

Abstract
The prognosis of most patients with AML is poor due to frequent disease relapse. The cause of relapse is thought to be from the persistence of leukemia initiating cells (LIC’s) following treatment. Here we assessed RNA based changes in LICs from matched patient diagnosis and relapse samples using single-cell RNA sequencing. Previous studies on AML progression have focused on genetic changes at the DNA mutation level mostly in bulk AML cells and demonstrated the existence of DNA clonal evolution. Here we identified in LICs that the phenomenon of RNA clonal evolution occurs during AML progression. Despite the presence of vast transcriptional heterogeneity at the single cell level, pathway analysis identified common signaling networks involving metabolism, apoptosis and chemokine signaling that evolved during AML progression and become a signature of relapse samples. A subset of this gene signature was validated at the protein level in LICs by flow cytometry from an independent AML cohort and functional studies were performed to demonstrate co-targeting BCL2 and CXCR4 signaling may help overcome therapeutic challenges with AML heterogeneity. It is hoped this work will facilitate a greater understanding of AML relapse leading to improved prognostic biomarkers and therapeutic strategies to target LIC’s.
INTRODUCTION
Acute myeloid leukemia (AML) is the most common type of acute leukemia affecting adults and responsible for the largest number of leukemia-related deaths, yet there has been no change in the standard therapy for most patients in over 40 years. Traditional chemotherapeutics have poor efficacy in patients over the age of 65, with a median survival of less than one year and only 20% surviving at least two years [1]. Though patients often enter remission after initial treatment, the majority succumb to their disease after relapse. It is hypothesized that AML relapse results from the persistence of AML initiating cells (LICs) that escape chemotherapy [2, 3]. LICs play a central role in disease progression and relapse due to their capacity for self-renewal and their ability to evade chemotherapy induced cell death due to their relative quiescence [4–13].
Despite the critical importance of LICs in AML, little is currently known about the heterogeneity of LICs and how they evolve during AML disease progression. Recently, the heterogeneity of molecular alterations at the single cell level in cancer has been demonstrated to be integral to the evasion from therapeutic interventions and the establishment of therapy-resistant populations. In one single cell study of metastatic breast cancer, the establishment of metastatic disease could be traced to a single founder drug-resistant cell [14]. Previous single cell sequencing studies of AML samples during disease progression have primarily focused on DNA mutations and none have focused on defining the biological processes that confer chemo-resistance to this crucial LIC population. These studies are important, as they have revealed that AML undergoes clonal evolution at the DNA level. DNA mutations are relatively rare in AML (average of ~13 per patient) as compared to other malignancies, therefore it is likely that transcriptomic and epigenetic based changes play crucial roles in the development and progression of the disease [15–17]. However, little is known about RNA expression changes during disease progression or changes specific to LICs [14, 18–24].
To better understand AML relapse to develop improved therapeutics and prognostic biomarkers, we performed single cell RNA sequencing of LICs from matched patient treatment naïve diagnosis and post-treatment relapse bone marrow samples. Through this single cell approach, we sought to understand the heterogeneity of LICs and to examine for the presence of RNA based evolution during disease progression.
RESULTS
Profiles of single leukemic initiating cells from patient bone marrow samples
We performed single cell RNA-seq on 813 LICs isolated from five AML patients’ matched diagnosis and relapse bone marrow samples (Supplementary Fig. 1A). Patients’ diagnosis samples were treatment naïve (Table 1). The patients’ average time to relapse was 242 days, with a range of 143–302 days (Table 1). All patients had favorable (inv (16), n = 2) or intermediate (normal, n = 2; t (1;8) n = 1) cytogenetic risk classification [25].
Table 1.
Clinical characteristics of patient cohort.
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | |
|---|---|---|---|---|---|
| Age at diagnosis | 7.5 mths | 51 yrs | 18.6 mths | 61 yrs | 73 yrs |
| Sex | Female | Female | Male | Female | Male |
| Event-free survival (days) | 277 | 259 | 228 | 143 | 302 |
| Overall survival (days) | 1464 | 1299 | 370 | 1460 | 431 |
| Drug treatment | NA | Cyta/Ida | NA | Cyta/Ida | Aza/Midostantin |
| Complete Remission end course 1 | CR | CR | CR | No | No |
| Complete Remission end course 2 | CR | CR | CR | CR | No |
| Stem cell transplant | No | No | No | No | No |
| Mutations identified clinically | FLT3, KIT | None | None | None | CEBPA |
| FAB subtype | M4 | M4 | M5 | M4 | M2 |
| Cytogenetics | inv (16) | inv (16) | Normal | t (1;8) | Normal |
| White blood cell count at diagnosis (per mm3) | 45.8 | 196 | 75.1 | 16 | 37 |
| Leukemic blasts at diagnosis (percent) | 62 | 88 | 82 | 80 | 15 |
| LIC at diagnosis (percent) | 0.08 | 0.07 | 0.8 | 0.006 | 0.1 |
| LIC at relapse (percent) | 0.4 | 0.16 | 0.8 | 0.1 | 0.2 |
Viable LICs were isolated by flow cytometry for single cell RNA-seq using a stringent panel of cell surface markers that define LICs (CD34+, CD38−, CD90−, CD45RA+, Lin−, and 7-AAD−) (Supplementary Fig. 1B) [26–28]. This panel was designed using previously established phenotypic markers and enabled the discrimination of LICs from normal hematopoietic stem cells, particularly within the CD34 + CD38− compartment. cDNA was prepared from the individual cells, followed by library preparation using a modified Smartseq2 protocol [29]. Data from 721 cells passed quality control and had a median of 717,531 reads per cell (range 155,405–2829,737) and a median library complexity of 2307 genes per cell (range 38–9360) (Supplementary Table 1 and Supplementary Fig. 2).
To establish we isolated LICs and not normal hematopoietic stem cells (HSC), we assessed the cells for the expression of known LIC and HSC signature genes [6, 30]. The sorted LICs had high expression of LIC and low expression of HSC signature genes (Supplementary Fig. 3A) (ex. CD47, CD99). Though LICs represent a highly specialized and rare cell type, single cell gene expression profiling revealed extensive cellular heterogeneity even within individual samples. We observed a clear clustering separation of the single LICs from the pooled AML LICs (Supplementary Fig. 3B). We assessed the gene expression correlation between single cells from each patient sample and found a wide range of cell-to-cell similarity (Pearson’s r; median: 0.36, range: 0.08–0.79) (Supplementary Fig. 3C). The relative stability of gene expression on the single-cell level was quantified using a Gamma Gaussian mixture-modeling computational framework [31]. This method can quantify heterogeneity by generating a stably expressed gene index (segIndex) as a value from 0 to 1. Housekeeping genes were stably expressed across patients (segIndex > 0.80; B2M, H3F3A, GAPDH, EEF1A, TPT1, FTL, EEF1G, PTMA, TMSB4X, CFL1, TMSB10, NACA, UBA52, EIF1; Supplementary File 1). Genes exhibiting higher levels of heterogeneity across single cells (segIndex < 0.25) were particularly enriched for genes that drive cell cycle progression (FDR q < 0.0001; ANAPC1, CDC14A, DCTN1, E2F4, LIG3, and SMC1A) and the innate immune system response (FDR q < 0.0001; CD84, COMM9, IRAK4, NFATC1, NLRC5, and PRKCQ).
Single cell sequencing reveals RNA-based evolution during AML disease progression
Following dimensionality reduction, a parallel consensus clustering approach was implemented in order to identify RNA cluster groups that are comprised of cells with similar transcriptional states. Importantly, every patient in the study demonstrated the presence of RNA cluster groups and RNA-based evolution during disease progression, though patterns of evolution varied (Figs. 1–2). Disease progression was largely characterized by the post-treatment loss of the cluster group that was dominant at diagnosis, along with the expansion or emergence of a relapse dominant cluster (Fig. 1 and Supplementary File 2). For example, in Patient 1 the diagnosis dominant RNA expression cluster encompassed 65% of LICs at diagnosis, but was not observed at relapse. Meanwhile, the relapse dominant cluster was observed in only 2% of diagnosis LICs, but expanded post-treatment to encompass 72% of relapse LICs (Fig. 1A).
Fig. 1. RNA clonal evolution.

A–E Patient 1–5, respectively. Left panel, visualizing disease progression with fish plots illustrates the distribution of single AML initiating cells (LICs) in RNA cluster groups during disease progression, where each cluster group 1–4 is illustrated, respectively, by the colors red, blue, yellow, and green. Relative cluster group prevalence within the LIC population is depicted on the Y axis in the plots. Center panel, multi-dimensional scaling plot shows the distribution of single diagnosis LICs, plotted as circle, and relapse LICs, plotted as triangles. LICs are plotted with distance between cells based on LogFC between top 500 most variant genes. The cells are colored according to k-means cluster group 1–4, respectively, by the colors red, blue, yellow, and green.
Fig. 2. RNA cluster group biomarkers.

A–E for patients 1–5, respectively, genes that significantly and accurately predicted a cell’s membership in a cluster group were identified for each patient cluster (Wilcoxon signed rank test p < 0.05, area under receiver operating characteristic curve (AUROC) > 0.75). The expression of the top ten predictive genes for each RNA cluster group are shown in the heatmap. Individual AML initiating cells are represented by columns and their log (count) expression is shown for cluster marker genes which are indicated in rows. A cell’s RNA cluster group membership and state (diagnosis or relapse) is indicated by color-coded labeling in horizontal bars above the heatmap.
A median of 269 genes per patient were significantly (q < 0.05) differentially expressed among RNA clusters (range 60–1047) out of a median of 8675 genes detected per patient (Supplementary File 2). Genes that significantly and accurately predicted a cell’s membership in a cluster group were identified for each patient cluster (Wilcoxon signed rank test p < 0.05, area under receiver operating characteristic curve (AUROC) > 0.75). A median of 125 genes (range 31–207) defined patients’ LIC membership in a particular RNA cluster (Supplementary File 2). Twenty two marker genes defining patients’ LIC membership in a particular cluster group overlapped in at least three out of five patients (Supplementary File 2). Cluster groups dominant at relapse exhibited low expression of KLF6, ENO1, TPI1, and TALDO1 and high expression of CD44, HLAs, and PTMA.
To reveal biological insights into the observed RNA clonal evolution, pathway analysis was performed to identify pathways enriched in the dominant RNA cluster group at diagnosis as compared to the RNA cluster groups present at relapse. In the majority of patients, we found that the dominant RNA expression cluster at relapse was significantly enriched for several potentially therapeutically targetable pathways including IL6/JAK/SAT3 and TNFα (Supplementary File 3).
AML initiating cells exhibit common changes in gene expression during disease progression
We next performed differential expression analysis between each patient’s diagnosis and relapse LICs to assess commonalities in disease progression across patients. We identified a total of 105 genes significantly (q < 0.05) downregulated in relapse LICs and 123 genes significantly (q < 0.05) upregulated in relapse LICs in at least four out of five patients (Supplementary File 4).
All patients exhibited broad and significant downregulation of histone expression during disease progression, particularly in Histone H1 family genes (Supplementary Fig. 4A). Histone H1 depleted cells are hyper-resistant to DNA damage and cell death, therefore, this downregulation may be an evolutionary mechanism to evade treatment [32–35]. The silencing of Histone H1 has been shown to promote cellular self-renewal by inducing downstream effectors of oncogenic pathways [36].
While Histone H1 downregulation was observed in LIC’s, similar downregulation was not observable in total AML cells as assessed in two independent RNA sequencing studies of matched patient diagnosis and relapse total AML samples that includes both LIC and non-LIC populations (TARGET (n = 31 patients) and Mason (n = 16 patients)) [17, 21]. In contrast to the downregulation of Histone H1, GADD45A and other DNA damage response genes, whose transcript levels are known to increase following treatment with many therapeutics including DNA-damaging agents, were upregulated in relapse LIC’s (Supplementary Fig. 5) [37].
A number of lncRNAs were also significantly upregulated during disease progression (AGPAT4-ITI, LINC01372, and LOC1005076) while the lncRNA MALAT1 was significantly downregulated in the relapse LICs in four out of five patients (Supplementary Fig. 4B). Finally, we also observed the upregulation of two phosphodiesterases (PDE4B and PDE1B) in relapse LICs (Supplementary Fig. 4C). PDE4B is a phosphodiesterase that has been shown to be an important marker in the progression from myelodysplastic syndrome to AML and a drug target for B cell tumors and colon cancer, but its role in AML relapse has not been previously reported [38–40].
Next, we compared all of the single cell LIC differential expression results to expression changes in the TARGET and Mason datasets of matched patient diagnosis and relapse bulk total AML samples. We identified only 40 out of 661 genes that were significantly differentially expressed in the Mason or TARGET total AML datasets and in the single cell LIC dataset. There was no significant change at the bulk AML cell level in the expression of the Histone H1 family, MALAT1, or PDE4B (Supplementary Fig. 4). We performed a transcriptome-wide correlation analysis of the fold-change between diagnosis and relapse states between our single cell LIC analysis and the bulk total AML samples in the Mason and TARGET databases (Supplementary File 5). There was an overall negative correlation of changes in gene expression during disease progression in the single LIC samples as compared to the Mason bulk total samples (Pearson’s r range −0.18 to 0.002) and TARGET bulk total samples (Pearson’s r range −0.13 to 0.02). This indicates that traditional analysis of bulk total AML cells can be a poor representative for LIC biology and demonstrates the importance of focused expression studies of this important cell type.
AML initiating cells downregulate glycolysis and upregulate fatty acid oxidation and amino acid metabolism during disease progression
An extension of the gene expression analysis to the pathway level revealed that disease progression in our patient set was characterized by significant changes in cell metabolism, apoptotic signaling and chemokine signaling across patients (Fig. 3A). Metabolic reprogramming, a known hallmark of cancer cells, presumably enables the LICs to maintain a more quiescent state and avoid chemotherapy induced cell death [41]. We confirmed the majority of LICs were quiescent (G0) utilizing a previously reported prediction method to classify cells into cell cycle phases based on the gene expression data [42].
Fig. 3. Metabolic dysregulation.
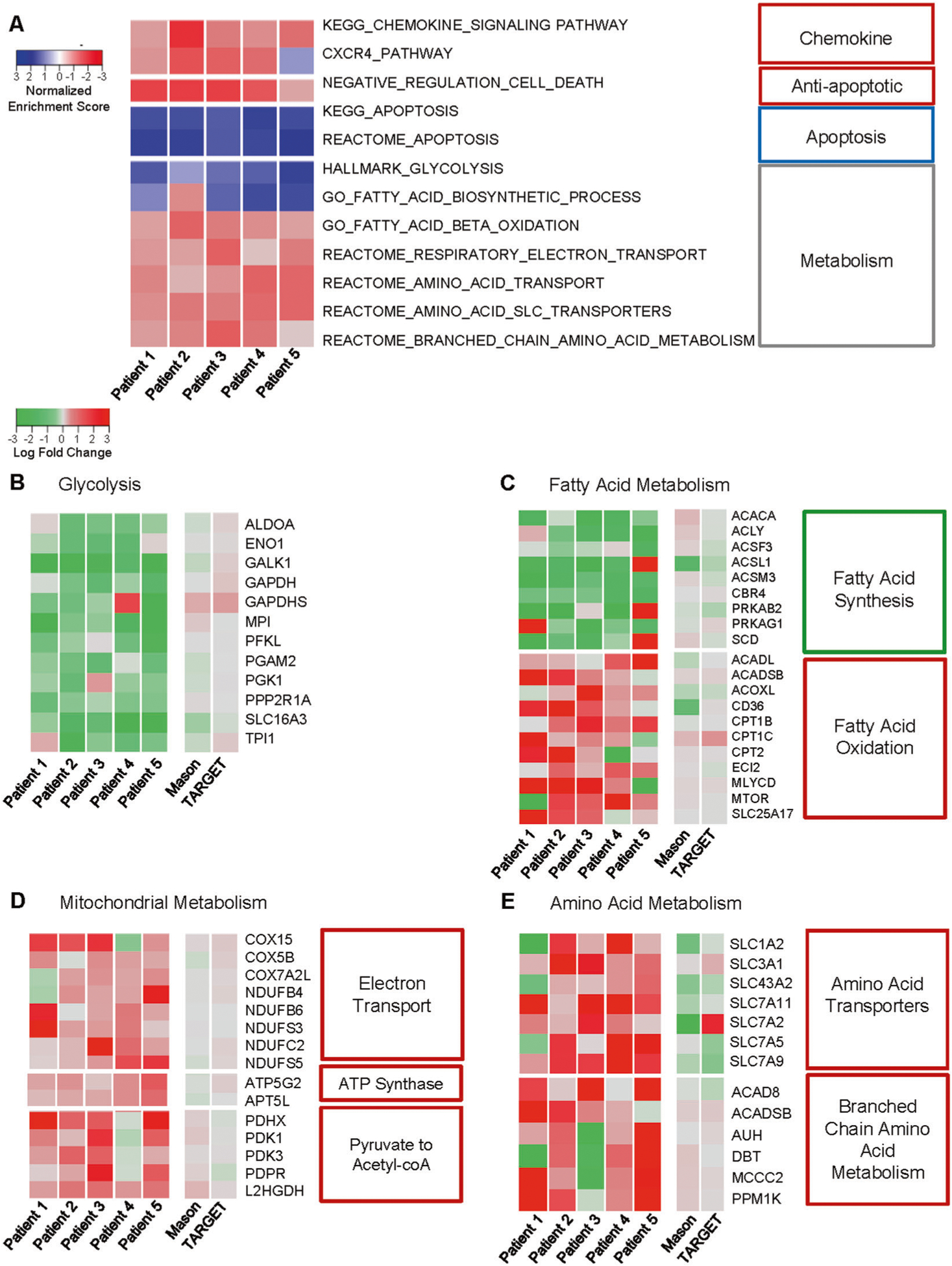
A Top pathways significantly and commonly (q < 0.05) dysregulated (as determined by GSEA) across Patients 1–5 are shown. The normalized enrichment scores (NES) are indicated, where negative NESs indicate pathway upregulation in the relapse state (red color) and positive NESs indicate pathway upregulation in the diagnosis state (blue color). The LogFC of leading edge genes commonly downregulated across patients in (B) glycolysis, (C) fatty acid metabolism, (D) mitochondrial metabolism, and (E) amino acid metabolism are shown for Patients 1–5 and patients from the TARGET and Mason databases. (−) LogFC indicates downregulation in relapse (green color) and (+) LogFC indicates upregulation in relapse (red color).
Global downregulation during disease progression in glycolysis (ex. ALDOA, ENO1, and GAPDH) and fatty acid synthesis was observed (Figs. 3B, C). In contrast, there was an upregulation in mitochondrial metabolism as well as in fatty acid oxidation and amino acid metabolism. Both fatty acid oxidation and amino acid metabolism have been reported to supply intermediates to the Krebs cycle in AML cells [43]. In the case of mitochondrial metabolism there was a particular increase, log-fold change >1.5, in the expression of electron transport chain genes (ex. COX and NDUF family genes) as well as the pyruvate dehydrogenase complex that catalyzes the conversion of pyruvate to acetyl-CoA for use in the Krebs cycle (Fig. 3D). Fatty acid oxidation and amino acid metabolism have both been reported to supply important intermediates to the Krebs cycle. Not only were genes involved in fatty acid oxidation itself upregulated across patients (ex. CPT1C, CPT2), but transporters of fatty acids such as CD36 were increased (Fig. 3C). In the case of amino acid metabolism, we identified an upregulation of a large number of SLC transporter genes, which transport exogenous amino acids into the cell (Fig. 3E). In addition, metabolism of branched-chain amino acids was observed to be upregulated in LICs during disease progression (Fig. 3E). While these metabolic pathways were observed to evolve as described during disease progression, there was significant heterogeneity observed in the expression of these metabolic genes (median segIndex 0.652; range 0.291–0.865) (Supplementary File 1). In summary, we observed decreased expression of glycolysis and fatty acid synthesis genes coupled with an upregulation in mitochondrial metabolism, fatty acid oxidation, and amino acid transport and metabolism.
Single cell sequencing reveals evolution of a common apoptotic signaling network in AML initiating cells during disease progression
Detailed examination of the apoptotic pathway revealed a network of significantly correlated co-expressed genes that evolved during disease progression at the single cell level. We found that anti-apoptotic interleukins (IL2/6) were upregulated in the relapse LICs (Fig. 4A). IL-2 and IL-6 have been shown in multiple cancer types to promote the upregulation of anti-apoptotic genes in response to DNA damage [44–47]. Transcription factor (TF) analysis revealed that anti-apoptotic transcription factors EP300 and JUND were upregulated during disease progression in the LICs, while the expression of pro-apoptotic transcription factors (BCLAF1, HMGB2, and TP53) were down-regulated (Fig. 4A). Using the ENCODE database we identified the target genes of these TFs and found that a subset of the downstream target anti-apoptotic genes, such as BCL2A1, c-FLIP, MCL1, and SOCS3, were upregulated in relapse LICs across patients (Fig. 4B). Conversely, pro-apoptotic target genes were down-regulated (ex. BAX, CASP1/2/6, and FAS) (Fig. 4B). The anti-apoptotic machinery is also controlled via interferon signaling, which we show to be upregulated during disease progression in the subsection below.
Fig. 4. Apoptotic dysregulation.

A LogFC of apoptotic transcription factors and apoptotic regulating interleukins are shown for patients 1–5 and patients from the TARGET and Mason databases, (−) LogFC indicates downregulation in relapse (green color) and (+) LogFC indicates upregulation in relapse (red color). B LogFC of apoptotic genes that are targets of the dysregulated apoptotic transcription factors Patients 1–5 and patients from the TARGET and Mason databases. For transcription factors (C) JUND, (D) EP300, (E) BCLAF1, (F) TP53, and (G) HGMB2 Pearson’s correlation correlograms shows the correlation between the apoptotic transcription factors and their target genes at the single cell level. Only significant (p < 0.05) associations are shown, with the size of circle indicating significance (larger circle indicates more significant). The color indicates Pearson’s r, with a red indicating a negative correlation and blue a positive correlation.
A correlation analysis demonstrated that the apoptotic regulating TFs (EP300, JUND, BCLAF1, HMGB2, and TP53) were positively and significantly correlated with the expression of their target genes at the single cell level across patients (Fig. 4C–G). The correlation analysis also demonstrated that anti-apoptotic genes (BCLA2A1, CFLAR, and MCL1) were significantly co-expressed at the single cell level across patients (Supplementary Fig. 6). We examined the heterogeneity of expression of the apoptotic regulating TFs and their target genes in single cell LICs across study patients. We found that apoptotic genes exhibited a heterogenous expression pattern at the single cell level (median segIndex 0.557; range 0.210–0.791) (Supplementary File 1).
Examination of apoptotic dysregulation also revealed potential novel AML biomarkers in the calcium-regulated membrane binding proteins of the annexin family, which have been shown to regulate p53 signaling [48, 49]. ANXA1/2 were downregulated during disease progression in the LICs in most study patients, identified as significant leading-edge genes in apoptotic pathway downregulation, and their expression was positively correlated with TP53 downregulation at the single cell level (Figs. 4B, F). There are reports that decreased ANXA1/2 can lead to drug resistance outside of the context of AML [50, 51]. Further, downregulation of ANXA1 in an erythroleukemia cell line must reported to lead to resistance to AML therapeutics [52].
Interferon, CXCR4, and Wnt signaling are upregulated during AML disease progression
Interferon regulatory factors (IRF) IRF4/8, which control the expression of downstream chemokines such as CXCR4, were upregulated in relapse LICs across study patients. In contrast, IRF5 which inhibits CXCR4 was downregulated (Fig. 5A) [53–55]. STATs, downstream of interferon signaling, have also been shown to regulate the expression of CXCR4. We found that expression levels of STAT3/5 were not significantly altered during disease progression. However, the activity of these important regulators is governed by phosphorylation, so we assessed STAT3/5 activity using a transcription factor analysis. Significant upregulation in the activity of STAT3 and STAT5A/5B (GSEA q < 0.05) during relapse was observed (Fig. 5B). Downstream target genes of IRFs and STAT3/5 involve anti-apoptotic machinery, metabolism, and migratory signaling, including but not limited to CXCR4 and Wnt/β-catenin signaling.
Fig. 5. Cytokine signaling dysregulation.
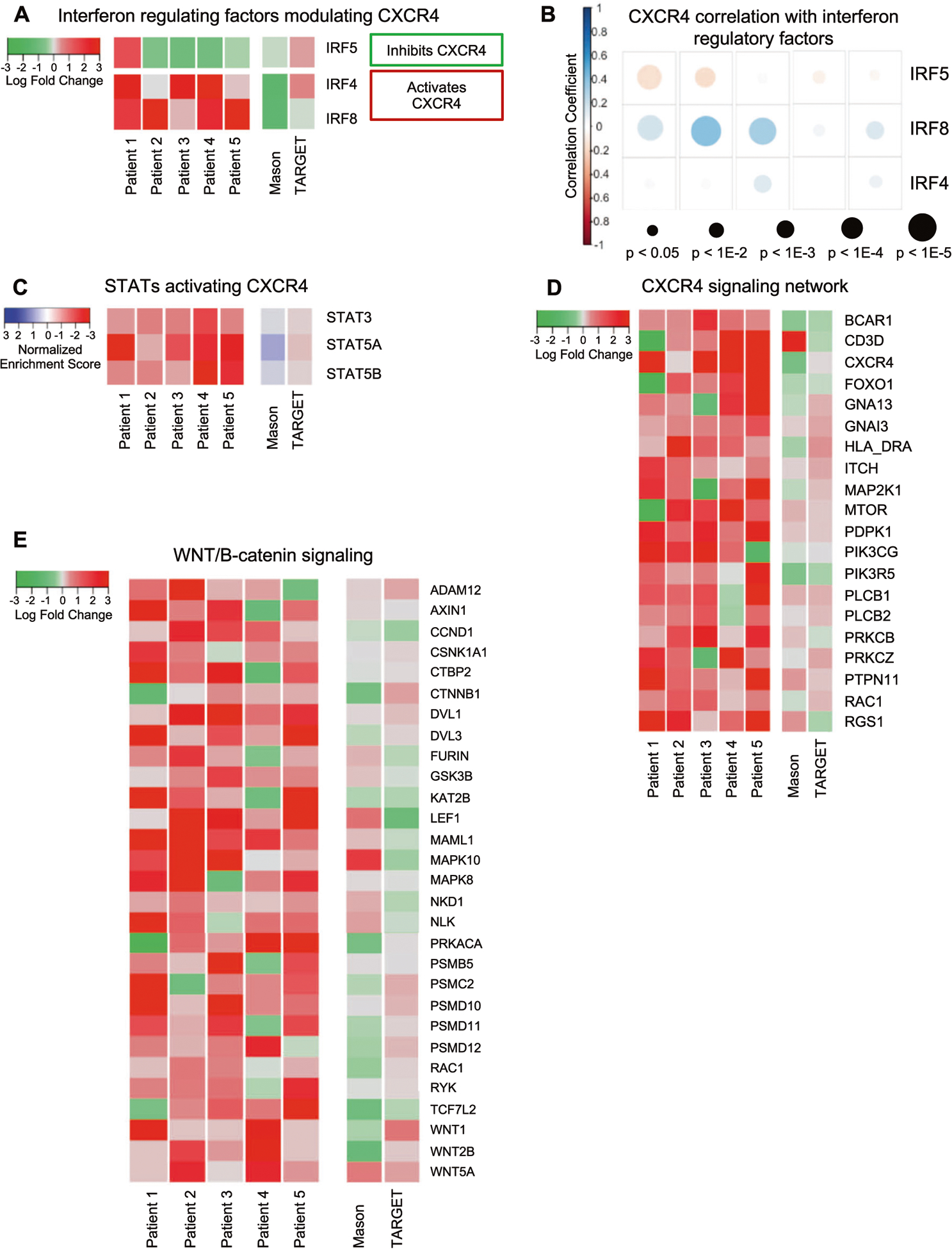
A LogFC of interferon regulatory factors are shown for patients 1–5 and patients from the TARGET and Mason databases, (−) LogFC indicates downregulation in relapse (green color) and (+) LogFC indicates upregulation in relapse (red color). B STAT mediated signaling transcription motif gene sets identified as significantly dysregulated across patients using GSEA are shown for Patients 1–5 and patients from the TARGET and Mason databases. The normalized enrichment scores (NES) are indicated, where negative NESs indicate pathway upregulation in the relapse state (red color) and positive NESs indicate pathway upregulation in the diagnosis state (blue color). C Pearson’s correlation correlogram shows the correlation between IRF4/5/8 and CXCR4 at the single cell level. Only significant (p < 0.05) associations are shown, with the size of circle indicating significance (larger circle indicates more significant). The color indicates Pearson’s r, with a red indicating a negative correlation and blue a positive correlation (D) LogFC of leading edge interferon and STAT transcription factor gene targets in the CXCR4 signaling network in Patients 1–5 and patients from the TARGET and Mason databases, (−) LogFC indicates downregulation in relapse. E LogFC of leading-edge genes upregulated in Wnt/B-catenin pathway in Patients 1–5 and patients from the TARGET and Mason databases, as determined by pathway analysis, commonly upregulated across patients.
In support of the connection between interferon and CXCR4 signaling, expression of CXCR4 was significantly positively correlated with the expression of IRF4/8 and negatively correlated with the expression of IRF5 (Fig. 5C). The upregulation of IRF8 in relapse LICs is consistent with previous human AML studies that demonstrated increased IRF8 expression is associated with decreased relapse free survival [56]. Furthermore, we found that not only were CXCR4 and its ligand, CXCL12, upregulated in relapse LIC’s, a number of genes in the CXCR4 signaling axis (ex. BCAR1, MTOR, MAP2K1, PKCs, and PTPN11) were also upregulated in relapse LICs across all patients (Fig. 5D). As described for the other pathways above, there was also significant heterogeneity at the single cell level in LSCs in the these IRF4/8 pathways (median segIndex 0.428; range 0.242–0.633) (Supplementary File 1).
CXCR4 signaling has been shown to affect numerous downstream signaling pathways including Wnt/β-catenin, which promotes self-renewal of LICs [10, 57–64]. We found that many Wnt/β-catenin pathway genes (ex. WNT1, WNT2B, WNT5A, DVL1/3, and LEF1) promoting cell proliferation in the bone marrow niche were also upregulated (Fig. 5E).
Flow cytometric analysis of AML initiating cells validates changes in selected markers during disease progression
Major genes associated with AML progression identified by single cell RNAseq were validated at the protein level by flow cytometry in an independent AML cohort of 32 patients (Clinical characteristics shown in Supplementary Table 2). The downstream analysis was performed in both the bulk AML cell population (CD34+) and LICs (Lin-CD34 + CD38-CD45RA+). A comprehensive unbiased analysis using TSNE (t-distributed stochastic neighbor embedding) was performed in both cell subsets comparing diagnosis and relapse subjects. Initially, the per cell level expression of chosen markers was plotted independently (MFI: Median of Intensity fluorescence) in total AML cells or LICs comparing both groups of patients. In addition, clustering analysis led to the identification of specific modulated clusters on those subjects. Flow panels were run and validated to assess the protein expression of GADD45, CXCR4, IRF8, BCL2 and β-catenin. Consistent with gene expression analysis of LICs, the levels of BCL2, IRF8 and β-catenin were significantly increased in relapse samples as compared to diagnosis samples in total AML cells (Fig. 6A). In contrast, GADD45A and CXCR4 that were highly expressed in relapse LIC samples, were instead found to be significantly increased in diagnosis samples in total AML cells. Importantly, when analyzing the LIC component of the AML samples, the expected phenotype found in single cell RNAseq analysis was supported by the protein analysis. GADD45A, CXCR4, IRF8, BCL2, and β-catenin (Fig. 6B) were all significantly upregulated in relapse samples, contributing to the long-term survival and stemness profile of these cells at relapse.
Fig. 6. Flow cytometry validation study.


A, B Median fluorescence intensity (MFI) for each of the indicated markers in total AML cells (CD34+) (A) or LICs samples (Lin−, CD34+, CD38−, CD45RA+) (B) for the 32 patients analyzed. The diagnosis cells are represented by black dots and relapse cells as red dots. White arrows on the plots point the direction of the increase or decrease. The median fluorescent intensity values are also shown for each sample (C–D) TSNE plots based on clustering analysis performed for both total AML (C) and LIC AML (D) populations. E–F Plots of the cluster frequencies from total AML (E) or LIC (F) cluster heatmaps showing the expression of the markers of interested on the highlighted clusters. On the top of each TSNE-heatmap cluster, the histogram compares the MFI of the specific clusters (color-coded) with the total number of clusters (gray-coded). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Next, we evaluated if there were any specific cell clusters among total AML or LIC cells that were significantly enriched between the groups. In total AML cells, we found that cell cluster 20 was significantly enriched in diagnosis samples when compared to relapse samples (p < 0.05 – Fig. 6D). This cell population presented significantly lower expression of GADD45A, IRF8, CXCR4 and BCL2, as shown in the cluster heatmaps and the histograms on the top of each plot (Fig. 6D). Interestingly, four clusters were significantly increased in diagnosis LIC cells (cluster 4, 10, 13, and 22 – Fig. 6E) when compared to relapse samples. These clusters presented significantly lower expression of all the aforementioned markers as shown on the heatmaps and histograms (Fig. 6F). In summary, key-proteins of the significantly enriched pathways at single cell RNAseq analysis are indeed modulated in LIC populations when comparing diagnosis and relapse. The importance of certain molecules such as GADD45A and CXCR4 are lost when considering total AML cells. In summary, the flow cytometry validation study supports the single cell RNAseq analysis.
Co-targeting BCL2 and CXCR4 signaling demonstrates enhanced AML efficacy in vitro and in vivo
As AML is highly heterogeneous at the single cell level, it is challenging to develop targeted therapies that are effective as single agents as well as to identify optimal combinations. Our RNAseq results and flow validation study reveal that CXCR4 signaling and BCL2 are both upregulated in LSCs during AML progression. Both of these molecules have been targeted in patients with AML with varying degrees of success, though never in combination. For example, Plerixafor has been used clinically to target CXCR4 and studies suggest it can both lead to direct apoptosis of target cells as well as mobilize AML cells to sensitize them to other agents. In addition, Venetoclax targets BCL2 leading to apoptosis and has shown promise in AML therapy [65, 66]. To explore the potential of this combination, we initially assessed the co-expression of BCL2 with activated CXCR4 signaling. Interestingly despite the fact that CXCR4 signaling and BCL2 can be upregulated during AML progression, there is a very poor correlation between expression of BCL2 and activation of the CXCR4 signaling pathway at the single cell level. For example, a nonparametric Kendall tau correlation comparing BCL2 and the CXCR4 signaling pathway per patient shown in Fig. 5D showed a poor correlation (mean −0.019; range −0.077 to 0.120; p = 0.656). Similarly, there was a poor correlation of BCL2 and CXCR4 (mean 0.025; range −0.095 to 0.128; p = 0.588). As these results suggest that co-targeting these pathways may lead to enhanced efficacy we assessed the effects of Plerixafor, Venetoclax or the combination on primary AML cells derived from relapsed AML patients. While Venetoclax had significant effects on the viability of most samples as a single agent, Plerixafor had little effect alone. Interestingly, there was a further decrease in cell viability with the combination regimen as opposed to single agent treatment in five out of six samples tested (Fig. 7A, B). To further explore the therapeutic potential of this combination, we assessed the impact of the agents using a previously characterized immunodeficient mouse model of circulating human AML that employs a PDX AML patient sample from an elderly patient with relapsed AML that exhibits EZH2 and TET2 mutations [67]. This model is known to lead to the replacement of the majority of normal hematopoietic cells in the bone marrow with AML cells in approximately six weeks. The mice were treated with either vehicle, Venetoclax (50 mg/kg), Plerixafor (5 mg/kg) or a combination and then assessed for survival. Plerixafor alone did not demonstrate any survival advantage in this model and Venetoclax alone led to only a modest benefit. However, the combination of Venetoclax and Plerixafor led to a marked increase in survival in this aggressive AML model system (log rank < 0.0001) (Fig. 7C).
Fig. 7. Co-targeting BCL2 and CXCR4 leads to enhanced efficacy against AML cells in vitro and in vivo.

A Characteristics of relapsed AML patient samples utilized in (B). B Relative viability of primary AML cells treated with 10 μM Plerixafor (P), 50 nM Venetoclax (V) or a combination after 72 hr as measured by the MTT assay. (C) Survival curve of NSG mice (n = 5 per group) injected with AML PDX cells and Plerixafor, Venetoclax or a combination. Plerixafor (2.5 mg/kg s.c). and Venetoclax (50 mg/kg oral gavage) were administered five days a week for three weeks starting five days after AML cell injection. *p < 0.05, **p < 0.001, NS not significant.
DISCUSSION
Here, we report that RNA cluster groups evolve during AML disease progression and demonstrated that an individual LIC’s membership in a specific RNA cluster group can be predicted with sensitivity and specificity based on cluster-specific gene expression markers. This is the first description of the phenomenon of RNA clonal evolution during cancer progression. Further, our studies reveal the vast transcriptional heterogeneity of LICs at the single cell level that may facilitate the evolution of these cells upon treatment with chemotherapeutics. These findings suggest that AML therapy selects for specific patterns of gene expression among AML initiating cells and that these gene expression changes facilitate treatment escape and resistance as LICs evolve at the RNA level. Of note, RNA expression pattern is dynamic and the analysis in this study is focused on a fixed transcriptional state of the cells. It will be of interest to assess the transition of cells between clusters in future studies. Due to the selective pressures from AML therapy, it is likely that the transition of transcriptional states present in cells from the relapse dominant clusters will not frequently revert back to those found in the diagnosis dominant clusters.
Through the study of serial AML patient samples, we identified that several pathways were commonly modulated during disease progression including signaling networks involving metabolism, apoptosis and chemokine signaling. Though many of these pathways can be targeted using existing therapeutics, LICs were found to be highly heterogeneous. Therefore, this work suggests curative treatment will require the simultaneous targeting of multiple pathways to fully eradicate LICs.
AML cells have previously been shown to have immense metabolic plasticity that enables them to utilize diverse fuels such as fatty acids and amino acids to support mitochondrial metabolism [43]. Oxidative phosphorylation, in particular, has been reported to be important for AML cells to become drug resistant [68]. Here we show for the first time, using cells from matched patient treatment-naïve and post-treatment bone marrow samples, how LICs evolve their metabolic programming over time in response to treatment. Across all patients, LICs became less dependent on glycolysis while upregulating mitochondrial metabolism. During disease progression, the LICs increased both fatty acid oxidation and amino acid metabolism, both of which are known to supply the Krebs cycle with intermediates [43]. In particular, branched-chain amino acid metabolism was found to be increased which supports previous studies demonstrating its importance in myeloid malignancy [69]. The upregulated expression of key transporters of fatty and amino acids such as CD36 and a large number of SLCs were also found to evolve during AML progression [70, 71]. Of note, LICs targeting fatty acid oxidation may be a desirable strategy as previous studies have demonstrated LICs may exploit fatty acid oxidation to evade chemotherapy [72].
Amino acid metabolism and transport has been previously shown to be upregulated in LICs as compared to the total leukemic cell population and amino acids in general have been found to be important alternative fuel sources for many types of cancer [73, 74]. For example, cysteine depletion has been reported to induce death in LICs. Our data, showing upregulation of the SLC7A11 transporter, which transports exogenous cysteine into the cell, suggests this approach as well as targeting other amino acid pathways may be promising strategies to target LICs in relapsed AML patients [75].
Of further clinical relevance, the heterogeneous expression of CXCR4 signaling in single LICs, which has not been previously described, has direct implications for therapeutic targeting of this pathway. Indeed, none of the agents inhibiting CXCR4 in AML have reached Phase III trials [76]. The heterogenous nature of CXCR4 expression suggests, CXCR4 inhibitors should be combined with other inhibitors of the leukemia-stroma interaction or other non-overlapping targets expressed in LICs [76, 77]. We also found that anti-apoptotic genes such as BCL2, c-FLIP, SOCS3, and MCL1 were upregulated and co-expressed in LICs. Recently, targeted treatments that inhibit the expression of many of these pro-apoptotic genes have been used to treat hematological cancers, but overall response rates have been highly variable in trials [78–83]. As BCL2 and CXCR4 signaling have clinically available targeted agents, were found to be upregulated during AML progression and demonstrate high levels of heterogeneity at the single cell level, we explored the combination of co-targeting these pathways. Combined targeting using Venetoclax and Plerixafor demonstrated potential that may be worthy of future clinical testing. Our work sheds light on the failure of single agent treatments targeting these genes in isolation and highlights the need for combination therapies to completely eradicate LICs.
Our work is distinctive in several crucial ways. First, to our knowledge, this study is the first to perform single cell RNA sequencing on matched treatment-naïve diagnosis and post-treatment relapse AML patient samples—enabling comprehensive assessment of clonal evolutionary changes at the transcriptome level. Second, our analysis is focused on stem-like AML-initiating cells, which compose only a small fraction of all AML cells, but drive AML relapse [2, 3, 10, 13, 84]. Finally, we have prioritized deep sequencing in an effort to thoroughly examine the LIC transcriptome to generate statistically significant differential expression and pathway analysis at the single cell level.
Recently, studies by Chen et al. [23] and van Galen et al. [21] have reported single cell sequencing results on AML samples. Chen et al. utilized matched patient samples, but their single cell DNA sequencing study investigated the transition from myelodysplastic syndrome to secondary AML [23]. Van Galen et al. analyzed a large number of cells (13,489 AML cells) at a low read depth (<5000 UMI counts per cell) that enabled the detection of <1000 genes expressed per cell which would preclude a detailed assessment of RNA-based evolution [24].
This study revealed new insights into the evolution of LICs during disease progression through single cell RNAseq of matched patient treatment-naïve diagnosis and post-treatment relapse samples. Despite variability in patient age, mutation status, and in some cases treatment regimens, this work still revealed commonalities in the biology of AML relapse. This is particularly important as there are needs for more effective therapeutic interventions among patients without known driver mutations.
METHODS
Please see Supplementary Materials.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to the patients who contributed to this study and thank the Children’s Oncology Group for providing some of the bone marrow samples used in this study. This study was supported by pilot funding from the Case Comprehensive Cancer Center (NCI P30CA0437093) and the Computational Genomic Epidemiology of Cancer Fellowship (NCI 5T32CA094186-17, L.S.). This research was also supported by the following Shared Resources of the Case Comprehensive Cancer Center (NCI P30CA0437093): Hematopoietic Cell Biorepository and Cellular Therapy, Integrated Genomics, and Cytometry & Imaging Microscopy. This work made use of the High Performance Computing Resources in the Core Facility for Advanced Research Computing at Case Western Reserve University. The authors thank Jose Cancelas, Folashade Otegbeye, Sheela Karunaithi, and Grace Lee for their scientific feedback and Caroline Perry and Kristin Waite for their critical reading and editing of the paper.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41375-021-01338-7.
REFERENCES
- 1.Estey EH. General approach to, and perspectives on clinical research in, older patients with newly diagnosed acute myeloid leukemia. Semin Hematol. 2006;43:89–95. [DOI] [PubMed] [Google Scholar]
- 2.Huntly BJ, Gilliland DG. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat Rev Cancer. 2005;5:311–21. [DOI] [PubMed] [Google Scholar]
- 3.Hanekamp D, Cloos J, Schuurhuis GJ. Leukemic stem cells: identification and clinical application. Int J Hematol. 2017;105:549–57. [DOI] [PubMed] [Google Scholar]
- 4.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–91. [DOI] [PubMed] [Google Scholar]
- 5.Lechman ER, Gentner B, Ng SW, Schoof EM, van Galen P, Kennedy JA. et al. miR-126 regulates distinct self-renewal outcomes in normal and malignant hematopoietic stem cells. Cancer cell. 2016;29:214–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P. et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17:1086–93. [DOI] [PubMed] [Google Scholar]
- 7.Jordan CT. The leukemic stem cell. Best Pr Res Clin Haematol. 2007;20:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Metzeler KH, Maharry K, Kohlschmidt J, Volinia S, Mrozek K, Becker H. et al. A stem cell-like gene expression signature associates with inferior outcomes and a distinct microRNA expression profile in adults with primary cytogenetically normal acute myeloid leukemia. Leukemia. 2013;27:2023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akinduro O, Weber TS, Ang H, Haltalli MLR, Ruivo N, Duarte D. et al. Proliferation dynamics of acute myeloid leukaemia and haematopoietic progenitors competing for bone marrow space. Nat Commun. 2018;9:519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hira VVV, Van Noorden CJF, Carraway HE, Maciejewski JP, Molenaar RJ. Novel therapeutic strategies to target leukemic cells that hijack compartmentalized continuous hematopoietic stem cell niches. Biochim Biophys Acta Rev Cancer. 2017;1868:183–98. [DOI] [PubMed] [Google Scholar]
- 11.Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saito Y, Uchida N, Tanaka S, Suzuki N, Tomizawa-Murasawa M, Sone A. et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol. 2010;28:275–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Essers MA, Trumpp A. Targeting leukemic stem cells by breaking their dormancy. Mol Oncol. 2010;4:443–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee MC, Lopez-Diaz FJ, Khan SY, Tariq MA, Dayn Y, Vaske CJ. et al. Single-cell analyses of transcriptional heterogeneity during drug tolerance transition in cancer cells by RNA sequencing. Proc Natl Acad Sci U.S.A. 2014;111:E4726–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ. et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl J Med 2013;368:2059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND. et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl J Med 2016;374:2209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolouri H, Farrar JE, Triche T Jr., Ries RE, Lim EL, Alonzo TA. et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. 2018;24:103–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS. et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes AE, Magrini V, Demeter R, Miller CA, Fulton R, Fulton LL. et al. Clonal architecture of secondary acute myeloid leukemia defined by single-cell sequencing. PLoS Genet. 2014;10:e1004462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paguirigan AL, Smith J, Meshinchi S, Carroll M, Maley C, Radich JP. Single-cell genotyping demonstrates complex clonal diversity in acute myeloid leukemia. Sci Transl Med. 2015;7:281re282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li S, Garrett-Bakelman FE, Chung SS, Sanders MA, Hricik T, Rapaport F. et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat Med. 2016;22:792–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan B, Hu Y, Ban KHK, Tiang Z, Ng C, Lee J. et al. Single-cell genomic profiling of acute myeloid leukemia for clinical use: a pilot study. Oncol Lett. 2017;13:1625–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J, Kao YR, Sun D, Todorova TI, Reynolds D, Narayanagari SR. et al. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat Med. 2019;25:103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Galen P, Hovestadt V, Wadsworth Ii MH, Hughes TK, Griffin GK, Battaglia S. et al. Single-cell RNA-seq reveals AML hierarchies relevant to disease progression and immunity. Cell. 2019;176:1265–81 e1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang ML, Bailey NG. Acute myeloid leukemia genetics: risk stratification and implications for therapy. Arch Pathol Lab Med. 2015;139:1215–23. [DOI] [PubMed] [Google Scholar]
- 26.Baum CM, Weissman IL, Tsukamoto AS, Buckle AM, Peault B. Isolation of a candidate human hematopoietic stem-cell population. Proc Natl Acad Sci U.S.A. 1992;89:2804–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kersten B, Valkering M, Wouters R, van Amerongen R, Hanekamp D, Kwidama Z. et al. CD45RA, a specific marker for leukaemia stem cell sub-populations in acute myeloid leukaemia. Br J Haematol. 2016;173:219–35. [DOI] [PubMed] [Google Scholar]
- 28.Majeti R, Park CY, Weissman IL. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell. 2007;1:635–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Picelli S, Bjorklund AK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smartseq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10:1096–8. [DOI] [PubMed] [Google Scholar]
- 30.Majeti R, Becker MW, Tian Q, Lee TL, Yan X, Liu R. et al. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc Natl Acad Sci U.S.A. 2009;106:3396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin Y, Ghazanfar S, Strbenac D, Wang A, Patrick E, Lin DM, et al. Evaluating stably expressed genes in single cells. Gigascience. 2019;8:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murga M, Jaco I, Fan Y, Soria R, Martinez-Pastor B, Cuadrado M. et al. Global chromatin compaction limits the strength of the DNA damage response. J Cell Biol. 2007;178:1101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu S, Kharbanda S, Stone RM. OncoHist®, an rh histone 1.3 is cytotoxic acute myeloid leuk cells results altered downstr signal. Blood. 2014;124:3604–4. [Google Scholar]
- 34.Su C, Gao G, Schneider S, Helt C, Weiss C, O’Reilly MA. et al. DNA damage induces downregulation of histone gene expression through the G1 checkpoint pathway. EMBO J. 2004;23:1133–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Izquierdo-Bouldstridge A, Bustillos A, Bonet-Costa C, Aribau-Miralbes P, Garcia-Gomis D, Dabad M. et al. Histone H1 depletion triggers an interferon response in cancer cells via activation of heterochromatic repeats. Nucleic Acids Res. 2017;45:11622–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Torres CM, Biran A, Burney MJ, Patel H, Henser-Brownhill T, Cohen AS, et al. The linker histone H1.0 generates epigenetic and functional intratumor heterogeneity. Science. 2016;353:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liebermann DA, Tront JS, Sha X, Mukherjee K, Mohamed-Hadley A, Hoffman B. Gadd45 stress sensors in malignancy and leukemia. Crit Rev Oncog. 2011;16:129–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chamseddine AN, Cabrero M, Wei Y, Ganan-Gomez I, Colla S, Takahashi K. et al. PDE4 differential expression is a potential prognostic factor and therapeutic target in patients with myelodysplastic syndrome and chronic myelomonocytic leukemia. Clin Lymphoma, Myeloma Leuk. 2016;16:S67–73. [DOI] [PubMed] [Google Scholar]
- 39.Smith PG, Wang F, Wilkinson KN, Savage KJ, Klein U, Neuberg DS. et al. The phosphodiesterase PDE4B limits cAMP-associated PI3K/AKT-dependent apoptosis in diffuse large B-cell lymphoma. Blood. 2005;105:308–16. [DOI] [PubMed] [Google Scholar]
- 40.Kim DU, Kwak B, Kim SW. Phosphodiesterase 4B is an effective therapeutic target in colorectal cancer. Biochem Biophys Res Commun. 2019;508:825–31. [DOI] [PubMed] [Google Scholar]
- 41.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 42.Scialdone A, Natarajan KN, Saraiva LR, Proserpio V, Teichmann SA, Stegle O. et al. Computational assignment of cell-cycle stage from single-cell transcriptome data. Methods. 2015;85:54–61. [DOI] [PubMed] [Google Scholar]
- 43.Kreitz J, Schonfeld C, Seibert M, Stolp V, Alshamleh I, Oellerich T. et al. Metabolic plasticity of acute myeloid leukemia. Cells. 2019;8:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Decker T, Bogner C, Oelsner M, Peschel C, Ringshausen I. Antiapoptotic effect of interleukin-2 (IL-2) in B-CLL cells with low and high affinity IL-2 receptors. Ann Hematol. 2010;89:1125–32. [DOI] [PubMed] [Google Scholar]
- 45.Lin MT, Juan CY, Chang KJ, Chen WJ, Kuo ML. IL-6 inhibits apoptosis and retains oxidative DNA lesions in human gastric cancer AGS cells through up-regulation of anti-apoptotic gene mcl-1. Carcinogenesis. 2001;22:1947–53. [DOI] [PubMed] [Google Scholar]
- 46.Chen Y, Zhang F, Tsai Y, Yang X, Yang L, Duan S. et al. IL-6 signaling promotes DNA repair and prevents apoptosis in CD133+ stem-like cells of lung cancer after radiation. Radiat Oncol. 2015;10:227. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Ahmed NN, Grimes HL, Bellacosa A, Chan TO, Tsichlis PN. Transduction of interleukin-2 antiapoptotic and proliferative signals via Akt protein kinase. Proc Natl Acad Sci U.S.A. 1997;94:3627–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zeng GQ, Yi H, Li XH, Shi HY, Li C, Li MY. et al. Identification of the proteins related to p53-mediated radioresponse in nasopharyngeal carcinoma by proteomic analysis. J Proteom. 2011;74:2723–33. [DOI] [PubMed] [Google Scholar]
- 49.Swa HL, Blackstock WP, Lim LH, Gunaratne J. Quantitative proteomics profiling of murine mammary gland cells unravels impact of annexin-1 on DNA damage response, cell adhesion, and migration. Mol Cell Proteom: MCP. 2012;11:381–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lim LH, Pervaiz S. Annexin 1: the new face of an old molecule. FASEB J. 2007;21:968–75. [DOI] [PubMed] [Google Scholar]
- 51.Guo C, Liu S, Sun MZ. Potential role of Anxa1 in cancer. Future Oncol. 2013;9:1773–93. [DOI] [PubMed] [Google Scholar]
- 52.Zhu F, Wang Y, Zeng S, Fu X, Wang L, Cao J. Involvement of annexin A1 in multidrug resistance of K562/ADR cells identified by the proteomic study. OMICS. 2009;13:467–76. [DOI] [PubMed] [Google Scholar]
- 53.Shukla V, Lu R. IRF4 and IRF8: governing the virtues of B Lymphocytes. Front Biol (Beijing). 2014;9:269–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mattei F, Schiavoni G, Sestili P, Spadaro F, Fragale A, Sistigu A. et al. IRF-8 controls melanoma progression by regulating the cross talk between cancer and immune cells within the tumor microenvironment. Neoplasia. 2012;14:1223–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bi X, Hameed M, Mirani N, Pimenta EM, Anari J, Barnes BJ. Loss of interferon regulatory factor 5 (IRF5) expression in human ductal carcinoma correlates with disease stage and contributes to metastasis. Breast Cancer Res. 2011;13:R111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pogosova-Agadjanyan EL, Kopecky KJ, Ostronoff F, Appelbaum FR, Godwin J, Lee H, et al. The prognostic significance of IRF8 transcripts in adult patients with acute myeloid leukemia. PloS ONE. 2013;8:e70812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao S, Wang J, Qin C. Blockade of CXCL12/CXCR4 signaling inhibits intrahepatic cholangiocarcinoma progression and metastasis via inactivation of canonical Wnt pathway. J Exp Clin Cancer Res. 2014;33:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu X, Shi W, Zhang Y, Wang X, Sun S, Song Z. et al. CXCL12/CXCR4 axis induced miR-125b promotes invasion and confers 5-fluorouracil resistance through enhancing autophagy in colorectal cancer. Sci Rep. 2017;7:42226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song ZY, Gao ZH, Chu JH, Han XZ, Qu XJ. Downregulation of the CXCR4/CXCL12 axis blocks the activation of the Wnt/beta-catenin pathway in human colon cancer cells. Biomed Pharmacother. 2015;71:46–52. [DOI] [PubMed] [Google Scholar]
- 60.Lin XL, Xu Q, Tang L, Sun L, Han T, Wang LW, et al. Regorafenib inhibited gastric cancer cells growth and invasion via CXCR4 activated Wnt pathway. PLoS ONE. 2017;12:e0177335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu TH, Yao Y, Yu S, Han LL, Wang WJ, Guo H. et al. SDF-1/CXCR4 promotes epithelial-mesenchymal transition and progression of colorectal cancer by activation of the Wnt/beta-catenin signaling pathway. Cancer Lett. 2014;354:417–26. [DOI] [PubMed] [Google Scholar]
- 62.Lu Y, Hu B, Guan GF, Chen J, Wang CQ, Ma Q. et al. SDF-1/CXCR4 promotes F5M2 osteosarcoma cell migration by activating the Wnt/beta-catenin signaling pathway. Med Oncol. 2015;32:194. [DOI] [PubMed] [Google Scholar]
- 63.Konopleva MY, Jordan CT. Leukemia stem cells and microenvironment: biology and therapeutic targeting. J Clin Oncol. 2011;29:591–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tabe Y, Konopleva M. Role of microenvironment in resistance to therapy in AML. Curr Hematol Malig Rep. 2015;10:96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cancilla D, Rettig MP, DiPersio JF. Targeting CXCR4 in AML and ALL. Front Oncol. 2020;10:1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pollyea DA, Amaya M, Strati P, Konopleva MY. Venetoclax for AML: changing the treatment paradigm. Blood Adv. 2019;3:4326–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gupta K, Stefan T, Ignatz-Hoover J, Moreton S, Parizher G, Saunthararajah Y. et al. GSK-3 inhibition sensitizes acute myeloid leukemia cells to 1,25D-mediated differentiation. Cancer Res. 2016;76:2743–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R. et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017;7:716–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hattori A, Tsunoda M, Konuma T, Kobayashi M, Nagy T, Glushka J. et al. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature. 2017;545:500–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bonen A, Campbell SE, Benton CR, Chabowski A, Coort SL, Han XX. et al. Regulation of fatty acid transport by fatty acid translocase/CD36. Proc Nutr Soc. 2004;63:245–9. [DOI] [PubMed] [Google Scholar]
- 71.Schweikhard ES, Ziegler CM. Amino acid secondary transporters: toward a common transport mechanism. Curr Top Membr. 2012;70:1–28. [DOI] [PubMed] [Google Scholar]
- 72.Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M. et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. 2016;19:23–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T. et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell. 2018;34:724–40 e724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tsun ZY, Possemato R. Amino acid management in cancer. Semin Cell Dev Biol. 2015;43:22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jones CL, Stevens BM, D’Alessandro A, Culp-Hill R, Reisz JA, Pei S. et al. Cysteine depletion targets leukemia stem cells through inhibition of electron transport complex II. Blood. 2019;134:389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Peled A, Klein S, Beider K, Burger JA, Abraham M. Role of CXCL12 and CXCR4 in the pathogenesis of hematological malignancies. Cytokine. 2018;109:11–6. [DOI] [PubMed] [Google Scholar]
- 77.Cho BS, Kim HJ, Konopleva M. Targeting the CXCL12/CXCR4 axis in acute myeloid leukemia: from bench to bedside. Korean J Intern Med. 2017;32:248–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Carter BZ, Gronda M, Wang Z, Welsh K, Pinilla C, Andreeff M. et al. Small-molecule XIAP inhibitors derepress downstream effector caspases and induce apoptosis of acute myeloid leukemia cells. Blood. 2005;105:4043–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cassier PA, Castets M, Belhabri A, Vey N. Targeting apoptosis in acute myeloid leukaemia. Br J Cancer. 2017;117:1089–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Phase I study of ABT-199 (GDC-0199) in patients with relapsed/refractory non-Hodgkin lymphoma: responses observed in diffuse large B-cell (DLBCL) and follicular lymphoma (FL) at higher cohort doses. Clin Adv Hematol Oncol. 2014; 12: 18–19. [PubMed] [Google Scholar]
- 81.Brinda B, Khan I, Parkin B, Konig H. The rocky road to personalized medicine in acute myeloid leukaemia. J Cell Mol Med. 2018;22:1411–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.DiNardo CD, Pratz KW, Letai A, Jonas BA, Wei AH, Thirman M. et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. 2018;19:216–28. [DOI] [PubMed] [Google Scholar]
- 83.Kayser S, Levis MJ. Advances in targeted therapy for acute myeloid leukaemia. Br J Haematol. 2018;180:484–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thomas D, Majeti R. Biology and relevance of human acute myeloid leukemia stem cells. Blood. 2017;129:1577–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.