

Abstract
Changes in placental function, in particular down-regulation of placental O-GlcNAc transferase (OGT) in response to maternal stress and increased placental secretion of serotonin into the fetal circulation following maternal infection, have been mechanistically linked to adverse neurodevelopment in mice. We hypothesized that mTOR signaling is a key regulator of trophoblast serotonin synthesis and OGT protein expression and that serotonin is secreted by the human placenta into the fetal circulation. Placental homogenates (n=46) from elective terminations at 8-22 weeks of gestation and from healthy term women were sexed and the protein levels of OGT and enzymes involved in serotonin synthesis was determined. Primary human trophoblast (PHT) cells were isolated from normal term placenta (n=27), cultured and transfected (n=8) with siRNA targeting a scramble sequence (control), raptor (inhibits mTOR Complex 1), or rictor (inhibits mTOR Complex 2). Subsequently, conditioned media and PHT cell lysates were collected. Free serotonin concentration was measured using ELISA in cell culture media and in platelet-depleted normal term umbilical vein and artery plasma (n=38). Both mTORC1 and mTORC2 inhibition down-regulated OGT levels in PHT cells. The level of serotonin synthesis enzyme TPH-1 was higher in early gestation female placentas and at term serotonin concentration was 3-fold higher in the umbilical vein than in the umbilical artery. Inhibition of mTORC2, but not mTORC1, increased cultured PHT cell serotonin secretion. Our data is consistent with the model that mTOR signaling is a key regulator of trophoblast serotonin synthesis and OGT expression.
Keywords: Neurodevelopmental Disorders, Fetal Development, Fetal Programming, 5-HT, Trophoblast, Stress, Pregnancy
Introduction
It is well established that a suboptimal intrauterine environment results in fetal adaptations with adverse consequences persisting after birth, throughout the lifespan and in future generations (1-5). An adverse intrauterine environment has been associated with the development of a wide range of non-communicable diseases, including obesity, diabetes and cardiovascular disease (2,6-8). Recent clinical and animal experimental evidence suggests that many neuropsychiatric diseases, such as schizophrenia (9-11), autism spectrum disorder (12,13), attention deficit/hyperactivity disorder (ADHD) and depression (14), also originate in utero (15-17). Furthermore, epidemiological studies have clearly linked maternal infection (11,17), stress (18,19) and depression (16) during pregnancy to poor birth outcomes, abnormal neurodevelopment and increased incidence of neuropsychiatric disease. Mechanisms underpinning these links may include epigenetic changes (20-22) that are hypothesized to influence the offspring’s neurodevelopment directly or by altering the offspring’s ability to respond to postnatal exposures. Reports of adverse maternal events occurring early in pregnancy, during the period of epigenome patterning, have the most substantial impact on neurodevelopment (12,23,24), underscoring the importance of timing of maternal complications on the developing fetal brain.
Throughout pregnancy the placenta represents the maternal-fetal interface performing an array of critical functions to support fetal development and growth, including oxygen and nutrient transfer, hormone production and serving as an immunological barrier (1,25-27). The placenta is therefore critical for maintaining an optimal intrauterine environment and is believed to be an important mediator of developmental programming in response to perturbations in the maternal compartment (25,28-30). Recent meta-analyses of patient genetic data demonstrate that a subset of the most significant genetic variants associated with schizophrenia are highly expressed in the placenta and variations in the expression of these placental genes largely explains the association between obstetric complications and schizophrenia in the offspring (31,32). Animal studies further support that the placenta directly influences fetal brain development and that changes in the placenta mediate the link between maternal obstetrical complications and abnormal neurodevelopment (33-37). For example, serotonin synthesized by the placenta in mid-gestation is critical for the development of the forebrain in mice (33,38) and maternal immune activation disrupts fetal neurodevelopment mediated by increased placental serotonin secretion to the fetus (34). Furthermore, placental O-GlcNAc transferase (OGT), the enzyme responsible for O--GlcNAcylation of proteins, mediates the impact of maternal stress in early pregnancy on fetal hypothalamic gene expression and neurocognitive development in the offspring in mice (35,36). Furthermore, the placenta of male fetuses has lower OGT levels than the female placenta, which has been suggested to protect the female against maternal stress (15,35,39) and may explain the higher frequency of some neurodevelopmental disorders in males. However, the signals linking maternal stress or immune activation to changes in placental function, such as OGT expression and serotonin release remain largely unknown.
OGT is the only known enzyme that catalyzes the addition of O-GlcNAc to serine and threonine residues of hundreds of target proteins in a UDP-GlcNAc-dependent manner (40). The pool of UDP-GlcNAc is generated from glucose metabolism through the hexosamine biosynthetic pathway and thus O-GlcNAcylation is often regarded as a broad marker of nutrient status (40). Cellular serotonin synthesis occurs after the conversion of tryptophan to serotonin (often referred to as 5 Hydroxy-Tryptophan or 5-HT) by the rate-limiting enzyme tryptophan hydroxylase (TPH). Of the two TPH isoforms 1 and 2, TPH-1 is believed to be the predominant isoform outside the brain (41). Serotonin can also be metabolized by monoamine oxidase enzymes (MAOA) in placenta, that together with synthesis enzymes, determines serotonin homeostasis at the fetal-maternal interface (42,43). The cellular machinery mediating the synthesis and metabolism of serotonin is present and active in rodent and human placenta (44-47). The regulation of human placental OGT expression and serotonin synthesis are poorly understood and changes in protein expression of placental OGT, TPH and MAOA across gestation is unknown.
Mechanistic target of rapamycin (mTOR) is a serine/threonine kinase that regulates cell survival, metabolism, growth and proliferation (48-51). mTOR exists in two complexes, mTOR complex (mTORC) 1 and 2, with the protein raptor associated to mTORC1 and rictor associated to mTORC2. mTORC1 regulates protein translation mediated by phosphorylation of ribosomal protein S6 kinase (S6K1) and eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) (48,51,52). mTORC2 regulates cell proliferation, anabolism and survival via phosphorylation of protein kinase B (Akt) and C (PKC) and serum and glucocorticoid-regulated kinase 1 (SGK1) (53,54). mTOR signaling has diverse actions in the placenta including serving as a positive regulator of trophoblast amino acid (55-58) and folate transport (59) as well as mitochondrial function (60).
Placental mTOR signaling is modulated by an array of upstream effectors, including oxygen, amino acids, glucose, energy, fatty acids, folate and growth factors (52,61,62). In many obstetric complications, the concentrations of these upstream effectors are altered in the maternal circulation and/or within the placenta. For example, placental mTOR is activated by growth factors in the maternal circulation (63) and placental mTOR signaling is inhibited in mothers with malaria and placental inflammation (64). Moreover, maternal cortisol, elevated in maternal stress, inhibits placental mTOR signaling (65). mTOR signaling therefore represents a master regulator of placental function that may link obstetrical complications, such as maternal infection/stress, to altered fetal neurodevelopment.
In the current study, we determine the protein expression patterns of OGT and enzymes responsible for serotonin synthesis in villous trophoblast tissue across gestation and determine the impact of fetal sex on protein expression patterns in early gestation compared to term samples. Furthermore, we tested the hypothesis that mTOR signaling is a key regulator of trophoblast serotonin synthesis and OGT expression and that serotonin is secreted by the human placenta into the fetal circulation.
Methods
Subjects and Tissue Collection
This study was approved by the Institutional Review Board at University of Colorado, Anschutz Medical Campus (COMIRB 06-1098 and 14-1073). All participants were recruited with written informed consent for collection of placental tissue and for use of their anonymous, protected health information. Placental tissue was collected from 46 women, with normal body mass index (BMI, 18-24.9 kg/m2) and no reported use of antidepressant drugs, undergoing elective termination of pregnancy (n=32; gestational age 8–22 weeks) at the University of Colorado Family Planning clinic or from normal term delivery (n=14; gestational age 37-40 weeks) were collected at University Colorado Hospital. In addition, term placenta (n=27) was obtained for PHT cell isolations from normal BMI women with no history of smoking recreational drug use, any form of maternal diabetes, pre-eclampsia, infection, or fetal anomaly. Gestational age was estimated by last menstrual period and confirmed by ultrasound. Demographic data, including age, parity, substance use, co-morbidities and recreational drug use and smoking status (Supplemental Table 1), was obtained from the patient's chart. Villous tissue across gestation was collected within 30 minutes of delivery, rinsed in ice- cold saline and transported to the laboratory where it was homogenized with a Polytron (15,000 rpm, 2 min) in ice-cold buffer D (250mM sucrose, 10mM Hepes, pH 7.4) supplemented with protease and phosphatase inhibitors (P8340, P2850, and P0044; Sigma-Aldrich, St Louis, MO), snap-frozen in liquid nitrogen and stored at −80°C (66,67). From term samples, arterial and venous umbilical cord plasma was collected (n=19 males, n=19 females) immediately after delivery and frozen at −80°C until batch analysis.
Isolation and Culture of Primary Human Trophoblasts (PHT)
Primary cytotrophoblasts were isolated from term placental villous tissue (13 males and 14 females) by trypsin digestion followed by discontinuous Percoll gradient separation, as described previously (55,68). In brief, isolated PHT cells were plated at 1.5 × 106 per well in six-well plates and cultured in a 1:1 mixture of Dulbecco’s modified Eagle’s medium (DMEM, 25 mM glucose) and Ham’s F12 medium (10 mM glucose) with 10% fetal bovine serum, 2 mM glutamine, 50 μg/mL gentamicin, 60 μg/mL penicillin and 100 μg/mL streptomycin. Cells were washed with warm PBS and the media was changed at 18 h followed by daily media changes. A sample of media was collected each day for measurement of human chorionic gonadotrophin (hCG) concentration by ELISA (IBL America, Minneapolis, MN, USA) to confirm differentiation and syncytialization (Supplemental Figure 1).
For silencing of raptor (mTORC1 inhibition) or rictor (mTORC2 inhibition), PHT cells were plated at 2.5 × 106 per well in six-well plates and cultured as described above. At 18 h culture, small interference RNAs (siRNAs) (Sigma) targeting raptor (100 nm; sense, 5′CAGUUCACCGCCAUCUACA); rictor (100 nm; sense, 5′ CGAUCAUGGGCAGGUAUUA) or a non-coding scramble control were added to the media along with DharmaFECT transfection reagent (ThermoFisher, Rockford, IL) and incubated for 24 h (57). Cells were then washed and returned to normal culture media. Media was collected between 66 and 90 hours culture for measurement of serotonin. At 90h PHT cells were collected in radioimmunoprecipitation assay buffer (RIPA buffer) supplemented with protease and phosphatase inhibitor cocktail (1:100; Sigma P8340, P2850, and P0044) and cell lysates were stored at −80°C.
Determination of Placental Sex
Sex of placental samples collected from first and second trimester pregnancies was determined by PCR detection of sex-determining region Y (SRY) transcripts from extracted genomic DNA. Briefly, DNA was extracted from frozen placental homogenate using a TissueLyser LT (Qiagen, Valencia, CA) and AllPrep DNA/RNA Mini kit (Qiagen) according to manufacturer’s instructions. Purity and concentration was determined by absorbance measurements at 260nm and 280nm wavelengths with a NanoDrop ND-1000 Spectrophotometer (NanoDrop, Wilmington, DE). Synthetic oligonucleotides (10 μM; Operon Eurofins, Louisville, KY) were used in the PCR with GoTaq Green Master Mix (Promega) and the products were separated by gel electrophoresis on 2% agarose gel with ethidium bromide and visualized. The presence of GAPDH confirmed reaction success and the presence of a band at the expected SRY product size without a band at the X-inactive specific transcript (XIST) product size established male sex (Primer sequences in supplemental Table 2).
Determination of Protein Levels
Protein concentrations were determined by bicinchoninic acid (BCA) protein assay (Thermo Fisher) for placental homogenate and PHT cell lysates. The protein levels in placental homogenate samples (1-2 μg) was determined using the automated Simple Western (Wes) capillary immunoblotting system (ProteinSimple, Santa Clara, CA), run according to manufacturer’s instructions (69). Primary antibodies (Supplemental Table 3) were validated using protein isolated from mouse adipose or skeletal muscle and optimized in human placenta (70). Immunocomplexes were detected with a goat anti-rabbit (and anti-mouse) horseradish peroxidase-conjugated secondary antibody (1:10,000; ProteinSimple). Bands for vinculin or ß-actin (Supplemental Table 3) were used to account for variation in protein loading. Relative protein abundance presented in figures is the protein abundance divided by the mean protein abundance of all female samples at term, which was arbitrarily assigned a value of 1.0.
The levels of OGT protein was determined in PHT cell lysates separated by SDS-PAGE and transferred by electrophoresis to a polyvinylidene fluoride membrane (Bio-Rad, Hercules, CA). Membranes were blocked in 5% BSA in Tris-buffered saline (10mM Tris-HCl, 150 mM NaCl, pH 8) with 0.05% Tween 20 (TBS-T) at room temperature for 1 h and then incubated with primary antibody at 4°C overnight. Immunocomplexes were detected with anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody (1:3,000; Sigma) then visualized with enhanced chemiluminescence (Millipore, Burlington, MA). Images were captured using G:Box with GeneSnap software (Syngene, Frederick, MD). Target densities were quantified and corrected for total protein loaded as measured by amido black stain. The mean density of the bands for control samples (cells transfected with the scramble siRNA) was assigned an arbitrary value of 1. All individual densitometry values were expressed relative to this mean.
Analysis of Serotonin Concentration
Serotonin concentrations were determined by ELISA in platelet-depleted umbilical vein and arterial plasma from male (n=19) and female (n=19) cord blood collected at term. Serotonin release from PHT cells was determined in culture media (66-90 h culture). ELISA (Immuno-Biological Laboratories Inc., IBL-America, Minneapolis, MN) was performed in duplicate with intra-assay coefficient of variation of 5.5 ± 2.1% and inter-assay coefficient of variation of 9.8 ± 3.8%. Data were plotted on 4-parametric curve and then concentration of serotonin (ng/mL) calculated per manufacturer’s instructions.
Statistical Analysis
All results were analyzed and plotted using PRISM (v9.0, GraphPad Software Inc, La Jolla, CA). The impact of fetal sex on placental protein levels across early gestation and term was determined using two-way ANOVA with multiple comparison corrections and correlated to gestational age with Pearson coefficient. Protein levels in PHT cells following silencing of mTOR complexes was analyzed with repeated measures ANOVA with Dunnett’s post hoc test. Serotonin concentration in umbilical circulation was analyzed by paired t-test and sex effect was determined by unpaired t-test. A significantly higher serotonin concentration in the umbilical vein as compared to the umbilical artery was interpreted as placental serotonin release into the fetal circulation. All data is presented as the mean ± SEM. Statistical significance was set at P < 0.05.
Results
Higher levels of OGT protein in female placentas across gestation.
OGT abundance in female placentas (n=11) was 78% higher (P < 0.05) than in male placentas (n=11) in first trimester samples (Figure 1). Similar differences were observed in second trimester (13-20 weeks) where placental OGT abundance was 57% higher (P < 0.05, n=6) in females than in males (n=6). At term, placental OGT abundance in females (n=7) tended to be higher than in males (n=7) but this difference did not reach statistical significance (Figure 1). The level of placental OGT did not change significantly across gestation in either females or males (Figure 1).
Figure 1: OGT levels in human placenta across gestation.
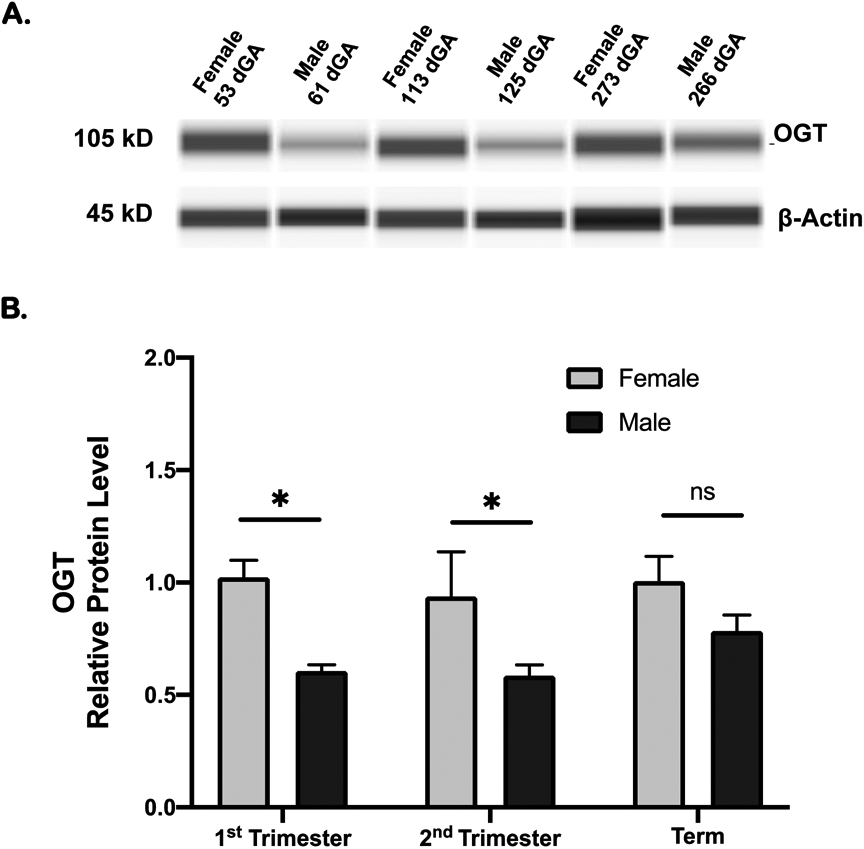
OGT protein levels in placental homogenate was determined by Simple Western and presented as mean ± SEM relative to female term group. A. Representative individual WES capillaries for OGT and loading control for samples from each gestational group; 1st trimester, 2nd trimester, and term. B. Summary data. 1st trimester: 11 females and 11 males; 2nd trimester 6 females and 6 males; term: 7 females and 7 males. Two-way ANOVA followed by multiple comparison Holm-Sidak correction; *P < 0.05; ns = not significant.
Inhibition of mTORC1 or mTORC2 reduced OGT levels in primary human trophoblast cells.
We silenced raptor to specifically inhibit mTORC1 and silenced rictor to specifically inhibit mTORC2 in cultured primary human trophoblast (PHT) cells. We have reported previously that silencing efficiency typically is ~50-70% when targeting either raptor or rictor in PHT cells (57,71). ADDRESS KNOCKDOWN EFFICIENCY OF CURRENT SAMPLES As compared to PHT cells transfected with scramble control siRNA, raptor silencing (n=8; 4 males, 4 females) reduced OGT levels by 30 % (P < 0.05, Figure 2) and rictor silencing decreased OGT levels by 27% (P < 0.05, Figure 2). Basal levels of OGT were similar in PHT cells isolated from male and female placentas and the effects of mTORC1 or mTORC2 inhibition on OGT protein levels were not different in PHT cells isolated from male and female placentas (data not shown).
Figure 2. Levels of OGT in PHT cells is reduced by mTOR inhibition.

Protein levels of OGT was determined using Western blot following inhibition of mTORC1 by raptor siRNA (Raptor KD) or mTORC2 by rictor siRNA (Rictor KD) in PHT cells. A. Representative Western blot for OGT. B. Summary data. n=8 (4 males and 4 females): Mean ±SEM Repeated measures ANOVA with Dunnett’s post hoc test, *P < 0.05 relative to PHT cells transfected with scramble siRNA (Control).
Enzymes involved in serotonin synthesis and breakdown are present throughout gestation in placenta and in cultured primary human trophoblast cells.
Tryptophan hydroxylase (TPH) represents the rate-limiting step in the synthesis of serotonin. TPH isoform 1 (TPH-1), the predominant TPH isoform in peripheral tissues, was detected in placenta as early as 6 weeks of gestation and levels decreased with increasing gestational age (Figure 3A). The negative correlation between gestational age and placental TPH-1 levels across early and mid-pregnancy was most pronounced in female placenta (R=0.64, P = 0.01) and did not reach significance in male placentas (Figure 3B). In the first trimester, placental TPH-1 protein level was 72% higher in female (n=9) than maIe placenta (n=11, P < 0.01, Figure 3D), whereas there was no significant sex difference in placental TPH-1 levels in second trimester or at term (Figure 3D).
Figure 3: Placental TPH-1 protein levels across gestation.
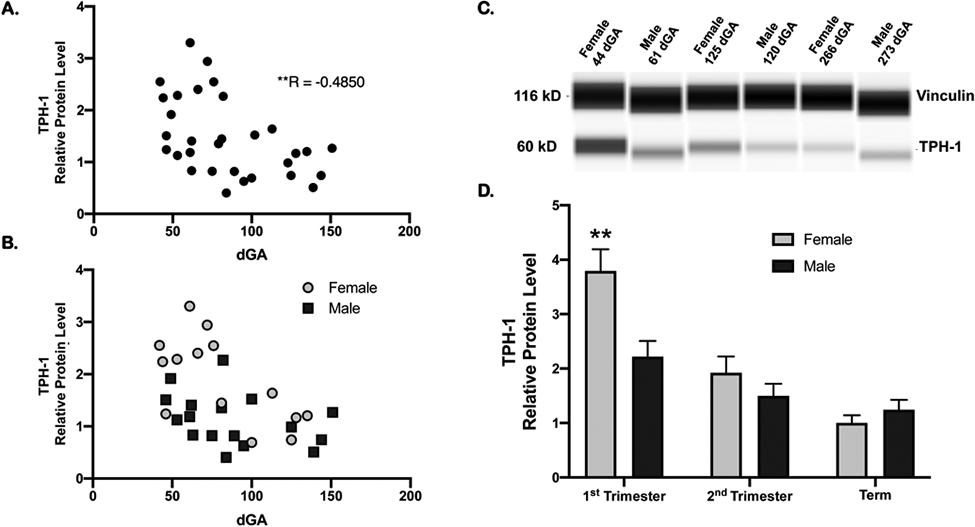
A. Placental TPH-1 levels in first and second trimester was negatively correlated to gestational age. R= −0.49, n=32, **P < 0.005. B. Placental TPH-1 protein levels in first and second trimester in male and female placentas. TPH-1 levels in female placentas (n=15, grey circles) was negatively correlated to gestational age (R= −0.6430, *P = 0.01) but not in males (n=17, black squares, R= −0.3677, p=0.15). C. Representative individual WES capillaries for TPH-1 and vinculin loading control for samples from each gestational age group; 1st trimester, 2nd trimester, and term. D. Placental TPH-1 levels in female and males across gestation relative to term female levels. 1st trimester: 9 females and 11 males; 2nd trimester 6 females and 6 males; term: 7 females and 7 males. Mean ± SEM Two-way ANOVA with Tukey’s multiple comparison test; **P < 0.01.
Monoamine oxidase A (MAOA), the key enzyme catalyzing the oxidation of serotonin, was also detected in placenta in early pregnancy and at term (Figure 4A), indicating placental capacity for serotonin metabolism. MAOA was more abundant (P < 0.01) in first half of pregnancy as compared to term (Figure 4B). There was no difference in MAOA levels between males and females at any gestational age group.
Figure 4: Placental MAOA protein levels across gestation.
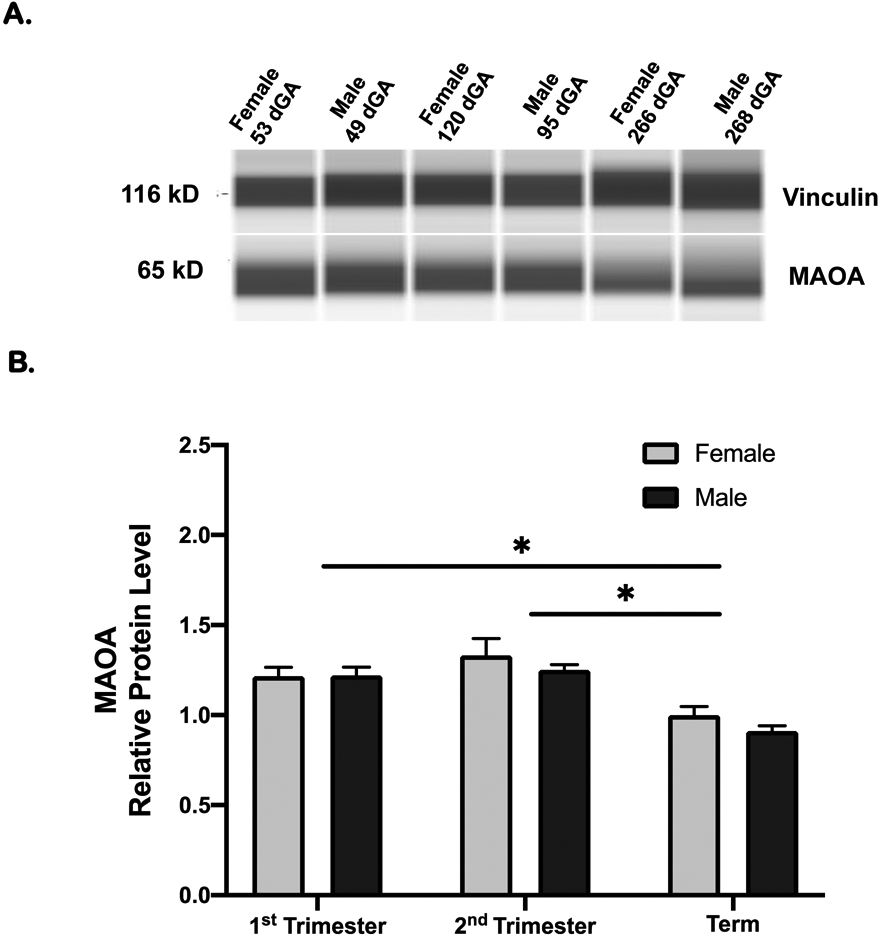
A. Representative individual WES capillaries for MAOA and vinculin loading control for samples from each gestational age group; 1st trimester, 2nd trimester, and term. B. Placental MAOA levels in females and males across gestation relative to term female levels. MAOA levels at term (7 females and 7 males) was lower than 1st trimester (8 females and 11 males) and 2nd trimester (6 females and 6 males). There was no significant difference between males and females. Mean ± SEM Two-way ANOVA followed by multiple comparison Holm-Sidak correction; *P < 0.05.
Term human placenta releases serotonin into the fetal circulation
Serotonin concentrations in platelet-depleted plasma were higher (P < 0.001) in the umbilical vein compared to umbilical artery when males and females were analyzed together (n=38; Figure 5A) indicating placental serotonin secretion into the fetal circulation in human pregnancy at term. This relationship was maintained when data was analyzed separately in males (Figure 5B) and females (Figure 5C). Placental sex did not influence the magnitude of venous-arterial concentration difference in serotonin across the feto-placental circulation (Figure 5D).
Figure 5. Serotonin concentrations in umbilical platelet-depleted plasma at term.

Serotonin concentrations from platelet depleted venous and arterial cord plasma. A. Males and females analyzed together, n= 38; (B) Males, n= 19; (C) Females n= 19. Mean ± SEM, ***P < 0.001, *P < 0.01, paired t-test. D. The venous to arterial concentration difference (V minus A) for serotonin was calculated for each fetus and plotted by sex as mean ± SEM.
Inhibition of mTORC2 increased serotonin release in primary human trophoblast cells.
PHT cells release serotonin under culture conditions as serotonin concentrations in culture media were higher in preparations with higher cell densities and undetectable when no cells were present (data not shown). We silenced raptor to specifically inhibit mTORC1 and rictor to specifically inhibit mTORC2 in PHT cells. As compared to PHT cells transfected with scramble control siRNA, rictor silencing increased serotonin release by 49% (n=7; 3 males, 4 females; Figure 6A). In contrast, raptor silencing did not significantly influence PHT cell serotonin release. Serotonin release was similar between PHT cells isolated from male and female placenta (Figure 6B).
Figure 6: Inhibition of mTORC2 increases serotonin release by PHT cells.
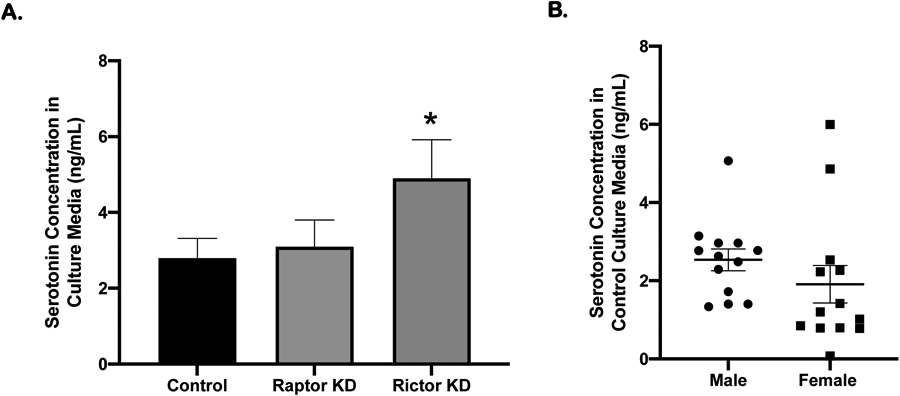
Serotonin concentrations in conditioned media, collected during 66-90 hrs culture. A. Media serotonin concentration following rictor silencing (mTORC2 inhibition) was higher than controls but serotonin concentration was not different following raptor silencing (mTORC1 inhibition). Mean ± SEM. Repeated measures ANOVA; n=7, 3 males and 4 females; *P < 0.05. B. Cultured PHT cells isolated from normal term pregnancies, males (n=13) and females (n=14) release similar amounts of serotonin (Unpaired t test).
Discussion
We show for the first time that mTOR signaling functions as a regulator of human placental OGT protein levels and serotonin release. Given previous animal experiments demonstrating that placental OGT represents a mechanistic link between maternal stress and impaired neurodevelopment and that the adverse effect of maternal infection on the fetal brain may be mediated by changes in placental serotonin release, this data implicates placental mTOR signaling as a critical hub linking changes in the mother to the development of the fetal brain. These findings will help us better understand placental mechanisms underlying the intrauterine origins of neurodevelopmental disorders and identify placental mTOR as a possible target for intervention.
In humans, stressors such as metabolic deficits or social/emotional distress result in increased risk for neuropsychiatric and mood disorders in the offspring (14), especially if the stress occurs early in pregnancy, during the critical window of fetal brain development (10,12,23,24). The enzyme OGT catalyzes the addition of O-linked N-acetylglucosamine (O-GlcNAc) to proteins, including epigenetic mediators such as histones and RNA polymerase, to coordinate cellular signaling events to nutrient availability (29,72). Placental OGT expression decreases in response to chronic variable stress throughout the first week of pregnancy in mice (36,73). Importantly, decreased placental OGT expression was mechanistically linked to poor neurodevelopmental outcomes, including hypothalamic mitochondrial dysfunction and HPA axis hypersensitivity, in elegant studies involving trophoblast specific gene targeting in mice (35). We found high placental OGT protein across gestation, suggesting a role of OGT as a placental nutrient sensor. Importantly, robust placental levels of OGT in the first and second trimester is consistent with the possibility that this enzyme links maternal stress in early pregnancy to poor neurodevelopment also in women.
Reports exploring placental synthesis and transfer of serotonin are limited but studies in human placental villous tissue and in mice (33,74) suggest that the placenta is an important source of fetal serotonin. Furthermore, in mice, serotonin from the placenta in early gestation is essential for proper forebrain development (33) and increased placental serotonin release to the mouse fetus mediates the adverse effects of maternal immune activation (i.e., infection) on fetal brain development (34,74). The clinical correlate to these observations is the well-established association between maternal viral infections in first and second trimester and poor neurodevelopmental outcomes, including schizophrenia (9,10).
The cellular machinery needed for serotonin synthesis and metabolism, including the rate-limiting enzyme responsible for serotonin synthesis, tryptophan hydroxylase, have been shown to be present and active in the human placenta (44-46). Yet, whether placental serotonin secretion in human pregnancy is required for normal fetal brain development is unknown. The current study provides further support for the concept that the human placenta synthesizes serotonin for release into the fetal circulation by demonstrating 1) placental protein expression of rate-limiting enzymes for serotonin synthesis (TPH-1) and degradation (MAOA) across early and mid-pregnancy and 2) pure preparations of primary human trophoblasts secrete serotonin into culture media in vitro and 3) that serotonin concentrations are higher in the umbilical vein than in the umbilical artery in vivo. We found TPH-1 and MAOA are more abundant in early gestation, indicating placenta in early gestation has a higher capacity for serotonin production than at term. These observations are consistent with the hypothesis from animal studies of a progressive switch from an early dependence on a placental source of serotonin to a later endogenous neuronal source (74).
The mTOR signaling complexes, mTORC1 and mTORC2, are established regulators of key placental functions such as amino acid and folate transport, mitochondrial respiration and protein synthesis (53,58-60,71). In the current study we add two new placental functions that mTOR regulates: OGT levels and serotonin synthesis. Specifically, inhibition of mTORC1 or 2 decrease OGT levels in cultured PHT cells. In other tissues, inhibition of mTOR signaling decreases O-GlcNAc activity by reducing the stability of OGT protein while hyperactivation of mTOR increases O-GlcNAcylation in cancer cells (75-77). Maternal stress is associated with increased circulating cortisol and catecholamine levels. Elevated maternal cortisol inhibits placental mTOR signaling in mice (65), catecholamines cause vasoconstriction in the uteroplacental circulation (78) leading to reduced blood flow, which results in inhibition of placental mTOR signaling (26). Thus, our findings are consistent with the possibility that mTOR inhibition mediates the link between maternal stress, down-regulation of placental OGT and poor neurodevelopment.
The expression of placental OGT and the responsiveness of this protein to maternal stress may explain the sexual dimorphism in the incidence of some neuropsychiatric disorders (15,35,36). In mice and women, placental OGT levels are higher in females than in males, which has been proposed to confer protection against maternal stress in females (28,35,39). We extend these observations by demonstrating that the male human placenta has lower OGT expression than the female placenta across gestation. These findings may have important postnatal consequences for boys, as lower placental OGT levels have been associated with postnatal behavioral complications in rodents, with greater impact on males than females (35).
We also demonstrated the inhibition of mTORC2 by silencing of rictor increased serotonin release by primary human trophoblast cells in culture. These findings are in general agreement with reports that SGK1, a mTORC2 downstream target, has been linked to serotonin production, release, and action. Specifically, in SGK1 knockout mouse platelets, the amount of serotonin and it’s ability to be released are significantly impaired compared to wild type (79). While the specific mechanisms linking mTORC2 to serotonin release in PHT cells is undefined, we demonstrate a novel role for mTOR as a key regulator of placental serotonin release and is an especially intriguing finding given the demonstration that increased placental serotonin following maternal inflammation disrupts normal fetal neurodevelopment in mice (34,74,80). Because we previously reported that one type of maternal infection, malaria with placental intervillositis, inhibits placental mTORC2 signaling (64) we speculate that inhibition mTORC2 links maternal infection to increased placental serotonin secretion.
The prevalence and onset of adult neuropsychiatric disorders related to serotonergic signaling differ markedly between males and females. For instance, ADHD and schizophrenia have a male bias while females have a higher prevalence for depression/anxiety (15,81-83). Our data demonstrates human placental TPH-1 protein was higher in females than in males in early gestation, suggesting that female placentas are more effective in supporting early brain development with serotonin. In contrast, we found no impact of sex on the level of serotonin metabolism enzyme MAOA, consistent with a previous study of term placenta (84). Thus, the synthesis of serotonin from the placenta in early gestation warrants further study as a factor in the sex difference observed in the developing neuropsychiatric disorders following adverse events during pregnancy.
mTORC1 and mTORC2 are two signaling pathways that are distinct with different upstream regulators and downstream targets, yet some cellular functions appear to be regulated by both. This is exemplified by mTOR regulation of mitochondrial biogenesis and amino acid transport, respectively, in the trophoblast, mTORC1, but not mTORC2, is a positive regulator of trophoblast mitochondrial biogenesis and oxidative phosphorylation (60). In contrast, activation of either mTORC1 or mTORC2 promotes trophoblast amino acid transport (57), albeit the molecular mechanisms are distinct (71,85). Thus, our observations in the present study that knockdown of either mTORC1 or 2 decreases OGT, but only knockdown of mTORC2 affects serotonin release are reminiscent of these previous reports. We can only speculate on the significance of these findings but they may be related to the importance of maintaining regulatory redundancy for certain critical cell functions, such as OGT levels, requiring regulation by both mTORC1 and 2. Alternatively, it may reflect that cellular functions only regulated by one arm of the mTOR signaling pathway, such as serotonin release, are also under regulated by other - currently unknown – pathways.
In summary, we propose that mTOR constitutes a mechanistic link between obstetrical complications, in particular maternal infections and stress, placental function and fetal neurodevelopment. A recent report of genetic risk for schizophrenia in a large human cohort revealed that multiple genes associated with risk for schizophrenia are abundantly expressed in placenta and were enriched for mTOR signaling, upstream regulators and downstream targets of mTOR signaling (32). This data implicates placental mTOR signaling as a critical hub linking changes in the maternal compartment to the development of the fetal brain. These findings will help us better understand the placental mechanisms underlying the intrauterine origins of neurodevelopmental disorders and identify placental mTOR as a possible target for intervention.
Supplementary Material
Clinical Perspectives.
Placental O-GlcNAc transferase (OGT) expression and serotonin synthesis have been reported to mechanistically link maternal stress/infection to impaired fetal neurodevelopment in mice. Placental mTOR signaling responds to an array of maternal signals including cortisol (elevated in maternal stress) and cytokines (increased in maternal infection). We propose that mTOR signaling is a key regulator of trophoblast serotonin synthesis and OGT expression.
OGT levels were stable across gestation in the human placenta, with lower levels in male compared to female fetuses. Silencing of mTORC1 or mTORC2 decreased trophoblast OGT protein levels. The human placenta secreted serotonin to the fetus and silencing of mTORC2, but not mTORC1, increased trophoblast secretion of serotonin.
Placental mTOR signaling may serve as a molecular link between maternal stress/infection and adverse neurodevelopment in the infant by modulating placental serotonin release and OGT expression.
Acknowledgements
We would like to thank the University of Colorado’s Perinatal Clinical and Translational Research Center (PN CTRC) and the OB research team (OBRT) for consent and collection of samples. A special thanks to Kathryn Erickson and Lana Madi for carrying out immunoblotting studies. Contents are the authors’ sole responsibility and do not necessarily represent official NIH views.
Funding:
NIH T32 HD007186, CTSA UL1 TR002535 and HD068370
Footnotes
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Barker DJP, Thornburg KL. Placental programming of chronic diseases, cancer and lifespan: a review. Placenta. 2013. Oct;34(10):841–5. [DOI] [PubMed] [Google Scholar]
- 2.Gluckman PD, Hanson MA. Living with the past: evolution, development, and patterns of disease. Science. 2004. Sep 17;305(5691):1733–6. [DOI] [PubMed] [Google Scholar]
- 3.Radford EJ, Ito M, Shi H, Corish JA, Yamazawa K, Isganaitis E, et al. In utero effects. In utero undernourishment perturbs the adult sperm methylome and intergenerational metabolism. Science. 2014. Aug 15;345(6198):1255903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barker DJ. In utero programming of chronic disease. Clin Sci Lond Engl 1979. 1998. Aug;95(2):115–28. [PubMed] [Google Scholar]
- 5.Limesand SW, Thornburg KL, Harding JE. 30th anniversary for the Developmental Origins of Endocrinology. J Endocrinol. 2019. May 1; [DOI] [PubMed] [Google Scholar]
- 6.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008. Jul 3;359(1):61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gluckman PD, Hanson MA. The developmental origins of the metabolic syndrome. Trends Endocrinol Metab TEM. 2004. Jun;15(4):183–7. [DOI] [PubMed] [Google Scholar]
- 8.Barker DJP. The origins of the developmental origins theory. J Intern Med. 2007. May;261(5):412–7. [DOI] [PubMed] [Google Scholar]
- 9.Patterson PH. Neuroscience. Maternal effects on schizophrenia risk. Science. 2007. Oct 26;318(5850):576–7. [DOI] [PubMed] [Google Scholar]
- 10.Brown AS. Exposure to prenatal infection and risk of schizophrenia. Front Psychiatry. 2011;2:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Estes ML, McAllister AK. Maternal immune activation: Implications for neuropsychiatric disorders. Science. 2016. Aug 19;353(6301):772–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beversdorf DQ, Manning SE, Hillier A, Anderson SL, Nordgren RE, Walters SE, et al. Timing of prenatal stressors and autism. J Autism Dev Disord. 2005. Aug;35(4):471–8. [DOI] [PubMed] [Google Scholar]
- 13.Careaga M, Murai T, Bauman MD. Maternal Immune Activation and Autism Spectrum Disorder: From Rodents to Nonhuman and Human Primates. Biol Psychiatry. 2017. 01;81(5):391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinstock M The potential influence of maternal stress hormones on development and mental health of the offspring. Brain Behav Immun. 2005. Jul;19(4):296–308. [DOI] [PubMed] [Google Scholar]
- 15.Bale TL. The placenta and neurodevelopment: sex differences in prenatal vulnerability. Dialogues Clin Neurosci. 2016. Dec;18(4):459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Donnell KJ, Meaney MJ. Fetal Origins of Mental Health: The Developmental Origins of Health and Disease Hypothesis. Am J Psychiatry. 2017. 01;174(4):319–28. [DOI] [PubMed] [Google Scholar]
- 17.Knuesel I, Chicha L, Britschgi M, Schobel SA, Bodmer M, Hellings JA, et al. Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol. 2014. Nov;10(11):643–60. [DOI] [PubMed] [Google Scholar]
- 18.Varcin KJ, Alvares GA, Uljarević M, Whitehouse AJO. Prenatal maternal stress events and phenotypic outcomes in Autism Spectrum Disorder. Autism Res Off J Int Soc Autism Res. 2017. Nov;10(11):1866–77. [DOI] [PubMed] [Google Scholar]
- 19.Walsh K, McCormack CA, Webster R, Pinto A, Lee S, Feng T, et al. Maternal prenatal stress phenotypes associate with fetal neurodevelopment and birth outcomes. Proc Natl Acad Sci U S A. 2019. 26;116(48):23996–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palma-Gudiel H, Córdova-Palomera A, Eixarch E, Deuschle M, Fañanás L. Maternal psychosocial stress during pregnancy alters the epigenetic signature of the glucocorticoid receptor gene promoter in their offspring: a meta-analysis. Epigenetics. 2015;10(10):893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Braithwaite EC, Kundakovic M, Ramchandani PG, Murphy SE, Champagne FA. Maternal prenatal depressive symptoms predict infant NR3C1 1F and BDNF IV DNA methylation. Epigenetics. 2015;10(5):408–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mattern F, Post A, Solger F, O’Leary A, Slattery DA, Reif A, et al. Prenatal and postnatal experiences associated with epigenetic changes in the adult mouse brain. Behav Brain Res. 2019. 01;359:143–8. [DOI] [PubMed] [Google Scholar]
- 23.Chan JC, Nugent BM, Bale TL. Parental Advisory: Maternal and Paternal Stress Can Impact Offspring Neurodevelopment. Biol Psychiatry. 2018. 15;83(10):886–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buss C, Davis EP, Shahbaba B, Pruessner JC, Head K, Sandman CA. Maternal cortisol over the course of pregnancy and subsequent child amygdala and hippocampus volumes and affective problems. Proc Natl Acad Sci U S A. 2012. May 15;109(20):E1312–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jansson T, Powell TL. Role of placental nutrient sensing in developmental programming. Clin Obstet Gynecol. 2013. Sep;56(3):591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dimasuay KG, Boeuf P, Powell TL, Jansson T. Placental Responses to Changes in the Maternal Environment Determine Fetal Growth. Front Physiol. 2016;7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carter AM. Placental Oxygen Consumption. Part I: In Vivo Studies—A Review. Placenta. 2000. Mar 1;21:S31–7. [DOI] [PubMed] [Google Scholar]
- 28.Bronson SL, Bale TL. The Placenta as a Mediator of Stress Effects on Neurodevelopmental Reprogramming. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. 2016. Jan;41(1):207–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nugent BM, Bale TL. The omniscient placenta: Metabolic and epigenetic regulation of fetal programming. Front Neuroendocrinol. 2015. Oct;39:28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thornburg KL, O’Tierney PF, Louey S. Review: The placenta is a programming agent for cardiovascular disease. Placenta. 2010. Mar;31 Suppl:S54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry. 2002. Jul;159(7):1080–92. [DOI] [PubMed] [Google Scholar]
- 32.Ursini G, Punzi G, Chen Q, Marenco S, Robinson JF, Porcelli A, et al. Convergence of placenta biology and genetic risk for schizophrenia. Nat Med. 2018;24(6):792–801. [DOI] [PubMed] [Google Scholar]
- 33.Bonnin A, Goeden N, Chen K, Wilson ML, King J, Shih JC, et al. A transient placental source of serotonin for the fetal forebrain. Nature. 2011. Apr 21;472(7343):347–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goeden N, Velasquez J, Arnold KA, Chan Y, Lund BT, Anderson GM, et al. Maternal Inflammation Disrupts Fetal Neurodevelopment via Increased Placental Output of Serotonin to the Fetal Brain. J Neurosci Off J Soc Neurosci. 2016. 01;36(22):6041–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Howerton CL, Bale TL. Targeted placental deletion of OGT recapitulates the prenatal stress phenotype including hypothalamic mitochondrial dysfunction. Proc Natl Acad Sci U S A. 2014. Jul 1;111(26):9639–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Howerton CL, Morgan CP, Fischer DB, Bale TL. O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc Natl Acad Sci U S A. 2013. Mar 26;110(13):5169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mikaelsson MA, Constância M, Dent CL, Wilkinson LS, Humby T. Placental programming of anxiety in adulthood revealed by Igf2-null models. Nat Commun. 2013;4:2311. [DOI] [PubMed] [Google Scholar]
- 38.Côté F, Fligny C, Bayard E, Launay J-M, Gershon MD, Mallet J, et al. Maternal serotonin is crucial for murine embryonic development. Proc Natl Acad Sci U S A. 2007. Jan 2;104(1):329–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pantaleon M, Steane SE, McMahon K, Cuffe JSM, Moritz KM. Placental O-GlcNAc-transferase expression and interactions with the glucocorticoid receptor are sex specific and regulated by maternal corticosterone exposure in mice. Sci Rep. 2017. 17;7(1):2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hardivillé S, Hart GW. Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metab. 2014. Aug 5;20(2):208–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walther DJ, Peter J-U, Bashammakh S, Hörtnagl H, Voits M, Fink H, et al. Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science. 2003. Jan 3;299(5603):76. [DOI] [PubMed] [Google Scholar]
- 42.Chen CH, Klein DC, Robinson JC. Monoamine oxidase in rat placenta, human placenta, and cultured choriocarcinoma. J Reprod Fertil. 1976. Mar;46(2):477–9. [DOI] [PubMed] [Google Scholar]
- 43.Karahoda R, Horackova H, Kastner P, Matthios A, Cerveny L, Kucera R, et al. Serotonin homeostasis in the materno-fetal interface at term: role of transporters (SERT/SLC6A4 and OCT3/SLC22A3) and monoamine oxidase A (MAO-A) in uptake and degradation of serotonin by human and rat term placenta. Acta Physiol Oxf Engl. 2020. Apr 20; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu K, Liu G, Fu C. The Tryptophan Pathway Targeting Antioxidant Capacity in the Placenta [Internet]. Oxidative Medicine and Cellular Longevity. 2018. [cited 2019 Jul 29]. Available from: https://www.hindawi.com/journals/omcl/2018/1054797/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laurent L, Deroy K, St-Pierre J, Côté F, Sanderson JT, Vaillancourt C. Human placenta expresses both peripheral and neuronal isoform of tryptophan hydroxylase. Biochimie. 2017. Sep;140:159–65. [DOI] [PubMed] [Google Scholar]
- 46.Ranzil S, Ellery S, Walker DW, Vaillancourt C, Alfaidy N, Bonnin A, et al. Disrupted placental serotonin synthetic pathway and increased placental serotonin: Potential implications in the pathogenesis of human fetal growth restriction. Placenta. 2019. 01;84:74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robson JM, Senior JB. The 5-hydroxytryptamine content of the placenta and foetus during pregnancy in mice. Br J Pharmacol Chemother. 1964. Apr;22:380–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017. 06;169(2):361–71. [DOI] [PubMed] [Google Scholar]
- 49.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004. Aug 15;18(16):1926–45. [DOI] [PubMed] [Google Scholar]
- 50.Jacinto E, Hall MN. Tor signalling in bugs, brain and brawn. Nat Rev Mol Cell Biol. 2003. Feb;4(2):117–26. [DOI] [PubMed] [Google Scholar]
- 51.Kim J, Guan K-L. mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol. 2019;21(1):63–71. [DOI] [PubMed] [Google Scholar]
- 52.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012. Apr 13;149(2):274–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005. Feb 18;307(5712):1098–101. [DOI] [PubMed] [Google Scholar]
- 54.Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008. Jul 23;27(14):1932–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roos S, Kanai Y, Prasad PD, Powell TL, Jansson T. Regulation of placental amino acid transporter activity by mammalian target of rapamycin. Am J Physiol Cell Physiol. 2009. Jan;296(1):C142–150. [DOI] [PubMed] [Google Scholar]
- 56.Roos S, Jansson N, Palmberg I, Säljö K, Powell TL, Jansson T. Mammalian target of rapamycin in the human placenta regulates leucine transport and is down-regulated in restricted fetal growth. J Physiol. 2007. Jul 1;582(Pt 1):449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rosario FJ, Kanai Y, Powell TL, Jansson T. Mammalian target of rapamycin signalling modulates amino acid uptake by regulating transporter cell surface abundance in primary human trophoblast cells. J Physiol. 2013. Feb 1;591(3):609–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roos S, Lagerlöf O, Wennergren M, Powell TL, Jansson T. Regulation of amino acid transporters by glucose and growth factors in cultured primary human trophoblast cells is mediated by mTOR signaling. Am J Physiol Cell Physiol. 2009. Sep;297(3):C723–731. [DOI] [PubMed] [Google Scholar]
- 59.Rosario FJ, Powell TL, Jansson T. Mechanistic target of rapamycin (mTOR) regulates trophoblast folate uptake by modulating the cell surface expression of FR-α and the RFC. Sci Rep. 2016. 26;6:31705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rosario FJ, Gupta MB, Myatt L, Powell TL, Glenn JP, Cox L, et al. Mechanistic Target of Rapamycin Complex 1 Promotes the Expression of Genes Encoding Electron Transport Chain Proteins and Stimulates Oxidative Phosphorylation in Primary Human Trophoblast Cells by Regulating Mitochondrial Biogenesis. Sci Rep. 2019. Jan 22;9(1):246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rosario FJ, Nathanielsz PW, Powell TL, Jansson T. Maternal folate deficiency causes inhibition of mTOR signaling, down-regulation of placental amino acid transporters and fetal growth restriction in mice. Sci Rep. 2017. 21;7(1):3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rosario FJ, Powell TL, Jansson T. mTOR folate sensing links folate availability to trophoblast cell function. J Physiol. 2017. 01;595(13):4189–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jansson N, Rosario FJ, Gaccioli F, Lager S, Jones HN, Roos S, et al. Activation of Placental mTOR Signaling and Amino Acid Transporters in Obese Women Giving Birth to Large Babies. J Clin Endocrinol Metab. 2013. Jan;98(1):105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dimasuay KG, Aitken EH, Rosario F, Njie M, Glazier J, Rogerson SJ, et al. Inhibition of placental mTOR signaling provides a link between placental malaria and reduced birthweight. BMC Med. 2017. Jan 3;15(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vaughan OR, Fisher HM, Dionelis KN, Jeffreys EC, Higgins JS, Musial B, et al. Corticosterone alters materno-fetal glucose partitioning and insulin signalling in pregnant mice. J Physiol. 2015. 01;593(5):1307–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rosario FJ, Sadovsky Y, Jansson T. Gene targeting in primary human trophoblasts. Placenta. 2012. Oct;33(10):754–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.James-Allan LB, Arbet J, Teal SB, Powell TL, Jansson T. Insulin stimulates GLUT4 trafficking to the syncytiotrophoblast basal plasma membrane in the human placenta. J Clin Endocrinol Metab. 2019. May 21; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kliman HJ, Nestler JE, Sermasi E, Sanger JM, Strauss JF. Purification, characterization, and in vitro differentiation of cytotrophoblasts from human term placentae. Endocrinology. 1986. Apr;118(4):1567–82. [DOI] [PubMed] [Google Scholar]
- 69.Harris VM. Protein detection by Simple Western™ analysis. Methods Mol Biol Clifton NJ. 2015;1312:465–8. [DOI] [PubMed] [Google Scholar]
- 70.Castillo-Castrejon M, Jansson T, Powell TL. No evidence of attenuation of placental insulin-stimulated Akt phosphorylation and amino acid transport in maternal obesity and gestational diabetes mellitus. Am J Physiol Endocrinol Metab. 2019. Dec 1;317(6):E1037–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rosario FJ, Dimasuay KG, Kanai Y, Powell TL, Jansson T. Regulation of amino acid transporter trafficking by mTORC1 in primary human trophoblast cells is mediated by the ubiquitin ligase Nedd4-2. Clin Sci Lond Engl 1979. 2016. Apr 1;130(7):499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bond MR, Hanover JA. O-GlcNAc cycling: a link between metabolism and chronic disease. Annu Rev Nutr. 2013;33:205–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mueller BR, Bale TL. Sex-specific programming of offspring emotionality after stress early in pregnancy. J Neurosci Off J Soc Neurosci. 2008. Sep 3;28(36):9055–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bonnin A, Levitt P. Fetal, maternal, and placental sources of serotonin and new implications for developmental programming of the brain. Neuroscience. 2011. Dec 1;197:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Park S, Pak J, Jang I, Cho JW. Inhibition of mTOR affects protein stability of OGT. Biochem Biophys Res Commun. 2014. Oct 17;453(2):208–12. [DOI] [PubMed] [Google Scholar]
- 76.Sodi VL, Khaku S, Krutilina R, Schwab LP, Vocadlo DJ, Seagroves TN, et al. mTOR/MYC Axis Regulates O-GlcNAc Transferase Expression and O-GlcNAcylation in Breast Cancer. Mol Cancer Res MCR. 2015. May;13(5):923–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Butkinaree C, Park K, Hart GW. O-linked beta-N-acetylglucosamine (O-GlcNAc): Extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim Biophys Acta. 2010. Feb;1800(2):96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jansson T Responsiveness to norepinephrine of the vessels supplying the placenta of growth-retarded fetuses. Am J Obstet Gynecol. 1988. May;158(5):1233–7. [DOI] [PubMed] [Google Scholar]
- 79.Walker B, Schmid E, Russo A, Schmidt E-M, Burk O, Münzer P, et al. Impact of the serum- and glucocorticoid-inducible kinase 1 on platelet dense granule biogenesis and secretion. J Thromb Haemost JTH. 2015. Jul;13(7):1325–34. [DOI] [PubMed] [Google Scholar]
- 80.Muller CL, Anacker AM, Rogers TD, Goeden N, Keller EH, Forsberg CG, et al. Impact of Maternal Serotonin Transporter Genotype on Placental Serotonin, Fetal Forebrain Serotonin, and Neurodevelopment. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. 2017;42(2):427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Castle DJ, Murray RM. The neurodevelopmental basis of sex differences in schizophrenia. Psychol Med. 1991. Aug;21(3):565–75. [DOI] [PubMed] [Google Scholar]
- 82.Sandman CA, Glynn LM, Davis EP. Is there a viability-vulnerability tradeoff? Sex differences in fetal programming. J Psychosom Res. 2013. Oct;75(4):327–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McGrath J, Saha S, Chant D, Welham J. Schizophrenia: a concise overview of incidence, prevalence, and mortality. Epidemiol Rev. 2008;30:67–76. [DOI] [PubMed] [Google Scholar]
- 84.Zhang H, Smith GN, Liu X, Holden JJA. Association of MAOA, 5-HTT, and NET promoter polymorphisms with gene expression and protein activity in human placentas. Physiol Genomics. 2010. Jun;42(1):85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jansson T, Castillo-Castrejon M, Gupta MB, Powell TL, Rosario FJ. Down-regulation of placental Cdc42 and Rac1 links mTORC2 inhibition to decreased trophoblast amino acid transport in human intrauterine growth restriction. Clin Sci Lond Engl 1979. 2020. 17;134(1):53–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.