

ABSTRACT
CPT1C, which is expressed in hippocampus, influences ceramide level, endogenous cannabinoid and oxidation process, as well as plays an important role in various brain functions such as learning. This study aimed to investigate the role of CPT1C in Alzheimer’s disease (AD) and its underlying mechanism. We established a model of Alzheimer’s disease in vitro by exposing primary hippocampal neurons to beta-Amyloid peptide fragment 25–35 (Aβ25-35). The cell viability, lactate dehydrogenase (LDH) level, expressions of reactive oxygen species (ROS), malondialdehyde (MDA) and superoxide dismutase (SOD) were detected using Cell Counting Kit-8 (CCK-8), LDH assay, ROS kits, malondialdehyde (MDA) kits and SOD kits, respectively. Moreover, the expression of oxidative stress-related proteins as well as the expressions of amyloid precursor protein (App), p-Tau andβ-site APP-cleaving enzyme1 (Bace-1) were measured using quantitative reverse transcription PCR (RT-qPCR) and western blot. Tunel and western blot were adopted to detect apoptosis as well as its related proteins. After the treatment of peroxisome proliferators-activated receptor alpha (PPARα), CPT1C expression was detected with the application of RT-qPCR and western blot. CPT1C expression was reduced in Aβ25-35-induced HT22 cells. Overexpression of CPT1C relieved cell viability and toxic injury as well as attenuated oxidative stress, apoptosis and expression levels of AD marker proteins. Moreover, higher doses of PPARα agonist activate the expression of CPT1C in Aβ25-35-induced HT22 cells. In conclusion, CPT1C alleviates Aβ25-35-induced oxidative stress, apoptosis and deposition of AD marker proteins in hippocampal neurons, suggesting that CPT1C has favorable effects on alleviating AD and participates in PPARα activation.
KEYWORDS: Aβ, CPT1C, Alzheimer’s disease, hippocampal neurons
Introduction
Alzheimer’s disease (AD) is referred to as progressive cognitive decline resulting in dementia [1]. According to recent statistics, around 35.6 million people have suffered from dementia in the globe, which may be partially due to the high prevalence of AD [2]. What is beyond people’s recognition is that neurodegenerative disease is not confined to the elder but is spreading quickly,, resulting in increased financial burden and lowering people’s life quality [3]. Current therapeutic methods, including treatment with cholinesterase inhibitors, cannot change the rising trend of illnesses [4].
Carnitine palmitoyl transferase (CPT) system, a multiprotein complex featuring catalytic properties is located in a core represented by CPT1 and CPT2 in the outer and inner membrane of the mitochondria, respectively [5]. Similar to the canonical CPT enzymes in the sequence of primary amino acid, CPT1C is located in the endoplasmic reticulum of neurons and exerts residual catalytic effects in vitro with palmitoyl-CoA [6,7]. As evidenced by previous studies, depletion of CPT1C made more palmitoyl-CoA available, and mice with CPT1C knockdown exhibited metabolic disturbance like impaired gluconeogenesis and muscle glucose uptake [8]. CPT1C, which is expressed in hippocampus, influences ceramide level, endogenous cannabinoid and oxidation process, and plays an important role in various brain functions such as learning [5]. In addition, some studies held that the deficiency of CPT1C on brain can cause motor dysfunction and behavioral defects [9]. CPT1C is considered to possibly involve in AD due to its role in energy homeostasis. The onset of AD is associated with insulin resistance, which is modulated by palmitate in the hypothalamus and implicated in increased production of reactive oxygen species (ROS) [5,10–12]. However, the role of CPT1C in AD has remained elusive, which constitutes the pivotal focus of our study.
In the present study, we hypothesize that CPT1C plays a role in AD and is involved in the regulation for oxidative stress, apoptosis and AD markers. The study was designed to decipher the role of CPT1C in an in vitro model of AD and figure out how CPT1C was implicated in AD.
Materials and methods
Cell culture and treatment
Mouse hippocampal neuron (HT22) was provided from Salk Institute (La Jolla, CA, USA). The cells were cultured in Dubelcco’s modified eagle medium (DMEM; Gibco, NY, USA) containing 10% fetal bovine serum (FBS; Gibco, NY, USA) and 1% penicillin/streptomycin at 37 C in a humidified incubator with 5% CO2. Aβ25-35 was diluted to 1 mM with sterilized saline water and then incubated at 37 C for 7 days before use, as previously described [13]. Thereafter, the HT22 cells were treated with 20 mol/L Aβ25-35 for 48 h. CPT1C overexpression plasmids (Ov-CPT1C, 1 µg/mL) and its empty vector (Ov-NC) were obtained from Shanghai Integrated Biotech Solutions Co., Ltd (Shanghai, China), and transfection was conducted for 24 h by Lipofectamine 3000 (Invitrogen). Gemfibrozil (Abcam, ab142883), a PPARa agonist [14], was used for the following experiments.
RT-qPCR
RNA from HT22 cells was extracted using a TRIzol® kit in accordance with the manufacturer’s instructions. The synthesization with complementary DNA (cDNA) was performed using a PrimeScript RT Master Mix kit (Takara Biotechnology, Co., Ltd.). Gene expression was quantified by quantitative real-time PCR (RT-qPCR) on ABI 7500 PCR system (ABI, USA) using SYBR Green PCR Master Mix. All samples were measured in triplicate and the mean value was calculated. Quantitative measurements were determined using 2−ΔΔCT method [15], and expression of GAPDH was considered as the internal control.
Western blot
Total proteins were isolated from HT22 cells using RIPA buffer (Beyotime, Shanghai, China). A BCA kit was used to quantify the concentration of protein samples. Subsequently, equal amounts of proteins were subjected to 10% SDS-PAGE gel, followed by the transfer to PVDF membranes (EMD Millipore, Billerica, MA, USA). After being blocked with 5% nonfat milk, the membranes were then incubated with primary antibodies (CPT1C, cat.no. DF12150; SOD1, cat.no. AF5198; SOD2, cat.no. AF5144; Bcl2, cat.no. AF6139; Bax, cat.no. AF0120; cleaved PARP, cat.no. BF9106; APAF-1, cat.no. AF0117; App, cat.no. AF6084; p-Tau, cat.no. AF3148. Affinity, USA) (Bace-1, cat.no. ab183612; GAPDH cat.no. ab8245. Abcam). Thereafter, these blots were incubated with a HRP-conjugated antibody (cat.no. #7074, Cell Signaling Technology, Inc.) at 37°C for 1 h. Proteins were visualized with an enhanced chemiluminescence (ECL) and the expression of protein was normalized to GAPDH.
CCK-8 assay
Briefly, cells were inoculated into 96-well plates at a concentration of 3 × 104/well for 12 h. After indicated treatment, CCK-8 reagent (abcam, England) was added to each well to incubate the cells for another 4 h. Then, the optical density (OD) value at 450 nm was determined using a microplate reader (Molecular Devices, San Jose, CA, United States).
Detection of Superoxide Dismutase (SOD) Activity, Malondialdehyde (MDA) Content, and LDH Release Assay
For the detection of cell SOD activity and MDA content, cells after treatment were cultured in 96-well plates, and then sonicated and centrifuged to obtain the supernatant. The SOD activity and MDA content were then measured according to the manufacturer’s protocol (Beyotime, Shanghai, China) and results were displayed as a multiple relative to the control group. The cell supernatant was collected and used for the determination of LDH activities using LDH Cytotoxicity Assay Kit according to manufacturer’s protocol (Beyotime, Shanghai, China).
Measurement of ROS
The fluorescent probe 2′,7′-dichlorfluorescein-diacetate (DCFH-DA) kit was used for the detection of ROS level in HT22 cells according to manufacturer’s guidance (Beyotime, Shanghai, China). Briefly, the cells were seeded in 96-well plates and then processed with working solution for 20 min at 37°C. After washing with serum-free medium for three times, the fluorescence density was detected using a fluorescence microplate (biosys, Germany).
TUNEL assay
The cell apoptosis rate was determined using TUNEL assay kits (Invitrogen; Thermo Fisher Scientific). Briefly, the collected cells were fixed with 4% paraformaldehyde for 30 min incubated with 0.3% Triton X-100 in PBS for 10 min according to the manufacturer’s protocol. After staining with DAPI for 30 min, the positive-apoptotic cells were counted under a magnification of 200 × .
Statistical analysis
Statistical analysis was performed using Graph Pad Prism 6 (Graph Pad Software, La Jolla, CA). The results were expressed as mean ± standard deviation (SD). One-way ANOVAwas used for comparison among multiple groups, followed by Tukey’s post hoc test between two groups. P < 0.05 indicated statistical significance. Each experiment was repeated at least three times.
Results
CPT1C expression was reduced in Aβ25-35-induced HT22 cells
Despite the fact that CPT1C is ubiquitously presented in various transformed cells and cancers [16], the current strategy we used for the following study is to detect its expression in vitro. Firstly, we induced the cells by 20 µM Aβ25-35, while the control group was not treated with Aβ25-35. Results from CCK-8 clearly implied that the cell viability of Aβ25-35-induced HT22 cells was decreased and the effects of A25-35 on HT22 cells were in a time-dependent manner (Figure 1(a)). As Figure 1(b) shown, the expression of CPT1C was decreased both at a trascript and at a protein level by Aβ25-35 over time.
Figure 1.
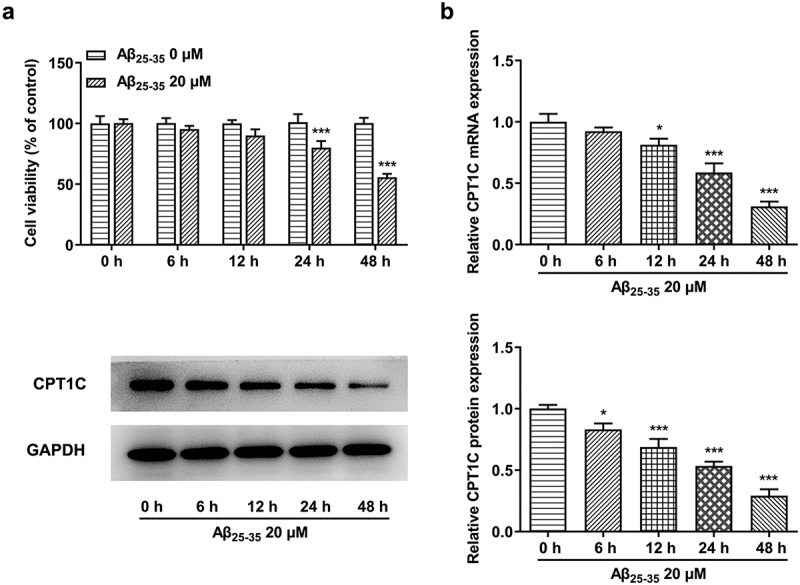
CPT1C expression was reduced in Aβ25-35-induced HT22 cells. HT22 cells were treated with Aβ25-35 (20 μM) for 6 h, 12 h, 24 h or 48 h, respectively
(a) The cell viability of Aβ25-35-induced HT22 cells was detected using CCK-8 assay. (b) mRNA and protein expressions of CPT1C in Aβ25-35-induced HT22 cells were measured using RT-qPCR and western blot, respectively. *P < 0.05 and ***P < 0.001 vs. Control group.
CPT1C overexpression attenuated cell viability and toxic injury in Aβ25-35-induced HT22 cells
To determine the role of CPT1C, we transfected Aβ25-35-induced HT22 cells with CPT1C overexpression plasmids to observe whether CPT1C expression could manipulate the cellular behaviors in hippocampal neurons. As Figure 2(a) indicated, the expression of CPT1C was greatly increased after Aβ25-35 induction. Results from CCK-8 assay indicated that the decreased cell viability of HT22 cells induced by Aβ25-35 was reverted by CPT1C overexpression (Figure 2(b)). Furthermore, the relative LDH level in HT22 cells was significantly increased after Aβ25-35 induction whileCPT1C overexpression reversed the promotive effects of Aβ25-35 (Figure 2(c)). The above results illustrated that the overexpression of CPT1C attenuated cell viability and toxic injury in Aβ25-35-induced HT22 cells.
Figure 2.
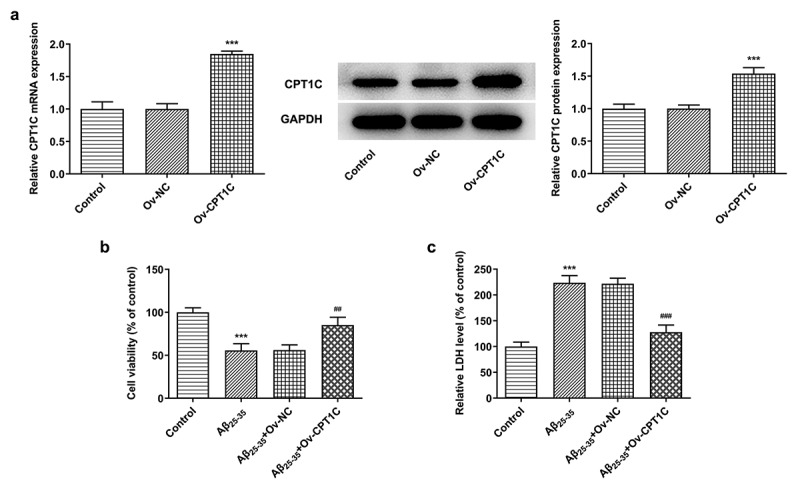
CPT1C overexpression attenuated cell viability and toxic injury in Aβ25-35-induced HT22 cells. HT22 cells were transfected with Ov-CPT1C or Ov-NC for 24 h, and then treated with Aβ25–35 for another 24 h
(a) mRNA and protein expressions of CPT1C were measured using RT-qPCR and western blot, respectively. ***P < 0.001 vs Ov-NC. (b) The cell viability of Aβ25-35-induced HT22 cells was detected using CCK-8. ***P < 0.001 vs. Control group, ##P < 0.01 vs Aβ25-35 + Ov-NC. (c) The relative LDH level of Aβ25-35-induced HT22 cells was checked using LDH assay. ***P < 0.001 vs. Control group, ###P < 0.001 vs Aβ25-35 + Ov-NC.
CPT1C overexpression attenuated oxidative stress in Aβ25-35-induced HT22 cells
We then observed if oxidative stress and cell apoptosis were changed after CPT1C was overexpressed in A25-35-induced HT22 cells. According to Figure 3(a), the greatly increased levels of ROS and MDA, as well as in Aβ25-35-induced HT22 cells was suppressed by CPT1C overexpression. In addition, CPT1C overexpression downregulated MDA levels in Aβ25-35-induced HT22 cells but enhanced SOD activities. Besides, SOD1 expression levels at transcription and translation levels were increased by CPT1C overexpression when compared with the cotreatment group of Aβ25-35 and Ov-NC. These suggested that CPT1C overexpression exhibited inhibitory effects on oxidative stress in Aβ25-35-induced HT22 cells (Figure 3(b,c)).
Figure 3.
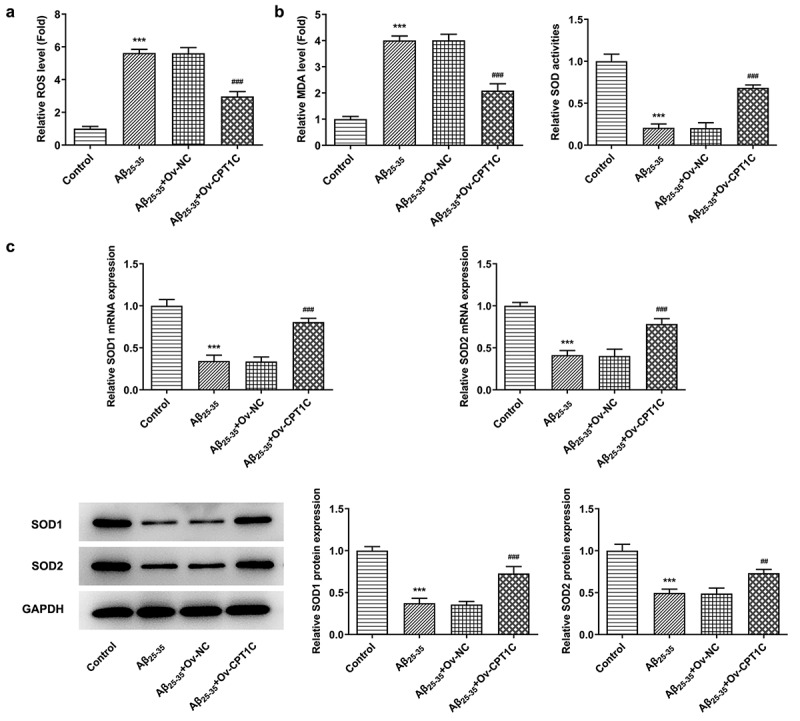
CPT1C overexpression attenuated oxidative stress in Aβ25-35-induced HT22 cells. Following transfection of Ov-CPT1C or Ov-NC for 24 h, HT22 cells were treated with Aβ25–35 for another 24 h
(a) ROS expression was detected using ROS kits. (b) MDA levels and SOD activity were measured using MDA and SOD kits, respectively. (c) The relative mRNA and protein expression of SOD1 and SOD2 in A25-35-induced HT22 cells were measured using RT-qPCR and western blot, respectively. ***P < 0.001 vs. Control group, ##P < 0.01 and ###P < 0.001 vs A25-35 + Ov-NC.
CPT1C overexpression decreased the apoptosis of Aβ25-35-induced HT22 cells
We further determined whether CPT1C regulated apoptosis in HT22 cells treated with Aβ25-35. As Figure 4(a) demonstrated, the apoptosis in HT22 cells was hugely increased by Aβ25-35 induction in comparison with the Aβ25-35 group. However, the increased apoptosis level was then decreased by CPT1C overexpression. Moreover, Aβ25-35 brought about Bcl2 downregulation and Bax, cleaved PARP, and APAF-1 upregulation, which were then reversed by CPT1C overexpression (Figure 4(b)). Therefore, we could conclude that overexpression of CPT1C attenuates the apoptosis in hippocampal neurons.
Figure 4.
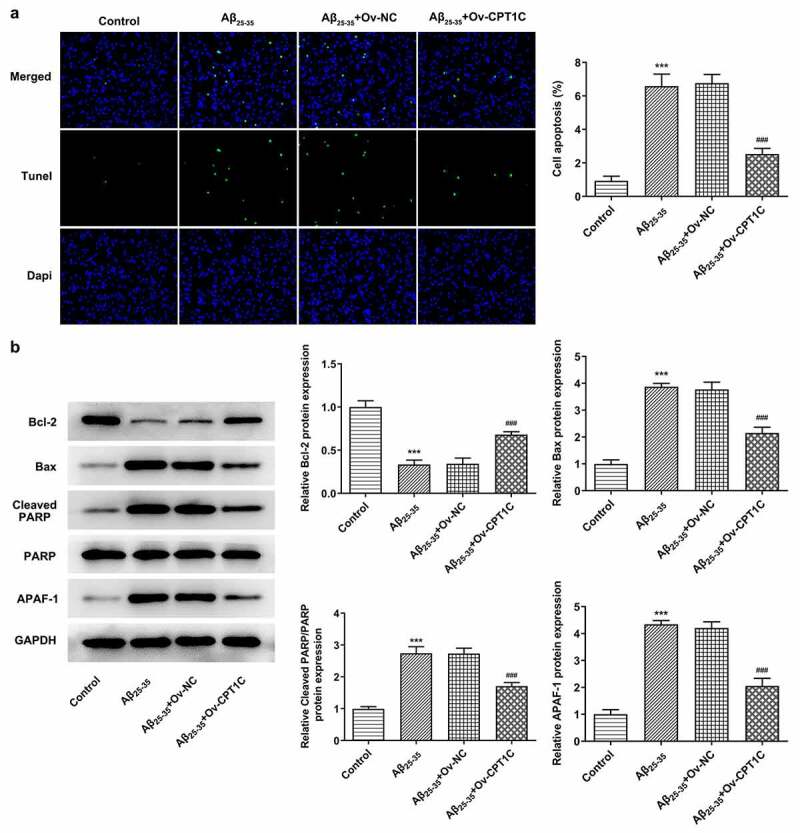
CPT1C overexpression decreased the apoptosis of Aβ25-35-induced HT22 cells. Following transfection of Ov-CPT1C or Ov-NC for 24 h, HT22 cells were treated with Aβ25–35 for another 24 h
(a) The apoptosis of Aβ25-35-induced HT22 cells was evaluated by TUNEL. (b) The expressions of Bcl2, Bax, cleaved PARP and APAF-1 were measured by western blot. ***P < 0.001 vs. Control group, ###P < 0.001 vs A25-35 + Ov-NC.
CPT1C overexpression decreased the deposition of AD marker proteins in Aβ25-35-induced HT22 cells
We next analyzed whether CPT1C overexpression could change the expression of AD markers in Aβ25-35-induced HT22 cells. With the application of RT-qPCR and western blot, the relative mRNA and protein expressions of App, p-Tau and Bace-1 were measured. Compared with Control, the expressions of App, p-Tau and Bace-1 were greatly upregulated by Aβ25-35, while CPT1C overexpression reversed the promotive effects of Aβ25-35 on deposition of AD marker proteins, evidenced by the downregulated expressions of App, p-Tau and Bace-1 in contrast with Aβ25-35 + Ov-NC (Figure 5(a,b)). PPARα activation could increase CPT1C expression in Aβ25-35-induced HT22 cells
Figure 5.
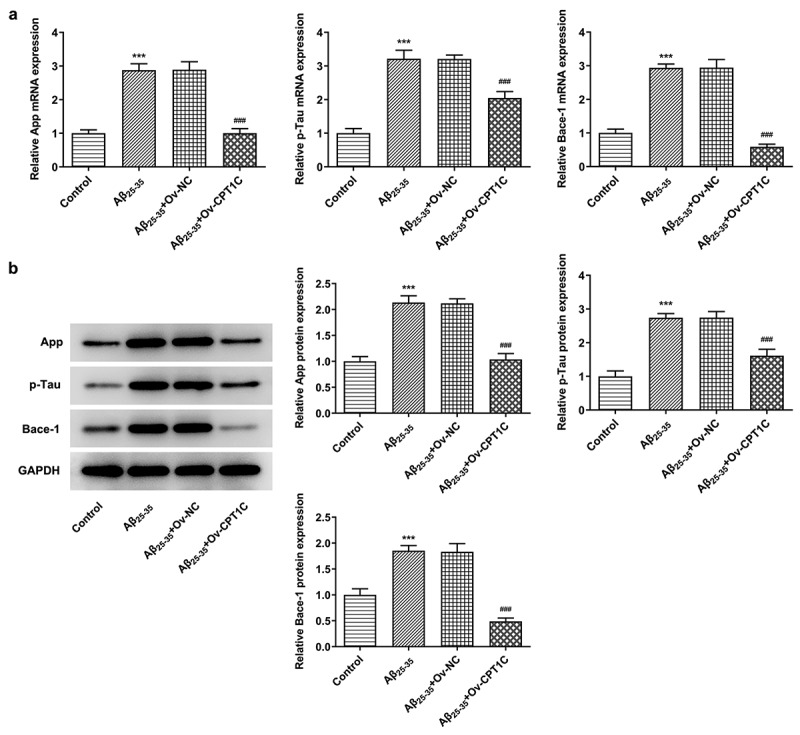
CPT1C overexpression decreased the deposition of AD marker proteins in Aβ25-35-induced HT22 cells. Following transfection of Ov-CPT1C or Ov-NC for 24 h, HT22 cells were treated with Aβ25–35 for another 24 h
(a) The mRNA expressions of App, p-Tau and Bace-1 were evaluated using RT-qPCR. (b) The protein expressions of App, p-Tau and Bace-1 were evaluated using western blot. ***P < 0.001 vs. Control group, ###P < 0.001 vs Aβ25-35 + Ov-NC.
In order to explore whether CPT1C could be activated by PPARα, gemfibrozil, an agonist of PPARα, was used to treat Aβ25-35-induced HT22 cells. According to Figure 6(a,b), the expression of CPT1C was significantly diminished by Aβ25-35 in comparison with Control. Studies have noted that PPARα regulates the malignant behaviors of tumor cells by targeting CPT1C, and PPARα activation can alleviate the amyloidosis and reverse memory deficits and anxiety in AD [14,17]. After the treatment of gemfibrozil with a dose of 250 μM, mRNA and protein expressions of CPT1C gained a huge growth, revealing that PPARα activation could increase CPT1C expression in Aβ25-35-induced HT22 cells.
Figure 6.
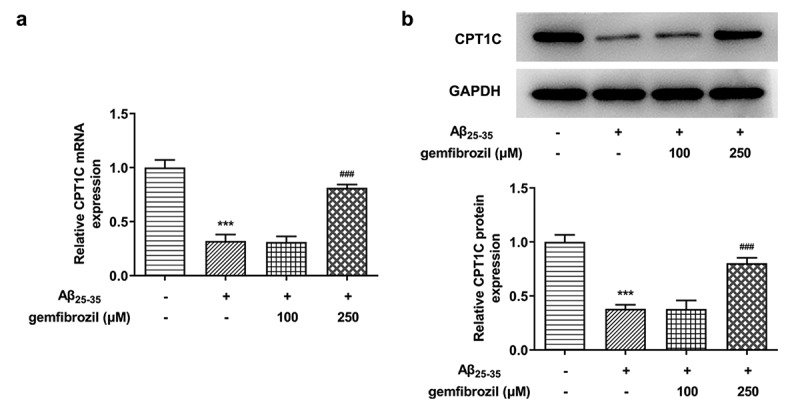
PPARα activation could increase CPT1C expression in Aβ25-35-induced HT22 cells. HT22 cells were co-treated with gemfibrozil 100 μM or 250 μM, and Aβ25-35 for 48 h
(a) The relative CPT1C mRNA expression in Aβ25-35-induced HT22 cells was detected using RT-qPCR. (b) The protein expression of CPT1C expression in Aβ25-35-induced HT22 cells was detected using western blot. ***P < 0.001 vs. Control group, ###P < 0.001 vs Aβ25-35+ gemfibrozil 100 μM.
Discussion
AD is a neurodegenerative disorder that affects the cognitive functions of human beings, especially the elderly [18]. The aggregation of Aβ, which has been considered as a major driver of AD progression, was linked to the severity of cognitive deficits [19,20]. Similar to the induction procedures conducted by previous studies, this paper used Aβ25-35 to construct the in vitro AD model.
The intricate pathology of AD has necessitated novel targeted treatment methods to fight against its invasion [21]. Oxidative stress is a major driver of AD pathophysiology, and antioxidant agents have been highlighted as optimal choices for the inhibition of AD progression [22]. Patients with AD are often found to suffer from oxidative damage to the neuronal tissues [23]. In this study, CPT1C overexpression led to increased SOD expression, and decreased MDA as well as ROS expression in Aβ25-35-induced HT22 cells. A previous study showed that the loss of CPT1C triggered increased sensitivity of cells to oxidative stress, as demonstrated by the elevation of ceramides, a major regulator of oxidative stress [24]. The findings in this study were consistent with the above-mentioned study. Mounting evidence has noted that apoptosis, which is also called programmed cell death, partakes in the AD-associated nerve cell death [25,26]. Thus, the changes of apoptosis were observed after CPT1C was overexpressed in Aβ25-35-induced HT22 cells. Importantly, the apoptosis elevated by Aβ25-35 was attenuated by CPT1C overexpression. Moreover, it was found that overexpression of CPT1C abolished the promotive effects of Aβ25-35 on enhancing the expression of AD markers. Thus, decreased CPT1C by Aβ25-35 treatment was able to induce oxidative stress and apoptosis, and CPT1C could be involved in AD pathology.
Peroxisome proliferators activated receptors (PPARs) are a family of ligand-regulated nuclear receptors regulating transcriptions via a complex mechanism [27–29]. Evidence has shown that apart from the modulation of mitochondria metabolism, PPARα participates in amyloid beta precursor protein (APP) in the brain and it may also influence Tau protein phosphorylation via A [30]. It is well acknowledged that PPARαplayed an essential role in neuronal cells [31]. Moreover, PPARα polymorphism may be deemed as a risk factor for AD [32]. Thus, we speculated that there might be a close connection between CPT1C and PPARα in regulating the progression of AD. The administration of gemfibrozil at higher dosage increased the expression of CPT1C inhibited by Aβ25-35, which tallied with the idea proposed by several experts that alteration of PPARα signaling may lead to activation of APP metabolism that contributed to AD pathogenesis [30]. Meanwhile, some held that the activation of PPARα, which was found to be downregulated in AD brain, may also alleviate the inducive effects of Aβ25-35 on AD [30]. Besides, the recognized association of PPARα and CPT1C in our study was also found by other study which revealed that PPARα was able to activate the transcription of CPT1C promoter to regulate cell proliferation [17]. Taken together, the inhibition of PPARα in HT22 cells under Aβ25-35 stimulation is the most cause of decreased CPT1C expression, further inducing oxidative stress and apoptosis.
Conclusion
Aβ25-35 induced decreased CPT1C, thereby leading to oxidative stress and apoptosis. The decreased CPT1C could be due to the inhibition of Aβ25-35 for PPARα. CPT1C could play a vital role in AD and may provide insight into AD treatment.
Limitation
The limitation is that current therapeutic strategies can only mildly slow but not halt the progression of AD and this paper has suggested favorable outcomes of CPT1C in alleviating AD and its underlying mechanism related to PPAR activation. It is noteworthy that further in vitro and vivo, and clinical studies regarding the role of CPT1C in AD are still needed for the better management of this disorder.
Highlights
1. HT22 cells expressed lower CPT1C levels when subjected to Aβ25-35 stimulation.
2. CPT1C participates in Aβ25-35-induced oxidative stress and apoptosis, along with reducing the expression levels of AD-related markers.
3. Aβ25-35 could inhibit PPARα to decrease CPT1C expression, thereby involving in AD pathology.
Disclosure statement
The authors declare that they have no competing interests.
References
- [1].Lawrence E, Vegvari C, Ower A, et al. A systematic review of longitudinal studies which measure Alzheimer’s disease biomarkers. J Alzheimers Dis. 2017;59(4):1359–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Crous-Bou M, Minguillon C, Gramunt N, et al. Alzheimer’s disease prevention: from risk factors to early intervention. Alzheimers Res Ther. 2017;9(1):71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wimo A, Winblad B, Jonsson L.. The worldwide societal costs of dementia: estimates for 2009. Alzheimers Dement. 2010;6(2):98–103. [DOI] [PubMed] [Google Scholar]
- [4].Weller J, Budson A.. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Res. 2018;7F1000 Faculty Rev-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Virmani A, Pinto L, Bauermann O, et al. The Carnitine Palmitoyl Transferase (CPT) system and possible relevance for neuropsychiatric and neurological conditions. Mol Neurobiol. 2015;52(2):826–836. . [DOI] [PubMed] [Google Scholar]
- [6].Lee J, Wolfgang MJ. Metabolomic profiling reveals a role for CPT1c in neuronal oxidative metabolism. BMC Biochem. 2012;13(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sierra AY, Gratacós E, Carrasco P, et al. CPT1c is localized in endoplasmic reticulum of neurons and has carnitine palmitoyltransferase activity. J Biol Chem. 2008;283(11):6878–6885. . [DOI] [PubMed] [Google Scholar]
- [8].Gao XF, Chen W, Kong XP, et al. Enhanced susceptibility of Cpt1c knockout mice to glucose intolerance induced by a high-fat diet involves elevated hepatic gluconeogenesis and decreased skeletal muscle glucose uptake. Diabetologia. 2009;52(5):912–920. . [DOI] [PubMed] [Google Scholar]
- [9].Carrasco P, Jacas J, Sahun I, et al. Carnitine palmitoyltransferase 1C deficiency causes motor impairment and hypoactivity. Behav Brain Res. 2013;256:291–297. [DOI] [PubMed] [Google Scholar]
- [10].Benoit SC, Kemp CJ, Elias CF, et al. Palmitic acid mediates hypothalamic insulin resistance by altering PKC-theta subcellular localization in rodents. J Clin Invest. 2009;119(9):2577–2589. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rönnemaa E, Zethelius B, Sundelöf J, et al. Impaired insulin secretion increases the risk of Alzheimer disease. Neurology. 2008;71(14):1065–1071. . [DOI] [PubMed] [Google Scholar]
- [12].Rhein V, Eckert A. Effects of Alzheimer’s amyloid-beta and tau protein on mitochondrial function – role of glucose metabolism and insulin signalling. Arch Physiol Biochem. 2007;113(3):131–141. [DOI] [PubMed] [Google Scholar]
- [13].Chen N, Wang J, He Y, et al. Trilobatin protects against Abeta25-35-Induced Hippocampal HT22 cells apoptosis through mediating ROS/p38/caspase 3-dependent pathway. Front Pharmacol. 2020;11:584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Luo R, Su LY, Li G, et al. Activation of PPARA-mediated autophagy reduces Alzheimer disease-like pathology and cognitive decline in a murine model. Autophagy. 2020;16(1):52–69. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta delta C(T)) method. Methods. 2001;25(4):402–408. [DOI] [PubMed] [Google Scholar]
- [16].Zaugg K, Yao Y, Reilly PT, et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011;25(10):1041–1051. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen Y, Wang Y, Huang Y, et al. PPARα regulates tumor cell proliferation and senescence via a novel target gene carnitine palmitoyltransferase 1C. Carcinogenesis. 2017;38(4):474–483. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kantarci K. Molecular imaging of Alzheimer disease pathology. AJNR Am J Neuroradiol. 2014;35(Supplement 6):S12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Song XY, Hu JF, Chu SF, et al. Ginsenoside Rg1 attenuates okadaic acid induced spatial memory impairment by the GSK3beta/tau signaling pathway and the Abeta formation prevention in rats. Eur J Pharmacol. 2013;710(1–3):29–38. . [DOI] [PubMed] [Google Scholar]
- [20].Fu AL, Zhou CY, Chen X. Thyroid hormone prevents cognitive deficit in a mouse model of Alzheimer’s disease. Neuropharmacology. 2010;58(4–5):722–729. [DOI] [PubMed] [Google Scholar]
- [21].Cassidy L, Fernandez F, Johnson JB, et al. Oxidative stress in alzheimer’s disease: a review on emergent natural polyphenolic therapeutics. Complement Ther Med. 2020;49:102294. [DOI] [PubMed] [Google Scholar]
- [22].Tarozzi A. Oxidative stress in neurodegenerative diseases: from preclinical studies to clinical applications. J Clin Med. 2020;Apr 24;9(4):1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12(10):1161–1208. [DOI] [PubMed] [Google Scholar]
- [24].Carrasco P, Sahun I, McDonald J, et al. Ceramide levels regulated by carnitine palmitoyltransferase 1C control dendritic spine maturation and cognition. J Biol Chem. 2012;287(25):21224–21232. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Behl C. Apoptosis and Alzheimer’s disease. J Neural Transm (Vienna). 2000;107(11):1325–1344. [DOI] [PubMed] [Google Scholar]
- [26].Sanvicens N, Cotter TG. Ceramide is the key mediator of oxidative stress-induced apoptosis in retinal photoreceptor cells. J Neurochem. 2006;98(5):1432–1444. [DOI] [PubMed] [Google Scholar]
- [27].Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53(1):409–435. [DOI] [PubMed] [Google Scholar]
- [28].Shang D, Liu Y, Zhang J, et al. Peroxisome proliferator-activated receptor γ (PPARγ) suppresses the proliferation and metastasis of patients with urothelial carcinoma after renal transplantation by inhibiting LEF1/β-catenin signaling. Bioengineered. 2020;11(1):1350–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhang JQ, Long XY, Xie Y, et al. Relationship between PPARα mRNA expression and mitochondrial respiratory function and ultrastructure of the skeletal muscle of patients with COPD. Bioengineered. 2017;8(6):723–731. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wojtowicz S, Strosznajder AK, Jezyna M, et al. The novel role of PPAR alpha in the brain: promising target in therapy of Alzheimer’s disease and other neurodegenerative disorders. Neurochem Res. 2020;45(5):972–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Warden A, Truitt J, Merriman M, et al. Localization of PPAR isotypes in the adult mouse and human brain. Sci Rep. 2016;6(1):27618. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Brune S, Kolsch H, Ptok U, et al. Polymorphism in the peroxisome proliferator-activated receptor alpha gene influences the risk for Alzheimer’s disease. J Neural Transm (Vienna). 2003;110(9):1041–1050. . [DOI] [PubMed] [Google Scholar]