular number and volume: Preliminary findings in a multiracial study of kidneys at autopsy. Kidney Int Suppl 63: S31–S37, 2003 [DOI] [PubMed] [Google Scholar]
- 6.Abdulkader RCRM, Burdmann EA, Lebrão ML, Duarte YAO, Zanetta DMT: Aging and decreased glomerular filtration rate: An elderly population-based study. PLoS One 12: e0189935, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denic A, Glassock RJ, Rule AD: Structural and functional changes with the aging kidney. Adv Chronic Kidney Dis 23: 19–28, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hommos MS, Glassock RJ, Rule AD: Structural and functional changes in human kidneys with healthy aging. J Am Soc Nephrol 28: 2838–2844, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang HC, Fogo AB. Fibrosis and renal aging. Kidney Int Suppl 4: 75–78, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coresh J, Selvin E, Stevens LA, Manzi J, Kusek JW, Eggers P, et al. : Prevalence of chronic kidney disease in the United States. JAMA 298: 2038–2047, 2007 [DOI] [PubMed] [Google Scholar]
- 11.Ge S, Nie S, Liu Z, Chen C, Zha Y, Qian J, et al. : Epidemiology and outcomes of acute kidney injury in elderly Chinese patients: A subgroup analysis from the EACH study. BMC Nephrol 17: 136, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cui Z, Zhao J, Jia XY, Zhu SN, Zhao MH: Clinical features and outcomes of anti-glomerular basement membrane disease in older patients. Am J Kidney Dis 57: 575–582, 2011 [DOI] [PubMed] [Google Scholar]
- 13.Reichert LJ, Koene RA, Wetzels JF: Prognostic factors in idiopathic membranous nephropathy. Am J Kidney Dis 31: 1–11, 1998 [DOI] [PubMed] [Google Scholar]
- 14.Gerstoft J, Balsløv JT, Brahm M, Brun C, Jørgensen F, Jørgensen HE, et al. : Prognosis in glomerulonephritis. II. Regression analyses of prognostic factors affecting the course of renal function and the mortality in 395 patients. Calculation of a prognostic model. Report from a Copenhagen study group of renal diseases. Acta Med Scand 219: 179–187, 1986 [PubMed] [Google Scholar]
- 15.Kim MG, Yang J, Ko YS, Lee HY, Oh SW, Cho WY, et al. : Impact of aging on transition of acute kidney injury to chronic kidney disease. Sci Rep 9: 18445, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurella M, Covinsky KE, Collins AJ, Chertow GM: Octogenarians and nonagenarians starting dialysis in the United States. Ann Intern Med 146: 177–183, 2007 [DOI] [PubMed] [Google Scholar]
- 17.Bowling CB, Sharma P, Fox CS, O’Hare AM, Muntner P: Prevalence of reduced estimated glomerular filtration rate among the oldest old from 1988–1994 through 2005–2010. JAMA 310: 1284–1286, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saran R, Robinson B, Abbott KC, Agodoa LYC, Bhave N, Bragg-Gresham J, et al. : US Renal Data System 2017 Annual Data Report: Epidemiology of kidney disease in the United States. Am J Kidney Dis 71[Suppl 1]: A7, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Sullivan ED, Hughes J, Ferenbach DA: Renal aging: Causes and consequences. J Am Soc Nephrol 28: 407–420, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmitt R, Melk A: Molecular mechanisms of renal aging. Kidney Int 92: 569–579, 2017 [DOI] [PubMed] [Google Scholar]
- 21.Hughson MD, Hoy WE, Bertram JF: Progressive nephron loss in aging kidneys: Clinical-structural associations investigated by two anatomical methods. Anat Rec (Hoboken) 303: 2526–2536, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iwamoto M, Nakahira Y, Kimpara H: Development and validation of the Total HUman Model for Safety (THUMS) toward further understanding of occupant injury mechanisms in precrash and during crash. Traffic Inj Prev 16: S36–S48, 2015 [DOI] [PubMed] [Google Scholar]
- 23.Sweetwyne MT, Pippin JW, Eng DG, Hudkins KL, Chiao YA, Campbell MD, et al. : The mitochondrial-targeted peptide, SS-31, improves glomerular architecture in mice of advanced age. Kidney Int 91: 1126–1145, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Regan CM: Rapid drug haptenization procedure: Application to gentamicin and quinidine. J Pharm Pharmacol 38: 834–836, 1986 [DOI] [PubMed] [Google Scholar]
- 25.Wiggins JE: Aging in the glomerulus. J Gerontol A Biol Sci Med Sci 67: 1358–1364, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson S, Brenner BM: Effects of aging on the renal glomerulus. Am J Med 80: 435–442, 1986 [DOI] [PubMed] [Google Scholar]
- 27.Floege J, Hackmann B, Kliem V, Kriz W, Alpers CE, Johnson RJ, et al. : Age-related glomerulosclerosis and interstitial fibrosis in Milan normotensive rats: a podocyte disease. Kidney Int 51: 230–243, 1997 [DOI] [PubMed] [Google Scholar]
- 28.Hodgin JB, Bitzer M, Wickman L, Afshinnia F, Wang SQ, O’Connor C, et al. : Glomerular aging and focal global glomerulosclerosis: A podometric perspective. J Am Soc Nephrol 26: 3162–3178, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puelles VG, Cullen-McEwen LA, Taylor GE, Li J, Hughson MD, Kerr PG, et al. : Human podocyte depletion in association with older age and hypertension. Am J Physiol Renal Physiol 310: F656–F668, 2016 [DOI] [PubMed] [Google Scholar]
- 30.Wiggins JE, Goyal M, Sanden SK, Wharram BL, Shedden KA, Misek DE, et al. : Podocyte hypertrophy, “adaptation,” and “decompensation” associated with glomerular enlargement and glomerulosclerosis in the aging rat: Prevention by calorie restriction. J Am Soc Nephrol 16: 2953–2966, 2005 [DOI] [PubMed] [Google Scholar]
- 31.Naik AS, Afshinnia F, Cibrik D, Hodgin JB, Wu F, Zhang M, et al. : Quantitative podocyte parameters predict human native kidney and allograft half-lives. JCI Insight 1: 86943, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneider RR, Eng DG, Kutz JN, Sweetwyne MT, Pippin JW, Shankland SJ: Compound effects of aging and experimental FSGS on glomerular epithelial cells. Aging (Albany NY) 9: 524–546, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dimke H, Maezawa Y, Quaggin SE: Crosstalk in glomerular injury and repair. Curr Opin Nephrol Hypertens 24: 231–238, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Romoli S, Angelotti ML, Antonelli G, Kumar Vr S, Mulay SR, Desai J, et al. : CXCL12 blockade preferentially regenerates lost podocytes in cortical nephrons by targeting an intrinsic podocyte-progenitor feedback mechanism. Kidney Int 94: 1111–1126, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Eng DG, Kaverina NV, Loretz CJ, Koirala A, Akilesh S, et al. : Global transcriptomic changes occur in aged mouse podocytes. Kidney Int 98: 1160–1173, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anton B, Vitetta L, Cortizo F, Sali A: Can we delay aging? The biology and science of aging. Ann N Y Acad Sci 1057: 525–535, 2005 [DOI] [PubMed] [Google Scholar]
- 37.Kane AE, Sinclair DA: Epigenetic changes during aging and their reprogramming potential. Crit Rev Biochem Mol Biol 54: 61–83, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Denic A, Ricaurte L, Lopez CL, Narasimhan R, Lerman LO, Lieske JC, et al. : Glomerular volume and glomerulosclerosis at different depths within the human kidney. J Am Soc Nephrol 30: 1471–1480, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melk A, Schmidt BM, Vongwiwatana A, Rayner DC, Halloran PF: Increased expression of senescence-associated cell cycle inhibitor p16INK4a in deteriorating renal transplants and diseased native kidney. Am J Transplant 5: 1375–1382, 2005 [DOI] [PubMed] [Google Scholar]
- 40.de Boer IH, Alpers CE, Azeloglu EU, Balis UGJ, Barasch JM, Barisoni L, et al. ; Kidney Precision Medicine Project : Rationale and design of the Kidney Precision Medicine Project. Kidney Int 99: 498–510, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lake BB, Chen S, Hoshi M, Plongthongkum N, Salamon D, Knoten A, et al. : A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nat Commun 10: 2832, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baylis C, Corman B: The aging kidney: Insights from experimental studies. J Am Soc Nephrol 9: 699–709, 1998 [DOI] [PubMed] [Google Scholar]
- 43.Zhang J, Hansen KM, Pippin JW, Chang AM, Taniguchi Y, Krofft RD, et al. : De novo expression of podocyte proteins in parietal epithelial cells in experimental aging nephropathy. Am J Physiol Renal Physiol 302: F571–F580, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee HJ, Gonzalez O, Dick EJ, Donati A, Feliers D, Choudhury GG, et al. : Marmoset as a model to study kidney changes associated with aging. J Gerontol A Biol Sci Med Sci 74: 315–324, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cianciolo RE, Benali SL, Aresu L: Aging in the canine kidney. Vet Pathol 53: 299–308, 2016 [DOI] [PubMed] [Google Scholar]
- 46.Kangawa A, Nishimura T, Nishimura T, Otake M, Enya S, Yoshida T, et al. : Spontaneous age-related histopathological changes in microminipigs. Toxicol Pathol 47: 817–832, 2019 [DOI] [PubMed] [Google Scholar]
- 47.Fox JG: The Mouse in Biomedical Research: Normative Biology, Husbandry, and Models, 2nd Ed., Amsterdam, Boston, Elsevier, 2007 [Google Scholar]
- 48.Dutta S, Sengupta P: Men and mice: Relating their ages. Life Sci 152: 244–248, 2016 [DOI] [PubMed] [Google Scholar]
- 49.Sengupta P: The laboratory rat: Relating its age with human’s. Int J Prev Med 4: 624–630, 2013 [PMC free article] [PubMed] [Google Scholar]
- 50.Rule AD, Amer H, Cornell LD, Taler SJ, Cosio FG, Kremers WK, et al. : The association between age and nephrosclerosis on renal biopsy among healthy adults. Ann Intern Med 152: 561–567, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ackert-Bicknell CL, Anderson LC, Sheehan S, Hill WG, Chang B, Churchill GA, et al. : Aging research using mouse models. Curr Protoc Mouse Biol 5: 95–133, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsaih SW, Pezzolesi MG, Yuan R, Warram JH, Krolewski AS, Korstanje R: Genetic analysis of albuminuria in aging mice and concordance with loci for human diabetic nephropathy found in a genome-wide association scan. Kidney Int 77: 201–210, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng F, Plati AR, Potier M, Schulman Y, Berho M, Banerjee A, et al. : Resistance to glomerulosclerosis in B6 mice disappears after menopause. Am J Pathol 162: 1339–1348, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schock-Kusch D, Geraci S, Ermeling E, Shulhevich Y, Sticht C, Hesser J, et al. : Reliability of transcutaneous measurement of renal function in various strains of conscious mice. PLoS One 8: e71519, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hard GC, Johnson KJ, Cohen SM: A comparison of rat chronic progressive nephropathy with human renal disease-implications for human risk assessment. Crit Rev Toxicol 39: 332–346, 2009 [DOI] [PubMed] [Google Scholar]
- 56.Schmitt R, Jacobi C, Susnik N, Broecker V, Haller H, Melk A: Ageing mouse kidney: Not always the SAME old story. Nephrol Dial Transplant 24: 3002–3005, 2009 [DOI] [PubMed] [Google Scholar]
- 57.Wanner N, Hartleben B, Herbach N, Goedel M, Stickel N, Zeiser R, et al. : Unraveling the role of podocyte turnover in glomerular aging and injury. J Am Soc Nephrol 25: 707–716, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kaverina NV, Eng DG, Miner JH, Pippin JW, Shankland SJ: Parietal epithelial cell differentiation to a podocyte fate in the aged mouse kidney. Aging (Albany NY) 12: 17601–17624, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lindeman RD, Goldman R: Anatomic and physiologic age changes in the kidney. Exp Gerontol 21: 379–406, 1986 [DOI] [PubMed] [Google Scholar]
- 60.Baylis C: Sexual dimorphism in the aging kidney: Differences in the nitric oxide system. Nat Rev Nephrol 5: 384–396, 2009 [DOI] [PubMed] [Google Scholar]
- 61.Baylis C: Changes in renal hemodynamics and structure in the aging kidney; sexual dimorphism and the nitric oxide system. Exp Gerontol 40: 271–278, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Westhoff JH, Schildhorn C, Jacobi C, Hömme M, Hartner A, Braun H, et al. : Telomere shortening reduces regenerative capacity after acute kidney injury. J Am Soc Nephrol 21: 327–336, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Helman A, Klochendler A, Azazmeh N, Gabai Y, Horwitz E, Anzi S, et al. : p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat Med 22: 412–420, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang J-H, Griffin PT, Vera DL, Apostolides JK, Hayano M, Meer MV, Salfati EL, et al. : Erosion of the epigenetic landscape and loss of cellular identity as a cause of aging in mammals. bioRxiv. 10.1101/808642 (Preprint posted October 19, 2019)
- 65.Sinclair DA, Guarente L: Extrachromosomal rDNA circles: A cause of aging in yeast. Cell 91: 1033–1042, 1997 [DOI] [PubMed] [Google Scholar]
- 66.Berkovich E, Monnat RJ Jr, Kastan MB: Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol 9: 683–690, 2007 [DOI] [PubMed] [Google Scholar]
- 67.McCord RA, Michishita E, Hong T, Berber E, Boxer LD, Kusumoto R, et al. : SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging (Albany NY) 1: 109–121, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ikezumi Y, Suzuki T, Karasawa T, Yamada T, Hasegawa H, Nishimura H, et al. : Low birthweight and premature birth are risk factors for podocytopenia and focal segmental glomerulosclerosis. Am J Nephrol 38: 149–157, 2013 [DOI] [PubMed] [Google Scholar]
- 69.Shankland SJ, Pippin JW, Reiser J, Mundel P: Podocytes in culture: Past, present, and future. Kidney Int 72: 26–36, 2007 [DOI] [PubMed] [Google Scholar]
- 70.Ni L, Saleem M, Mathieson PW: Podocyte culture: Tricks of the trade. Nephrology (Carlton) 17: 525–531, 2012 [DOI] [PubMed] [Google Scholar]
- 71.Campisi J: The biology of replicative senescence. Eur J Cancer 33: 703–709, 1997 [DOI] [PubMed] [Google Scholar]
- 72.Olovnikov AM: A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol 41: 181–190, 1973 [DOI] [PubMed] [Google Scholar]
- 73.Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C, et al. : A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol 18: e3000599, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harbo M, Koelvraa S, Serakinci N, Bendix L: Telomere dynamics in human mesenchymal stem cells after exposure to acute oxidative stress. DNA Repair (Amst) 11: 774–779, 2012 [DOI] [PubMed] [Google Scholar]
- 75.von Zglinicki T, Pilger R, Sitte N: Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic Biol Med 28: 64–74, 2000 [DOI] [PubMed] [Google Scholar]
- 76.von Zglinicki T, Saretzki G, Döcke W, Lotze C: Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: A model for senescence? Exp Cell Res 220: 186–193, 1995 [DOI] [PubMed] [Google Scholar]
- 77.Petrova NV, Velichko AK, Razin SV, Kantidze OL: Small molecule compounds that induce cellular senescence. Aging Cell 15: 999–1017, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen J, Huang X, Halicka D, Brodsky S, Avram A, Eskander J, et al. : Contribution of p16INK4a and p21CIP1 pathways to induction of premature senescence of human endothelial cells: Permissive role of p53. Am J Physiol Heart Circ Physiol 290: H1575–H1586, 2006 [DOI] [PubMed] [Google Scholar]
- 79.Gava AL, Freitas FP, Meyrelles SS, Silva IV, Graceli JB: Gender-dependent effects of aging on the kidney. Braz J Med Biol Res 44: 905–913, 2011 [DOI] [PubMed] [Google Scholar]
- 80.Wheeler HE, Metter EJ, Tanaka T, Absher D, Higgins J, Zahn JM, et al. : Sequential use of transcriptional profiling, expression quantitative trait mapping, and gene association implicates MMP20 in human kidney aging. PLoS Genet 5: e1000685, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Melk A, Mansfield ES, Hsieh SC, Hernandez-Boussard T, Grimm P, Rayner DC, et al. : Transcriptional analysis of the molecular basis of human kidney aging using cDNA microarray profiling. Kidney Int 68: 2667–2679, 2005 [DOI] [PubMed] [Google Scholar]
- 82.Rodwell GE, Sonu R, Zahn JM, Lund J, Wilhelmy J, Wang L, et al. : A transcriptional profile of aging in the human kidney. PLoS Biol 2: e427, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ransick A, Lindström NO, Liu J, Zhu Q, Guo JJ, Alvarado GF, et al. : Single-cell profiling reveals sex, lineage, and regional diversity in the mouse kidney. Dev Cell 51: 399–413.e7, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang Y, Eng DG, Pippin JW, Gharib SA, McClelland A, Gross KW, et al. : Sex differences in transcriptomic profiles in aged kidney cells of renin lineage. Aging (Albany NY) 10: 606–621, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wu H, Lai CF, Chang-Panesso M, Humphreys BD: Proximal tubule translational profiling during kidney fibrosis reveals proinflammatory and long noncoding RNA expression patterns with sexual dimorphism. J Am Soc Nephrol 31: 23–38, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Menezes LF, Lin CC, Zhou F, Germino GG: Fatty acid oxidation is impaired in an orthologous mouse model of autosomal dominant polycystic kidney disease. EBioMedicine 5: 183–192, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Oxburgh L, Carroll TJ, Cleaver O, Gossett DR, Hoshizaki DK, Hubbell JA, et al. : (Re)Building a kidney. J Am Soc Nephrol 28: 1370–1378, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kuppe C, Perales-Patón J, Saez-Rodriguez J, Kramann R: Experimental and computational technologies to dissect the kidney at the single-cell level [published online ahead of print December 17, 2020] Nephrol Dial Transplant 2020 [DOI] [PubMed] [Google Scholar]
- 89.Chung JJ, Goldstein L, Chen YJ, Lee J, Webster JD, Roose-Girma M, et al. : Single-cell transcriptome profiling of the kidney glomerulus identifies key cell types and reactions to injury. J Am Soc Nephrol 31: 2341–2354, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Puelles VG, Moeller MJ: Postnatal podocyte gain: Is the jury still out? Semin Cell Dev Biol 91: 147–152, 2019 [DOI] [PubMed] [Google Scholar]
- 91.Kim YH, Goyal M, Kurnit D, Wharram B, Wiggins J, Holzman L, et al. : Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int 60: 957–968, 2001 [DOI] [PubMed] [Google Scholar]
- 92.Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, et al. : Podocyte depletion causes glomerulosclerosis: Diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 16: 2941–2952, 2005 [DOI] [PubMed] [Google Scholar]
- 93.Fukuda A, Chowdhury MA, Venkatareddy MP, Wang SQ, Nishizono R, Suzuki T, et al. : Growth-dependent podocyte failure causes glomerulosclerosis. J Am Soc Nephrol 23: 1351–1363, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kaplan C, Pasternack B, Shah H, Gallo G. Age-related incidence of sclerotic glomeruli in human kidneys. Am J Pathol 80: 227–234 1975 [PMC free article] [PubMed] [Google Scholar]
- 95.McLachlan MS. The ageing kidney. Lancet 2: 143–145 1978 [DOI] [PubMed] [Google Scholar]
- 96.Glassock RJ, Rule AD: The implications of anatomical and functional changes of the aging kidney: with an emphasis on the glomeruli. Kidney Int 82: 270–277, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Roeder SS, Stefanska A, Eng DG, Kaverina N, Sunseri MW, McNicholas BA, et al. : Changes in glomerular parietal epithelial cells in mouse kidneys with advanced age. Am J Physiol Renal Physiol 309: F164–F178, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McNicholas BA, Eng DG, Lichtnekert J, Rabinowitz PS, Pippin JW, Shankland SJ: Reducing mTOR augments parietal epithelial cell density in a model of acute podocyte depletion and in aged kidneys. Am J Physiol Renal Physiol 311: F626–F639, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pippin JW, Glenn ST, Krofft RD, Rusiniak ME, Alpers CE, Hudkins K, et al. : Cells of renin lineage take on a podocyte phenotype in aging nephropathy. Am J Physiol Renal Physiol 306: F1198–F1209, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Puelles VG, Douglas-Denton RN, Cullen-McEwen LA, Li J, Hughson MD, Hoy WE, et al. : Podocyte number in children and adults: Associations with glomerular size and numbers of other glomerular resident cells. J Am Soc Nephrol 26: 2277–2288, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Goyal VK: Changes with age in the human kidney. Exp Gerontol 17: 321–331, 1982 [DOI] [PubMed] [Google Scholar]
- 102.Wiggins J: Podocytes and glomerular function with aging. Semin Nephrol 29: 587–593, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Musso CG, Oreopoulos DG: Aging and physiological changes of the kidneys including changes in glomerular filtration rate. Nephron, Physiol 119: 1–5, 2011 [DOI] [PubMed] [Google Scholar]
- 104.Dodane V, Chevalier J, Bariety J, Pratz J, Corman B: Longitudinal study of solute excretion and glomerular ultrastructure in an experimental model of aging rats free of kidney disease. Lab Invest 64: 377–391, 1991 [PubMed] [Google Scholar]
- 105.Kang DH, Anderson S, Kim YG, Mazzalli M, Suga S, Jefferson JA, et al. : Impaired angiogenesis in the aging kidney: vascular endothelial growth factor and thrombospondin-1 in renal disease. Am J Kidney Dis 37: 601–611, 2001 [DOI] [PubMed] [Google Scholar]
- 106.Kikuchi M, Wickman L, Rabah R, Wiggins RC: Podocyte number and density changes during early human life. Pediatr Nephrol 32: 823–834, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Camici M, Carpi A, Cini G, Galetta F, Abraham N: Podocyte dysfunction in aging-related glomerulosclerosis. Front Biosci (Schol Ed) 3: 995–1006, 2011. PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pippin J, Kumar V, Stein A, Jablonski P, Shankland SJ, Davis CL: The contribution of podocytes to chronic allograft nephropathy. Nephron, Exp Nephrol 111: e1–e10, 2009 [DOI] [PubMed] [Google Scholar]
- 109.Naik AS, Afshinnia F, Aqeel J, Cibrik DM, Samaniego M, Wickman L, et al. : Accelerated podocyte detachment early after kidney transplantation is related to long-term allograft loss of function. Nephrol Dial Transplant 34: 1232–1239, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lee JH, Jung KJ, Kim JW, Kim HJ, Yu BP, Chung HY: Suppression of apoptosis by calorie restriction in aged kidney. Exp Gerontol 39: 1361–1368, 2004 [DOI] [PubMed] [Google Scholar]
- 111.Tower J. Programmed cell death in aging. Ageing Res Rev 23: 90–100, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Braun F, Becker JU, Brinkkoetter PT: Live or let die: Is there any cell death in podocytes? Semin Nephrol 36: 208–219, 2016 [DOI] [PubMed] [Google Scholar]
- 113.Li X, Chuang PY, D’Agati VD, Dai Y, Yacoub R, Fu J, et al. : Nephrin preserves podocyte viability and glomerular structure and function in adult kidneys. J Am Soc Nephrol 26: 2361–2377, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang Y, Li H, Song SP: β-arrestin 1/2 aggravates podocyte apoptosis of diabetic nephropathy via Wnt/β-catenin pathway. Med Sci Monit 24: 1724–1732, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schiffer M, Bitzer M, Roberts IS, Kopp JB, ten Dijke P, Mundel P, et al. : Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest 108: 807–816, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sasaki T, Kanematsu K: Studies on morphine-like compounds. VI. Transformation of thebaine to benzomorphan analogues by ozonolysis. Chem Pharm Bull (Tokyo) 15: 1247–1250, 1967 [DOI] [PubMed] [Google Scholar]
- 117.Bergsbaken T, Fink SL, Cookson BT: Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7: 99–109, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yu D, Petermann A, Kunter U, Rong S, Shankland SJ, Floege J: Urinary podocyte loss is a more specific marker of ongoing glomerular damage than proteinuria. J Am Soc Nephrol 16: 1733–1741, 2005 [DOI] [PubMed] [Google Scholar]
- 119.Petermann AT, Pippin J, Krofft R, Blonski M, Griffin S, Durvasula R, et al. : Viable podocytes detach in experimental diabetic nephropathy: Potential mechanism underlying glomerulosclerosis. Nephron, Exp Nephrol 98: e114–e123, 2004 [DOI] [PubMed] [Google Scholar]
- 120.Tharaux PL, Huber TB: How many ways can a podocyte die? Semin Nephrol 32: 394–404, 2012 [DOI] [PubMed] [Google Scholar]
- 121.Cho CR, Lumsden CJ, Whiteside CI: Epithelial cell detachment in the nephrotic glomerulus: A receptor co-operativity model. J Theor Biol 160: 407–426, 1993 [DOI] [PubMed] [Google Scholar]
- 122.Fukuda A, Wickman LT, Venkatareddy MP, Sato Y, Chowdhury MA, Wang SQ, et al. : Angiotensin II-dependent persistent podocyte loss from destabilized glomeruli causes progression of end stage kidney disease. Kidney Int 81: 40–55, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ichikawa I, Ma J, Motojima M, Matsusaka T: Podocyte damage damages podocytes: Autonomous vicious cycle that drives local spread of glomerular sclerosis. Curr Opin Nephrol Hypertens 14: 205–210, 2005 [DOI] [PubMed] [Google Scholar]
- 124.Shankland SJ, Freedman BS, Pippin JW: Can podocytes be regenerated in adults? Curr Opin Nephrol Hypertens 26: 154–164, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lasagni L, Lazzeri E, Shankland SJ, Anders HJ, Romagnani P: Podocyte mitosis: A catastrophe. Curr Mol Med 13: 13–23, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pippin JW, Durvasula R, Petermann A, Hiromura K, Couser WG, Shankland SJ: DNA damage is a novel response to sublytic complement C5b-9-induced injury in podocytes. J Clin Invest 111: 877–885, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kriz W: Podocyte is the major culprit accounting for the progression of chronic renal disease. Microsc Res Tech 57: 189–195, 2002 [DOI] [PubMed] [Google Scholar]
- 128.Garg P: A review of podocyte biology. Am J Nephrol 47: 3–13, 2018 [DOI] [PubMed] [Google Scholar]
- 129.Fukasawa H, Bornheimer S, Kudlicka K, Farquhar MG: Slit diaphragms contain tight junction proteins. J Am Soc Nephrol 20: 1491–1503, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Itoh M, Nakadate K, Horibata Y, Matsusaka T, Xu J, Hunziker W, et al. : The structural and functional organization of the podocyte filtration slits is regulated by Tjp1/ZO-1. PLoS One 9: e106621, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shirato I: Podocyte process effacement in vivo. Microsc Res Tech 57: 241–246, 2002 [DOI] [PubMed] [Google Scholar]
- 132.Kriz W, Shirato I, Nagata M, LeHir M, Lemley KV: The podocyte’s response to stress: The enigma of foot process effacement. Am J Physiol Renal Physiol 304: F333–F347, 2013 [DOI] [PubMed] [Google Scholar]
- 133.Shankland SJ: The podocyte’s response to injury: Role in proteinuria and glomerulosclerosis. Kidney Int 69: 2131–2147, 2006 [DOI] [PubMed] [Google Scholar]
- 134.Kakinuma N, Zhu Y, Wang Y, Roy BC, Kiyama R: Kank proteins: Structure, functions and diseases. Cell Mol Life Sci 66: 2651–2659, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liu M, Liang K, Zhen J, Zhou M, Wang X, Wang Z, et al. : Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting Notch signaling. Nat Commun 8: 413, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Eremina V, Baelde HJ, Quaggin SE: Role of the VEGF: A signaling pathway in the glomerulus: Evidence for crosstalk between components of the glomerular filtration barrier. Nephron, Physiol 106: 32–37, 2007 [DOI] [PubMed] [Google Scholar]
- 137.van Deursen JM: The role of senescent cells in ageing. Nature 509: 439–446, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Prata LGPL, Ovsyannikova IG, Tchkonia T, Kirkland JL: Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin Immunol 40: 101275, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, et al. : The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev 31: 172–183, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mosteiro L, Pantoja C, de Martino A, Serrano M: Senescence promotes in vivo reprogramming through p16INK4a and IL-6. Aging Cell 17: e12711 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. : Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530: 184–189, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sis B, Tasanarong A, Khoshjou F, Dadras F, Solez K, Halloran PF: Accelerated expression of senescence associated cell cycle inhibitor p16INK4A in kidneys with glomerular disease. Kidney Int 71: 218–226, 2007 [DOI] [PubMed] [Google Scholar]
- 143.Verzola D, Gandolfo MT, Gaetani G, Ferraris A, Mangerini R, Ferrario F, et al. : Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. Am J Physiol Renal Physiol 295: F1563–F1573, 2008 [DOI] [PubMed] [Google Scholar]
- 144.Zhang L, Zhou F, Yu X, Zhu Y, Zhou Y, Liu J, et al. : C/EBPα deficiency in podocytes aggravates podocyte senescence and kidney injury in aging mice. Cell Death Dis 10: 684, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chuang PY, Cai W, Li X, Fang L, Xu J, Yacoub R, et al. : Reduction in podocyte SIRT1 accelerates kidney injury in aging mice. Am J Physiol Renal Physiol 313: F621–F628, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hamatani H, Eng DG, Kaverina NV, Gross KW, Freedman BS, Pippin JW, et al. : Lineage tracing aged mouse kidneys shows lower number of cells of renin lineage and reduced responsiveness to RAAS inhibition. Am J Physiol Renal Physiol 315: F97–F109, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wiggins JE, Patel SR, Shedden KA, Goyal M, Wharram BL, Martini S, et al. : NFkappaB promotes inflammation, coagulation, and fibrosis in the aging glomerulus. J Am Soc Nephrol 21: 587–597, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kaneko Y, Cho T, Sato Y, Goto K, Yamamoto S, Goto S, et al. : Attenuated macrophage infiltration in glomeruli of aged mice resulting in ameliorated kidney injury in nephrotoxic serum nephritis. J Gerontol A Biol Sci Med Sci 73: 1178–1186, 2018 [DOI] [PubMed] [Google Scholar]
- 149.Shen H, Kreisel D, Goldstein DR: Processes of sterile inflammation. J Immunol 191: 2857–2863, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zindel J, Kubes P: DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol 15: 493–518, 2020 [DOI] [PubMed] [Google Scholar]
- 151.Balaban RS, Nemoto S, Finkel T: Mitochondria, oxidants, and aging. Cell 120: 483–495, 2005 [DOI] [PubMed] [Google Scholar]
- 152.Ventura-Clapier R, Garnier A, Veksler V: Transcriptional control of mitochondrial biogenesis: The central role of PGC-1alpha. Cardiovasc Res 79: 208–217, 2008 [DOI] [PubMed] [Google Scholar]
- 153.LeBleu VS, O'Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, et al. : PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol 16: 992–1003, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Brinkkoetter PT, Bork T, Salou S, Liang W, Mizi A, Özel C, et al. : Anaerobic glycolysis maintains the glomerular filtration barrier independent of mitochondrial metabolism and dynamics. Cell Rep 27: 1551–1566.e5, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kampe K, Sieber J, Orellana JM, Mundel P, Jehle AW: Susceptibility of podocytes to palmitic acid is regulated by fatty acid oxidation and inversely depends on acetyl-CoA carboxylases 1 and 2. Am J Physiol Renal Physiol 306: F401–F409, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chugh SS: Transcriptional regulation of podocyte disease. Transl Res 149: 237–242, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, et al. : Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat Genet 48: 838–847, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhong F, Wang W, Lee K, He JC, Chen N: Role of C/EBP-α in Adriamycin-induced podocyte injury. Sci Rep 6: 33520, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Magassa S, Aron L, Hoguin C, Isnard P, Terzi F, Legendre C, et al. : REST and stress resistance in the aging kidney. J Am Soc Nephrol 32: 1974–1986, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Salminen A, Kaarniranta K: SIRT1: Regulation of longevity via autophagy. Cell Signal 21: 1356–1360, 2009 [DOI] [PubMed] [Google Scholar]
- 161.Gomes P, Fleming Outeiro T, Cavadas C: Emerging role of sirtuin 2 in the regulation of mammalian metabolism. Trends Pharmacol Sci 36: 756–768, 2015 [DOI] [PubMed] [Google Scholar]
- 162.Huang W, Liu H, Zhu S, Woodson M, Liu R, Tilton RG, et al. : Sirt6 deficiency results in progression of glomerular injury in the kidney. Aging (Albany NY) 9: 1069–1083, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Vazquez BN, Thackray JK, Simonet NG, Kane-Goldsmith N, Martinez-Redondo P, Nguyen T, et al. : SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. EMBO J 35: 1488–1503, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lenoir O, Tharaux PL, Huber TB: Autophagy in kidney disease and aging: Lessons from rodent models. Kidney Int 90: 950–964, 2016 [DOI] [PubMed] [Google Scholar]
- 165.Hartleben B, Gödel M, Meyer-Schwesinger C, Liu S, Ulrich T, Köbler S, et al. : Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest 120: 1084–1096, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Fischer DC, Jacoby U, Pape L, Ward CJ, Kuwertz-Broeking E, Renken C, et al. : Activation of the AKT/mTOR pathway in autosomal recessive polycystic kidney disease (ARPKD). Nephrol Dial Transplant 24: 1819–1827, 2009 [DOI] [PubMed] [Google Scholar]
- 167.Bedsaul JR, Carter NM, Deibel KE, Hutcherson SM, Jones TA, Wang Z, et al. : Mechanisms of regulated and dysregulated CARD11 signaling in adaptive immunity and disease. Front Immunol 9: 2105, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ladiges W, Liggitt D: Testing drug combinations to slow aging. Pathobiol Aging Age Relat Dis 8: 1407203, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zullo A, Simone E, Grimaldi M, Musto V, Mancini FP: Sirtuins as mediator of the anti-ageing effects of calorie restriction in skeletal and cardiac muscle. Int J Mol Sci 19: 928, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Szeto HH: First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol 171: 2029–2050, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Fouron JC: Pulmonary edema with upper airway obstruction. Am J Dis Child 139: 331, 1985 [DOI] [PubMed] [Google Scholar]
- 172.Cornel MC, Swagemakers ML, te Meerman GJ, Haayer EJ, ten Kate LP: [EUROCAT registration of congenital abnormalities and multiple births; aims, methods and results of the Dutch share of the project 1981–1983]. Ned Tijdschr Geneeskd 130: 1233–1237, 1986 [PubMed] [Google Scholar]
- 173.Escribano-López I, de Marañon AM, Iannantuoni F, López-Domènech S, Abad-Jiménez Z, Díaz P, et al. : The mitochondrial antioxidant SS-31 modulates oxidative stress, endoplasmic reticulum stress, and autophagy in type 2 diabetes. J Clin Med 8: E1322, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Puelles VG, van der Wolde JW, Wanner N, Scheppach MW, Cullen-McEwen LA, Bork T, et al. : mTOR-mediated podocyte hypertrophy regulates glomerular integrity in mice and humans. JCI Insight 4: 99271, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.O’Hare T, Walters DK, Stoffregen EP, Jia T, Manley PW, Mestan J, et al. : In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res 65: 4500–4505, 2005 [DOI] [PubMed] [Google Scholar]
- 176.Russo M, Spagnuolo C, Tedesco I, Bilotto S, Russo GL: The flavonoid quercetin in disease prevention and therapy: Facts and fancies. Biochem Pharmacol 83: 6–15, 2012 [DOI] [PubMed] [Google Scholar]
- 177.Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. : The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14: 644–658, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Palmer AK, Xu M, Zhu Y, Pirtskhalava T, Weivoda MM, Hachfeld CM, et al. : Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 18: e12950, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ortmann J, Amann K, Brandes RP, Kretzler M, Münter K, Parekh N, et al. : Role of podocytes for reversal of glomerulosclerosis and proteinuria in the aging kidney after endothelin inhibition. Hypertension 44: 974–981, 2004 [DOI] [PubMed] [Google Scholar]
- 180.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, et al. : The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 313: 1929–1935, 2006 [DOI] [PubMed] [Google Scholar]
- 181.Grgic I, Hofmeister AF, Genovese G, Bernhardy AJ, Sun H, Maarouf OH, et al. : Discovery of new glomerular disease-relevant genes by translational profiling of podocytes in vivo. Kidney Int 86: 1116–1129, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Szabó C, Biser A, Benko R, Böttinger E, Suszták K: Poly(ADP-ribose) polymerase inhibitors ameliorate nephropathy of type 2 diabetic Leprdb/db mice. Diabetes 55: 3004–3012, 2006 [DOI] [PubMed] [Google Scholar]
- 183.Lasagni L, Angelotti ML, Ronconi E, Lombardi D, Nardi S, Peired A, et al. : Podocyte regeneration driven by renal progenitors determines glomerular disease remission and can be pharmacologically enhanced. Stem Cell Reports 5: 248–263, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Appel D, Kershaw DB, Smeets B, Yuan G, Fuss A, Frye B, et al. : Recruitment of podocytes from glomerular parietal epithelial cells. J Am Soc Nephrol 20: 333–343, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tabula Muris C; Tabula Muris Consortium : A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583: 590–595, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.van Heemst D, Beekman M, Mooijaart SP, Heijmans BT, Brandt BW, Zwaan BJ, et al. : Reduced insulin/IGF-1 signalling and human longevity. Aging Cell 4: 79–85, 2005 [DOI] [PubMed] [Google Scholar]
- 187.Wiggins J, Bitzer M: Slowing the aging process. Clin Geriatr Med 29: 721–730, 2013 [DOI] [PubMed] [Google Scholar]