

ABSTRACT
Retinoblastoma is the most common intraocular malignancy during childhood. Currently, there is no effective treatment for metastatic retinoblastoma. We investigated potential biomarkers of retinoblastoma by utilizing three datasets from a public database. Functional enrichment analysis, including gene ontology, Kyoto encyclopedia of genes and genomes, gene set enrichment analysis and variation analysis, suggested that differentially expressed genes in retinoblastoma were enriched in accelerated cell cycle events. Protein-protein interaction analysis constructed a network consisting of six hub genes, including benzimidazoles 1 (BUB1), cyclin dependent kinase 1 (CDK1), centromere protein E (CENPE), kinesin family member 20A (KIF20A), PDZ binding kinase (PBK), and targeting protein for xklp2 (TPX2). Drug sensitivity analysis showed that nelarabine was positively correlated with five hub genes. All six genes were expressed differently in six immune subtypes and were positively correlated with stemness indices in most human cancer types. Since CENPE is the least known hub gene in retinoblastoma, we further analyzed the potential non-coding RNAs and transcription factors that regulate CENPE and built interaction networks of competing endogenous RNA and transcription factors. Immune cell infiltration, especially by plasma and B cells, was enhanced in samples with high CENPE expression. Pan-cancer analysis illustrated that CENPE was highly expressed in a wide range of human tumors. In vitro validation revealed that CENPE was significantly upregulated at both the mRNA and protein levels in retinoblastoma cells. In conclusion, CENPE, along with other hub genes, could serve as a potential biomarker and intervention target for retinoblastoma.
KEYWORDS: Retinoblastoma, centromere protein E (CENPE), biomarker, Gene Expression Omnibus (GEO), bioinformatics, RT-qPCR, immunoblotting
1. Introduction
Retinoblastoma is the most common primary intraocular malignancy during childhood. Although it has been reported that the disease-free survival rate of retinoblastoma is nearly 100% in developed countries[1], its prognosis in developing countries is not optimistic due to poor accessibility to health care and delayed diagnosis[2]. A predictive study demonstrated that the incidence of retinoblastoma in Asia, accounting for 53% of all patients, is the highest in the world[3]. The foremost aim of retinoblastoma treatment is to preserve life without eyeball enucleation and to preserve vision and quality of life as subordinate goals[3]. Precise and timely diagnosis at an early stage is critical to improve patient survival and ocular salvage[4]. Therefore, investigating novel diagnostic biomarkers and therapeutic targets of retinoblastoma would be beneficial for overcoming this disease.
Human tumors vary in their tissue origin, in addition to individual biological and genetic backgrounds, and are highlighted by variations in gene expression. Profiling tumor-specific gene expression might offer a new theoretical basis for the categorization of tumors instead of morphological appearance[5]. With the rapid development and extensive application of high-throughput sequencing technologies, it has become feasible to explore and understand gene expression profiles at a comprehensive level, rather than at the traditional gene-by-gene level[6]. As a multidisciplinary field, integrative bioinformatic analysis, combining computational biology, informatics, statistics, and mathematics, has emerged as a promising and increasingly important method for investigating promising targets.
Research into novel biomarkers is an effective way to improve the probability of satisfactory outcomes, including the prognosis and quality of life in patients with retinoblastoma. Although new molecular biomarkers for retinoblastoma have been revealed in recent years [7–11], they are still in the experimental stage and have not been approved for clinical use[12]. Currently, there is no gold standard biomarker for the diagnosis and treatment of retinoblastoma. Therefore, further investigation of feasible biomarker candidates for retinoblastoma is necessary, which would also lead to a deeper understanding of the disease. Integrative bioinformatics analysis offers hopeful support for screening and discovery of novel targets, which is the principal approach in our study.
The purpose of this study was to identify potential biomarkers of retinoblastoma. We screened out differentially expressed genes for retinoblastoma by analyzing three retinoblastoma gene expression matrices from the Gene Expression Omnibus (GEO) database. Subsequently, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses, gene set enrichment analysis (GSEA), gene set variation analysis (GSVA), and protein-protein interaction analysis were employed to explore the molecules and key hub genes involved in the pathogenesis of retinoblastoma. Among the identified genes, the centromere protein E (CENPE) gene, the least known hub gene in retinoblastoma, was extensively analyzed, including in terms of immune infiltration and interaction networks with non-coding RNAs and transcription factors. We validated CENPE expression in two retinoblastoma cell lines using immunoblotting and reverse transcription-quantitative polymerase chain reaction (RT-qPCR).
2. Materials and methods
2.1. Data sources
The study was conducted in accordance with the tenets of the Declaration of Helsinki. GSE24673[13], GSE97508[14], and GSE110811 [15] were retrieved from the GEO database (https://www.ncbi.nlm.nih.gov/geo/), an international public repository of microarray profiles. The platform for GSE24673 was GPL6244 (Affymetrix Human Gene 1.0 ST array; Thermo Fisher Scientific, Waltham, MA), which included 9 retinoblastoma samples and 2 normal control retinas. The platform for GSE97508 was GPL15207 (Affymetrix Human Gene Expression Array; Thermo Fisher Scientific), which included 6 retinoblastoma samples and 3 control retinas. The platform for GSE110811 was GPL16686 (Affymetrix Human Gene 2.0 ST array; Thermo Fisher Scientific), which included 28 retinoblastoma samples and 3 control tissues. The H (hallmark) gene sets, C2 curated gene sets, and C5 ontology gene sets were downloaded from the molecular signatures database (MSigDB, https://www.gsea-msigdb.org/gsea/msigdb/)[16]. Expression profiles, immune subtypes, stem cell transcriptomes, and stem cell DNA methylation data were downloaded from the Xena Functional Genomics Explorer (https://xena.ucsc.edu/)[17].
2.2. Identification of differentially expressed genes
The Affy package in R software 4.0.3 (R Foundation for Statistical Computing, Vienna, Austria) was used to preprocess the raw data of these three datasets[18], and the corresponding gene expression profiles were obtained. The ‘combat’ function of the ‘sva’ package was used to remove batch effects from the three datasets and generate a box plot of the data distribution after correction[19]. Principal component analysis was performed using the ‘princomp’ function in R, and 3D principal component analysis of the samples was performed using the ‘scatterplot3d’ R package[20].
Differentially expressed genes that met the criteria of |logFC| (an absolute log2 value in the fold change of gene expression) >1 and P value <0.05 were screened and filtered using the ‘limma’ R package[21]. Screened differentially expressed genes were visualized using the ‘ggplot2’ R package to create volcano plots and the ‘pheatmap’ R package was used to draw clustered heatmaps[22]. A Venn diagram utilizing an online tool (http://bioinformatics.psb.ugent.be/webtools/Venn/) was generated to determine common differentially expressed genes among the three datasets.
2.3. Enrichment analysis
GO functional enrichment analysis, including terms related to biological process (BP), cell components (CC), and molecular function (MF), was used to identify the characteristic biological features[23]. KEGG functional analysis was performed to identify pathways involved in various cellular processes and organismal systems[24]. GO term and KEGG pathway analyses of the above common differentially expressed genes were performed and visualized using clusterProfiler V3.14.3 [25] and the Goplot package [26] in R software. Only terms with both false discovery rate (FDR) < 0.05 and P value <0.05 were considered significantly enriched.
2.4. GSEA and GSVA
GSEA was performed on the differentially expressed genes of GSE110811 with the largest sample size using clusterProfiler V3.14.3[25], and C2 curated gene sets of the MSigDB were used as the reference. Gene sets were considered significantly enriched if they had a P value <0.05, FDR <0.25, and normalized enrichment score (|NES|) >1. The ten pathways with the most significant enrichment scores were selected for visualization using the R packages ‘gseaplot2’ and ‘ggridges’.
The R package ‘GSVA’[27] was utilized to explore the differences in enrichment pathways in GSE110811, and five different MSigDB gene sets were used as references, including the Hallmark, GO_BP, GO_CC, GO_MF, and curated KEGG gene sets. A logFC value <0.05 and a P value < 0.05 were used as the cutoffs to identify significantly enriched pathways.
2.5. Protein-protein interaction network construction
To further investigate the interactions between of the above common differentially expressed genes, a protein-protein interaction network of differentially expressed genes was analyzed using the Search tool for the retrieval of interacting genes (STRING database, https://string-db.org/, version 11.0b). [28] An interaction with a combined score higher than 0.4, which is a widely used threshold, was considered statistically significant. Cytoscape software (version 3.8.2; Cytoscape Consortium, San Diego, CA) [29] was used to visualize the regulatory relationship of differentially expressed genes. The Molecular Complex Detection (MCODE) [30] plugin of Cytoscape was employed to detect significant modules (MCODE score ≥4). In addition, cytoHubba[31], another plugin of Cytoscape, was utilized to explore essential nodes and screen out the top six weighted hub genes in the protein-protein interaction network using the maximal clique centrality (MCC) algorithm.
2.6. Drug sensitivity analysis of hub genes
To assess the correlation between potential drugs and hub gene expression, drug sensitivity and genomic data were obtained from CellMiner (https://discover.nci.nih.gov/cellminer)[32]. Drugs that were significantly positively correlated with hub genes were then screened out and visualized using the R package ‘corrplot’.
2.7. Pan-cancer analysis of hub genes
To further explore the role and significance of these five hub genes in cancer, we analyzed their expression profiles in six cancer immune subtypes and their relevant co-expression across cancer types. To measure the stemness of tumors, mRNA expression-based stemness score (RNAss) and DNA methylation-based stemness score (DNAss) were evaluated using pan-cancer data from The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/). All results were visualized using the R packages ‘ggplot2’ and ‘corrplot’.
2.8. Construction of the CENPE-microRNA-long non-coding RNA network and CENPE-transcription factor network
Since CENPE, one of the six hub genes, has rarely been studied in retinoblastoma, we further focused on CENPE and analyzed its potential regulatory microRNAs (miRNAs). Three databases, including the Encyclopedia of RNA Interactomes (ENCORI, http://starbase.sysu.edu.cn/)[33], MicroRNA target prediction database (miRDB, http://mirdb.org/)[34], and miRWalk database (version 3.0, http://mirwalk.umm.uni-heidelberg.de/)[35], were integrated, and we analyzed their intersection. Moreover, the interaction between the predicted miRNAs and long-non-coding RNAs (lncRNAs) was estimated using the ENCORI database. The CENPE mRNA-miRNA-lncRNA competing endogenous RNA (ceRNA) network was constructed using Cytoscape software.
Furthermore, relevant transcription factors of CENPE were predicted using ChIP-X enrichment analysis version 3 (ChEA3, https://maayanlab.cloud/chea3/) [36] and visualized using Cytoscape software. Classification of transcription factors was performed using the Human Transcription Factor Database (HumanTFDB, http://bioinfo.life.hust.edu.cn/HumanTFDB/#!/)[37].
2.9. Analysis of immune infiltration
Immune infiltration analysis of the three datasets was performed using CIBERSORT (https://cibersort.stanford.edu/)[38], an online tool that utilizes 547 gene expression profiles to estimate the relative proportions of 22 immune cell types in a mixed cell population with a deconvolution algorithm. The different fractions of infiltrating immune cells in each sample of the three datasets were obtained and visualized using ‘ggplot2’ through bar plots. The comparison of infiltrating immune cell expression between retinoblastoma and normal retinas was visualized using ‘ggplot2’ through heatmaps and violin plots.
The infiltration levels of immune cells under different CENPE expression levels were also investigated, and samples were divided into high and low CENPE expression groups as previously described. Box plots were generated using ‘ggplot2’.
2.10. Pan-cancer analysis of CENPE expression
The correlation between CENPE and the other five hub genes in the 33 tumor types from TCGA were investigated. In addition, pathway enrichment analysis was performed between high and low CENPE expression groups across various types of cancer and visualized using ‘ggplot2’. The correlation between CENPE expression and the prognosis of patients with various tumor types was explored based on Gene Expression Profiling Interactive Analysis (GEPIA, http://gepia.cancer-pku.cn/)[39]. CENPE expression profiles in tumor and normal samples were obtained from GEPIA and Oncomine (http://www.oncomine.org). CENPE mutation analysis was performed and acquired from the cBioPortal for Cancer Genomics (http://www.cbioportal.org/)[40].
2.11. Cell culture
The retinal pigment epithelium cell line Adult retinal pigment epithelium-19 (ARPE-19, cat. no. CRL-2302), the low-invasive human retinoblastoma cell line WERI-Rb-1 (cat. no. HTB-169), and the high-invasive retinoblastoma cell line Y79 (cat. no. HTB-18) were purchased from the American Type Culture Collection (ATCC, Manassas, VA). ARPE-19 cells were cultivated in Dulbecco’s modified Eagle’s medium/nutrient mixture F-12 (DMEM/F-12; Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific). WERI-RB-1 and Y79 cells were cultured in modified Roswell Park Memorial Institute (RPMI)-1640 medium (Thermo Fisher Scientific) containing 10% FBS. All cells were incubated in a humidified atmosphere containing 5% CO2 at 37°C.
2.12. RT-qPCR
CENPE mRNA expression in retinoblastoma cell lines was evaluated by qRT-PCR. Total RNA was extracted from cells using an RNA simple total RNA kit (Tiangen Biotech, Beijing, China) according to the manufacturer’s protocol. The isolated RNA was quantitated by spectrophotometry at 260 nm using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). qRT-PCR was performed according to the manufacturer’s protocol using the QuantiTect SYBR Green PCR Kit and QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany). The primers for CENPE and β-actin were as follows: CENPE forward 5ʹ- GATTCTGCCATACAAGGCTACAA −3ʹ; reverse, 5ʹ- TGCCCTGGGTATAACTCCCAA −3ʹ; β-actin forward, 5ʹ- CTCGCCTTTGCCGATCC −3ʹ; and everse, 5ʹ- TCTCCATGTCGTCCCAGTTG −3ʹ. The thermal cycling conditions were 95°C for 5 min, followed by 40 cycles at 95°C for 15 s and 60°C for 30 s. Experiments were performed in triplicate for each datapoint. Relative CENPE expression was quantified using the 2ΔΔCt method and normalized to β-actin.
2.13. Immunoblotting
Cell lysates were prepared from cell lines using a radioimmunoprecipitation lysis and extraction buffer kit (Thermo Fisher Scientific), and the procedure was performed as described previously[41]. Primary antibodies for CENPE (ab124733, RRID:AB_10974754; Abcam, Cambridge, UK) and β-actin (cat. no. 3779, RRID:AB_735551; ProSci, Fort Collins, CO) were diluted at 1:1,000, and the corresponding secondary antibodies (Proteintech catalog number SA00001-2, RRID:AB_2722564, Proteintech, Rosemont, IL) were diluted 1:2,000.
2.14. Statistical analysis
Statistical analyses in the bioinformatic analysis work were performed with R software, while statistical analyses of in vitro experiments were performed by SPSS Statistics 23.0 (IBM, Armonk, NY). The in vitro validation experiments were performed in triplicate. Data are presented as means ± standard deviation (SD). Differences among multiple groups were analyzed by one-way analysis of variance (ANOVA) with Tukey’s post hoc test. Statistical significance was set at P < 0.05.
3. Results
In the present study, potential biomarkers of retinoblastoma were explored using bioinformatics analysis. In addition, in vitro validation was performed for CENPE expression, which is the least known hub gene in relation to retinoblastoma identified in our study. First, we retrieved three datasets from the GEO database and overlapping upregulated DEGs were screened out. Functional enrichment analysis suggested that differentially expressed genes in retinoblastoma were enriched in accelerated cell cycle events. Protein-protein interaction analysis constructed a network consisting of six hub genes. Drug sensitivity analysis showed that nelarabine was positively correlated with five hub genes. All six genes were expressed differently in the six immune subtypes and were positively correlated with stemness indices in most human cancer types. Furthermore, we built interaction networks of ceRNAs and TFs. Immune cell infiltration, especially by plasma and B cells, was enhanced in samples with high CENPE expression. Pan-cancer analysis illustrated that CENPE was highly expressed in a wide range of human tumors. Finally, RT-qPCR and immunoblotting revealed that CENPE was significantly upregulated at both the mRNA and protein levels in two retinoblastoma cell lines.
3.1. Identification of differentially expressed genes in retinoblastoma by analyzing three datasets
The expression profiles of the three datasets after standardization are shown as box plots in Figure 1(a-c). The median values of gene expression for each sample were similar (Figure 1(a-c)). As shown in the 3D principal component analysis plot (Figure 1(d-f)), samples in the same group (retinoblastoma/normal control) were close to each other, while samples in different groups were far apart, indicating that tumor and normal samples could be clearly distinguished and that the data sources were qualified for further analysis.
Figure 1.
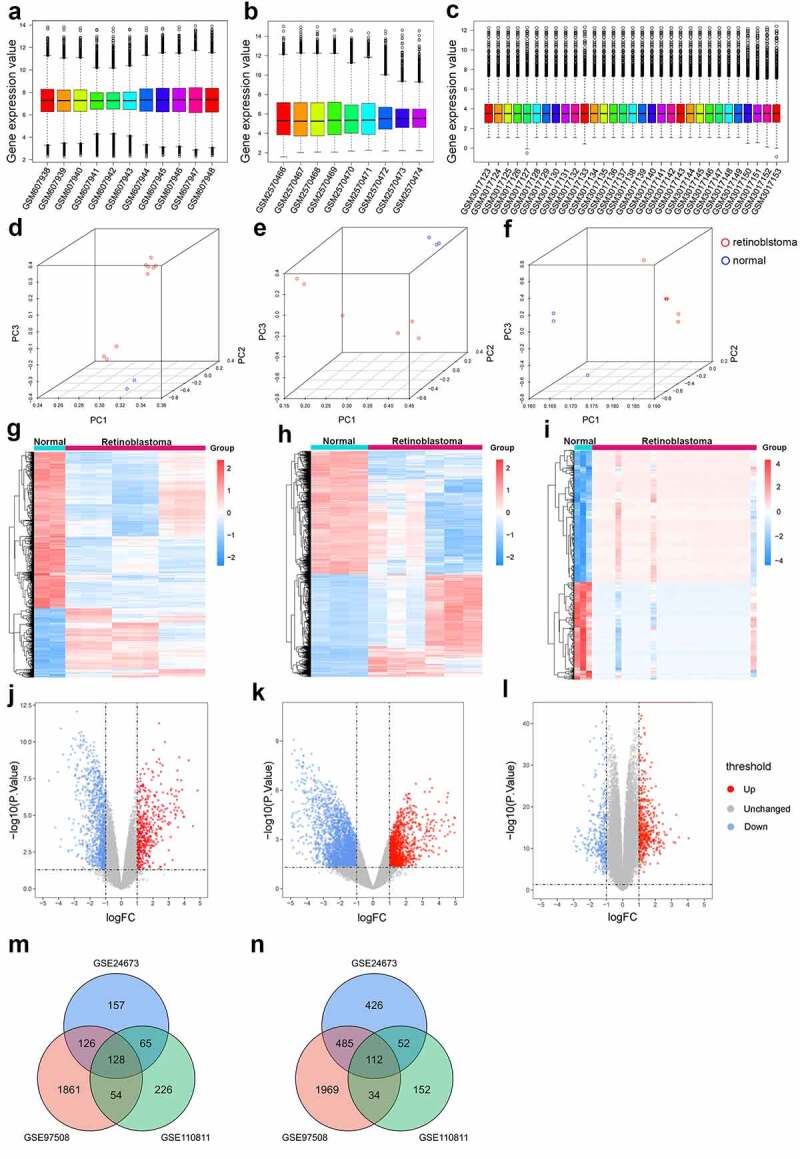
Identification of differentially expressed genes in retinoblastoma. (a-c) Box plots of gene expression profiles after data standardization in GSE24673 (a), GSE97508 (b), and GSE110811 (c). (d-f) Principal component analysis of retinoblastoma samples and normal control samples in GSE24673 (d), GSE97508 (e), and GSE110811 (f). (g-i) Heatmap of differentially expressed genes identified in GSE24673 (g), GSE97508 (h), and GSE110811 (i). (j-l) Volcano plot of differentially expressed genes identified in GSE24673 (j), GSE97508 (k), and GSE110811 (l). (m-n) Venn diagram of overlapping upregulated (m) and downregulated (n) differentially expressed genes from the three datasets
Based on the defined criteria (|logFC|> 1 and P value <0.05), upregulated and downregulated genes are displayed by heatmap plots and volcano plots in Figure 1(g-l), where red represents upregulated genes and blue represents downregulated genes. A total of 5847 differentially expressed genes were identified, of which GSE24673 contributed 476 upregulated genes and 1075 downregulated genes, GSE97508 contributed 2169 upregulated genes and 2600 downregulated genes, and GSE110811 contributed 473 upregulated genes and 350 downregulated genes (Figure 1(g-i); Supplementary Tables 1–3). The relative expression of differentially expressed genes between the three datasets was significantly different (Figure 1(j-l)). Moreover, a total of 240 overlapping differentially expressed genes were identified, including 128 upregulated and 112 downregulated genes. The distribution of overlapping genes is illustrated in Venn diagrams in Figure 1(m-n).
3.2. Functional enrichment analysis of differentially expressed genes showed increased cell cycle and proliferation in retinoblastoma
GO and KEGG pathway analysis of the differentially expressed genes was performed to identify significant signaling pathways. As shown in Figure 2(a-e), GO enrichment analysis demonstrated that for biological process, differentially expressed genes were significantly enriched in organelle fission, nuclear division, light stimulus, and chromosome segregation, which are critical for the mitotic cell cycle. Regarding cell components, differentially expressed genes were significantly enriched in chromosomal regions, spindles, and microtubules, which play pivotal roles in cell proliferation. For molecular function, differentially expressed genes were significantly enriched in tubulin binding, microtube binding, and activity, which are mostly associated with the mitotic cell cycle and proliferation. Collectively, GO enrichment analysis showed that cell cycle–related GO terms were significantly enriched in differentially expressed genes associated with retinoblastoma.
Figure 2.
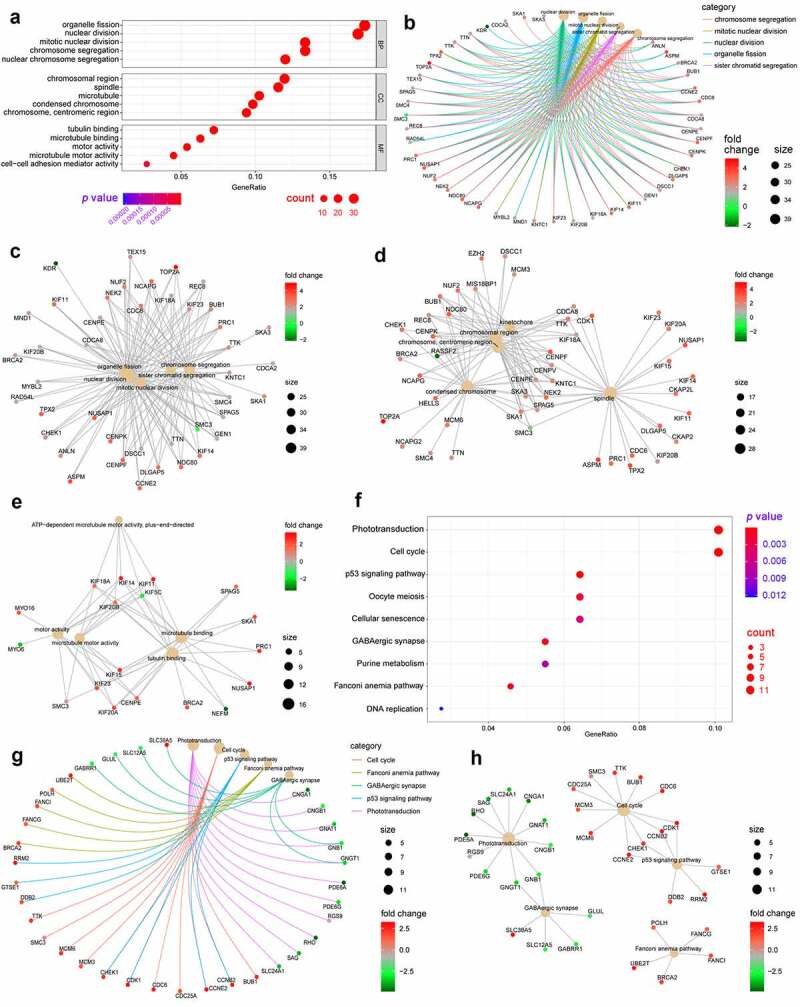
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of differentially expressed genes in retinoblastoma. (a) Bubble chart displaying GO enrichment terms of differentially expressed genes in retinoblastoma, including biological processes (BP), cellular components (CC), and molecular functions (MF). (b) Enriched network of differentially expressed genes and enriched GO terms. (c-e) ClueGO network of differentially expressed genes and enriched GO terms of biological process (c), cell components (d), and molecular function (e). (f) Bubble chart displaying the enrichment of differentially expressed genes in KEGG pathways in retinoblastoma. (g) Enriched network of differentially expressed genes and enriched KEGG pathways. (h) ClueGO network of differentially expressed genes and enriched KEGG pathways
Additionally, 9 KEGG pathways and 5 KEGG modules were significantly enriched in retinoblastoma samples. ‘Phototransduction’ and ‘cell cycle’ were the two most significantly enriched KEGG pathways, which were relevant to visual perception, retinal function, and cell proliferation (figure 2(f-h)).
3.3. GSEA and GSVA demonstrated cell cycle dysregulation and visual cell impairment in retinoblastoma
GSEA was performed on GSE110811, which was the largest sample among the three datasets, to identify the possible mechanisms involved in the pathogenesis of retinoblastoma. Five significantly upregulated pathways and five significantly downregulated pathways are shown in Figure 3(a-c) and Table 1, indicating cell cycle dysregulation and visual cell impairment during retinoblastoma development.
Figure 3.
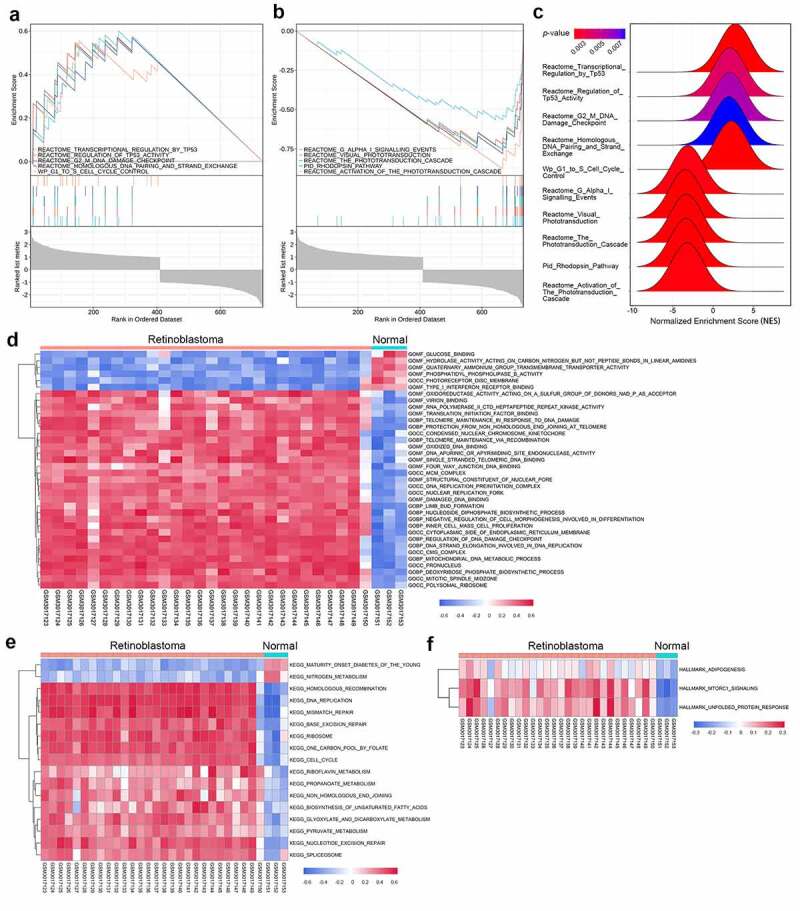
Gene set enrichment analysis (GSEA) and gene set variation analysis (GSVA) in the GSE110811 dataset. (a-b) Multi-GSEA plot showing the five most significantly upregulated pathways (a) and the five most significantly downregulated pathways (b) in GSE110811 samples. (c) Ridge plot displaying the normalized enrichment score (NES) of the above ten pathways. (d-f) Heatmap of GSVA results showing GSVA scores of the GO gene set (d), KEGG gene set (e), and hallmark gene set (f) enriched in GSE110811
Table 1.
Significantly upregulated and downregulated pathways through gene set enrichment analysis
| Description | Enrichment score (ES) | Normalized enrichment score (NES) | P-value | False discovery rate (FDR) |
|---|---|---|---|---|
| Reactome_Transcriptional_Regulation_by_ Tumor Protein P53 | 0.600427 | 2.767542 | 0.001988 | 0.004122 |
| Reactome_Regulation_of_ Tumor Protein P53_Activity | 0.572310 | 2.066904 | 0.003816 | 0.006433 |
| Reactome_G2_M_DNA_Damage_Checkpoint | 0.570086 | 2.058872 | 0.005725 | 0.008753 |
| Reactome_Homologous_DNA_Pairing_and_ Strand_Exchange | 0.569061 | 1.972560 | 0.007692 | 0.010738 |
| Wp_G1_to_S_Cell_Cycle_Control | 0.555013 | 2.257859 | 0.001972 | 0.004122 |
| Reactome_G_Alpha_I_Signalling_Events | −0.566715 | −3.133801 | 0.002118 | 0.004122 |
| Reactome_Visual_Phototransduction | −0.727044 | −3.426219 | 0.002000 | 0.004122 |
| Reactome_The_Phototransduction_Cascade | −0.754767 | −3.314062 | 0.002008 | 0.004122 |
| Pid_Rhodopsin_Pathway | −0.780250 | −3.364626 | 0.002012 | 0.004122 |
| Reactome_Activation_of_The_Phototransduction_Cascade | −0.876170 | −3.201726 | 0.002092 | 0.004122 |
Furthermore, we performed GSVA, a non-parametric approach to facilitate the evaluation of gene set enrichment variation of each expression profile on GSE110811. The H hallmark gene sets, C2 curated gene sets, KEGG subsets, and C5 ontology gene sets including GO_BP, GO_CC, and GO_MF subsets were used as reference sets. The most significantly upregulated pathways in retinoblastoma were related to cell cycle events such as DNA replication, homologous recombination, regulation of DNA damage checkpoints, and base excision repair (Figure 3(d-f)), which were in line with the GSEA results.
3.4. Network analysis and hub gene selection narrows down to CENPE
As illustrated in Figure 4(a), the protein-protein interaction network of 240 differentially expressed genes was constructed based on the STRING database and gathered as a cluster consisting of 186 nodes and 2482 edges. The MCODE plugin was used to determine the most significant module that contained 70 nodes and 2042 edges (Figure 4(b), MCODE score ≥30), which were all upregulated differentially expressed genes. The clusters obtained were further explored using the cytoHubba plugin by the MCC method (score ≥5000), and the top six hub genes, including budding uninhibited by benzimidazoles 1 (BUB1), cyclin dependent kinase 1 (CDK1), CENPE, kinesin family member 20A (KIF20A), PDZ binding kinase (PBK), and targeting protein for xklp2 (TPX2), were screened out (Figure 4(c)).
Figure 4.
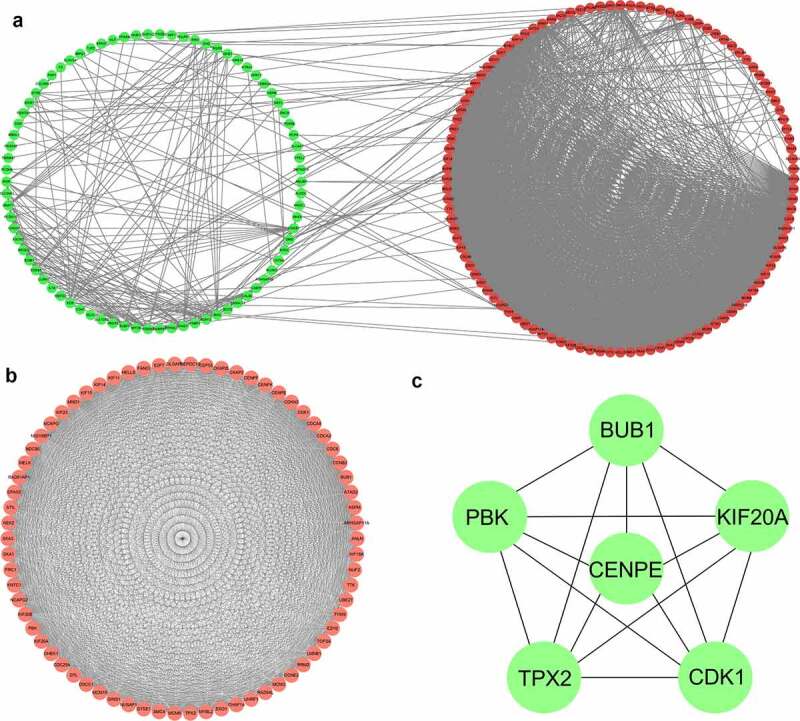
Protein-protein interaction network and the most significant modules of differentially expressed genes. (a) The protein-protein interaction network was analyzed using the STRING database. There were 186 nodes and 2482 edges in the network. (b) The most significant module identified by the MCODE plugin (score ≥30) consists of 70 nodes and 2042 edges. (c) The cytoHubba plugin identified the top six genes as hub genes by the maximum correlation criteria method (score ≥5000)
3.5. Drug sensitivity analysis showed that nelarabine is a potential drug for retinoblastoma targeting multiple hub genes
CellMiner is a database and online query tool that facilitates the integration and research of molecular and pharmacological data for NCI-60, a group of 60 multiple human cancerous cell lines, to evaluate more than 100,000 chemical compounds and natural products. As illustrated in Figure 5(a,b,d and e), chelerythrine and nelarabine had the most significant positive correlations with CENPE and TPX2, indicating that both CENPE and TPX2 are targeted by chelerythrine and nelarabine. Similar correlations were found for allopurinol and nelarabine for BUB1 (Figure 5(g-h)), irofulven for KIF20A (Figure 5(j)), nelarabine for PBK (Figure 5(k)), and nelarabine and PX-316 for CDK1 (Figure (5l-m)).
Figure 5.
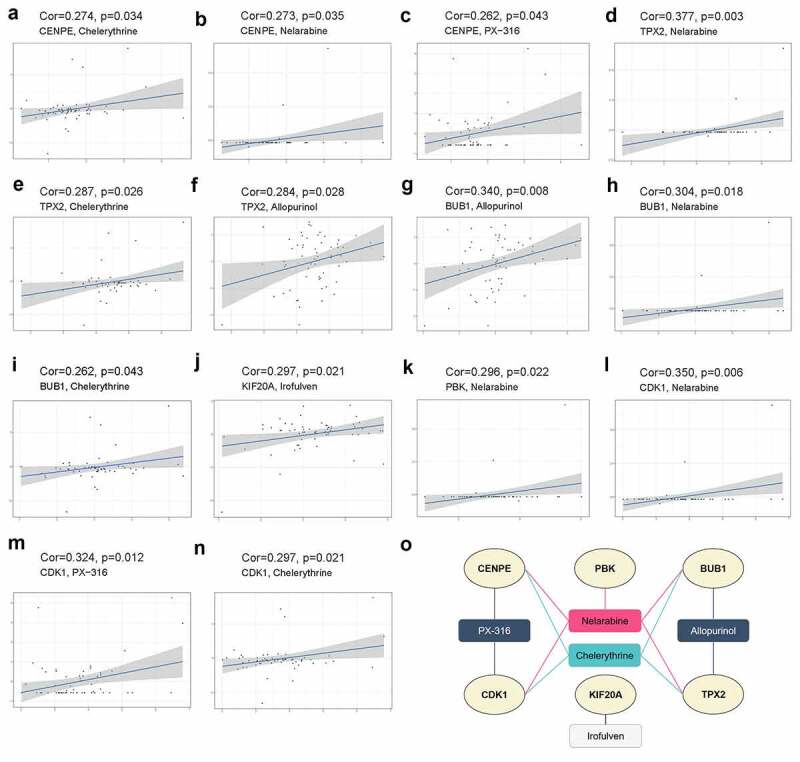
Drug sensitivity analysis of six hub genes using NCI‐60 cell line data in the CellMiner database. (a-n) Scatter plots displaying the most significant positive correlations between hub genes CENPE (a-c), TPX2 (d-f), BUB1 (g-i), KIF20A (j), PBK (k), CDK1 (l-n) and drug sensitivity based on Pearson correlation. (o) Network of the six hub genes and sensitive drugs. Ellipses indicate hub genes in light yellow, and rectangles represent sensitive drugs targeting these hub genes
Interestingly, nelarabine, an anticancer chemotherapy drug used to treat T-cell leukemia and lymphoma, was found to be positively correlated with five hub genes (BUB1, CDK1, CENPE, PBK, and TPX2), suggesting its potential use in the treatment of retinoblastoma (Figure 5(o)).
3.6. Pan-cancer analysis of hub genes
Pan-cancer analysis provides a panoramic view of the oncogenic processes that contribute to human cancer. Thorsson et al [42]. classified more than 10,000 tumors into 6 immune subtypes by immunogenomic analyses, including C1 (wound healing), C2 (IFN-γ dominant), C3 (inflammatory), C4 (lymphocyte-depleted), C5 (immunologically quiet), and C6 (TGF-β dominant), which correspond to different immune response patterns. As indicated in Figure 6(a), immunosubtyping analysis showed that hub gene expression differed significantly among these six pan-cancer immune subtypes. All six hub genes were highly expressed in the C1 and C2 subtypes and lowly expressed in the C3 and C5 subtypes.
Figure 6.
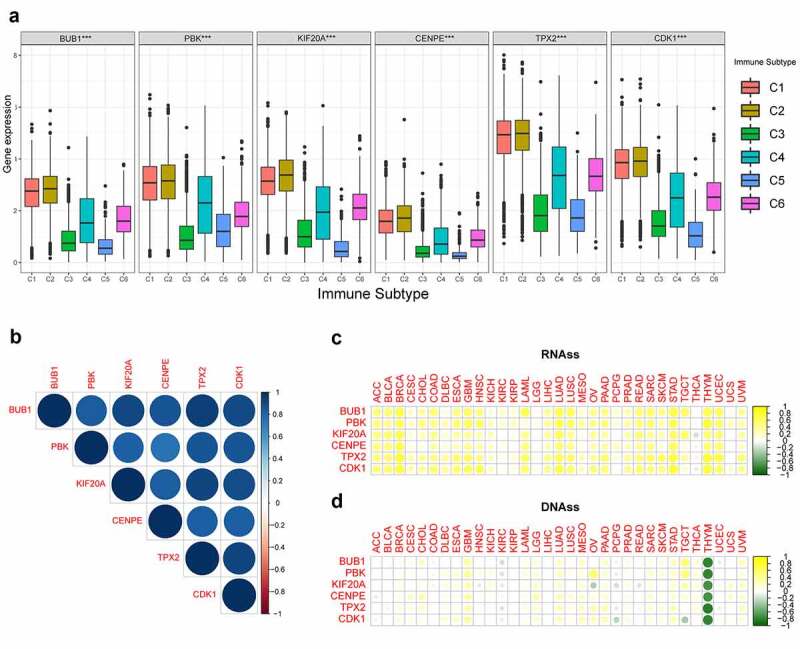
Correlation analysis of expression levels of the six hub genes with immune infiltration subtypes and cancer stemness in pan‐cancer. (a) The distribution of expression levels of the six hub genes with immune infiltration subtypes across all 33 cancer types. C1 (wound healing); C2 (IFN-gamma dominant); C3 (inflammatory); C4 (lymphocyte-depleted); C6 (TGF-β dominant). ***, P < 0.001. (b) Correlation matrix based on Pearson’s correlation test among the six hub genes. The circle color intensity is proportional to the correlation coefficient. Blue represents positive correlations and red represents negative correlations. (c-d) Heatmaps showing the correlation between the expression levels of the six hub genes and the RNAss (c) and DNAss (d) stemness indices in 33 TCGA cancer types. DNAss, DNA methylation-based stemness score; RNAss, RNA-based stemness score. Yellow points represent a positive correlation while green points represent a negative correlation. ACC, adrenocortical carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD, colon adenocarcinoma; DLBC, lymphoid neoplasm diffuse large B-cell lymphoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LAML, acute myeloid leukemia; LGG, brain lower grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; MESO, mesothelioma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TGCT, testicular germ cell tumors; THCA, thyroid carcinoma; THYM, thymoma; UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma; UVM, uveal melanoma
Additionally, the expression levels of all six hub genes were highly positively correlated with each other in pan-cancer (Figure 6(b)). mRNA expression-based stemness analysis showed that, except for a negative correlation in thyroid carcinoma, all hub genes were positively correlated with stemness index in the other 32 tumors (Figure 6(c)). DNA methylation-based stemness analysis demonstrated that the six hub genes were negatively correlated only with thymoma (Figure 6(d)).
3.7. CENPE is a critical point in the ceRNA and TF interaction network
Currently, little is known regarding the role of CENPE in retinoblastoma; thus, we focused on CENPE for further analysis. The ceRNA hypothesis suggests that some RNAs possessing miRNA-binding sites can competitively bind to target miRNAs as molecular sponges and subsequently alter the expression of genes encoding proteins[43]. As shown in Figures 7(A), 2 miRNAs and 141 lncRNAs were identified as nodes in the network. hsa-miR-642a-3p and hsa-miR-556-3p were predicted to interact with 95 and 50 lncRNAs, respectively, of which opa interacting protein 5-antisense RNA 1 (OIP5-AS1), nuclear paraspeckle assembly transcript 1 (NEAT1), poly(RC) binding protein 1-antisense RNA 1 (PCBP1-AS1), and noncoding RNA activated by DNA damage (NORAD) were shared by the two miRNAs.
Figure 7.
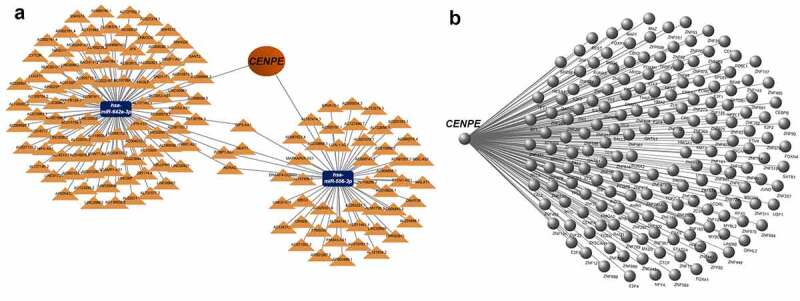
The CENPE-miRNA-lncRNA competing endogenous RNA (ceRNA) network and CENPE-transcription factor interaction network. (a) ceRNA network. Orange triangles represent lncRNAs and blue rectangles represent miRNAs. (b) Transcription factor network for CENPE. The right part of the diagram indicates the transcription factors predicted to interact with CENPE.
Transcription factors regulate gene expression by binding and unbinding near coding sequences to control the rate of gene transcription. In this study, a transcription factors regulatory network was established to identify potential transcription factors involved in retinoblastoma. As shown in Figure 7(b), CENPE was modulated by 172 transcription factors. Among these, 100 transcription factors belonged to the zinc-coordinating group, while 36 transcription factors were categorized as helix-turn-helix (Supplementary Table 4).
3.8. Immune cell infiltration, especially by B cells and plasma cells, was enhanced in samples with high CENPE expression
The compositions and proportions of twenty-two types of immune cells in each sample in the three datasets are shown in Figure 8(a-c). The correlation of immune cells is illustrated by heatmaps (Figure 8(d-f)). The populations with the most significantly positive relationship were monocytes and CD8+ T cells (r = 0.95) in GSE24673, monocytes and resting mast cells (r = 0.96) in GSE97508, and memory B cells and plasma cells or activated mast cells (r = 0.97) in GSE110811. The populations with the most significant negative relationship included monocytes and activated natural killer (NK) cells (r = −0.93) in GSE24673; monocytes and naïve B cells (r = −0.87) in GSE97508, and monocytes and resting mast cells (r = −0.75) in GSE110811.
Figure 8.
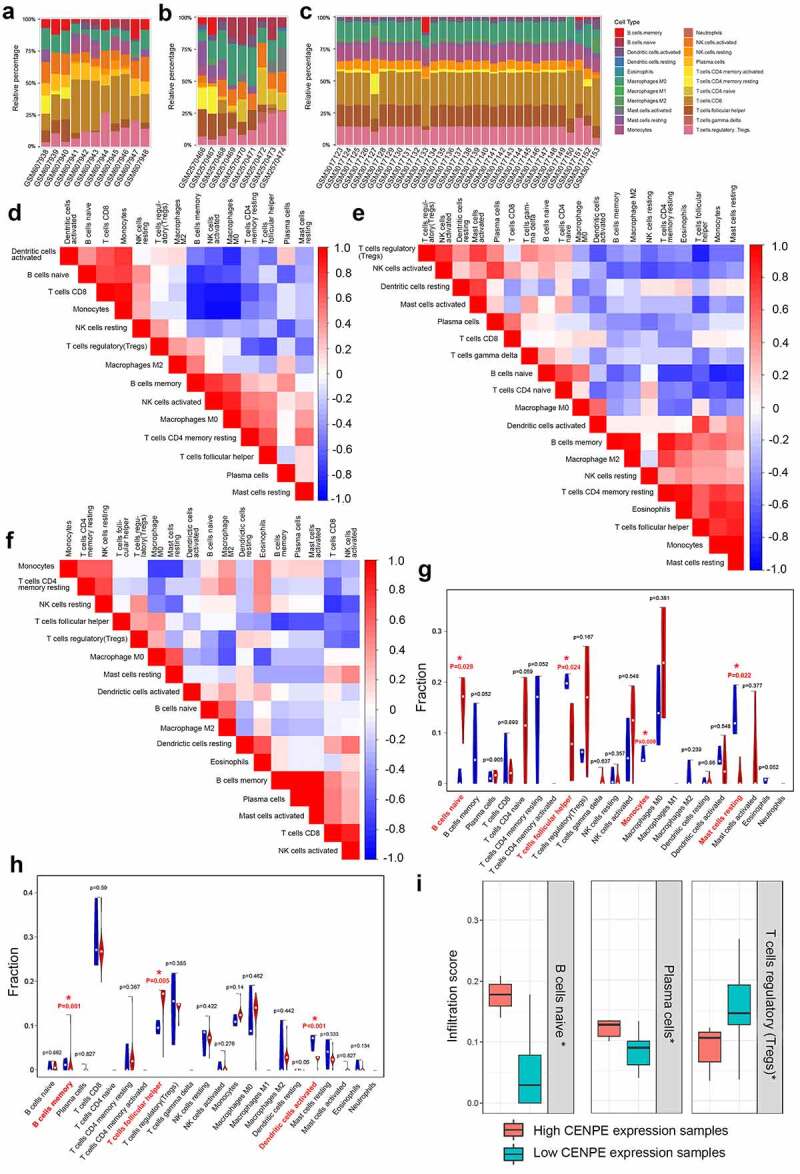
Profile of immune infiltration in retinoblastoma and normal retinas. (a-c) The proportions of immune cells in each sample of GSE24673 (a), GSE97508 (b), and GSE110811 (c) are displayed with different colors, and the lengths of the bars in the stacked bar chart indicate the proportion of each immune cell population. (d-f) Correlation matrices of immune cell proportions in GSE24673 (d), GSE97508 (e), and GSE110811 (f). A positive correlation is indicated in red, while a negative correlation is indicated in blue. The darker the color, the stronger the correlation. (g-h) Differences in the proportion of each immune cell in retinoblastoma and normal samples of GSE97508 (g) and GSE110811 (h). Blue represents normal retinas, and red represents retinoblastoma tissues. * P < 0.05. (i) Comparison of immune cells in samples with high and low CENPE expression. * P < 0.05
Next, we compared the immune cell expression between retinoblastoma and normal retinas. T follicular helper cells were significantly downregulated in retinoblastoma in GSE97508 but upregulated in retinoblastoma in GSE110811. Monocytes and naïve B cells were significantly upregulated, while resting mast cells were downregulated in GSE97508 (Figure 8(g)). Memory B cells and activated dendritic cells were downregulated in the GSE110811 cells (Figure 8(h)). No significant changes in the immune cell fraction were detected in GSE24673 (Supplementary Fig. 1). Moreover, naïve B cells and plasma cells were significantly upregulated, while regulatory T cells were significantly downregulated in the sample with high CENPE expression (Figure 8(i)).
3.9. Expression profiles and mutation features of CENPE in pan-cancer
Co-expression analysis showed that CENPE was positively correlated with the other five hub genes in most of the thirty-three cancer types (Figure 9(a)). In most cases, CENPE was remarkably upregulated in tumors in human organs or tissues such as colorectal, cervical, and gastric cancer, whereas it was downregulated in leukemia (Figure 9(b)). Moreover, CENPE expression was significantly elevated in thirty-two cancer types from the GEPIA database, which computed TCGA and Genotype-Tissue Expression (GTEx) profiles in the form of transcripts per million, whereas it was decreased in acute myeloid leukemia (Figure 9(c)). The Oncomine database also revealed that CENPE mRNA expression was relatively higher in breast cancer, lung cancer, and sarcoma, but less expressed in leukemia (Figure 9(d)). All the genetic alterations of CENPE were mutations, with no amplifications, deletions, or fusions detected. The highest mutation frequency of CENPE (>6%) occurred in acral melanoma and melanoma of unknown origin (Figure 9(e)). Missense mutations of CENPE were the main type of genetic alteration, such as a P121S alteration in the kinesin domain could lead to mistranslation from P (Proline) to S (Serine) at the 121 site of CENPE protein (figure 9(f-g)).
Figure 9.
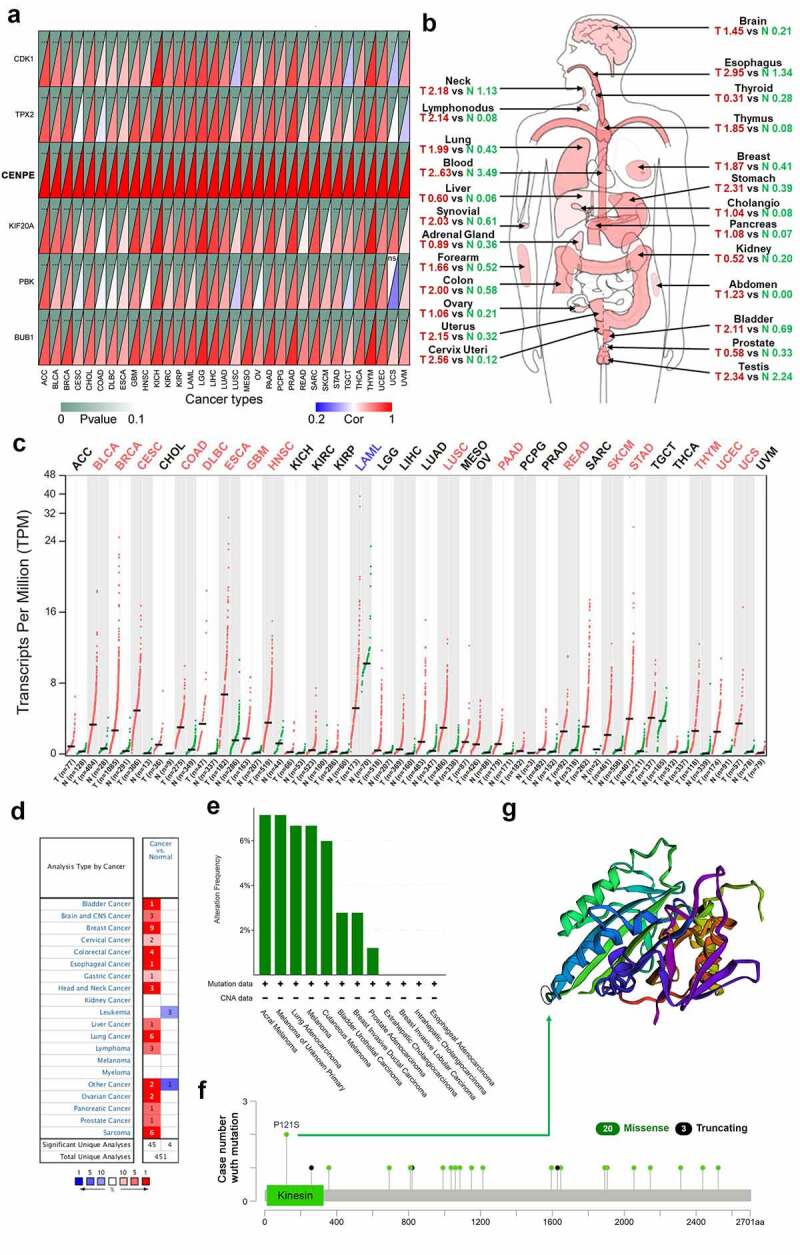
Expression profiles and mutation features of CENPE in different types of human cancers. (a) Correlation analysis of CENPE expression and the other five hub genes across thirty-three cancer types. The lower right rectangles in red color represent positive correlations whereas blue rectangles represent negative correlations. The darker the color, the stronger the correlation. The upper left rectangles represent the P value. As the P value approaches zero, the color of the triangle is closer to dark green. ***P < 0.001, **P < 0.01. (b) The median expression of tumor and normal samples in a body map. A darker red color corresponds to a higher gene expression level. T, tumor (red); N, normal (green). (c) Dot plot showing the expression profile of CENPE across thirty-three tumor samples and paired normal tissues. Red dots represent tumor samples while green dots represent normal samples. Tumor annotations in red represent significant CENPE upregulation and blue represents significant CENPE downregulation. (d) Expression of CENPE in different types of cancers compared with normal samples in the Oncomine database. (e-f) The alteration frequency with mutation type (e) and site (f) of CENPE in pan-cancers based on the cBioPortal database. (g) The mutation site with the highest alteration frequency (P121S) is marked with a circle in the crystal structure of CENPE
3.10. CENPE upregulation was validated in retinoblastoma cell lines
CENPE expression was further validated by RT-qPCR and immunoblotting in two different retinoblastoma cell lines, Y79 and WERI-RB-1. As illustrated in Figure 10(a), RT-qPCR assays revealed that CENPE expression was significantly higher in Y79 and WERI-RB-1 cells than in ARPE-19 cells by approximately 77.8% and 61.8%, respectively. Simultaneously, immunoblotting also showed upregulated CENPE expression in two retinoblastoma cell lines compared to ARPE-19 cells (Figure 10(b)). Both differences were statistically significant (P < 0.05, Figure 10(c)).
Figure 10.
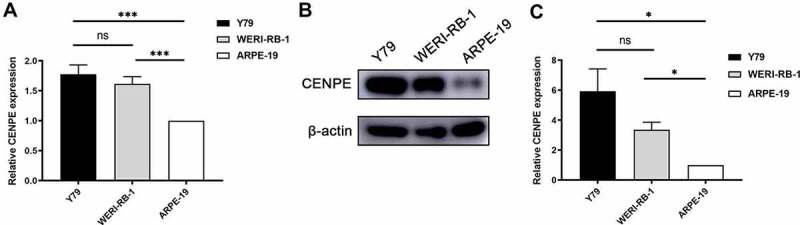
In vitro study of CENPE expression in retinoblastoma cells. (a) Evaluation of CENPE mRNA expression using RT-qPCR in two human retinoblastoma cell lines, WERI-Rb-1 and Y79, with ARPE-19 cells as a normal control. ***P < 0.001. (b) Representative image of CENPE protein expression in the three groups using immunoblotting. (c) Statistical analysis of CENPE protein expression in the three groups. *P < 0.05
4. Discussion
Retinoblastoma, with an estimated mortality rate of 70% in juvenile patients in developing countries[44], might metastasize to the brain along the optic nerve or via hematogenous metastasis to the bones, bone marrow, or liver[45]. Early diagnosis would offer pediatric patients opportunities for treatment. Although imaging examinations such as magnetic resonance imaging and computerized tomography are effective approaches for diagnosing malignancy, they still have some limitations. Therefore, exploration of new biomarkers could facilitate a more thorough understanding of the molecular mechanisms involved in retinoblastoma and offer novel insights into the development of effective treatments for patients[46]. Although diverse biomarkers associated with the pathogenesis of retinoblastoma have been recognized, there is still a deficit in the identification of expression biomarkers involved in retinoblastoma tumorigenesis[47].
In this study, we performed integrative bioinformatics analysis based on three GEO datasets to investigate potential biomarkers of retinoblastoma. Functional enrichment analysis, including GO, KEGG, GSEA, and GSVA, suggested that differentially expressed genes in retinoblastoma were significantly enriched in various mitotic procedure events, which were related to uncontrolled cell growth. Previous publications reported that loss of proliferative regulation is a pivotal characteristic of tumors and that disordered cell organelles and nuclear division lead to excessive proliferation of tumor cells[48]. The progression of retinoblastoma is intimately associated with accelerated and improper proliferation and the capacity to survive mitotic infidelity[49]. Knockdown of integrin-linked kinase (ILK), a protein that mediates microtubule dynamics and centrosome clustering in the cell cycle process and tumor transformation, results in mitotic arrest[50]. Therefore, our present study is consistent with previous studies, suggesting that dysregulation of mitosis and imbalanced cell proliferation play essential roles in the progression of retinoblastoma.
Six hub genes (BUB1, CDK1, CENPE, KIF20A, PBK, and TPX2) were determined using protein-protein interaction network assays and Cytoscape software. All hub genes were upregulated in retinoblastoma, and five (BUB1, CDK1, CENPE, PBK, and TPX2) were predicted to be sensitive and positively related to nelarabine, an antineoplastic medicine used to treat T-cell acute lymphoblastic leukemia and lymphoma. This is well-explained by the pharmacological mechanism of nelarabine being demethylated to its active form (9-beta-D-arabinosylguanine) which exerts its S-phase-specific cytotoxic potency by suppressing DNA synthesis and inducing tumor cell death[51], as the identified hub genes are mainly involved in the cell cycle.
Previous studies suggest that these six hub genes exert tumorigenic effects in various cancers through different mechanisms. BUB1, initially identified in budding yeast, encodes a serine/threonine-protein kinase that plays a crucial role in mitosis [52,53]. BUB1 expression is significantly upregulated in advanced-stage multiple myeloma, resulting in aggravated mitotic segregation errors and chromosome instability[54]. Aberrantly high BUB1 expression was detected in three retinoblastoma tissues via RNA sequencing[55]. Suppression of BUB1 could alleviate the progression of osteosarcoma by inhibiting phosphoinositide 3-kinase (PI3K)/AKT serine/threonine kinase (AKT) and extracellular signal-regulated kinase (ERK) signaling pathways[56]. CDK1 is a member of the serine/threonine-specific protein kinase family and is fundamental for G1/S and G2/M phase transitions of the cell cycle[57]. Nishida et al. reported that CDK1 was upregulated and correlated with poor prognosis in gastric cancer[58]. A pilot quantitative phosphoproteomics study revealed that CDK1 was hyperphosphorylated in retinoblastoma[59]. CDK1 is upregulated in the nuclear factor-kappa b (NF-κB) dependent pattern in glioblastoma[60]. KIF20A is generally located in the center of mitotic spindles and engages in procedures driving mitosis[61]. Upregulation of KIF20A in renal carcinoma promotes tumor progression and is related to an adverse clinical prognosis[62]. KIF20A promotes colorectal cancer tumor progression through the janus kinase (JAK)/signal transducer and activator of transcription 3 (STAT3) signaling pathway[63]. PBK was initially identified as a serine/threonine protein kinase for mitogen-activated protein kinase and is associated with cytokinesis and spermatogenesis[64]. PBK has been suggested as a promising prognostic predictor for patients with oral squamous cell carcinoma treated with radiotherapy[65]. PBK can enhance aggressive phenotypes of cervical cancer by regulating the ERK/c-Myc signaling pathway[66]. TPX2 is a microtubule-relevant protein that modulates the formation of mitotic spindles during the cell cycle[67]. A recent breast cancer cohort study demonstrated an intense correlation between upregulation of TPX2 and invasive cancer behaviors[68]. Inhibition of TPX2 could suppress the progression of hepatocellular carcinoma through the PI3K/AKT signaling pathway[69]. CENPE is a mitotic motor protein that accumulates in the G2 phase of cell division, and its inactivation obstructs the spindle assembly checkpoint[70]. High CENPE expression in non-small cell lung cancer samples is related to poor prognosis[71]. To date, the mechanism of CENPE in cancers has rarely been reported, and the role of CENPE in retinoblastoma remains unclear.
Additionally, we categorized tumor samples by C1-C6 immune signature subtypes and analyzed the expression of the six hub genes from C1 to C6, which all showed significant differences. These immune characteristics, as well as extracellular matrix, stromal cells, and blood vessels, comprise the tumor microenvironment[72], the heterogeneity of which greatly affects therapeutic efficacy and clinical prognosis[73]. Stemness is used to evaluate the stem cell-like features of tumors, including self-renewal and dedifferentiation[74]. Miranda et al [75]. proposed that tumor cells would acquire stem cell-like characteristics during tumor progression. We evaluated mRNA expression-based and DNA methylation-based stemness scores in thirty-three TCGA cancer types and correlated them with transcriptional signatures of the six hub genes, and found that the higher the expression of the hub genes, the more poorly differentiated the tumor cells in thirty-two cancers (not in thyroid carcinoma), suggesting that these hub genes might contribute to stemness maintenance.
Through an online publication search, we failed to retrieve any literature regarding CENPE in either retinoblastoma or pan-cancer analysis. Therefore, we preliminarily validated CENPE expression in two retinoblastoma cell lines and found that CENPE was significantly upregulated at both the mRNA and protein levels. Interestingly, the mean expression level in highly invasive Y79 cells was higher than that in less invasive WERI-Rb-1 cells, suggesting that CENPE might be positively correlated with retinoblastoma invasion behavior. We also assessed CENPE in multiple tumor types based on TCGA, GEPIA, and Oncomine databases. Comprehensive pan-cancer bioinformatics analysis demonstrated that CENPE is highly expressed in a wide range of human tumors. All the above findings indicate that CENPE plays a pivotal role in the pathogenesis of retinoblastoma, although the mechanisms exerted by CENPE merit further exploration.
Moreover, we comprehensively analyzed CENPE in 33 different tumors based on the profiles of TCGA, GEPIA, Oncomine, and cBioPortal databases. CENPE was upregulated in most cancers but downregulated in leukemia, as indicated in the GEPIA and Oncomine databases. It has been reported that approximately 80% of patients with acute lymphoblastic leukemia manifested low expression of CENPE in bone marrow samples[76]. Alternative splicing of the CENPE transcript might contribute to this contradictory role in different cancers.
The present study has some limitations that warrant consideration. First, the patient samples enrolled in this study were from India and the United States, and we did not ascertain the effect of ethnic heterogeneity on our research. Second, due to the limited sample size and incomplete clinical information, it is challenging to establish a prediction model for the prognosis of patients with retinoblastoma. Third, we measured CENPE expression only in vitro, and the molecular mechanisms involved in retinoblastoma remain to be elucidated.
5. Conclusion
To the best of our knowledge, our work provides a multidimensional analysis of potential intervention targets for retinoblastoma. We identified the differentially expressed genes in retinoblastoma, revealed accelerated cell cycle events as possible functional pathways, constructed a protein-protein interaction network comprising six hub genes associated with mitosis, and predicted potential sensitive therapeutic drugs. In addition, we suggest manipulating CENPE as a novel biomarker and therapeutic target for retinoblastoma. These findings may facilitate the development of targeted therapies for retinoblastoma. Further research is required to validate and explore the underlying mechanisms.
Supplementary Material
Acknowledgements
The authors thank “Taylor & Francis Editing Services” for language editorial assistance.
Funding Statement
This work was supported by the [National Key R&D Program] under Grant [2017YFA0105301]; the [National Natural Science Foundation of China] under Grant [81730026, 81760176, and 81960158]; the [Science and Technology Commission of Shanghai Municipality] under Grant [20Z11900400]; the [Shanghai Hospital Development Center] under Grant [SHDC2020CR2040B]; the [Cultivation Program of National Natural Science Foundation for Outstanding Youth] under Grant [20202ZDB01014]; and the [Key Program of Youth Science Foundation of Jiangxi Province] under Grant [20202ACBL216009].
Research highlights
CENPE, BUB1, CDK1, KIF20A, PBK, and TPX2 are potential biomarkers of retinoblastoma.
CENPE expression is positively related with immune cell infiltration.
CENPE and the five other hub genes are highly expressed in various other cancers.
Nelarabine may be a potential antitumor agent in the treatment of retinoblastoma.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The datasets (GSE24673, GSE97508, and GSE110811) analyzed for this study can be found in the Gene Expression Omnibus (GEO) database [https://www.ncbi.nlm.nih.gov/geo/].
Author contributions
Ke Shi performed bioinformatic analysis and wrote the manuscript. Xinyue Zhu conducted in vitro experiments and statistical analysis. Jingfa Zhang, Jiali Wu and Yuhong Chen revised the manuscript for important content. Xiaodong Sun designed of the study and revised the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Fabian ID, Abdallah E, Abdullahi SU, et al. Global Retinoblastoma Study G . Global retinoblastoma presentation and analysis by national income level. JAMA Oncol. 2020;6:685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chawla B, Hasan F, Azad R, et al. Clinical presentation and survival of retinoblastoma in Indian children. Br J Ophthalmol. 2016;100(2):172–178. [DOI] [PubMed] [Google Scholar]
- [3].Munier FL, Beck-Popovic M, Chantada GL, et al. Conservative management of retinoblastoma: challenging orthodoxy without compromising the state of metastatic grace. “Alive, with good vision and no comorbidity”. Prog Retin Eye Res. 2019;73:100764. [DOI] [PubMed] [Google Scholar]
- [4].Lin FY, Chintagumpala MM.. Neonatal retinoblastoma. Clin Perinatol. 2021;48(1):53–70. [DOI] [PubMed] [Google Scholar]
- [5].Stransky B, Barrera J, Ohno-Machado L, et al. Modeling cancer: integration of “omics” information in dynamic systems. J Bioinform Comput Biol. 2007;5(4):977–986. [DOI] [PubMed] [Google Scholar]
- [6].Tao Z, Shi A, Li R, et al. Microarray bioinformatics in cancer- a review. J BUON. 2017;22:838–843. [PubMed] [Google Scholar]
- [7].Wu C, Shang XQ, You ZP, et al. TRIM59 promotes retinoblastoma progression by activating the p38-MAPK signaling pathway. Invest Ophthalmol Vis Sci. 2020;61(10):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xu Y, Fu Z, Gao X, et al. Long non-coding RNA XIST promotes retinoblastoma cell proliferation, migration, and invasion by modulating microRNA-191-5p/brain derived neurotrophic factor. Bioengineered. 2021;12(1):1587–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Guo L, Bai Y, Ni T, et al. MicroRNA1533p suppresses retinoblastoma cell growth and invasion via targeting the IGF1R/Raf/MEK and IGF1R/PI3K/AKT signaling pathways. Int J Oncol. 2021;59(1). DOI: 10.3892/ijo.2021.5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zeng Q, Wang S, Tan J, et al. The methylation level of TFAP2A is a potential diagnostic biomarker for retinoblastoma: an analytical validation study. PeerJ. 2021;9:e10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sun Z, Zhang A, Hou M, et al. Circular RNA hsa_circ_0000034 promotes the progression of retinoblastoma via sponging microRNA-361-3p. Bioengineered. 2020;11(1):949–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Martinez-Sanchez M, Hernandez-Monge J, Rangel M, et al. Retinoblastoma: from discovery to clinical management. FEBS J. 2021. DOI: 10.1111/febs.16035 [DOI] [PubMed] [Google Scholar]
- [13].Nalini V, Segu R, Deepa PR, et al. Molecular insights on post-chemotherapy retinoblastoma by microarray gene expression analysis. Bioinform Biol Insights. 2013;7:289–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hudson LE, Mendoza P, Hudson WH, et al. Distinct gene expression profiles define anaplastic grade in retinoblastoma. Am J Pathol. 2018;188(10):2328–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Danda R, Ganapathy K, Sathe G, et al. Membrane proteome of invasive retinoblastoma: differential proteins and biomarkers. Proteomics Clin Appl. 2018;12(5):e1700101. [DOI] [PubMed] [Google Scholar]
- [16].Liberzon A, Birger C, Thorvaldsdottir H, et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Goldman MJ, Craft B, Hastie M, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38(6):675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gautier L, Cope L, Bolstad BM, et al. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307–315. [DOI] [PubMed] [Google Scholar]
- [19].Leek JT, Johnson WE, Parker HS, et al. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ligges U, Mächler M.. scatterplot3d- an R package for visualizing multivariate data. J Stat Softw. 2003;8(11):1–20. [Google Scholar]
- [21].Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Khomtchouk BB, Van Booven DJ, Wahlestedt C. HeatmapGenerator: high performance RNAseq and microarray visualization software suite to examine differential gene expression levels using an R and C++ hybrid computational pipeline. Source Code Biol Med. 2014;9(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25(1):25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yu G, Wang LG, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Walter W, Sanchez-Cabo F, Ricote M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics. 2015;31(17):2912–2914. [DOI] [PubMed] [Google Scholar]
- [27].Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chin CH, Chen SH, Wu HH, et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Reinhold WC, Sunshine M, Liu H, et al. CellMiner: a web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res. 2012;72(14):3499–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Li JH, Liu S, Zhou H, et al. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42(D1):D92–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48(D1):D127–D31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Dweep H, Gretz N. miRWalk2.0: a comprehensive atlas of microRNA-target interactions. Nat Methods. 2015;12(8):697. [DOI] [PubMed] [Google Scholar]
- [36].Keenan AB, Torre D, Lachmann A, et al. ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 2019;47(W1):W212–W24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hu H, Miao YR, Jia LH, et al. AnimalTFDB 3.0: a comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 2019;47(D1):D33–D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Newman AM, Steen CB, Liu CL, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37(7):773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tang Z, Li C, Kang B, et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):l1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].You ZP, Zhang YL, Shi K, et al. Suppression of diabetic retinopathy with GLUT1 siRNA. Sci Rep. 2017;7(1):7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Thorsson V, Gibbs DL, Brown SD, et al. The immune landscape of cancer. Immunity. 2018;48:812–30 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Salmena L, Poliseno L, Tay Y, et al. A ceRNA hypothesis: the Rosetta stone of a hidden RNA language? Cell. 2011;146(3):353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dimaras H, Kimani K, Dimba EA, et al. Retinoblastoma. Lancet. 2012;379(9824):1436–1446. [DOI] [PubMed] [Google Scholar]
- [45].Gunduz K, Muftuoglu O, Gunalp I, et al. Metastatic retinoblastoma clinical features, treatment, and prognosis. Ophthalmology. 2006;113(9):1558–1566. [DOI] [PubMed] [Google Scholar]
- [46].de Carvalho IN, de Freitas RM, Vargas FR. Translating microRNAs into biomarkers: what is new for pediatric cancer? Med Oncol. 2016;33(5):49. [DOI] [PubMed] [Google Scholar]
- [47].Golabchi K, Soleimani-Jelodar R, Aghadoost N, et al. MicroRNAs in retinoblastoma: potential diagnostic and therapeutic biomarkers. J Cell Physiol. 2018;233(4):3016–3023. [DOI] [PubMed] [Google Scholar]
- [48].Marshall AE, Roes MV, Passos DT, et al. RB1 deletion in retinoblastoma protein pathway-disrupted cells results in DNA damage and cancer progression. Mol Cell Biol. 2019;39(16). DOI: 10.1128/MCB.00105-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Manning AL, Dyson NJ. RB: mitotic implications of a tumour suppressor. Nat Rev Cancer. 2012;12(3):220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sikkema WK, Strikwerda A, Sharma M, et al. Regulation of mitotic cytoskeleton dynamics and cytokinesis by integrin-linked kinase in retinoblastoma cells. PLoS One. 2014;9(6):e98838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rodriguez CO Jr., Stellrecht CM, Gandhi V. Mechanisms for T-cell selective cytotoxicity of arabinosylguanine. Blood. 2003;102(5):1842–1848. [DOI] [PubMed] [Google Scholar]
- [52].Hoyt MA, Totis L, Roberts BT. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 1991;66(3):507–517. [DOI] [PubMed] [Google Scholar]
- [53].Kim T, Gartner A. Bub1 kinase in the regulation of mitosis. Anim Cells Syst (Seoul). 2021;25(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fujibayashi Y, Isa R, Nishiyama D, et al. Aberrant BUB1 overexpression promotes mitotic segregation errors and chromosomal instability in multiple Myeloma. Cancers (Basel). 2020;12(8):2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Nie C, Ma H, Gao Y, et al. RNA sequencing and bioinformatic analysis on retinoblastoma revealing that cell cycle deregulation is a key process in retinoblastoma tumorigenesis. Ophthalmologica. 2021;244:51–59. [DOI] [PubMed] [Google Scholar]
- [56].Huang Z, Wang S, Wei H, et al. Inhibition of BUB1 suppresses tumorigenesis of osteosarcoma via blocking of PI3K/Akt and ERK pathways. J Cell Mol Med. 2021. DOI: 10.1111/jcmm.16805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Roskoski R Jr. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol Res. 2019;139:471–488. [DOI] [PubMed] [Google Scholar]
- [58].Nishida T, Matsushima T, Tsujimoto M, et al. Cyclin-dependent kinase activity correlates with the prognosis of patients who have gastrointestinal stromal tumors. Ann Surg Oncol. 2015;22(11):3565–3573. [DOI] [PubMed] [Google Scholar]
- [59].Selvan LDN, Danda R, Madugundu AK, et al. Phosphoproteomics of retinoblastoma: a pilot study identifies aberrant kinases. Molecules. 2018;23(6):1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Voce DJ, Bernal GM, Cahill KE, et al. CDK1 is up-regulated by temozolomide in an NF-kappaB dependent manner in glioblastoma. Sci Rep. 2021;11(1):5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lai F, Godley LA, Joslin J, et al. Transcript map and comparative analysis of the 1.5-Mb commonly deleted segment of human 5q31 in malignant myeloid diseases with a del(5q). Genomics. 2001;71(2):235–245. [DOI] [PubMed] [Google Scholar]
- [62].Ren X, Chen X, Ji Y, et al. Upregulation of KIF20A promotes tumor proliferation and invasion in renal clear cell carcinoma and is associated with adverse clinical outcome. Aging (Albany NY). 2020;12(24):25878–25894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhang Q, Di J, Ji Z, et al. KIF20A predicts poor survival of patients and promotes colorectal cancer tumor progression through the JAK/STAT3 signaling pathway. Dis Markers. 2020;2020:2032679. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [64].Gaudet S, Branton D, Lue RA. Characterization of PDZ-binding kinase, a mitotic kinase. Proc Natl Acad Sci U S A. 2000;97(10):5167–5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Yu WN, Lin HF, Lee YI, et al. PBK expression is associated with prognosis of patients with oral squamous cell carcinoma treated with radiotherapy: a retrospective study. Anticancer Res. 2021;41:2177–2182. [DOI] [PubMed] [Google Scholar]
- [66].Ma H, Han F, Yan X, et al. PBK promotes aggressive phenotypes of cervical cancer through ERK/c-Myc signaling pathway. J Cell Physiol. 2021;236(4):2767–2781. [DOI] [PubMed] [Google Scholar]
- [67].Kufer TA, Sillje HH, Korner R, et al. Human TPX2 is required for targeting Aurora-A kinase to the spindle. J Cell Biol. 2002;158(4):617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Matson DR, Denu RA, Zasadil LM, et al. High nuclear TPX2 expression correlates with TP53 mutation and poor clinical behavior in a large breast cancer cohort, but is not an independent predictor of chromosomal instability. BMC Cancer. 2021;21(1):186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Huang DH, Jian J, Li S, et al. TPX2 silencing exerts antitumor effects on hepatocellular carcinoma by regulating the PI3K/AKT signaling pathway. Int J Mol Med. 2019;44:2113–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Rao CV, Yamada HY, Yao Y, et al. Enhanced genomic instabilities caused by deregulated microtubule dynamics and chromosome segregation: a perspective from genetic studies in mice. Carcinogenesis. 2009;30(9):1469–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Hao X, Qu T. Expression of CENPE and its prognostic role in non-small cell lung cancer. Open Med (Wars). 2019;14(1):497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Anderson NM, Simon MC. The tumor microenvironment. Curr Biol. 2020;30(16):R921–R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501(7467):346–354. [DOI] [PubMed] [Google Scholar]
- [74].Friedmann-Morvinski D, Verma IM. Dedifferentiation and reprogramming: origins of cancer stem cells. EMBO Rep. 2014;15(3):244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Miranda A, Hamilton PT, Zhang AW, et al. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc Natl Acad Sci U S A. 2019;116(18):9020–9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Jimenez-Avila CE, Villegas-Ruiz V, Zapata-Tarres M, et al. Centromere-associated protein E expresses a novel mRNA isoform in acute lymphoblastic leukemia. Int J Mol Epidemiol Genet. 2018;9:43–54. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets (GSE24673, GSE97508, and GSE110811) analyzed for this study can be found in the Gene Expression Omnibus (GEO) database [https://www.ncbi.nlm.nih.gov/geo/].