

Abstract
The global expansion of COVID-19 and the mutations of severe acute respiratory syndrome coronavirus necessitate quick development of treatment and vaccination. Because the androgen-responsive serine protease TMPRSS2 is involved in cleaving the SARS-CoV-2 spike protein allowing the virus to enter the cell, therefore, direct TMPRSS2 inhibition will inhibit virus activation and disease progression which make it an important target for drug discovery. In this study, a homology model of TMPRSS2 protein was initially developed. Then, we used the fragment-based drug design (FBDD) technique to develop effective TMPRSS2 inhibitors. Over a half-million fragments from the enamine database were screened for their binding ability to target protein, and then best-scoring fragments were linked to building new molecules with a good binding affinity. XP docking and MM-GBSA studies revealed 10 new formed molecules with docking score ≤ −14.982 kcal/mol compared to ambroxol (control) with a docking score of −6.464 kcal/mol. Finally, molecular dynamics (MD) and density functional theory (DFT) were calculated for the top 3 molecules.
Keywords: SARS-CoV-2, TMPRSS2, Fragment-based drug design, Homology modeling, Docking, Molecular dynamics, Quantum mechanics
1. Introduction
COVID-19 started in Wuhan, China, and quickly spread around the world. This pandemic, which is triggered by the SARS-Corona Virus-2 virus, is an international health crisis [[1], [2], [3], [4]]. COVID-19 has been linked to approximately 240 million illnesses and over 5 million fatalities as of October 2021 and the cases continue to increase as there is no effective treatment available [5]. This outbreak had a significant burden on healthcare systems and the worldwide economy, which demand the development of effective treatments to avoid the spread and reduce the severity of the disease [6,7].
SARS-Corona Virus-2 belongs to the Coronaviridae family, which together with Arteriviridae and Roniviridae, is a large viral family with several hundred species that makes up the order Nidovirales [8]. The SARS-Corona Virus-2 is a class of beta coronavirus that shares seventy-nine percent sequence identity with the SARS-Corona virus, the agent that initiated the 2003 SARS infection [[9], [10], [11], [12]]. Recent studies advocate that both SARS-Corona Virus and SARS-Corona Virus-2 utilize the angiotensin-converting enzyme2 (ACE2) receptor for cell entry, and Transmembrane protease serine type2 (TMPRSS2) to cleave the viral S glycoprotein, enhancing viral activation, but transmission is much higher with SARS-CoV-2 [[13], [14], [15]]. TMPRSS2 and ACE2 are present in the GI tract, kidney, liver, heart, and other organs [[16], [17], [18]], thus SARS-Corona Virus-2 can infect these organs and as a result increase disease severity, which makes TMPRSS2 a crucial target to control the virus entry and spread [[19], [20], [21], [22], [23], [24]].
TMPRSS2 is less important than ACE2 in body homeostasis and its inhibition will not affect the vital processes in the body [25,26]. Binding of the S1 of spike protein (S) with a cell receptor requires its priming by intracellular proteases, a process that involves cleavage at the (S1/S2) and S2 sites and increases viral and cellular membrane fusion, therefore enhancing viral invasion into target cells [10,15,[27], [28], [29], [30], [31]].
TMPRSS2 processing is indeed one of the critical stages in activating the membrane function of the SARS-Corona virus-2 S protein [32]. As a result, drugs that suppress its proteolytic activity are needed to avoid SARS-Corona Virus-2 membrane fusion. Notably, TMPRSS2 is a human protease, and as a therapeutic target, it'll not induce drug resistance like viral protein targets [33]. So, TMPRSS2 is among the most promising anti-SARS-Corona virus-2 therapeutic targets [34,35]. Like many other protease inhibitors, TMPRSS2 inhibitor has been reported and demonstrated to block virus entrance into host cells [[36], [37], [38], [39], [40]].
Simulating nature through computational structure-based drug development is among the most major and newest techniques to investigating pharmacological action. Compounds are tested into the chosen binding sites in 3-dimensional models of the protein targets utilizing computer software. By using physics-based calculations to determine binding energies, the interaction of the examined molecules with the binding pocket could be evaluated.
When experimental techniques fail or there is a lack in data of structural information of a protein, homology modeling is effective in silico tool for generating “a tertiary structure” of the protein of interest which is essential in structure-based drug discovery [41]. Fragment-based drug design approach (FBDD) has a track record of helping to uncover approved and advanced investigational medications, the primary approach for finding small-molecule ligands [42], saves experimental cost, and can be used in several ways to develop new drugs [43], and have an important role in target-based drug discovery [44]. It used fragments that have a low-binding affinity, low complexity in their structures and low molecular weight (<300 Da) to build the new compounds [45].
In our previous studies, TMPRSS2 inhibitors were investigated using in silico drug repurposing and pharmacophore modeling approaches [46,47]. In this article, we used a fragment-based drug design approach to discover novel TMPRSS2 inhibitors to combat COVID-19.
2. Materials and methods
All computational studies were carried out using maestro v 12.8 of Schrodinger and academic Desmond v6.5 by D.E. Shaw Research for molecular dynamics. The workflow of this study is summarized in Fig. 1 .
Fig. 1.

Study workflow.
2.1. Homology modeling and active site detection
The model of TMPRSS2 protein was built using the Schrodinger program. Initially, the TMPRSS2 protein sequence was obtained from Universal Protein Knowledgebase (UniProtKB-O15393) [48]. Then, using BLASTp in NCBI [49], the sequence of target protein was compared to other proteins to find regions of identity, and the protein with the highest percentage was selected as a template. After that, using structure prediction wizard in Schrodinger suit, the sequences of amino acids of both TMPRSS2 protein and template were imported and alignment was performed using a single template alignment (STA).
Then, using the "protein preparation wizard" tool, the modeled protein was preprocessed to correct the order of all bonds in the structure and through Epik [50], proper ligand ionization and tautomeric state were confirmed in the specified pH range. Also, the model was minimized using the OPLS3e force field.
Finally, the Ramachandran was used to identify residues that accumulating in disallowed regions of protein dihedrals which indicate whether the model is suitable for further processes or not, so that adjustments can be made to the protein geometry and make it appropriate [51]. Also, a protein reliability report was used to show a simple graphical depiction of different measures such as Average B-factors, backbone dihedrals, binding site packing, bond angle deviations, bond length deviations, missing atoms, missing loops, Side-chain dihedrals, Side-chain planarity, and steric clashes, which indicating a protein structure's reliability or quality.
Then, the SiteMap tool [52] was used to preview and evaluate active sites within the Protein Homology Model, set a minimum site point number required for defining a Site15, the number of sites to be reported in the output was 5, and select a more restrictive hydrophobicity definition excluding points at the border of too many assigned points that have too few neighbors that are classified as phobic or inside the protein, or that border on too many points that occupy free space.
2.2. Fragment library
Over a half-million fragments, structures were downloaded in the form of .sdf files from the enamine database (https://enamine.net/). These fragments were prepared using ligprep [53] and their ionization states were generated at pH 7.0 (±2.0) using Epik ionizer.
2.3. Fragment screening
The "receptor grid generation" of the Schrodinger suite was used to generate the receptor grid on TMPRSS2 to define interaction grids for molecular docking. Then the prepared fragments were docked against the binding site of the target using two glide docking filters "high throughput virtual screening HTVS" and "standard precision SP".
2.4. Fragment linking
Fragments with high docking scores were combined in three rounds using the combine fragments of the Schrodinger suite. The panel links the fragments by detecting feasible bonds that formed between the fragments. The maximum bond angle deviation was 15°. The maximum atom-atom distance was 1 Å, while the minimum fragments centroid distance was 2 Å, and the maximum number of fragment atoms was 200 atoms. The newly formed molecules were prepared using the ligprep before docking.
2.5. Molecular docking
Using the Glide module [54], newly designed molecules were docked into the TMPRSS2 active site via "high throughput virtual screening HTVS", which screen a very large number of ligands rapidly. Then for top docking molecules, "extra precision XP" docking was performed. In these two docking methods, ligands were docked flexibly, in which conformations were generated internally throughout the docking procedure. Since the ligands have been prepared with Epik for ionization and tautomerization, Epik sanctions have been added to the docking ratings for the adoption of higher-energy states (which also include those with metals). The results were compared against approved TMPRSS2 inhibitors ambroxol and nafamostat.
2.6. Binding free energy calculations of the docked complex by MM-GBSA
Prime molecular mechanics-generalized Born and surface area (MM-GBSA) panel in Schrodinger suit [55], was used to calculate free binding energies, which determine the length of binding time and thus the ability to exert pharmacologic action, for top-100 docking fragments that have the best docking score in XP docking. For the refinement, VSGB was selected as a solvation model; water was used as a solvent and OPLS3e force field was applied. The results were also compared against approved TMPRSS2 inhibitors ambroxol and nafamostat.
2.7. Molecular dynamics (MD) simulation
Molecular dynamics (MD) simulation is performed to mimic the physiological environment in the human body. In the present study, we performed MD simulations to analyze the protein-ligand interaction between the TMPRSS2 and top-3 docking molecules using Desmond v6.5 [56]. The system was neutralized by the addition of Na+ and Cl-ions, and solvated using the TIP3P water model. The system was situated at a distance of 10 Å from the edge of the orthorhombic box (10 × 10 x 10) and LBFGS minimization was conducted with 3 vectors and minimum 10 steepest descent steps until a gradient threshold of 25 kcal/mol/Å was achieved. The maximum iterations during minimization were 2000 and convergence was set at 1.0 kcal/mol/Å. For long-range electrostatic interactions Smooth Particle Mesh Ewald method was used at a tolerance of 1e-09 and a cut-off radius of 9 Å was selected for short-range electrostatic interactions. The MD simulation was performed for 100 ns via NPT ensemble, under temperature (300 K) and atm pressure (1 bar); which are maintained constant throughout the simulation process using a Nose-Hoover thermostat and a Martyna-Tobias-Klein barostat. In the MD simulation system, the desired protein-ligand combination was saturated, and partial charges were computed. Also, the OPLS3e force field was used for energy minimization [57]. During the simulations, 1000 frames were recorded for each system. Finally, Plots of RMSD, RMSF, and hydrogen bonds were created beside dynamic simulation.
2.8. Density functional theory (DFT) calculations
We use the optimization panel of the Jaguar program [58] to perform DFT calculations for top-3 docking fragments. We chose 6-31G**as the basis set and B3LYP as the level of theory as it is the most used basis set for small organic compounds [46,47,59,60]. Standard Poisson-Boltzmann (PBF) was used as the solvation model in which water was selected as a solvent, which is included in the calculation as a dielectric continuum with a cavity for the molecule, water is the solvent and the geometries were optimized 100 times. The accuracy for self-consistent field (SCF) calculations is quick, which uses mixed pseudospectral grids with loose cutoffs (iacc = 3). Here, we calculate the electron densities, electrostatic potential, molecular orbitals for the molecules. Similarly, the results were compared against approved TMPRSS2 inhibitors ambroxol and nafamostat.
3. Results
3.1. Homology modeling
The 3-dimensional structure of TMPRSS2 is not yet available in the protein databank. Here, we use human plasma kallikrein (PDB ID: 5TJX) [61] as the model template. The results of BLASTp alignment showed that the sequence identity of TMPRSS2 and human plasma kallikrein is 42.21%, and the sequence similarity is 47%. So, TMPRSS2 and the human plasma kallikrein sequences were aligned. TMPRSS2 model was subjected to model refinement and energy minimization to remove steric clashes from their structural geometry using protein preparation wizard (Fig. 2 A). The resulting model was validated using the Ramachandran plot and protein reliability report. Ramachandran plot displayed that 82.7% of the residues are located in the most favored area, 16.3% of the residues are located in the additional allowed area, 1% of the residues are located in the generously allowed area, and 0% are located in the disallowed area Fig. 1B. The protein reliability report indicated that there were no missing loops, missing atoms, and no improper torsions as depicted in Fig. 2C.
Fig. 2.

Protein structures, Ramachandran plot, and sequence alignment. A. TMPRSS2 homology. B. Ramachandran plot. C. protein reliability report.
3.2. Active site prediction
The active site was predicated using the sitemap tool. Using advanced search and analysis tools, this program offers data on the binding site's features. An initial search process in SiteMap analysis is to find one or more areas on the protein surface that may be appropriate for binding ligands to the receptor using grid points. The next part of the analysis is the evaluation, which consists of rating each site by computing different features such as Site Score, a favorable binding site has an average SiteScore of 1.0. It also determines the protein's druggability by determining its ability to efficiently bind passively absorbed small compounds. Five possible sites were determined, each with a quantitative site score and a D score. The first site was chosen because it had a high site score of 1.016 and a high D score of 1.067, as well as containing the catalytic residues His 296 Asp 345, and Ser 441, and the substrate-binding residues Asp435, Ser460, Gly462 [26] as displayed in Table 1 and Fig. 3 . The active site has the following amino acid residues 296, 299, 300, 337, 340, 341, 342, 343, 345, 389, 418, 419, 420, 424,436,437,438,441,460,461,462,463,464,465,473.
Table 1.
D scores, site scores, and volume for the five predicted sites.
| Site number | Site score | D score | Volume |
|---|---|---|---|
| Site 1 | 1.016 | 1.067 | 262.738 |
| Site 2 | 0.951 | 0.955 | 43.904 |
| Site 3 | 0.875 | 0.860 | 169.785 |
| Site 4 | 0.866 | 0.857 | 282.289 |
| Site 5 | 0.727 | 0.518 | 95.354 |
Fig. 3.

Binding site and the catalytic residues of TMPRSS2.
3.3. Fragment database screening and linking
Over a half-million fragments from the enamine library, were prepared by ligprep resulting in one million and a half confirmations, and then screened using two docking modes of Glide. First, HTVS mode was used and the top 321,045 high-scoring fragments were further docked in the SP mode. 1800 fragments with high docking scores (≤7 kcal/mol) were then combined using combine fragments of the Schrodinger suite to produce 4270 newly formed molecules. The compounds were prepared to give 69016 confirmations. The docking score for the top-100 fragments were listed in Table 1 in supplementary data.
3.4. Docking of the newly designed compounds, MM-GBSA and ADMET prediction
In this study, we aim to find novel inhibitors of TMPRSS2 through molecular docking. The strength and affinity of the generated compounds for the target could be predicted using molecular docking, as well as the binding site residues of the drug in the target protein and their interactions.
The prepared compounds were first docked to the TMPRSS2 using HTVS mode, then the top 2955 docked poses were further coupled to the XP mode.
The top-10 high-docking score compounds were displayed in Table 2 and Fig. 4 . Most of the hydrophobic interactions of compounds and TMPRSS2 were formed with the residues ASN418, ASN343, THR341, HIS296, SER441, SER460, THR495, and SER436, including the three major catalytic residues. TRP46 contributes to all of the pi-cation interaction. While most of the salt bridged interactions were with PO4501, ASP345, and ASP435.
Table 2.
Docking score, MM-GBSA results, and interactions of the top 10 compounds and controls with TMPRSS2.
| Name | Docking score | MM-GBSA dG Bind | Pi-cation interaction | Hydrogen bonding interaction | Hydrophobic interaction | Salt bridges |
|---|---|---|---|---|---|---|
| Nafamostat | −5.424 | −59.76 | LYS300 | GLU299, GLY464, ASP435, PO4501 | CYS437, TRP461, VAL473,CYS465,ALA466, PRO301 | ASP435, GLU299 |
| Ambroxol | −6.464 | −55.48 | HIS296 | SER460, THR431 | TYR337, TRP461, CYS437, CYS465 | – |
| Combine-1 | −14.982 | −88.97 | TRP461 | ASN418, LYS340, THR341, PO4501, SER460 | SER441, GLN438, SER436, THR459, SER460, ASN418, THR341, HIS296 | PO4501, ASP345 |
| Combine-2 | −14.758 | −88.56 | TRP461 | GLY464, ASN343, THR341, ASN418 | SER441, GLN438, SER436, THR459, SER460, ASN418, THR341, HIS296, ASN343 | ASP345, ASP435 PO4501, |
| Combine-3 | −14.561 | −87.97 | TRP461 | THR341, PO4501, ASP345 | SER441, GLN438, SER436, THR459, SER460, ASN418, THR341, HIS296, ASN343 | ASP345, ASP435,PO4501 |
| Combine-4 | −14.558 | −88.06 | TRP461 | THR341, GLY464, ASP345 | SER441, GLN438, SER436, THR459, SER460, ASN418, THR341, HIS296 | ASP345, ASP435 PO4501 |
| Combine-5 | −14.541 | −79.18 | TRP461 | LYN342, ASP435, ASP345, THR341 | SER441, GLN438, SER436, THR459, SER460, ASN418, THR341, HIS296, ASN343 | ASP345, ASP435 PO4501 |
| Combine-6 | −14.499 | −94.37 | TRP461 | THR341, ASP435, ASP345 | SER441, GLN438, SER436, THR459, SER460, ASN418, THR341, HIS296, ASN343 | ASP345, ASP435, PO4501 |
| Combine-7 | −14.499 | −86.18 | TRP461 | ASP435, ASP345, SER460, GLY464, TRP461, TYR337 | SER441, GLN438, SER436, THR459, SER460, ASN418, THR341, HIS296 | ASP345, ASP435, PO4501 |
| Combine-8 | −14.453 | −75.85 | TRP461 | ASP435, SER436, TYR337, LYS340 | SER441, GLN438, SER436, THR459, SER460, ASN418, THR341, HIS296, ASN343 | ASP345, ASP435, PO4501 |
| Combine-9 | −14.441 | −93.21 | TRP461 | SER460, ASN418, LYN342, THR341, PO4501 | SER441, GLN438, SER436, THR459, SER460, ASN418, THR341, HIS296 | ASP345, ASP435, PO4501 |
| Combine-10 | −14.42 | −87.11 | TRP461 | THR341, ASP435, ASP345, ASN343, GLY464 | SER441, GLN438, SER436, THR459, SER460, ASN418, THR341, HIS296,ASN343 | ASP345, ASP435 PO4501 |
Fig. 4.

Chemical structures of the 10 compounds that showed promising binding affinity against TMPRSS2 protein.
Hydrogen Bonds play a significant role in ligand binding. Because of their substantial effect on drug selectivity, metabolism, and adsorption, hydrogen-bonding characteristics should be considered in drug design. The residues ASN418, LYS340, THR341, PO4501, and SER460 in combine-1, GLY464, ASN343, THR341, and ASN418 in combine-2, and THR341, PO4501, and ASP345 in combine-3 showed hydrogen bond interaction with the ligand as illustrated in Fig. 4.
The top 10 compounds were further subjected to MM-GBSA. MM/GBSA calculates the ligand strain energy by placing the ligand in a solvent thus giving more reliable results. The MM-GBSA results were for the 10 compounds were in the range from −75.85 to −94.37 kcal/mol.
3.5. Molecular dynamic stimulation
MD simulation was conducted on the compounds that demonstrated stronger ligand-protein interactions, namely combine-1, combine-2, and combine-3 following XP-docking, and the results are illustrated in Fig. 5, Fig. 6, Fig. 7, Fig. 8 . The changes in structural conformation were monitored in terms of RMSD and RMSF. Also, the protein interactions with the ligand were monitored throughout the simulation.
Fig. 5.

The 2D and 3D interactions of the best three compounds with TMPRSS2.
Fig. 6.

RMSD plot of TMPRSS2 protein-ligand interaction of top three complexes: (A) Combine 1 RMSD (B) Combine 2 RMSD (C) Combine 3 RMSD.
Fig. 7.

RMSF plot of protein and ligand interaction of top 3 complexes: (A) Combine 1 (B) Combine 2 (C) Combine 3.
Fig. 8.

Histogram of ligand interaction of top three complexes: (A) Combine 1 (B) Combine 2 (C) Combine 3.
Throughout the 100-nsec of MD all ligand-TMPRSS2 complexes, showed a low RMSD average for protein structures (≤2.96 Å) and ligand (≤3.32 Å) as shown in Table 4 . Also, they showed a low RMSF average for both protein (≤1.23 Å) and ligand (≤3.36 Å) which indicate that the conformations obtained from MD simulations were structurally stable and ideal for further computational analysis. All the average RMSD and RMSF values are provided in Table 4.
Table 4.
RMSD and RMSF average of the best three compounds.
| Name | RMSD |
RMSF |
||
|---|---|---|---|---|
| Cα | ligand | Cα | ligand | |
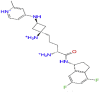 |
2.96 ± 0.25 | 2.51 ± 0.43 | 1.23 ± 0.89 | 0.94 ± 0.38 |
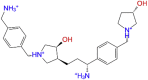 |
2.96 ± 0.25 | 2.09 ± 0.30 | 1.23 ± 0.89 | 3.36 ± 2.49 |
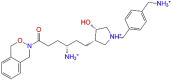 |
2.96 ± 0.25 | 3.23 ± 0.36 | 1.23 ± 0.89 | 1.17 ± 0.39 |
Combine 1-TMPRSS2 docked complex showed strong direct hydrogen bond interaction with the catalytic residue ASP345 mainly with the hydrogen atom of the 2-methylpyridine ring in the ligand, moderate interaction with catalytic residues SER441 and HIS296, and with other active site residues namely THR459 and TRP46. Bridged hydrogen bonds with the catalytic residue SER441, the substrate-binding residue GLY462, and other active site residues THR459 AND GLY439 were noticed. The TRP461, TYR47, and the catalytic residue HIS296 showed hydrophobic interaction. TRP461 residue contributes to pi-cation interaction with the ligand. Also, the residue SER441 showed ionic interaction with the nitrogen atom of the 2-methylpyridine ring in the ligand.
Combine2-TMPRSS2 docked complex showed weak direct hydrogen bond interactions with the residues GLN438 and SER463 mainly with the hydrogen atom of the phenylmethanaminium and pyrrolidine-3-ol moiety respectively. Bridged hydrogen bonds with active site residues namely GLY439, GLY438, CYS437, THR459, and GLU389 were noticed. ASP417, GLY4 464, LYS300, GLN438, LU389, and the catalytic residue SER441 residues also showed ionic interactions with the nitrogen atom of phenylmethanaminium moiety in the ligand.
Combine3-TMPRSS2 docked complex showed strong direct hydrogen bond interaction with the catalytic residue ASP345 mainly with the hydrogen atom of the phenylmethanaminium moiety in the ligand and moderate interactions with other active site residues GLY464 and SER436. Bridged hydrogen bonds with GLY439, HIS296, ASP435, SER436, THR461 and GLU299 AND GLY464 were noticed. SER441 showed additional ionic interaction with the nitrogen atom of phenylmethanaminium moiety in the ligand.
The low RMSD, RMSF, and simulation duration imply that the TMPRSS2 protein's 3D structural model is accurate and the three complexes were structurally stable and equilibrated.
3.6. DFT calculations
Molecular orbitals (MOs) typically play a major role in giving data on optical and electrical properties, as well as quantum chemistry, in molecular systems, and are used to comprehend the many forms of conjugated system processes according to molecular orbital theory. The main principle of this theory is that the maximum positive overlap between LUMO and HOMO orbitals is required for a simple reaction pathway. The reactivity of combine 1, 2, and 3 was studied using the DFT approach (B3LYP/6-31G* level). The results are summarized in Table 3 and Fig. 9 . For HOMO and LUMO orbitals, a consistent negative value was observed. For examined drugs, the measured HOMO and LUMO range from −0.187 to −0.056 kcal/mol. HOMO and LUMO findings were both compared to the reference drugs nafamostat and ambroxol.
Table 3.
Quantum chemical proprieties of the references and the best three compounds.
| Drug | HOMO kcal/mol | LUMO kcal/mol | HLG kcal/mol | Solvation Energy kcal/mol |
|---|---|---|---|---|
| Ambroxol | −0.22955 | −0.04444 | −0.185 | −55.25 |
| Nafamostat | −0.23571 | −0.07482 | −0.160 | −148.16 |
| Combine-1 | −0.24351 | −0.05609 | −0.187 | −450.49 |
| Combine-2 | −0.26510 | −0.04341 | −0.222 | −321.84 |
| Combine-3 | −0.24607 | −0.04834 | −0.198 | −294.84 |
Fig. 9.

A. Combine-1 B. Combine-2 C. Combine-3.
In combine-1 HOMO orbitals (Fig. 7A) are located essentially on 4, 6-difluoro-2, 3-dihydro-1H-indene moiety. While LUMO orbitals are located on 4-azaniumyl-2-methyl pyridine-1-ium moiety in which N24 interacts with TRP461. The HLG in combine-1 is −0.187 kcal/mol. In combine-2 HOMO orbitals (Fig. 7B) are located essentially on the 1, 4-xylene moiety and N41 and C50. LUMO orbitals are located on phenylmethanaminium moiety in which N1 interacts with GLU388. The HLG in combine-2 is −0.225 kcal/mol. While in combine-3, HOMO orbitals (Fig. 7 B) are situated essentially on 3,4-dihydro-1H-2,3-benzoxazine moiety. LUMO orbitals are found on phenylmethanaminium moiety in which N1 interacts with GLU389 and the benzene ring interacts with TRP461. The HLG in combine-3 is −0.198 kcal/mol.
Moreover, the HOMO, LUMO, and solvation energy results were compared with the reference drugs. HOMO molecular orbitals in ambroxol are located on the amino group, the two bromine atoms, and the benzene ring of the 2-amino-3, 5-dibromophenyl moiety. The LUMO molecular orbitals are located on nitrogen number 11 of the methylamino moiety and the benzene ring of the 2-amino-3, 5-dibromophenyl moiety. In nafamostat, HUMO is located on oxygen atom number 14, between carbon 7 and carbon 8, between carbon 9 and 10, between carbon 1 and carbon 4, between carbon 2 and 3 all displayed in 6-[amino(iminiumyl)methyl]naphthalen-2-olate moiety. Whereas LUMO is situated in nitrogen atoms number 12 and 13 those interact with ASP435, oxygen atom number 14, carbon atoms number 3, 4,7and 9, between carbon 5 and carbon 10, and between carbon 2 and 11 of 6-[amino (iminiumyl) methyl] naphthalene-2-olate moiety, also in oxygen number 17, carbon atoms number 18, 20, 22 and 24 and between carbon 15 and 16 of 4-(diamino methylidene amino) moiety as shown in Fig. 10 .
Fig. 10.

HOMO and LUMO representation of reference drugs.
The molecular electrostatic potential is a well-known method for investigating molecule reactive properties and characterizing intermolecular interactions. It allows us to compare a molecule's most reactive nucleophilic and electrophilic sites to physiologically reactive potentials.
As it can be seen in Fig. 9, the MEP map of combine-1 range from −110 to 275 kcal/mol. The maximum positive region is found on N1 that interacts with GLU299 and PO4501 and N38 that interacts with ASP345 which are all located in the 1-(methyl carbamoyl)propane-1-aminium moiety. The maximum negative region is situated on the two fluorine atoms F13 and F16 in the 4,6-difluoro-2,3-dihydro-1H-indene moiety. In combine-2, the MEP map is ranging from −81.44 to 269.4 kcal/mol. The maximum positive region is located on N34 of pentane-2-aminium moiety that interacts with GLU299 and the maximum negative region is found on O45 of the pyrrolidine-3-ol moiety. While, in combine-3, the MEP map is ranging from −78.5 to 239.7 a maximum positive region is localized on N1of the phenylmethanaminium that interacts with GLU389, N8 of the pyrrolidine-3-ol that interacts with SER440 and N34 of the heptane-4-aminium moiety. A maximum negative region is situated on the benzene ring of the 3,4-dihydro-1H-2,3-benzoxazine.
These findings of the above-mentioned interactions are closely related to the docking interactions and support docking results and the potential of these three compounds to act as anti-TMPRSS2.
4. Discussion
The globe has been afflicted by the SARS-Cov-2 pandemic, a condition unprecedented in human history that has produced a global medical crisis [1,2]. There are insufficient safety precautions in place, and there are no licensed medications for the COVID-19 illness [62]. As a result, it is critical to design a particular inhibitor for COVID-19. For coronaviruses like SARS-CoV-2, several therapeutic targets have been proposed. The human protein TMPRSS2, which aids coronavirus life cycles by cleaving viral spike proteins, is the topic of this study. Non-viral drug targets, such as TMPRSS2, offer the benefit of not allowing the virus to acquire resistance mutations that lower the medication's affinity for the target [63]. As a drug target TMPRSS2 is less important than ACE2 in body homeostasis and its inhibition will not affect the vital processes in the body which means less or no side effects will develop [25,26] and also TMPRSS2 has the additional advantage that the drug discovery community has significant experience in developing drugs targeting serine proteases.
We chose the protease TMPRSS2 as our target of interest in this investigation for the fragment-based development of molecules with optimum binding to TMPRSS2 in order to act as novel anti-COVID-19. The three-dimensional structure of TMPRSS2 is not known so we have generated a model of the TMPRSS2 from human plasma kallikrein. The resulting model was validated using the Ramachandran plot and protein reliability report. Over a half-million fragments from the enamine database were screened for their potential to inhibit TMPRSS2. The structures were subjected to energy minimization, followed by molecular docking with the TMPRSS2. Top fragment hits with docking score < _7.00 were retained from SP fragment-docking calculations for further design. The ‘combine fragments’ panel from the library design module was used for direct joining of the fragments prepositioned at different regions of the TMPRSS2 binding site to design new compounds. The newly designed compounds were further subjected to docking to confirm their affinity to TMPRSS2. Molecular docking generates and ranks the receptor-ligand poses according to their interaction energies.
The active site of the TMPRSS2 showed hydrogen bonding with the residues SER460, ASN418, LYN342, THR341, PO4501, GLU299, GLY464, ASP435, ASN343, ASP345, LYS342, TYR337 which played a significant role in binding with the ligands as analyzed from the binding interaction analysis. Similar results with different molecules were shown in different computational and experimental studies [36,46,[63], [64], [65], [66], [67], [68], [69], [70], [71]]. In our previous work, GLU299, GLY464, ASP435, PO4501, SER460, SER436, LYS342, ASN418, LYS340, TRP461, PO4 501, THR341 showed H-bond with the ligand [46,64]. In a work done by Idris M. et al. group LYN342, Asp435, SER460, SER436, TRP461, GLY464, LYN342 showed H-bond with the ligand [69] also Asp435, SER436, TRP461, GLY464, GLU 299, LYS342 showed the same interactions in a work conducted by Kumar V. et al. group [68].
The docking interaction energy, showed the highest interaction energy than the reference drugs Nafamostat and Ambroxol as shown in Table 1. These findings were additionally confirmed by determining the free binding energy by the MMGBSA method. MM-GBSA calculations inferred that our selected compounds had the most favorable energy with the active site of TMPRSS2 Even better than Nafamostat and Ambroxol.
Despite the excellent results of the docking investigation, which supported our design reasoning, MD studies were carried out for additional confirmation and validation of the whole work. We did three dynamic simulations intending to identify and study the nature of the TMPRSS2 dynamic and to provide insights for future lead optimization. The RMSD is a prominent measure used to analyze the structural stability of protein structures using MD simulations. The low RMSD values obtained during the whole simulation in the RMSD interpretation of MD simulations suggested the stability for both protein and its inhibitors complex. Furthermore, the minimal changes in RMSF values suggested that protein-ligand complexes were structurally stable. MD simulations produced structurally stable conformations that were suited for further computational study.
Also, DFT calculations supported the docking results. We noticed that the designed compounds have lower HLG (−0.222 to 0–187 kcal/mol) as the reference drugs Nafamostat and Ambroxol (−0.160 to −0.185 kcal/mol) which indicate their stability. Also, they have low solvation energy (−450 to – 294 kcal\mol) compared to the reference drugs Nafamostat and Ambroxol (−55.25 to −148.16) indicating their good water solubility.
5. Conclusion
In this study we designed novel compounds for the catalytic site of TMPRSS2 of SARS-CoV-2 using in silico fragment based drug design. These hit compounds have favorable predicted binding scores and free binding energies with the catalytic binding site compared to reference drugs Nafamostat and Ambroxol. Also, three of the compounds showed stable interactions with the target using molecular dynamics studies as well as favorable pharmacokinetic properties. These hits could be used as starting points for lead optimization. At present, we cannot examine these hits experimentally, we think these designed compounds can be valuable to other research groups.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
We acknowledge Schrodinger for support and help.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.imu.2022.100870.
Appendix A. Supplementary data
The following is the supplementary data to this article:
References
- 1.Wang W., Xu Y., Gao R., Lu R., Han K., Wu G., et al. Detection of SARS-CoV-2 in different types of clinical specimens. JAMA, J Am Med Assoc. 2020;323:1843–1844. doi: 10.1001/jama.2020.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morens D.M., Fauci A.S. Emerging pandemic diseases: how we got to COVID-19. Cell. 2020;182:1077–1092. doi: 10.1016/j.cell.2020.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–733. doi: 10.1056/nejmoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D.M.E., Fouchier R.A.M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367:1814–1820. doi: 10.1056/nejmoa1211721. [DOI] [PubMed] [Google Scholar]
- 5.WHO coronavirus (COVID-19) dashboard | WHO coronavirus (COVID-19) dashboard with vaccination data, (n.d.). https://covid19.who.int/(accessed June 15, 2021).
- 6.Mckee D.L., Sternberg A., Stange U., Laufer S., Naujokat C. Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol Res. 2020;157:104859. doi: 10.1016/j.phrs.2020.104859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Q., Wang Z., Zheng Q., Liu S. Potential clinical drugs as covalent inhibitors of the priming proteases of the spike protein of SARS-CoV-2, Comput. Struct. Biotechnol. J. 2020;18:2200–2208. doi: 10.1016/j.csbj.2020.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Modrow S., Falke D., Truyen U., Schätzl H. Mol Virol. 2013 doi: 10.1007/978-3-642-20718-1. [DOI] [Google Scholar]
- 9.Xia S., Zhu Y., Liu M., Lan Q., Xu W., Wu Y., Ying T., Liu S., Shi Z., Jiang S., Lu L. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein, Cell. Mol Immunol. 2020;17:765–767. doi: 10.1038/s41423-020-0374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li C., Yang Y., Ren L. Genetic evolution analysis of 2019 novel coronavirus and coronavirus from other species. Infect Genet Evol. 2020;82:1–3. doi: 10.1016/j.meegid.2020.104285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mollica V., Rizzo A., Massari F. The pivotal role of TMPRSS2 in coronavirus disease 2019 and prostate cancer. Future Oncol. 2020;16:2029–2033. doi: 10.2217/fon-2020-0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou P., Yang X., Wang X., Hu B., Zhang L., Zhang W., Guo H., Jiang R., Liu M., Chen Y., Shen X., Wang X., Zhan F., Wang Y., Xiao G., Shi Z. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579 doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheng W.H. Coronavirus disease 2019 (covid-19) J Intern Med Taiwan. 2020;31:61–66. doi: 10.6314/JIMT.202004_31(2).01. [DOI] [Google Scholar]
- 14.Thunders M., Delahunt B. Gene of the month: TMPRSS2 (transmembrane serine protease 2) J Clin Pathol. 2020;73:773–776. doi: 10.1136/jclinpath-2020-206987. [DOI] [PubMed] [Google Scholar]
- 15.Kuhn J.H., Li W., Choe H., Farzan M. vol. 61. 2004. pp. 2738–2743. (Cellular and molecular life sciences angiotensin-converting enzyme 2 : a functional receptor for SARS coronavirus). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Antalis T.M., Thomas H. first ed. Elsevier Inc.; 2011. Membrane-anchored serine proteases in health and disease I . Introduction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma J., Bhardwaj V.K., Singh R., Rajendran V., Purohit R., Kumar S. An in-silico evaluation of different bioactive molecules of tea for their inhibition potency against non structural protein-15 of SARS-CoV-2. Food Chem. 2021;346 doi: 10.1016/J.FOODCHEM.2020.128933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glycoproteins B.S. vol. 90. 2016. pp. 5586–5600. (Identification of the fusion peptide-containing region in). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glowacka I., Bertram S., Muller M.A., Allen P., Soilleux E., Pfefferle S., Steffen I., Tsegaye T.S., He Y., Gnirss K., Niemeyer D., Schneider H., Drosten C., Pohlmann S. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol. 2011;85:4122–4134. doi: 10.1128/jvi.02232-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou Y., Vedantham P., Lu K., Agudelo J., Carrion R., Nunneley J.W., et al. Protease inhibitors targeting coronavirus and filovirus entry. Antivir Res. 2015;116:76–84. doi: 10.1016/j.antiviral.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gierer S., Bertram S., Kaup F., Wrensch F., Heurich A., Kramer-Kuhl A., Welsch K., Winkler M., Meyer B., Drosten C., Dittmer U., von Hahn T., Simmons G., Hofmann H., Pohlmann S. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can Be activated by TMPRSS2, and is targeted by neutralizing antibodies. J Virol. 2013;87:5502–5511. doi: 10.1128/jvi.00128-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong M., Zhang J., Ma X., Tan J., Chen L., Liu S., Xin Y., Zhuang L. ACE2, TMPRSS2 distribution and extrapulmonary organ injury in patients with COVID-19, Biomed. Pharma. 2020;131:110678. doi: 10.1016/j.biopha.2020.110678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsuyama S., Nao N., Shirato K., Kawase M., Saito S., Takayama I. Enhanced isolation of SARS-CoV-2 by TMPRSS2- expressing cells. 2020. 1-3. [DOI] [PMC free article] [PubMed]
- 24.Zhou H., Fang Y. Potential therapeutic targets and promising drugs for combating SARS-CoV-2. 2020. 3147-3161. [DOI] [PMC free article] [PubMed]
- 25.Kobayashi L.C., O'Shea B.Q., Kler J.S., Nishimura R., Palavicino-Maggio C.B., Eastman M.R., Vinson Y.R., Finlay J.M. Cohort profile: the COVID-19 Coping Study, a longitudinal mixed-methods study of middle-aged and older adults' mental health and well-being during the COVID-19 pandemic in the USA. BMJ Open. 2021;11 doi: 10.1136/bmjopen-2020-044965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Idris M.O., Yekeen A.A., Alakanse O.S., Durojaye O.A. Computer-aided screening for potential TMPRSS2 inhibitors: a combination of pharmacophore modeling, molecular docking and molecular dynamics simulation approaches. J Biomol Struct Dyn. 2020:1–19. doi: 10.1080/07391102.2020.1792346. 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuyama S., Nagata N., Shirato K., Takeda M., Taguchi F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2 ᰔ. 2010. [DOI] [PMC free article] [PubMed]
- 28.Heurich A., Hofmann-winkler H., Gierer S., Liepold T., Jahn O. Proteolysis by TMPRSS2 augments entry driven by the severe. Acute. 2014;88:1293–1307. doi: 10.1128/JVI.02202-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simmons G., Zmora P., Gierer S., Heurich A., Pöhlmann S. 2020. Since January 2020 Elsevier has created a COVID-19 resource centre with free information in English and Mandarin on the novel coronavirus COVID- 19 . The COVID-19 resource centre is hosted on Elsevier Connect , the company ’ s public news and information. [Google Scholar]
- 30.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A., Müller M.A., Drosten C., Pöhlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280. doi: 10.1016/j.cell.2020.02.052. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang T., Bidon M., Jaimes J.A., Whittaker G.R., Daniel S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antivir Res. 2020;178:104792. doi: 10.1016/j.antiviral.2020.104792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stopsack K.H., Mucci L.A., Mph S., Antonarakis E.S., Nelson P.S., Kantoff P.W. TMPRSS2 and COVID-19 : serendipity or opportunity for intervention ? 2020. [DOI] [PMC free article] [PubMed]
- 33.Shen L.W., Mao H.J., Wu Y.L., Tanaka Y., Zhang W. TMPRSS2: a potential target for treatment of influenza virus and coronavirus infections. Biochimie. 2017;142:1–10. doi: 10.1016/j.biochi.2017.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iwata-yoshikawa N., Okamura T., Shimizu Y., Hasegawa H., Takeda M., Nagata N., Health G. TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. 2019. [DOI] [PMC free article] [PubMed]
- 35.Zhu H., Du W., Song M., Liu Q., Herrmann A., Huang Q. Spontaneous binding of potential COVID-19 drugs (Camostat and Nafamostat) to human serine protease TMPRSS2. Comput Struct Biotechnol J. 2021;19:467–476. doi: 10.1016/j.csbj.2020.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hussain M., Jabeen N., Amanullah A., Baig A.A., Aziz B., Shabbir S., Raza F., Uddin N. Molecular docking between human tmprss2 and sars-cov-2 spike protein: conformation and intermolecular interactions. AIMS Microbiol. 2020;6:350–360. doi: 10.3934/microbiol.2020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Touret F., Gilles M., Barral K., Nougairède A., Van Helden J., Decroly E., De Lamballerie X., Coutard B. In vitro screening of a FDA approved chemical library reveals potential inhibitors of SARS - CoV - 2 replication. Sci Rep. 2020;1–8 doi: 10.1038/s41598-020-70143-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elmezayen A.D., Al-Obaidi A., Şahin A.T., Yelekçi K. Drug repurposing for coronavirus (COVID-19): in silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J Biomol Struct Dyn. 2020:1–13. doi: 10.1080/07391102.2020.1758791. 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hempel T., Raich L., Olsson S., Azouz N.P., Klingler A.M., Hoffmann M., Pöhlmann S., Rothenberg M.E., Noé F. Molecular mechanism of inhibiting the SARS-CoV-2 cell entry facilitator TMPRSS2 with camostat and nafamostat. Chem Sci. 2021;12:983–992. doi: 10.1039/d0sc05064d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Repurposing approved drugs as potential inhibitors of 3CL-protease of SARS-CoV-2 : virtual screening and structure based drug design. Comput Biol Chem. 2020;88:107351. doi: 10.1016/j.compbiolchem.2020.107351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vyas V.K., Ukawala R.D., Ghate M., Chintha C. Homology modeling a fast tool for drug discovery: current perspectives. Indian J Pharmaceut Sci. 2012;74:1–17. doi: 10.4103/0250-474X.102537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shepherd C.A., Hopkins A.L., Navratilova I. Fragment screening by SPR and advanced application to GPCRs. Prog Biophys Mol Biol. 2014;116:113–123. doi: 10.1016/j.pbiomolbio.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 43.Erlanson D.A., Fesik S.W., Hubbard R.E., Jahnke W., Jhoti H. Twenty years on: the impact of fragments on drug discovery. Nat Rev Drug Discov. 2016;15:605–619. doi: 10.1038/nrd.2016.109. [DOI] [PubMed] [Google Scholar]
- 44.Harner M.J., Frank A.O., Fesik S.W. Fragment-based drug discovery using NMR spectroscopy. J Biomol NMR. 2013;56:65–75. doi: 10.1007/s10858-013-9740-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Doak B.C., Norton R.S., Scanlon M.J. The ways and means of fragment-based drug design. Pharmacol Ther. 2016;167:28–37. doi: 10.1016/j.pharmthera.2016.07.003. [DOI] [PubMed] [Google Scholar]
- 46.Elbadwi F.A., Khairy E.A., Alsamani F.O., Mahadi M.A., Abdalrahman S.E., Alsharf Z., Ahmed M., Elsayed I., Ibraheem W., Alzain A.A. Informatics in Medicine Unlocked Identification of novel transmembrane Protease Serine Type 2 drug candidates for COVID-19 using computational studies. Informatics Med. Unlocked. 2021;26:100725. doi: 10.1016/j.imu.2021.100725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sajid M., McKerrow J.H., Hansell E., Mathieu M.A., Lucas K.D., Hsieh I., Greenbaum D., Bogyo M., Salter J.P., Lim K.C., Franklin C., Kim J.-H., Caffrey C.R. Functional expression and characterization of Schistosoma mansoni cathepsin B and its trans-activation by an endogenous asparaginyl endopeptidase. Mol Biochem Parasitol. 2003;131:65–75. doi: 10.1016/s0166-6851(03)00194-4. [DOI] [PubMed] [Google Scholar]
- 48.TMPRSS2 - transmembrane protease serine 2 precursor - Homo sapiens (Human) - TMPRSS2 gene & protein, (n.d.). https://www.uniprot.org/uniprot/O15393 (accessed September 21, 2021).
- 49.Protein BLAST: search protein databases using a protein query, (n.d.). https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins (accessed September 21, 2021).
- 50.Greenwood J.R., Calkins D., Sullivan A.P., Shelley J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J Comput Aided Mol Des. 2010;24:591–604. doi: 10.1007/s10822-010-9349-1. [DOI] [PubMed] [Google Scholar]
- 51.Hollingsworth S.A., Karplus P.A. A fresh look at the Ramachandran plot and the occurrence of standard structures in proteins. Biomol Concepts. 2010;1:271–283. doi: 10.1515/BMC.2010.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.TA H. Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model. 2009;49:377–389. doi: 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- 53.LigPrep . LLC; New York, NY: 2020. Schrödinger. (n.d.) [Google Scholar]
- 54.Friesner R.A., Murphy R.B., Repasky M.P., Frye L.L., Greenwood J.R., Halgren T.A., Sanschagrin P.C., Mainz D.T. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 55.Jacobson M.P., Friesner R.A., Xiang Z., Honig B. On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol. 2002;320:597–608. doi: 10.1016/S0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 56.SC ’06: proceedings of the 2006 ACM/IEEE conference on supercomputing. Association for Computing Machinery; New York, NY, USA: 2006. [Google Scholar]
- 57.Schüttelkopf A.W., Van Aalten D.M.F. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr Sect D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 58.Bochevarov A.D., Harder E., Hughes T.F., Greenwood J.R., Braden D.A., Philipp D.M., Rinaldo D., Halls M.D., Zhang J., Friesner R.A. Jaguar: a high-performance quantum chemistry software program with strengths in life and materials sciences. Int J Quant Chem. 2013;113:2110–2142. doi: 10.1002/qua.24481. [DOI] [Google Scholar]
- 59.Prabhu S.V., Singh S.K. E-pharmacophore-based screening of mGluR5 negative allosteric modulators for central nervous system disorder. Comput Biol Chem. 2019;78:414–423. doi: 10.1016/j.compbiolchem.2018.12.016. [DOI] [PubMed] [Google Scholar]
- 60.Noureddine O., Issaoui N., Al-dossary O. DFT and molecular docking study of chloroquine derivatives as antiviral to coronavirus COVID-19 DFT and molecular docking study of chloroquine derivatives as antiviral to coronavirus COVID-19. J King Saud Univ - Sci. 2020:101248. doi: 10.1016/j.jksus.2020.101248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li Z., Partridge J., Silva-Garcia A., Rademacher P., Betz A., Xu Q., Sham H., Hu Y., Shan Y., Liu B., Zhang Y., Shi H., Xu Q., Ma X., Zhang L. Structure-guided design of novel, potent, and selective macrocyclic plasma kallikrein inhibitors. ACS Med Chem Lett. 2017;8:185–190. doi: 10.1021/acsmedchemlett.6b00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huggins D.J. Structural analysis of experimental drugs binding to the SARS-CoV-2 target TMPRSS2. J Mol Graph Model. 2020;100:107710. doi: 10.1016/j.jmgm.2020.107710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Durdaği S. Virtual drug repurposing study against SARS-CoV-2 TMPRSS2 target. Turkish J Biol. 2020;44:185–191. doi: 10.3906/biy-2005-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alzain A.A., Elbadwi F.A. Identification of novel TMPRSS2 inhibitors for COVID-19 using e-pharmacophore modelling, molecular docking, molecular dynamics and quantum mechanics studies. Informatics Med. Unlocked. 2021;26:100758. doi: 10.1016/j.imu.2021.100758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baby K., Maity S., Mehta C.H., Suresh A., Nayak U.Y., Nayak Y. SARS-CoV-2 entry inhibitors by dual targeting TMPRSS2 and ACE2: an in silico drug repurposing study. Eur J Pharmacol. 2021;896 doi: 10.1016/j.ejphar.2021.173922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.M P., Reddy G.J., Hema K., Dodoala S., Koganti B. Unravelling high-affinity binding compounds towards transmembrane protease serine 2 enzyme in treating SARS-CoV-2 infection using molecular modelling and docking studies. Eur J Pharmacol. 2021;890 doi: 10.1016/j.ejphar.2020.173688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Istifli E.S., Şihoğlu Tepe A., Netz P.A., Sarikürkcü C., Kiliç İ.H., Tepe B. Determination of the interaction between the receptor binding domain of 2019-nCoV spike protein, TMPRSS2, cathepsin B and cathepsin L, and glycosidic and aglycon forms of some flavonols. Turkish J. Biol. = Turk Biyol. Derg. 2021;45:484–502. doi: 10.3906/biy-2104-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kumar V., Dhanjal J.K., Bhargava P., Kaul A., Wang J., Zhang H., Kaul S.C., Wadhwa R., Sundar D. Withanone and Withaferin-A are predicted to interact with transmembrane protease serine 2 (TMPRSS2) and block entry of SARS-CoV-2 into cells. J Biomol Struct Dyn. 2022;40:1–13. doi: 10.1080/07391102.2020.1775704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Idris M.O., Yekeen A.A., Alakanse O.S., Durojaye O.A. Computer-aided screening for potential TMPRSS2 inhibitors: a combination of pharmacophore modeling, molecular docking and molecular dynamics simulation approaches. J Biomol Struct Dyn. 2021;39:5638–5656. doi: 10.1080/07391102.2020.1792346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sonawane K., Barale S.S., Dhanavade M.J., Waghmare S.R., Nadaf N.H., Naik N.M. Homology modeling and docking studies of TMPRSS2 with experimentally known inhibitors camostat mesylate , nafamostat and bromhexine hydrochloride to control SARS-coronavirus-2. 2020. [DOI] [PMC free article] [PubMed]
- 71.Istifli E.S., Netz P.A., Sihoglu Tepe A., Husunet M.T., Sarikurkcu C., Tepe B. In silico analysis of the interactions of certain flavonoids with the receptor-binding domain of 2019 novel coronavirus and cellular proteases and their pharmacokinetic properties. J Biomol Struct Dyn. 2020:1–15. doi: 10.1080/07391102.2020.1840444. 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.