

Abstract
Variants of concern (VOC) of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), including alpha, beta, gamma, delta, and omicron, threaten to prolong the pandemic, leading to more global morbidity and mortality. Genome sequencing is the mainstay of tracking the evolution of the virus, but is costly, slow, and not easily accessible. Multiplex quantitative RT-PCR assays for SARS-CoV-2 have been developed that identify all VOCs as well as other mutations of interest in the viral genome, nine mutations in total, using single-nucleotide discriminating molecular beacons. The presented variant molecular beacon assays showed a limit of detection of 50 copies of viral RNA, with 100% specificity. Twenty-six SARS-CoV-2–positive patient samples were blinded and tested using a two-tube assay. When testing patient samples, the assay was in full agreement with results from deep sequencing with a sensitivity and specificity of 100% (26 of 26). We have used our design methodology to rapidly design an assay that detects the new omicron variant. This omicron assay was used to accurately identify this variant in 17 of 33 additional patient samples. These quantitative RT-PCR assays identify all currently circulating VOCs of SARS-CoV-2, as well as other important mutations in the spike protein coding sequence. These assays can be easily implemented on broadly available five-color thermal cyclers and will help track the spread of these variants.
The coronavirus disease-2019 (COVID-19) pandemic has changed the world, leading to at least 5.3 million deaths and many permanent injuries, with more than 271 million cases worldwide as of December 20, 2021 (https://www.who.int/emergencies/diseases/novel-coronavirus-2019, last accessed December 20, 2021). Despite advances in diagnosis and vaccination, the emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants threaten to keep the pandemic going.1 The World Health Organization and the CDC (https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html, last accessed December 20, 2021) have identified SARS-CoV-2 variants of concern (VOCs), which lead to increased disease severity (https://www.gov.uk/government/publications/nervtag-paper-on-covid-19-variant-of-concern-b117, last accessed December 20, 2021), increased transmission,2 and immune/vaccine evasion.3, 4, 5 Variants of interest also have been identified that present theoretical risks because they possess mutations similar to the mutations in the VOC. Specific frequently occurring mutations also have been identified that can affect therapeutic antibody treatments for patients infected with such variants.6 These new variants and substitutions necessitate new tools for their detection and tracking.7
Currently, the most common technique to identify, classify, and track variants of SARS-CoV-2 is deep sequencing.8,9 Although sequencing is accurate and can identify each mutation present in a sample, it is costly, slow, and requires specialized instruments and interpretation when compared with other genotyping techniques, such as PCR.1 Although PCR assays target preselected mutations, they are cheaper and more accessible than sequencing for the genotyping of SARS-CoV-2, and they yield faster results. These methods may permit better tracking of variants because they spread throughout populations over time, especially in resource-limited settings.1
Several SARS-CoV-2 variant genotyping PCR assays have been developed. They use TaqMan probes (Thermo Fisher Scientific, Waltham, MA), sloppy molecular beacon probes, followed by melting temperature analysis,10 and several other strategies such as PKamp (PerkinElmer, Waltham, MA), and PANDAA qDx (Aldatu Biosciences, Watertown, MA). However, most of these assays either target single mutations per reaction or lack the appropriate number of targets to thoroughly characterize SARS-CoV-2. Simple assays that detect multiple mutations at the same time are needed for identification of circulating VOCs, variants of interest, and for the identification of specific mutations.
Our approach is to generate a multiplex assay for SARS-CoV-2 variants by exploiting the superior selectivity and self-quenching characteristics of molecular beacons. Molecular beacons can be designed to selectively bind to a mutant target sequence while avoiding the wild-type sequence, which differs from the mutant by a single-nucleotide substitution. The interaction of a molecular beacon with its target is inherently more specific than linear oligonucleotides that do not have a stem-and-loop structure because target binding requires the dissociation of the stem, which is thermodynamically costly.11 The utility of this discriminatory power has been shown previously in real-time PCR for a number of applications.12,13
This manuscript presents a two-tube multiplex quantitative RT-PCR (qRT-PCR) assay that identifies current VOCs by detecting eight different mutations in the SARS-CoV-2 spike protein. Mutations that have been shown to increase immune escape, avoid neutralization, and increase transmissibility were selected. Targeting these causative mutations is fruitful because these strains may mutate and lose certain coincidental mutations, but mutations that increase transmission and immune evasion are likely to be maintained. In addition, by targeting these types of mutations, it is possible to detect new variants that arise from combinations of previously confirmed mutations, as is the case for the VOC omicron. This manuscript also describes a molecular beacon design process that can be used to rapidly produce allele-discriminating assays for new variants as they arise. As proof of this design process, we generated a new assay within 2 weeks for accurately identifying the omicron variant.
Materials and Methods
Synthetic RNA Targets
RNA standards of the SARS-CoV-2 whole genome and variant spike proteins were provided by Exact Diagnostics through Bio-Rad Laboratories (Hercules, CA; catalog numbers COV019, COV019CE, COVA, COVB, COVE, and COVG). Additional spike protein RNA sequences, such as those containing the E484Q and T478K mutations and the omicron spike RNA, were generated from a double-stranded synthetic DNA (gBlock) from Integrated DNA Technologies (Coralville, IA). A 5′-T7 promoter was appended to it, which enabled transcription by T7 RNA polymerase, which was performed using the HiScribe T7 High-Yield RNA Synthesis Kit (New England Biolabs, Ipswich, MA). The RNA produced was purified with a 50-μg Monarch RNA Cleanup Kit (New England Biolabs), and by denaturing polyacrylamide gel electrophoresis, eluting the band of correct size in gel elution buffer [400 mmol/L NaCl, 20 mmol/L Tris-HCl (pH 8.0), and 1 mmol/L EDTA]. The concentration of the eluted RNAs was determined by a NanoDrop spectrophotometer (Thermo Fisher Scientific).
Primer Design
Primers were designed by using the primer3 server (https://dev.primer3plus.com/index.html, last accessed December 20, 2021).14 Two hundred nucleotides surrounding the mutation of interest on either side of the SARS-CoV-2 genome was used as input, the target-binding region of the molecular beacon was used as the hybridization probe, and the amplicon size was set between 50 and 300 nucleotides. The sequences of all the primers that were used in the assay are shown in Table 1.
Table 1.
Primers and Probes Used in Multiplex Assay
| Reaction | Oligo | Sequence | Concentration, nmol/L |
|---|---|---|---|
| Tube 1 | K417f | 5′-GGTGATGAAGTCAGACAAATCG-3′ | 100 |
| S.K417N-MB | 5′-FAM-CCGGCCGCAAACTGGAAATATTGCTGATTATGGCCGG-BHQ1-3′ | 250 | |
| S.K417T-MB | 5′-Q705-CCGGCTCAAACTGGAACGATTGCTGATAGCCGG-BHQ2-3′ | 250 | |
| K417r | 5′-GCAGCCTGTAAAATCATCTGG-3′ | 500 | |
| L452Rf | 5′-AGGCTGCGTTATAGCTTGGA-3′ | 100 | |
| S.L452R-MB | 5′-Q670-CGTCGCGGGTAATTATAATTACCGGTATAGATTGTTTAGCGCGACG-BHQ2-3′ | 250 | |
| L452Rr | 5′-TCAGTTGAAATATCTCTCTCAAAAGGT-3′ | 500 | |
| d69-70f | 5′-GAACTCAATTACCCCCTGCAT-3′ | 100 | |
| S.d69-70-MB | 5′-CFG-CGCTCGGTTCCATGCTAT CTCTGGGACCACGAGCG-BHQ1-3′ | 150 | |
| d69-70r | 5′-TGGTAGGACAGGGTTATCAAA-3′ | 500 | |
| CDC-N1-F | 5′-GACCCCAAAATCAGCGAAAT-3′ | 50 | |
| N1-MB | 5′-CFR-CGCGAGACCCCGCATTACGTTTGGTGGACCCTCGCG-BHQ2-3′ | 50 | |
| CDC-N1-R | 5′-TCTGGTTACTGCCAGTTGAATCTG-3′ | 250 | |
| Tube 2 | T478Kf | 5′-TTGTTTAGGAAGTCTAATCTCAAACC-3′ | 100 |
| S.T478K-MB | 5′-HEX-CCGCGACCGGTAGCAAACCTTGTAATTCGCGG-BHQ1-3′ | 250 | |
| S.E484K-MB | 5′-CFR-CGCAGGCTTGTAATGGTGTTAAAGGTTTTAATTGCCTGCG-BHQ2-3′ | 250 | |
| S.E484Q-MB | 5′-FAM-CGCGGGCTTGTAATGGTGTTCAAGGTTTTAATTGCCCGCG-BHQ1-3′ | 250 | |
| S.N501Y-MB | 5′-Q670-GCCAACCCACTTATGGTGTTGGC-BHQ2-3′ | 250 | |
| N501Yr | 5′-ACAAACAGTTGCTGGTGCAT-3′ | 500 |
Underlined text identifies the hairpins of the molecular beacons, bold letters indicate the point mutation nucleotides being identified by that molecular beacon, and a wide central space indicates where the deletion is for the d69-70 molecular beacon.
BHQ, Black Hole Quencher; CFG, Cal Fluor Gold 540; CFR, Cal Fluor Red 610; FAM, fluorescein; HEX, hexachlorofluorescein; Q670, Quasar 670; Q705, Quasar 705.
Design and Characterization of Single-Nucleotide Discriminating Molecular Beacons
A key consideration in the design of single-nucleotide discriminating molecular beacons is that the melting temperature (Tm) of the probe-target hybrid with the perfect target (in the present case, the mutant sequence) is minimally higher, by only 3°C to 7°C, than the annealing temperature of the PCR (which is the temperature at which fluorescence will be monitored). This makes it so that the mismatched hybrid (in the present case, the wild-type sequence) will not appreciably bind to the molecular beacon at the fluorescence monitoring temperature. Furthermore, the Tm of the stem of the molecular beacon should be higher than the detection temperature.
The primers were designed so that they will function at the PCR annealing temperature of 58°C. A region of sequence around the site of each mutation was selected such that the Tm of the perfect probe-target hybrid was higher than the annealing temperature, but the mismatched single-nucleotide polymorphism had a Tm that was lower than the annealing temperature. These Tms were predicted by using the DINAMelt (http://www.unafold.org/Dinamelt/applications/two-state-melting-hybridization.php, last accessed December 20, 2021)15 two-state melting hybridization server with the following parameters: 0.25 μmol/L oligonucleotide concentration, [Na+] = 60 mmol/L, [Mg2+] = 1.5 mmol/L, for DNA at 58°C.
After deciding on the target binding region, a hairpin stem was generated by appending five or six nucleotides on either side (with four to five G:C pairs) such that they were complementary to each other, but not complementary to the target. In addition, it was ensured that the 5′ nucleotide was not a guanosine because that could lead to fluorescence quenching as a result of the 5′ position of the fluorophore. These sequences then were ordered from either LGC Biosearch Technologies (Petaluma, CA) or from Eurofins (Luxembourg City, Luxembourg). They were purified by high-performance liquid chromatography through a PRP-3 Reversed-Phase High-Performance Liquid Chromatography Column (Hamilton Company, Reno, NV) in a Beckman System Gold High-Performance Liquid Chromatography System (Fullerton, CA). The desired fractions then were ethanol-precipitated and resuspended in water, and their concentration was determined by a NanoDrop spectrophotometer. The sequences of all molecular beacons that were used in the assay are shown in Table 1.
To determine if the designed sequences could sufficiently discriminate the alleles at the annealing temperature of the PCR assay, the fluorescence of the molecular beacons was measured in the presence of either a mutant target, a wild-type target, or no target, under the buffer conditions that are expected to be present during PCR. Each 25-μL thermal denaturation reaction contained 20 mmol/L Tris-HCl (pH 8.4), 50 mmol/L KCl, 1.5 mmol/L MgCl2, 1 μmol/L oligonucleotide target, and 0.4 μmol/L molecular beacon. The fluorescence intensity of each reaction was measured at every degree in the appropriate fluorescence channel as the temperature was lowered from 95°C to 30°C (decreasing 1°C/10 seconds) in a Bio-Rad CFX96 Touch Real-Time PCR Detection System.
Melt curve signals were normalized for each molecular beacon. The difference between a normalization point and the other melt curve fluorescent signals was subtracted from the raw fluorescent intensity values and then all values in that channel were divided by the maximum intensity for that molecular beacon to derive the relative fluorescence units, which range between 0 and 1. These data are shown in Supplemental Figure S1.
Optimizing the Concentrations of the qRT-PCR Multiplex Primers and Molecular Beacons
qRT-PCR was performed with TaqMan Fast Virus 1-Step Master Mix (No ROX) to optimize conditions, and TaqPath 1-Step Multiplex Master Mix (No ROX) (both from Applied Biosystems, Waltham, MA) for final experiments and patient samples.
Asymmetric PCR was performed for each amplicon, in which the primer generating the strand complementary to the molecular beacon was in 5-fold excess over the other primer. All molecular beacons were designed in the same forward strand direction. G:T mismatches can be discriminated from each other by switching the strand being investigated to the complementary strand.16 The molecular beacon and primer concentrations were adjusted by trial and error for sufficient amplification and signal. The final working concentrations of each of the components for each tube are listed in Table 1, along with their sequences.
Each reaction had a final volume of 20 μL comprising TaqPath or TaqMan Master 1-Step Mix (No ROX), water, and primers and probes (as listed in Table 1, Table 2, Table 3). This complete master mix was pipetted (15 μL) into each PCR tube, and then 5 μL of sample was added and mixed before running on the thermal cycler.
Table 2.
Listing Sequences of Oligonucleotides Used in the SARS-CoV-2 Multiplex Screening Assay Presented in Supplemental Figure S2
| Oligonucleotide name | Sequence |
|---|---|
| CoV-RdRP forward | 5′-GTGARATGGTCATGTGTGGCGG-3′ |
| CoV-RdRP reverse | 5′-CARATGTTAAASACACTATTAGCATA-3′ |
| CoV-RdRP molecular beacon | 5′-FAM-CGCAGGGTGGAACCTCATCAGGAGATGCCTGCG-BHQ1-3′ |
| CoV-N forward | 5′-GACCCCAAAATCAGCGAAAT-3′ |
| CoV-N reverse | 5′-TCTGGTTACTGCCAGTTGAATCTG-3′ |
| CoV-N molecular beacon | 5′-CFR-CGCGAGACCCCGCATTACGTTTGGTGGACCCTCGCG-BHQ2-3′ |
| ACTB forward | 5′-CCCAGCACAATGAAGATCAAGATC-3′ |
| ACTB reverse | 5′-AAGCATTTGCGGTGGACGAT-3′ |
| ACTB molecular beacon | 5′-Q705-CGCCCGGCAAGCAGGAGTATGACGAGTCCGGCGGGCG-BHQ2-3′ |
Underlined nucleotides in molecular beacon probes indicate the 5′ and 3′ arm regions of the probe.
BHQ, black hole quencher; CFR, Cal Fluor Red 610; CoV, coronavirus; FAM, fluorescein; Q705, Quasar 705.
Table 3.
Primers and Probes Used in Omicron Assay
| Oligo | Sequence | Concentration, nmol/L |
|---|---|---|
| T478Kf | 5′-TTGTTTAGGAAGTCTAATCTCAAACC-3′ | 100 |
| S.E484A-MB | 5′-Cy5.5-CCGCCTTTGTAATGGTGTTGCAGGTTTTAATTAGGCGG-BHQ2-3′ | 250 |
| N501Yr | 5′-ACAAACAGTTGCTGGTGCAT-3′ | 500 |
| CDC-N1-F | 5′-GACCCCAAAATCAGCGAAAT-3′ | 50 |
| N1-MB | 5′-CFR-CGCGAGACCCCGCATTACGTTTGGTGGACCCTCGCG-BHQ2-3′ | 50 |
| CDC-N1-R | 5′-TCTGGTTACTGCCAGTTGAATCTG-3′ | 250 |
Underlined text identifies the hairpins of the molecular beacons, the bold letter indicates the point mutation nucleotide being identified.
BHQ, black hole quencher; CFR, Cal Fluor Red 610.
Assay Sensitivity and Specificity
The sensitivity of the assay was determined by preparing dilution series of in vitro transcribed targets that were diluted 10-fold from 1000 copies/μL to 0.1 copies/μL. Five microliters of each of these dilutions was combined with 15 μL completed master mix per reaction. The tubes were sealed, shaken briefly, spun down at 800 × g for 1 minute, and then run on a Bio-Rad CFX96 Touch Real-Time PCR Detection System with the TaqPath or TaqMan Fast specified thermal cycler conditions, with an annealing temperature of 58°C. Specificity of the assay was determined by comparing the known sequences of the in vitro transcribed targets with the signals in these dilution series. For patient samples, sensitivity and specificity were determined by comparing this assay's results with sequencing results, which are the gold standard.
Collection of Patient Samples and Sequencing
This study used archived, deidentified samples left over from clinical testing. The study received expedited approval with a full waiver of consent, Advarra Institutional Review Board approval Pro00058476. Patient midnasal swabs were collected for clinical testing in 1 mL DNA/RNA Shield Reagent (Zymo, Irvine, CA). Aliquots (400 μL) were used for RNA extraction using the Kingfisher Flex magnetic bead extraction system (Thermo Fisher Scientific) and the Quick RNA/DNA Magbead kit (Zymo) per the manufacturer's instructions. qRT-PCR screening was performed using 2 μL purified RNA, TaqPath 1-step Multiplex Master Mix (No ROX), and an in-house validated multiplex screening assay. Samples were run on a Quantstudio 7 (Thermo Fisher Scientific). Amplicon sequencing was performed using the Ion AmpliSeq SARS-CoV-2 Insight Research Panel (Ion Torrent, Guilford, CT). Each library contained six samples, one positive control sample, and one no template control. Barcoded libraries were prepared using the Ampliseq Kit for Chef DL8 and the Ion 530 Kit-Chef (Ion Torrent). Sixteen libraries were combined to run on a single Ion 530 chip. Templates were prepared using the Ion 530 Chef Reagents template kit. Sequencing was performed on the Ion S5XL instrument with Ion S5 Sequencing Solutions and Reagents (Ion Torrent). Sequence data were analyzed for on-target and mean depth of coverage using the SARS_COV_2_Coverage Analysis plug-in v1.3.0.2 (Ion Torrent). FASTA files were generated using the IRMA (iterative refinement meta-assembler) report plug-in v1.3.0.2 (Ion Torrent). Sequence data and corresponding metadata were uploaded to Mendeley data and are available at the following link: https://data.mendeley.com/datasets/v3n2dzt8st/1 (last accessed January 13, 2022). Aliquots of purified RNA samples no longer needed for diagnosis were deidentified and transferred to Rutgers laboratory (Newark, NJ) for initial testing of the multiplex variant molecular beacon assay.
SARS-CoV-2 Screening Assay
The SARS-CoV-2 screening assay detects conserved sequences in the N and RdRp genes of SARS-CoV-2, along with human ACTB (β-actin) mRNA, with high sensitivity. The primers and molecular beacons targeting regions in the RdRp, N, and human ACTB genes were modified and adapted from previously validated assays17 (https://www.cdc.gov/coronavirus/2019-ncov/lab/rt-pcr-panel-primer-probes.html, last accessed December 7, 2021), and are listed in Table 2.
The RdRp and N genes were chosen as targets to detect lowly and highly expressed transcripts generated by SARS-CoV-2, as well as genomic copies of the SARS-CoV-2 virus. Real-time RT-PCR assays were performed in a 20-μL volume that contained 1 × TaqPath 1-step RT-qPCR Master Mix (A28521; Thermo Fisher Scientific), 100 nmol/L CoV-RdRP forward primer, 500 nmol/L CoV-RdRP reverse primer, 250 nmol/L CoV-RdRP molecular beacon probe, 100 nmol/L CoV-N forward primer, 500 nmol/L CoV-N reverse primer, 250 nmol/L CoV-N molecular beacon probe, 100 nmol/L ACTB forward primer, 500 nmol/L ACTB reverse primer, and 250 nmol/L ACTB molecular beacon probe. In Supplemental Figure S2, each reaction was initiated with 2 to 5 μL RNA template. The PCR assays were performed in 200-μL white polypropylene PCR tubes (USA Scientific, Ocala, FL) in a CFX96 Touch real-time PCR detection system (Bio-Rad Laboratories). The thermal cycler was programmed to incubate the reaction mixtures for 10 minutes at 53°C to generate cDNA, followed by 2 minutes at 95°C to activate the DNA polymerase, and by 45 thermal cycles that consisted of DNA denaturation at 95°C for 15 seconds and primer annealing and elongation at 58°C for 60 seconds. Molecular beacon fluorescence intensity was monitored during the 58°C annealing and chain elongation stage of each thermal cycle.
RNA Purification from Inactivated SARS-CoV-2 Virus
Ten thousand copies of each inactivated VOC of SARS-CoV-2 virus from ZeptoMetrix [Buffalo, NY; catalog: NATSARS(COV2)-VP] was treated with proteinase K in a 210-μL reaction (1% SDS, 25 mmol/L EDTA, 10 mmol/L Tris pH 8.3, and 20 μg proteinase K; catalog: 25530049; Thermo Fisher) at 45°C for 30 minutes, to decrosslink the protein matrix entrapping the viral RNA. After the addition of 1 to 2 μg yeast carrier tRNA (catalog: 10109495001; Roche, Basel, Switzerland), RNA was purified using the QIAgen (Düsseldorf, Germany) RNeasy Mini kit (catalog: 74104).
Multiplex qRT-PCR with Samples
RNA purified patient and inactivated virus samples were tested in a similar manner to how the assay's sensitivity and specificity was tested. Five microliters of each sample was combined with 15 μL each completed master mix using TaqPath and run as mentioned in Optimizing the Concentrations of the qRT-PCR Multiplex Primers and Molecular Beacons.
qRT-PCR Analysis and Plotting
Data generated from the CFX96 Touch Real-Time PCR Detection System were analyzed with Bio-Rad's CFX Maestro Software version 2.0, which corrects for fluorescence drift, subtracts the baseline, and fits curves to each qRT-PCR signal. Threshold cycles were determined via a manually specified single threshold of our analyzed data, which was clearly greater than all negative control signals. For the specificity plots, fluorescent signals were normalized by dividing all values by the highest fluorescent value in each channel for the respective tubes.
Results
Design of a Two-Tube, Eight Mutation, Multiplex qRT-PCR Assay for SARS-CoV-2 Variants
In our assay, SARS-CoV-2 variants were identified by probing the coding region of the spike protein transcript for the presence of eight different point mutations, as detailed in Figure 1. Because only the mutants are expected to yield a positive signal, an additional set of primers and molecular beacons for a conserved region of the viral N gene were included in tube one as a positive control for the presence of the virus. Because the multiplexing capacity of common thermal cyclers is four or five colors, the assay was divided into two tubes. The colors of the molecular beacons and the locations of the primers are indicated in Figure 1A. In tube one, there were three amplicons; in tube two, all four targets were present in the same amplicon.
Figure 1.

A: A schematic representation of the primers and molecular beacons used in the assay. A small black x on the molecular beacons target binding regions indicates that the molecular beacon is a single-nucleotide discriminant. B: Identification of key mutations present in each variant. The mutations detected in the assay are shown in the rows and variants are shown in the columns and each of the variants of concern (VOCs) can be identified by these mutation combinations. A black X indicates that the specified variant possesses the indicated mutation, and the ± symbol indicates that the variant may or may not have the indicated mutation. CFG, Cal Fluor Gold 540; CFR, Cal Fluor Red 610; FAM, fluorescein; HEX, hexachlorofluorescein; Q670, Quasar 670; Q705, Quasar 705.
Even though individual isolates of each variant may possess other mutations, sets of mutations that are functionally the most important were studied. By using in vitro binding and epidemiologic data of how these mutations affect immune evasion and binding to the ACE2 receptor,2, 3, 4, 5,18,19 the mutations shown in Figure 1, d69-70, K417N/T, L452R, T478K, E484K/Q, and N501Y, were chosen for identification. These mutations were also chosen such that a single mutation in tube one is sufficient to identify the VOCs, which of course is subject to change as the virus evolves. The mutations targeted in tube two complete the identification of the VOCs and identify other common mutations, which are useful for identifying other variants.
The Variant Molecular Beacon Assay Is Specific for the Identification of Common SARS-CoV-2 Variant Mutations
Before assembling the multiplex assay, molecular beacons were designed for each targeted mutation, their thermal denaturation profiles determined, and they were tested in monoplex PCRs. To determine whether the molecular beacons were allele discriminating at the annealing temperature, the thermal denaturation profiles of the molecular beacons were determined by themselves, and together with mutant or wild-type targets. In these experiments, the targets were synthetic oligonucleotides, and they were used in a molar excess greater than the molecular beacon concentration. A representative profile is shown in Figure 2, and the profiles of all the molecular beacons are shown in Supplemental Figure S2. Although the window of discrimination varies somewhat from molecular beacon to molecular beacon, all of our molecular beacons permitted single-nucleotide discrimination (Figure 3A). After qualifying the molecular beacons in this manner, a set of monoplex PCR assays were performed for each mutation, using wild-type targets and mutant targets in alternate reactions.
Figure 2.
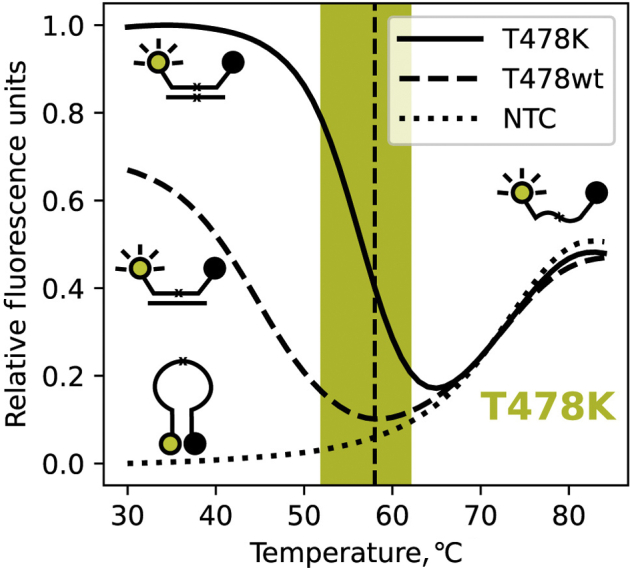
Fluorescence of the T478K discriminating molecular beacon, as a function of temperature, in the presence and absence of targets. When no target is present, indicated by the dotted line, the fluorescence increases as temperature increases, as a result of the helical order of the stem giving way to a random-coil configuration, separating the fluorophore from the quencher, and resulting in fluorescence. This reaction is labeled as no template control (NTC). When the molecular beacon binds to its target its stem dissociates, which turns on its fluorescence. Because of the higher binding strength of the perfect probe-target hybrid, indicated by the solid line, compared with the imperfect probe–target hybrid, indicated by the dashed line, the imperfect probe–target hybrid dissociates at a lower temperature. The PCR monitoring temperature (58°C) is indicated by the vertical dashed line and the window of good discrimination is indicated by the shaded area.
Figure 3.

Demonstration of the specificity and sensitivity of the variant molecular beacon assay. A: Each panel shows the amplification curves of a single multiplex reaction using either the tube one or tube two specified primers and probes (columns), and the indicated in vitro transcribed RNA targets (rows). The wild-type template is RNA of the original strain of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) entire genome. The labeled strains contain the RNA sequences of the S gene from the respective SARS-CoV-2 variants and the sample labeled kappa/delta uses an RNA corresponding to (positions 1251 to 1630 nt in the spike protein coding sequence) and containing the E484Q and T478K mutations, which are from the B.1.617 (kappa) and B.1.617.2 (delta) variants. This kappa/delta spike RNA fragment was generated and used because of the lack of commercially available in vitro–transcribed RNAs at the time. B: Sensitivity analysis was run by diluting templates containing the specified mutations from 5000 copies down to 0.5 copies per reaction. The threshold cycle (CT) values are plotted against the template copy number and the correlation coefficients of the natural log of the copy number to the CT are greater than 0.98 for all amplification products, with an approximate 3 CT delay between 10-fold dilutions, both as expected.
When all the monoplex reactions indicated successful amplification and allele discrimination, two multiplex reactions that included all of the primers and molecular beacons were assembled, as detailed in Figure 1. To demonstrate the specificity of the multiplex assays, six pairs of reactions were performed in which each reaction received 5000 copies of either the wild type, B.1.1.7 (alpha)-, B.1.351 (beta)-, P.1 (gamma)-, B.1.427 (epsilon)-, or E484Q and T478K (kappa/delta)-containing spike RNA sequences. The targets were in vitro–transcribed RNAs corresponding to a portion of the spike protein, or commercially available RNA standards, as detailed in Materials and Methods (Figure 3). These data show that when the wild-type SARS-CoV-2 genome is present, only the N gene positive control signal occurs. When testing spike RNAs containing the variant mutations, the correct mutations were identified. For example, the beta variant (B.1.351) contains not only the uniquely identifying K417N mutation, but also the N501Y and E484K mutations.
The Variant Molecular Beacon Assay Can Detect as few as Five Copies of Viral RNA
To demonstrate the sensitivity of the variant molecular beacon assay, PCR assays were initiated with serial dilutions of each target (Figure 3B). Each dilution series tested from 5000 copies to 0.5 copies. Where a clear signal was present, the threshold cycle was determined by a manually identified single threshold for each channel. Reactions starting with as little as five copies yielded a detectable signal in each case. The correlation coefficients between the logarithm of the copy number and the threshold cycles in each case are each greater than 0.98, showing a very strong exponential relationship between the two. These results also indicate that in tube one, which contains four pairs of primers, the amplification of multiple different amplicons does not interfere with each other, which would diminish sensitivity.
Inactivated Virus and Patient Sample Strains of SARS-CoV-2 Are Identified Accurately
After demonstrating the specificity and sensitivity of the variant molecular beacon assay with in vitro–transcribed RNAs, this assay was performed on RNA purified from inactivated virus obtained from ZeptoMetrix. The assay identified each of the mutations as listed in Figure 1B for the respective VOC (Figure 4A). A set of patient samples collected before May 18, 2021 (RNA extracted from midnasal swabs) that were SARS-CoV-2 positive, as identified by an in-house multiplex screening assay (Supplemental Figure S2) were tested next. After being identified as positive for SARS-CoV-2 by this assay, the samples were sequenced to identify all mutations present in the strain genomes (sequencing data are available online at https://data.mendeley.com/datasets/v3n2dzt8st/1, last accessed January 13, 2022). Afterward, the RNA extracted from the samples were provided to Rutgers in a blinded manner for variant identification.
Figure 4.

Threshold cycle (CT) data for virus and patient samples using the variant molecular beacon assay. A: Virus samples. All variant of concern (VOC) virus samples tested were identified accurately as shown in Figure 1B. B: Patient samples. All samples tested positive for the N gene control. For the other targets, a CT value less than 40 indicated the presence of the specified mutation, which could be used to identify the variants of the virus. For example, the presence of the T478K and L452R in Patient 19 indicated that this virus is the delta variant. All the specified mutations shown in this figure have been sequence-confirmed by deep sequencing. SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
These RNA samples were tested using the two-tube variant molecular beacon assay, as described in Multiplex qRT-PCR with Samples. Figure 4B shows the threshold cycles for each molecular beacon target in each sample. All samples tested positive for the N gene positive control signal. Two samples were identified as the alpha variant (B.1.1.7 with d69-70 and N501Y); two samples were identified as the epsilon variant (B.1.427/429 with only L452R); eight samples were identified as the delta variant (B.1.617.2 with L452R and T478K); 10 samples had the E484K mutation, although not associated with any VOCs; and four samples had none of the targeted mutations and therefore are not any of the currently identified VOCs.
After strain identification was performed with the variant molecular beacon assay, the results were compared with sequencing data, which were in complete agreement. Our results indicate 100% sensitivity and specificity of the assay as tested to date. It also is notable that one of the samples possessed an L452Q mutation, which is similar to the L452R mutation, but the L452R molecular beacon did not show a fluorescent signal owing to the L452Q mutation, further confirming the specificity of the results of our single-nucleotide discriminating molecular beacons.
Design and Implementation of an Omicron-Identifying Molecular Beacon Assay
The recent emergence of the omicron variant, which possesses more than 30 mutations in the spike protein, necessitates the design of a new identifying single-nucleotide–discriminating molecular beacon. Because a number of omicron mutations occur in the target regions of the two-tube assay, it is not feasible to incorporate uniquely omicron-specific molecular beacons into the two-tube assay discussed in the previous section in a straightforward manner. Therefore, a new stand-alone two-color, omicron-specific assay was designed by targeting the E484A mutation, which is unique in the omicron variant (Figure 5A), along with N gene detection. Previous studies also have shown that this mutation is important for immune evasion.20 The melt curve for the designed molecular beacon shows high specificity for its intended target at the annealing temperature of the assay of 58°C (Figure 5). Next, the sensitivity was tested using in vitro–transcribed omicron spike fragment RNA, similar to what was performed previously. The results in Figure 5C show the sensitivity to detect 50 copies of the RNA target. The sequences of probes and primers are listed in Table 3, and the amplicon schematic is shown in Figure 5A. This omicron assay was tested against the other VOC sequences and the results show clear identification of the omicron variant, with no signals from any other target sequences (Figure 5D). Thirty-three patient samples collected between November 1 and December 8, 2021 (when omicron was first detected in the United States) were then tested and 17 of them were identified as omicron (Figure 5E). These samples also were analyzed using the TaqPath COVID-19 Multiplex Diagnostic Solution assay (Thermo Fisher Scientific), in which the omicron variant failed to produce a S gene–specific signal because one of the primers binds in the omicron variant's d69-70 mutation. This S gene dropout data were provided to Rutgers in a blinded manner, and on comparison it was found to be in complete agreement, indicating high accuracy of our assay. However, in contrast with the TaqPath COVID-19 Multiplex Diagnostic Solution assay, our assay yielded a positive signal for a unique mutation in the omicron variant.
Figure 5.

Duplex assay for the omicron variant. A: Schematic showing the locations of primers and molecular beacons. A small black x on the molecular beacons target binding region indicates that the molecular beacon is a single-nucleotide discriminant. B: Thermal denaturation profile of the molecular beacon with various target sequences. The PCR monitoring temperature (58°C) is indicated by the vertical dashed line and the window of good discrimination is indicated by the shaded area. The reaction without a template for the beacon to bind to is labeled as no template control (NTC). C: Linearity of detection of in vitro–transcribed RNA corresponding to a portion of the omicron S gene. D: Specificity of the omicron assay using synthetic targets (described in Figure 3). E: Analysis of 33 recently collected severe acute respiratory syndrome coronavirus 2 samples by the duplex omicron assay.
Discussion
Molecular beacons designed to differentiate point mutations are an ideal tool for genotyping on a global scale owing to their ease of design and production. In the case of newer emerging variants, molecular beacons can be synthesized quickly and validated for that variant's identification, which can help to monitor strains and improve patient care via epidemiologic tracking. By using the outlined strategy, molecular beacons for new targets can be designed and added to the assay in 2 or 3 weeks. This assay not only enables the identification of the four VOCs and many variants of interest, but because it targets many known mutations that confer increased transmission and immune evasion, it will be able to identify new variants that recombine with existing mutations. An example of the assay's ability to pick up newer variants lies in the delta variant's mutations, which include the L452R mutation that also is present in the epsilon variant (Figure 1B).
Because of the specificity of the molecular beacons, other mutations in the target region do not elicit false-positive signals. Evidence of this is found in the lack of signal in one of the samples that contained the L452Q mutation, as identified by sequencing. More evidence of the specificity is in the clean differentiation of the K417T and K417N mutations, as well as the E484Q, E484K, and E484A mutations, because these signals only become positive in the presence of their respective mutations. This specificity enables precise identification, but also comes with the downside that mutations in the binding region of the molecular beacon could result in false-negative signals. Despite this, if other mutations do arise near the uniquely identifiable mutations, such as in the case of the omicron variant, this can be accounted for by either designing different molecular beacons, or by introducing degenerate nucleotides into the current molecular beacons.
This study was limited by the number of patient samples tested. In the cases in which a variant harbored multiple mutations in the target regions of molecular beacons of the two-tube assay, false negatives may occur as a result of the high specificity of molecular beacons. However, in such situations a completely new assay can be assembled easily with equal ease, as was shown in the case of the omicron variant (Figure 5).
This assay introduces the sequences of many useful molecular beacons for the identification of point mutations in SARS-CoV-2. These variant molecular beacon assays are most useful as a screening tool for positive SARS-CoV-2 patient samples, which can identify all major VOCs (Figure 1B). The primers and molecular beacons also can be combined in varying combinations to test for other pathogens, they can be added to current assays, and they can be used with human RNA controls as were added in the multiplex COVID-19 multiplex screening assay (Supplemental Figure S2). These assays then can be used to test for the most common variants in a region or to directly test human samples as a single pathogen/COVID-19 variant identification test in general practice.
Here, we describe multiplexed molecular beacon assays for the classification of SARS-CoV-2 variants via their identifying functional mutations. The assay clearly differentiates between the different SARS-CoV-2 variant sequences, and accurately classifies patient samples that have been identified by deep sequencing technologies and S gene dropout assays. All primer and probe sequences used in this assay are listed in Tables 1 and 3 to facilitate SARS-CoV-2 variant genotyping on a larger scale, and to address the public need for variant tracking.
Acknowledgment
We thank Diana Y. Vargas for help designing the SARS-CoV-2 multiplex screening assay.
Footnotes
Supported by NIH grant R01 CA227291 (S.T.) and a ResourcePath, LLC, grant (S.T. and S.A.E.M.).
Disclosures: D.A.H. is the Medical Director of ResourcePath, LLC, and A.P.F., A.B., and A.C. are employed by ResourcePath, LLC. ResourcePath, LLC, partly funded this work through a grant to Rutgers. R.J.D., S.A.E.M., and S.T. declare no conflicts of interests.
Supplemental material for this article can be found at http://doi.org/10.1016/j.jmoldx.2022.01.004.
Contributor Information
Ryan J. Dikdan, Email: rjd254@njms.rutgers.edu.
D. Ashley Hill, Email: hill@resourcepath.net.
Author Contributions
R.J.D., S.A.E.M., and S.T. conceptualized the study and designed methodology; D.A.H., A.B., A.C., and A.P.F. provided the patient samples that were tested, and the sequencing results they generated were used to validate the assay used; R.J.D. wrote the original draft; all authors reviewed and edited the manuscript; and S.T. and S.A.E.M. supervised R.J.D. in running experiments.
Supplemental Data
Supplemental Figure S1.

Melt curves for each molecular beacon with the on- and off-target oligonucleotides. The windows of discrimination are highlighted in the appropriate color and the annealing temperature for the assay (58°C) is indicated via the dashed vertical line. The on-target sequence oligonucleotide reactions are indicated by the solid lines, the off-target sequence oligonucleotide reactions are indicated by the dashedand dot-dashed lines and the no template control (NTC) reactions are indicated by the dottedlines. The differences in these melting temperatures between on- and off-target binding are what allows each of these molecular beacons to become fluorescent only in the presence of the target sequence.
Supplemental Figure S2.

Molecular beacon–based real-time RT-PCR assay of a serial dilution (100,000 copies to 0 copies) of a severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA transcript in a background of RNA isolated from 100 HeLa cells. Left: Amplification curves of the SARS-CoV-2 RdRp gene (greenlines), the SARS-CoV-2 N gene (redlines), and the human ACTB gene (bluelines). Right: Inverse linear relationship between the logarithm of the number of SARS-CoV-2 constructs (green line with closed circles for the RdRp gene and red line with closed circles for the N gene) and the number of thermal cycles that are required to generate a visible fluorescent signal. The dashed blue line indicates the threshold cycle of the human ACTB gene in each reaction. The plots indicate that as few as one copy SARS-CoV-2 RNA in a background of RNA from 100 human cells can be reliably detected and identified.
References
- 1.Fontanet A., Autran B., Lina B., Kieny M.P., Karim S.S.A., Sridhar D. SARS-CoV-2 variants and ending the COVID-19 pandemic. Lancet. 2021;397:952–954. doi: 10.1016/S0140-6736(21)00370-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies N.G., Abbott S., Barnard R.C., Jarvis C.I., Kucharski A.J., Munday J.D., Pearson C.A.B., Russell T.W., Tully D.C., Washburne A.D., Wenseleers T., Gimma A., Waites W., Wong K.L.M., van Zandvoort K., Silverman J.D., Diaz-Ordaz K., Keogh R., Eggo R.M., Funk S., Jit M., Atkins K.E., Edmunds W.J. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science. 2021;372:eabg3055. doi: 10.1126/science.abg3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deng X., Garcia-Knight M.A., Khalid M.M., Servellita V., Wang C., Morris M.K., et al. Transmission, infectivity, and antibody neutralization of an emerging SARS-CoV-2 variant in California carrying a L452R spike protein mutation. medRxiv. 2021 doi: 10.1101/2021.03.07.21252647. [Preprint] [DOI] [Google Scholar]
- 4.Wang P., Casner R.G., Nair M.S., Wang M., Yu J., Cerutti G., Liu L., Kwong P.D., Huang Y., Shapiro L., Ho D.D. Increased resistance of SARS-CoV-2 variant P.1 to antibody neutralization. Cell Host Microbe. 2021;29:747–751.e4. doi: 10.1016/j.chom.2021.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weisblum Y., Schmidt F., Zhang F., DaSilva J., Poston D., Lorenzi J.C.C., Muecksch F., Rutkowska M., Hoffmann H.H., Michailidis E., Gaebler C., Agudelo M., Cho A., Wang Z., Gazumyan A., Cipolla M., Luchsinger L., Hillyer C.D., Caskey M., Robbiani D.F., Rice C.M., Nussenzweig M.C., Hatziioannou T., Bieniasz P.D. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife. 2020;9:e61312. doi: 10.7554/eLife.61312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lusvarghi S., Wang W., Herrup R., Neerukonda S.N., Vassell R., Bentley L., Eakin A.E., Erlandson K.J., Weiss C.D. Key substitutions in the spike protein of SARS-CoV-2 variants can predict resistance to monoclonal antibodies, but other substitutions can modify the effects. bioRxiv. 2021 doi: 10.1101/2021.07.16.452748. [Preprint] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bandoy D.J.D.R., Weimer B.C. Analysis of SARS-CoV-2 genomic epidemiology reveals disease transmission coupled to variant emergence and allelic variation. Sci Rep. 2021;11:7380. doi: 10.1038/s41598-021-86265-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nasir J.A., Kozak R.A., Aftanas P., Raphenya A.R., Smith K.M., Maguire F., Maan H., Alruwaili M., Banerjee A., Mbareche H., Alcock B.P., Knox N.C., Mossman K., Wang B., Hiscox J.A., McArthur A.G., Mubareka S. A comparison of whole genome sequencing of SARS-CoV-2 using amplicon-based sequencing, random hexamers, and bait capture. Viruses. 2020;12:895. doi: 10.3390/v12080895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Izquierdo-Lara R., Elsinga G., Heijnen L., Oude Munnink B.B., Schapendonk C.M.E., Nieuwenhuijse D., Kon M., Lu L., Aarestrup F.M., Lycett S., Medema G., Koopmans M.P.G., De Graaf M. Monitoring SARS-CoV-2 circulation and diversity through community wastewater sequencing, the Netherlands and Belgium. Emerg Infect Dis. 2021;27:1405–1415. doi: 10.3201/eid2705.204410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banada P., Green R., Banik S., Chopoorian A., Streck D., Jones R., Chakravorty S., Alland D. A simple reverse transcriptase PCR melting-temperature assay to rapidly screen for widely circulating SARS-CoV-2 variants. J Clin Microbiol. 2021;59 doi: 10.1128/JCM.00845-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonnet G., Tyagi S., Libchaber A., Kramer F.R. Thermodynamic basis of the enhanced specificity of structured DNA probes. Proc Natl Acad Sci USA. 1999;96:6171–6176. doi: 10.1073/pnas.96.11.6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marras S.A.E., Kramer F.R., Tyagi S. Multiplex detection of single-nucleotide variations using molecular beacons. Genet Anal. 1999;14:151–156. doi: 10.1016/s1050-3862(98)00018-7. [DOI] [PubMed] [Google Scholar]
- 13.Kostrikis L.G., Tyagi S., Mhlanga M.M., Ho D.D., Kramer F.R. Spectral genotyping of human alleles. Science. 1998;279:1228–1229. doi: 10.1126/science.279.5354.1228. [DOI] [PubMed] [Google Scholar]
- 14.Untergasser A., Cutcutache I., Koressaar T., Ye J., Faircloth B.C., Remm M., Rozen S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Markham N.R., Zuker M. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res. 2005;1:W577–W581. doi: 10.1093/nar/gki591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tyagi S., Bratu D.P., Kramer F.R. Multicolor molecular beacons for allele discrimination. Nat Biotechnol. 1998;16:49–53. doi: 10.1038/nbt0198-49. [DOI] [PubMed] [Google Scholar]
- 17.Corman V.M., Landt O., Kaiser M., Molenkamp R., Meijer A., Chu D.K., Bleicker T., Brünink S., Schneider J., Schmidt M.L., Mulders D.G., Haagmans B.L., van der Veer B., van den Brink S., Wijsman L., Goderski G., Romette J.-L., Ellis J., Zambon M., Peiris M., Goossens H., Reusken C., Koopmans M.P., Drosten C. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020;25:2000045. doi: 10.2807/1560-7917.ES.2020.25.3.2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim S., Liu Y., Lei Z., Dicker J., Cao Y., Zhang X.F., Im W. Differential interactions between human ACE2 and spike RBD of SARS-CoV-2 variants of concern. bioRxiv 2021, [Preprint] 2021 doi: 10.1021/acs.jctc.1c00965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan A., Zia T., Suleman M., Khan T., Ali S.S., Abbasi A.A., Mohammad A., Wei D.Q. Higher infectivity of the SARS-CoV-2 new variants is associated with K417N/T, E484K, and N501Y mutants: an insight from structural data. J Cell Physiol. 2021;236:7045–7057. doi: 10.1002/jcp.30367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harvey W.T., Carabelli A.M., Jackson B., Gupta R.K., Thomson E.C., Harrison E.M., Ludden C., Reeve R., Rambaut A., Peacock S.J., Robertson D.L. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol. 2021;19:409–424. doi: 10.1038/s41579-021-00573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]