

Abstract
The epigenetic state of chromatin is altered by regulators which influence gene expression in response to environmental stimuli. While several post-translational modifications contribute to chromatin accessibility and transcriptional programs, our understanding of the role that specific phosphorylation sites play is limited. In cancer, kinases and phosphatases are commonly deregulated resulting in increased oncogenic signaling and loss of epigenetic regulation. Aberrant epigenetic states are known to promote cellular plasticity and the development of therapeutic resistance in many cancer types, highlighting the importance of these mechanisms to cancer cell phenotypes. Protein Phosphatase 2A (PP2A) is a heterotrimeric holoenzyme that targets a diverse array of cellular proteins. The composition of the PP2A complex influences its cellular targets and activity. For this reason, PP2A can be tumor suppressive or oncogenic depending on cellular context. Understanding the nuances of PP2A regulation and its effect on epigenetic alterations can lead to new therapeutic avenues that afford more specificity and contribute to the growth of personalized medicine in the oncology field. In this review, we summarize the known PP2A-regulated substrates and potential phosphorylation sites that contribute to cancer cell epigenetics and possible strategies to therapeutically leverage this phosphatase to suppress tumor growth.
Graphical Abstract
Graphical Abstract.
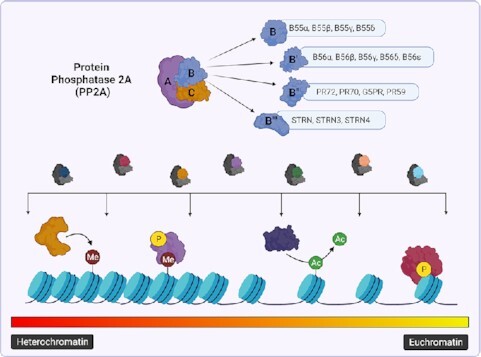
The diverse family of PP2A holoenzymes dynamically regulates the cancer epigenome.
INTRODUCTION
Aberrant epigenetic regulation of the cancer genome contributes to a wide range of phenotypes, including therapeutic resistance and cellular plasticity (1). In normal cells, control of gene expression through epigenetic regulatory mechanisms allows for rapid cellular responses to various microenvironmental signals. However, oncogenic mutations can significantly alter the epigenetic state of cells, leading to extensive chromatin remodeling, aberrant activation of oncogenes or repression of tumor suppressor genes. These alterations can further contribute to therapeutic resistance and increased survival under unfavorable conditions (2). Further, epigenetic plasticity (either restrictive or permissive) can significantly influence tumorigenic phenotypes, including cell state (e.g. epithelial-to-mesenchymal transition [EMT]) and intratumoral heterogeneity (2,3). Given that epigenetic dysregulation frequently occurs in tumors and significantly contributes to oncogenesis, the therapeutic inhibition of epigenetic pathways that control genome accessibility has shown great clinical promise (4). However, many questions remain regarding the mechanisms that contribute to cancer cell epigenetic states.
In addition to specific DNA sequences that influence gene transcription, there exists a higher order structure to the genome that provides an additional layer of regulation to maintain cellular homeostasis. The nucleosome comprises DNA wrapped around a histone octamer (two copies of H2A, H2B, H3 and H4), which shields the associated DNA from the transcriptional machinery. Reversible epigenetic marks, such as DNA methylation or posttranslational modifications (PTMs) on histones, allow for dynamic changes in the genome without altering the DNA sequence. In response to repressive epigenetic marks, histones will condense to form heterochromatin, or ‘silent’ chromatin regions. Active epigenetic marks help direct remodeling complexes to specific sites of DNA to displace histones and expose critical coding regions, providing additional security that only genes in euchromatic, ‘active’ chromatin regions are readily expressed. Densely packed regions of chromatin are moved to the nuclear lamina to make room for the transcriptional machinery within areas of open chromatin. This dynamic spatial organization, or 3D architecture, generates topologically associated domains (TADs) within the nucleus. During tumorigenesis, the epigenetic regulation of these various chromatin states is frequently altered, significantly contributing to cancer phenotypes and therapeutic response (5–7). However, the mechanisms that regulate this balance are highly complex and context dependent, making the identification of specific epigenetic alterations challenging.
There is a diverse array of cellular complexes that balance the deposition, reading, and removal of epigenetic marks to rapidly alter gene expression based on changes in the cellular environment. These epigenetic modifiers can be divided into three main categories: ‘readers’, ‘writers’ and ‘erasers’. Epigenetic ‘writers’ and ‘erasers’ deposit or remove epigenetic marks, such as methylation, on DNA and histones (5,8). Epigenetic ‘readers’ interpret deposited marks and either recruit transcriptional co-factors or additional chromatin remodeling complexes to specific sites of DNA (5). Together, these modifiers function in a delicate balance to maintain normal gene expression. Epigenetic dysregulation disrupts this balance, contributing to the abnormal activation of oncogenic signaling networks and initiation of tumorigenesis (9). There is a growing body of research indicating that the epigenetic state of cancer cells is significantly impacted by protein phosphorylation (10–13). Kinases and phosphatases rapidly and reversibly modify proteins in response to cellular stimuli, ultimately altering protein activity, localization and binding partners. Critical phosphorylation sites have been identified on epigenetic modifiers and histones that impact their function and/or cellular localization. However, the specific contribution of these phosphorylation sites is still poorly understood.
Kinase inhibitors have been a primary focus for anti-tumor therapeutics contributing to the rise in personalized medicine (14). However, phosphatases play a significant and underappreciated role in suppressing oncogenic signaling and are emerging as novel targets for therapeutic compounds (15,16). Protein Phosphatase 2A (PP2A) is a serine/threonine phosphatase that accounts for approximately 50% of global phosphatase activity (17). The PP2A holoenzyme is composed of three subunits: the scaffolding (A), regulatory (B) and catalytic (C) subunits. While the A and C subunits only contain two different isoforms, the B subunit contributes to a majority of PP2A’s functional diversity with 16 characterized B subunits, many of which contain additional splice variants (15). This array of subunits enables the formation of over 90 distinct and potentially functionally diverse holoenzymes (18). As a whole, the PP2A holoenzyme is categorized as a tumor suppressor due to its negative regulation of many common oncogenes including c-MYC, BCL2, ERK and AKT (19–22). Consistent with this role, PP2A activity is often suppressed in cancer (23,24). Of all the PP2A subunits, PPP2R1A, the PP2A Aα scaffolding subunit, has the highest mutation rate occurring in ∼1% of all cancers (25). These mutations suppress global PP2A activity by disrupting B or C subunit binding and have been shown to drive transformation and tumor growth (26,27). Similarly, the epigenetic silencing of the PP2A B subunit, B55β (PPP2R2B), through DNA hypermethylation results in similar phenotypes, further supporting an important role for PP2A in oncogenesis (28,29). While genetic/epigenetic loss of PP2A function does occur and represents an interesting biomarker for aggressive disease, it is relatively rare and not the primary mechanism by which cancer cells inhibit PP2A activity (24). Instead, tumor cells negatively regulate the PP2A holoenzyme through aberrant expression of endogenous inhibitors and PTMs, which alter PP2A activity, composition, and subcellular localization (30). As a variety of epigenetic regulators have been identified as PP2A targets (Table 1), the dysregulation of PP2A can significantly impact gene transcription, genomic instability and post-translational signaling during oncogenesis. In light of these findings, the therapeutic re-activation of PP2A has emerged as a novel anti-tumor strategy to mitigate oncogenic signaling on multiple levels. Several compounds have been identified to have indirect PP2A activating properties (e.g. FTY720), and small molecules that directly target the PP2A holoenzyme (e.g. SMAPs and iHAPs) are quickly taking the spotlight. However, recent studies have identified pro-tumorigenic roles for specific PP2A subunits, indicating that PP2A inhibitors (e.g. LB100) may also have a place in the clinic (Table 2).
Table 1.
Epigenetic targets of PP2A.
| PP2A Target | Associated B-Subunit | S/T Residue | Opposing Kinase(s) | Effect of Dephosphorylation | References |
|---|---|---|---|---|---|
| PRR14 | B56a contains SLIM motif and may interact with other B56 family members | S242, T266, T270, S277 | Not determined | Promotion of proper localization of heterochromatin to nuclear periphery | (39) |
| H3 | Not determined | S10 | AURKA, AURKB, IKKα, JNK, AKT1, MAP3K8, MSK1/2, PIM1 | Decreased output of MYC and MYC-related gene targets; Decrease in cell proliferation | (54,57,185) |
| Lamin A/C | Possibly B56 family members as phosphorylation status changes in response to CIP2A expression | S22, S628 | CDK1/CyclinB | Head-to-tail polymerization of Lamin A/C, proper Lamin distribution at nuclear envelope | (41–43) |
| BRD4 | Not determined | S484/S488 | CK2 | Nuclear localization; Negative regulation of BRD4-associated gene transcription | (68,69) |
| SWI/SNF | Possibly B55α, only identified in C. elegans | Not determined | ERK1 | Activation of SWI/SNF complex activity, Mitotic Exit | (186,187) |
| HDAC2 | Not determined | S394 | CK2α1 | Negative regulation of hypertrophic response in cardiomyocytes | (188) |
| HDAC4 | B55α | S298, S246, S467, S632 | PKCϵ, SIK1, SIK2, CaMKII, PKD | Negative regulation of interaction with 14-3-3; Nuclear import of HDAC4; Fibroblast differentiation; Increased glucose uptake; amelioration of neuropathic allodynia; promotes neuronal apoptosis | (76,95,101,189–194) |
| B56 | Not determined | Not determined | Ensures proper chromosomal segregation during mitosis in p53-null cells | (77) | |
| HDAC5 | B55α | S259, S279, S498 | SIK1, CaMKII, PKD, PRK1 | Nuclear localization; Negative regulation of interaction with 14-3-3 | (78,81,189,193–196) |
| HDAC7 | B55α | S155, S181, S321, S449 | PRK1 | Negative regulation of interaction with 14-3-3; Proper endothelial vessel formation | (75,80,196) |
| PRMT1 | Not determined | S297 | Not determined | Inhibition of PRMT1 activity; Decreased methylation of H4 (H4R3me2); HBV-mediated inhibition of INF-α response | (113–115,197) |
| PRMT5 | Not determined | S355 | Not determined | Inactivation of PRMT5 methyltransferase activity | (120) |
| H2A.X | B56ϵ | S129 | ATM | Antagonizes DNA damage repair initiation; promotes DNA damage resolution | (115,116,198,199) |
| TET2 | B55α | S99 | AMPK | Antagonize stability of TET2 | (128) |
| RNA Pol II | Integrator INTS8, INTS6 | S2, S5, S7 | CDK9 | Negative regulation of transcriptional initiation and elongation | (131,132) |
| SPT5 | Integrator INTS8 | S666 | CDK7, CDK9 | Impaired ‘pause, release’ mechanism of RNA Pol II | (141) |
Table 2.
Therapeutic modulators of PP2A activity.
| PP2A modulators | Target | Phosphatase activity | Ongoing Clinical Trials | Application | References |
|---|---|---|---|---|---|
| Ceramide | SET | Activating | NCT04716452: Phase I | Acute Myeloid Leukemia | (150,151,200) |
| FTY720 (Fingolimoid) | SET | Activating | NCT03941743: Phase I | Breast Cancer | (201,202) |
| FDA Approved | Multiple Myeloma | ||||
| FDA Approved | Mantle Cell Lymphoma | ||||
| NCT05137860: Phase IV | Acute Lymphoblastic Leukemia | ||||
| Bortezomib | CIP2A | Activating | NCT01371981: Phase III | Acute Myeloid Leukemia | (203–205) |
| NCT03509246: Phase II | Ovarian Cancer | ||||
| NCT01142401: Phase II | Breast Cancer | ||||
| NCT00479128: Phase I | Urothelial Cancer | ||||
| LB100 | PP2Ac | Inhibiting | NCT03027388: Phase II | Glioblastoma | (174,206) |
| NCT04560972: Phase I | Small Cell Lung Cancer | ||||
| OP499 | SET | Activating | Pre-clinical | (50,201,207) | |
| iHAP1 | PP2A-B56ϵ | Activating | Pre-clinical | (169) | |
| DT061 | PP2A-B56α, PP2A-B55α | Activating | Pre-clinical | (169–171,173,208,209) |
The function of PP2A and its contribution to tumorigenesis is highly dependent on the composition of the PP2A holoenzyme. Here, we review the complex mechanisms by which PP2A regulates the epigenetic state of cancer cells and potential ways to leverage this activity for anti-cancer therapeutics.
REGULATION OF HISTONE AND CHROMATIN DYNAMICS BY PP2A
The loss of normal chromatin regulation leads to irregular nuclear morphologies, heterochromatin redistribution and deregulated transcriptional programs, emphasizing the importance of understanding how chromatin states are dynamically regulated (31). Chromatin remodeling requires a coordinated effort between epigenetic regulators and transcriptional complexes and is largely regulated through epigenetic marks within the c-terminal tails of core histones. These marks include acetylation, methylation, ubiquitination and phosphorylation; However, the full impact of protein phosphorylation on the dynamics of chromatin architecture and chromatin reorganization is understudied and still being explored in the field (13).
Chromatin architecture
Heterochromatic regions are formed in response to repressive histone modifications, such as histone H3 lysine 9 di- and trimethylation (H3K9me2/me3) (32,33). These marks promote the sequestration and anchoring of heterochromatin to the nuclear lamina and is driven through the interaction between chromatin and intermediate filament proteins or nuclear Lamins (Lamin A, Lamin C, Lamin B1 and Lamin B2) (34). Importantly, this higher-order structure plays a critical role in the regulation of gene transcription.
Functionally, nuclear Lamins are critical for maintaining proper nuclear architecture and tethering heterochromatin. Genes located within these Lamin-associated domains at the nuclear periphery exhibit significantly reduced transcriptional levels compared to genes at more central locations (35). Recently, the Proline-Rich Protein 14 (PRR14) was discovered to have a Lamin binding domain (LBD) and associate with heterochromatin protein 1 (HP1) to sequester heterochromatin to the nuclear lamina during interphase and mitotic exit (36). Genetic knockdown of PRR14 results in abnormal nuclear morphologies similar to those seen in cancer cells, suggesting that the localization of heterochromatin to the nuclear lamina through PRR14–HP1 interaction is critical for proper chromatin organization. Dunlevy et al. demonstrated that PRR14 contains a conserved short linear motif (SLiM) consistent with PP2A-B56 substrates. This SLiM motif drives the association of PRR14 and PP2A complexes containing B56α (PP2A-B56α) (37,38). PRR14 interaction with PP2A-B56α results in the dephosphorylation of the PRR14 LBD, allowing PRR14–HP1 interaction and localization of heterochromatic regions to the nuclear lamina (Figure 1A) (37,39). Ablation of the PP2A SLiM resulted in a redistribution of PRR14 to the nucleoplasm, suggesting that PP2A is a key regulator of heterochromatin spatial organization within the nucleoplasm.
Figure 1.
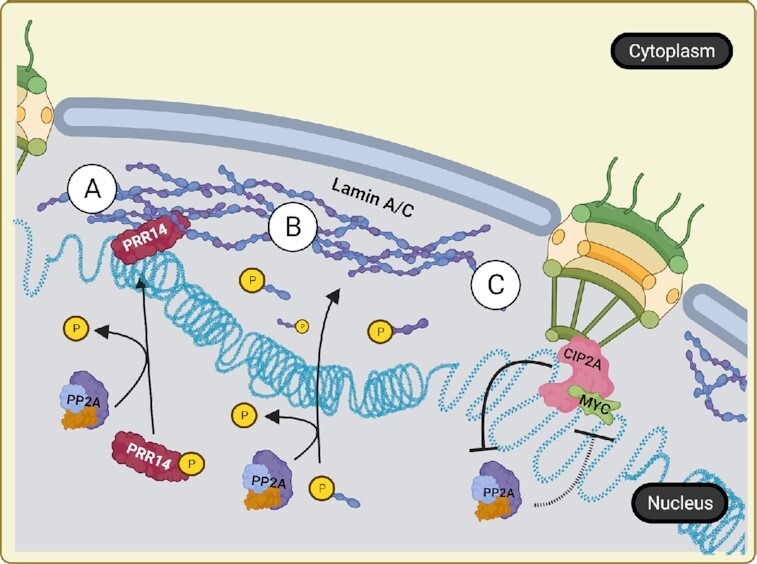
PP2A regulates heterochromatin localization and prevents euchromatic gene expression. (A) PP2A dephosphorylates heterochromatin-bound PRR14 to allow HP1–PRR14 interaction and sequestration of heterochromatin to the nuclear lamina. (B) PP2A dephosphorylates free nucleoplasmic Lamins to promote Lamin polymerization at the nuclear periphery. (C) CIP2A and MYC bind nuclear pore proteins to promote the rapid expression of MYC-dependent genes within associated euchromatin. CIP2A is a known inhibitor of PP2A activity, thereby repressing the ability of PP2A to regulate MYC.
Additionally, direct phosphorylation of Lamin proteins during mitosis promotes depolymerization and breakdown of the nuclear lamina so that cells may properly undergo cell division (40). Both PP2A and protein phosphatase 1 (PP1) are required for the dephosphorylation of Lamin proteins and the subsequent reassembly of the nuclear envelope (40,41). Phosphorylation of Lamin A/C at serine 22 (pS22) by protein kinases, such as Protein Kinase C and Cyclin Dependent Kinase 1, results in Lamin depolymerization and nucleoplasmic localization (40). This dynamic turnover has been implicated in several cancer processes independent of mitosis (40). Interestingly, nucleoplasmic Lamin has been shown to localize to specific promoters to increase gene transcription (42). This unique cellular function is regulated by the Lamin phosphorylation state, suggesting that there exists a more complex role for Lamins in cancer where phosphatases and kinases are dysregulated. Knockdown of the PP2A A subunit in HeLa cells results in a ten-fold increase in two major phosphorylation sites of Lamin A/C, pS22 and pS628 (41), resulting in nucleoplasmic localization (43). Correct Lamin distribution is important for proper sequestration and silencing of heterochromatic regions; therefore, these results indicate that PP2A suppression may lead to a spatial redistribution of chromatin and expanded gene transcription (Figure 1B). Cancer cells often exhibit atypical nuclear morphologies including increased nuclear to cytoplasmic ratio and irregular nuclear shape as a result of abnormal distribution of lamina and chromatin (44). Given the importance of phosphorylation in the breakdown and reassembly of nuclear lamina, phosphatase deregulation may significantly contribute to these abnormal nuclear morphologies.
Chromatin organization and transcriptional regulation
In addition to anchoring heterochromatic regions, Lamins also contribute to the distribution of nuclear pore complexes (NPCs) across the membrane and the transcriptional regulation of genes associated with these structures. Although the majority of DNA associated with the nuclear lamina is heterochromatic, the regions surrounding the NPC are commonly euchromatic and highly transcribed (45). NPC-associated genes have been implicated as immediate-early genes (IEGs) which are rapidly transcribed in response to cellular stimulus and are often oncogenic in nature (46). The proto-oncogene c-MYC (MYC) is a transcription factor and an IEG that regulates a wide range of genes. The active, phosphorylated form of MYC has been shown to associate with several NPC proteins, including nucleoporins TPR and Nup153 (47,48). The localization of MYC to this region is associated with a more accessible chromatin state and increased expression of genes involved in migration, proliferation and survival (48). Aberrant MYC expression and/or activation can drive tumorigenesis in most tissue types, highlighting the importance of MYC regulatory pathways. Therefore, to prevent MYC from becoming oncogenic, its activity and expression are highly regulated at the transcriptional, translational and post-translational levels (49). PP2A-B56α negatively regulates a critical MYC phosphorylation site serine 62, which is responsible for MYC’s post-translational stability and activity (20,48,50). Further complicating the role of phosphatases in MYC function, PP2A-B55α positively regulates MYC protein activity and stability through threonine 58 (51). As PP2A likely localizes to the nuclear lamina, PP2A may be an essential regulator of MYC activity at the NPC (Figure 1C) (47,48). Consistent with this hypothesis, the PP2A inhibitor, Cancerous Inhibitor of PP2A (CIP2A), has demonstrated co-localization with MYC and Lamin A/C at the NPC (47). However, CIP2A has been identified to only interact with PP2A complexes that contain B56 family subunits (52). Therefore, only PP2A-B56α, not PP2A-B55α, can be sequestered through CIP2A binding. The presence of CIP2A and MYC at the NPC signifies that the tumor suppressive activity of PP2A-B56α is likely inhibited, potentially promoting oncogenic signaling though a PP2A-B55α-MYC axis. Moreover, knockdown of CIP2A preferentially reduces phosphorylated, active MYC at the NPC but has no effect on MYC phosphorylation within the nucleoplasm (47). These studies set up a dynamic interplay between PP2A and MYC, where PP2A-B56 inhibition allows for the rapid transcription of MYC target genes at the NPC. This phenomenon implicates PP2A in regulating the spatial organization of subnuclear transcriptional domains.
Together, these studies support a model where CIP2A-mediated inhibition of PP2A at the nuclear periphery to facilitate the oncogenic activities of MYC. However, the PP2A subunits that are involved in the process and the contribution of Lamin A/C phosphorylation to association with oncogenic phenotypes need to be further explored.
Histone-mediated transcription
Phosphorylation of histone H3 (H3) at the serine 10 residue (pH3S10) provides a docking site for chromatin modifiers and increases the acetylation of neighboring residues (53). This PTM is commonly found at sites of active transcription and contributes to the expression of IEGs including c-FOS, c-JUN and MYC, indicating that phosphorylation at this site may contribute to tumorigenesis (54–56). Phosphorylation of the H3S10 residue is elevated in RAS-transformed cells and is associated with poor outcome in multiple cancers (57). Several kinases are known to contribute to the phosphorylation of H3S10, including Aurora kinases, Mitogen and Stress activated protein Kinase 1/2 (MSK1/2), and Proviral Integration site for Moloney murine leukemia virus (PIM-1) kinase (Table 1) (57,58). In particular, PIM-1 has been implicated in cancer proliferation, therapeutic resistance, and contributes to tumorigenesis in multiple tissues (59). Phosphorylation of H3S10 by PIM-1 significantly increases the transcriptional activation of MYC, upregulating approximately 20% of MYC transcriptional targets and leading to oncogenic transformation (58,60,61). Given the importance of pH3S10 to oncogenic transcription and transformation, therapeutic methods to reduce this modification could have significant clinical benefit. Both PP1 and PP2A dephosphorylate the H3S10 site, providing an essential balance to this PTM (54,60). Consistent with these findings, inhibition of PP2A/PP1 by Okadaic Acid (OA) or knockdown of A and C components of the PP2A holoenzyme promote chemical carcinogenesis by preventing PP2A from dephosphorylating the H3S10 residue (54). In addition to H3S10 dephosphorylation, the PP2A-B56β holoenzyme also negatively regulates the stability of PIM-1 (60). Knockdown of B56β leads to a 4-fold increase in PIM-1 half-life, implicating PP2A as a direct and indirect regulator of the H3S10 phosphorylation site (60).
INTERPLAY OF PP2A AND CHROMATIN REMODELING COMPLEXES
PP2A is reported to alter the activity of chromatin remodeling complexes that regulate or ‘read’ histone epigenetic marks in addition to the direct dephosphorylation of histones. Aberrant regulation of PP2A holoenzyme composition and activity alters the precise signaling that methylation- and acetylation-associated complexes provide, further contributing to oncogenesis by indirectly influencing euchromatic region accessibility.
PP2A regulation of acetylation
Histone acetylation primarily results in the relaxation of the chromatin and increased transcription of nearby genes. Acetylation marks are added and removed by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively, which function in a coordinated effort to maintain normal transcriptional programs. While increased acetylation is often associated with cancer, elevated levels of HDAC activity can similarly contribute to tumor phenotypes by silencing key tumor suppressor genes, such as p53 (5,62,63). Despite the critical role of acetylases in regulating gene transcription, therapeutic inhibition of acetylases has shown limited clinical success as single agents (64). This lack of efficacy is likely due to heterogeneous expression (65) and function of chromatin remodelers (66). The cellular localization and activity of acetylation complexes are controlled through phosphorylation, adding an additional layer of regulation to global transcriptional programs and a potential mechanism by which cells can become resistant to HAT/HDAC inhibition (67). Therefore, the use of HAT/HDAC inhibitors may benefit from combinatorial treatment with specific phosphatases that aid in the regulation of acetylases.
‘Readers’ of acetylation
PP2A also regulates the activity of lysine acetylation ‘readers’, including the Bromodomain and Extraterminal (BET) protein, Bromodomain-containing 4 (BRD4). BRD4 accumulates at euchromatic regions to promote gene transcription through the recruitment of transcription factors to the transcription start site or as a part of super-enhancer complexes (65). Phosphorylation of BRD4 by casein kinase II (CK2) is reported to increase the association of BRD4 with activating acetylation marks, making hyperphosphorylation of BRD4 a correlate of poor prognosis in multiple cancers (68–70).
Given the prominent role that BET proteins play in gene transcription, a considerable amount of effort has gone into the development of BET inhibitors with varying success. In 2011, Delmore et al. unexpectedly demonstrated that rather than global transcriptional repression, the BET inhibitor, JQ1, only modified the expression of a limited number of genes, indicating that specific genes displayed unique susceptibility to BET inhibition. Of the affected targets, the MYC super-enhancer was acutely reliant on BRD4-dependent transcriptional activation (71). MYC contains large intrinsically disordered regions and lacks tangible binding pockets, making the direct therapeutic targeting of this potent transcription factor challenging (72). Thus, BET inhibitors opened a door to new MYC therapeutic strategies (71).
While BET inhibitors have shown efficacy across various cancers, development of resistance was an immediate concern. As phosphorylation of BRD4 is counteracted by PP2A (Figure 2A), suppression of PP2A can increase BRD4 activation and resistance to BET inhibitors, including JQ1 (41,73). Therefore, restoring PP2A activity may be important for maximizing the therapeutic efficacy of BET inhibitors. For example, the combination of the PP2A-activating compound, perphenazine and JQ1 resulted in a synergistic loss of triple negative breast cancer cell viability (69,73). In addition to the transcriptional control of MYC, PP2A also directly post-translationally regulates MYC protein stability (20). In cancer, PP2A activity is commonly suppressed, leading to increased MYC phosphorylation and transcriptional activity (74). Together, these studies place PP2A as a critical mediator of both epigenetic and post-translational regulation of MYC during tumor progression.
Figure 2.

PP2A regulates proteins that contribute to acetylation and methylation of epigenetic targets. (A) PP2A dephosphorlates BRD4, preventing BRD4 from binding to acetylated residues and promoting gene transcription. (B) PP2A dephosphorylates HDAC 4/5/7 and prevents 14-3-3 binding, allowing HDAC nuclear localization and subsequent deacetylation of histones. (C) High PP2A activity is correlated with low PRMT1/5 methylase activity at the H4R3me2 methylation site. The impact of this regulation on gene transcription is cancer dependent. (D) PP2A dephosphorylates TET2 and reduces TET2 stability. This action antagonizes efficient methylcytosine removal.
‘Erasers’ of acetylation
Histone deacetylase (HDAC) complexes play an important role in suppressing gene transcription by removing acetylation marks and promoting formation of heterochromatin. Given that deregulation of HDAC activity can contribute to either tumor suppressive or oncogenic signaling, proper HDAC nuclear localization is extremely important for cellular homeostasis (11). Class IIa HDACs: HDAC4, HDAC5 and HDAC7 have all been reported to be substrates of PP2A (75–78). Phosphorylation of these complexes creates binding sites for the 14-3-3 chaperone proteins, sequestering HDAC 4/5/7 to the cytosol (78–81). Dephosphorylation of these sites by PP2A promotes the rapid nuclear localization of HDACs and the suppression of respective target genes (Figure 2B).
PP2A-mediated regulation of HDAC complexes greatly depends on the composition of the PP2A holoenzyme (24). In contrast to the known tumor suppressive role of the B56α subunit, a growing number of studies indicate that the B55α subunit may play a pro-oncogenic role (51,82–86). Consistent with these findings, the PP2A-B55α holoenzyme has been shown to regulate angiogenesis by increasing yes-associated protein 1 (YAP1) transcription in response to vascular endothelial growth factor receptor (VEGFR) signaling and drive the accumulation of hypoxia inducible factor 1 subunit alpha (HIF1a) (84,87). Additionally, PP2A-B55α has been implicated in promoting endothelial vascular integrity and proper lumen architecture by positively regulating HDAC7 nuclear import and promoting HDAC7-dependent transcriptional programs (75,80). Although these results implicate B55α in the positive regulation of angiogenesis, treatment with the small molecule inhibitor of PP2A, LB100, resulted in increased angiogenesis and vascular permeability in hepatocellular carcinoma and pancreatic cancer (88,89). As LB100 is a ubiquitous catalytic inhibitor of PP2A, this discrepancy highlights the importance of understanding the individual contribution of PP2A B subunits to tumor phenotypes.
HDAC4 signaling contributes to several cancer phenotypes, including increased proliferation and suppression of differentiation transcriptional programs (90). Although HDAC4 nuclear localization by PP2A has been shown in other cell types, the contribution of this interaction to tumorigenesis is not well characterized. HDAC4 promotes proper chromosomal segregation during mitosis in p53-null cancer cells (77). This function is in part dependent on the interaction between HDAC4 and PP2A-B56α, a key regulator of the metaphase-to-anaphase checkpoint of mitosis (91–94). Consistent with PP2A’s regulation of HDAC function, loss of PP2A-B56α expression resulted in chromosomal segregation defects and phenocopied HDAC4 knockdown. However, the direct impact of altered PP2A-B56α function on HDAC4 localization and transcriptional programs is unknown. It is plausible that decreased PP2A activity combined with genetic loss of p53 may lead to the propagation of cells with a high degree of genomic instability, ultimately promoting tumorigenesis, but further studies are needed.
Regulation of HDAC4 by PP2A has also been shown to impact the differentiation state of fibroblasts (95). Reprogramming of fibroblasts by tumor cells has emerged as a critical component of tumor biology, with infiltration and activation of fibroblasts contributing to proliferation, metastasis and therapeutic resistance in several cancer types (96–99). In response to extracellular signals such as transforming growth factor beta (TGFβ), fibroblasts can take on a myofibroblastic phenotype, displaying increased expression of alpha smooth muscle actin (αSMA) and extracellular matrix proteins (100). This reprogramming is dependent on the nuclear localization of HDAC4, as knockdown of HDAC4 prevents TGFβ−induced αSMA expression and the suppression of PP2A rescues this phenotype (95,100,101). PP2A activating compounds have been shown to reduce oncogenic signaling in cancer cells; however, this phenomenon may not be true in fibroblasts. PP2A activation in cancer associated fibroblasts (CAFs) may further promote oncogenic activities of HDAC regulation given that activation of CAFs can positively contribute to fibrosis. This event can further complicate therapeutic treatment and stresses the importance of understanding PP2A holoenzyme- and tissue-specific function.
PP2A regulation of methylation
Histone methylation marks are extremely diverse in function and can be associated with promotion or inhibition of gene transcription. The role of each methylation mark depends on the genomic site, the presence of other neighboring histone PTMs or epigenetic marks, as well as the number of methyl groups deposited on any given residue (mono-, di-, tri-methylation) (8). Similar to phosphorylation and acetylation, the methylation of various histone residues is regulated by methyltransferases and demethylases that work in tandem to maintain homeostatic gene expression. Importantly, mutation or dysregulation of these complexes can lead to transformation and tumorigenesis (102,103).
‘Writers’ of histone methylation
While methylation of lysine residues (K) is more common, the field of arginine (R) methyltransferases has significantly expanded within the past decade. There are three subclasses of protein arginine methyltransferases (PRMTs): (i) asymmetric dimethylarginine (ADMA), (ii) symmetric dimethylarginine (SDMA) and (iii) monomethyl arginine (MMA) (103). All PRMTs deposit monomethyl marks, but symmetrical or asymmetrical dimethylation is dependent on the specific PRMT class, adding another layer of complexity to the signaling (104). PRMTs dynamically contribute to the co-activation or co-repression of genes by promoting the recruitment of transcription factors or other epigenetic complexes (105).
PRMT1 is the predominant ADMA methyltransferase, accounting for approximately 85% of total arginine methyltransferase activity (106). PRMT1 primarily asymmetrically dimethylates the H3R4 residue (H3R4me2a), which is associated with transcriptional activation (107,108). High levels of H3R4 methylation have been positively correlated within tumor grade and risk of recurrence in prostate cancer (109). Additionally, PRMT1 has oncogenic roles in colorectal cancer, breast cancer and pancreatic cancer (110–112). PP2A has been reported to negatively regulate PRMT1 through dephosphorylation at S297, supporting a possible tumor suppressive role for PP2A in the context of PRMT1 regulation (113). However, in Hepatitis C (HepC)-induced hepatocellular carcinoma (HCC), PP2A activity is upregulated in the presence of the HepC virus, aiding in viral replication and paradoxically promoting tumorigenesis of HepC-HCC (114). The combination of high PP2A activity and low PRMT1 activity in HepC-HCC has also been implicated in promoting anchorage independent growth and clonogenic proliferation through dysregulation of histone H4R3 methylation (Figure 2C) (115,116). Further, a follow-up study determined that the inhibition of PP2A with LB100 enhanced the effect of chemotherapies in HepC-HCC, potentially pointing to a unique role for PP2A in this tumor type (115). Although the specific PP2A B subunits responsible for these phenotypes were not explored, these results implicate PP2A in a novel pro-tumorigenic role in HepC-HCC.
PRMT5 is the predominant SDMA methyltransferase and is generally associated with gene repression (103). There is an abundance of evidence suggesting that PRMT5 plays an oncogenic role in many cancers such as prostate cancer, pancreatic cancer, and colorectal cancer (117–119). In adult T-cell leukemia (ATL), PP2A has been reported to negatively regulate PRMT5 through the dephosphorylation of S355. This interaction is mediated by N-MYC downstream regulated gene (NDRG2), which acts as an adaptor between PRMT5 and PP2A (120,121). When NDRG2 expression is low, PRMT5 is reported to promote cell growth and prevent apoptosis (120). Therefore, PP2A-activating compounds may be effective in cancers with functional NDRG2 and may synergize with PRMT5 inhibiting compounds, while NDRG2 deletions may render this strategy ineffective. Conversely, in glioblastoma, PRMT5 exhibits a tumor suppressive role, while NDRG2 is reported to have oncogenic properties (122,123). Consistent with the findings, treatment of glioblastoma with PP2A-inhibitor LB100 has an anti-tumor effect in the context of PRMT5 regulation (122). Therefore, while the mechanism by which PP2A regulates PRMT5 is the similar, the biological outcome of this signaling is contradictory between these two cancers and supports the use of alternative therapeutic approaches.
‘Erasers’ of DNA methylation
In addition to histones, cytosine nucleotides can also be directly methylated. Methylated cytosine results in the eventual formation of large stretches of CpG islands, or areas of dense DNA methylation, normally found at promoters of actively repressed genes. Reversal of methylated cytosine is a process that requires multiple oxidative reactions by ten-eleven translocation (TET) proteins and conversion to an abasic site that can be recognized for base excision repair (124). The TET proteins predominantly function as tumor suppressors and are important for sequential oxidation of 5’-methylcytosine for restoration of the unmodified cytosine base (125). The stability of TET proteins is regulated by phosphorylation (126). AMP activated kinase (AMPK) is reported to phosphorylate TET2 at serine 99 (S99), driving the interaction with 14-3-3 and increasing global DNA 5-hydroxymethylcytosine levels. PP2A dephosphorylates TET2 at S99, antagonizing TET2 stability and activity (Figure 2D) (127,128). The PP2A-TET2 interaction is mediated specifically through the PP2A-B55α complex, consistent with an oncogenic role for this PP2A B subunit (128). The importance of this phospho-regulation is underscored by the fact that mutations in TET2 often occur in the S99 region (129), causing aberrant formation of CpG islands and silencing of genes that are important for homeostatic regulation.
Direct transcriptional regulation
PP2A-integrator complex (INTAC)
The canonical PP2A heterotrimeric complex consists of an A/C dimer paired with a regulatory B subunit, which dictates substrate specificity. Recently, multiple papers have been published describing a non-canonical PP2A holoenzyme called INTAC in which the 14-subunit Integrator complex (130) displaces canonical B subunits and directly associates with the PP2A A/C heterodimer (131–133). Consistent with the characterized role of the Integrator complex (130,134), INTAC localizes to RNA polymerase II (RNA Pol II)-bound chromatin and exhibits both endonuclease and phosphatase activity (131). Knockdown of the PP2A C subunit resulted in a genome-wide increase in RNA Pol II phosphorylation at Serine 2, 5 and 7 within the c-terminal tail and increased the expression of genes targeted by INTAC activity (131). Based on the canonical function of these residues, the phosphatase activity of the INTAC complex negatively regulates both transcriptional initiation and elongation (Figure 3) (131). The role of phosphatases and PP2A-integrator function on transcriptional machinery was well summarized by Cossa et al. earlier this year (135).
Figure 3.
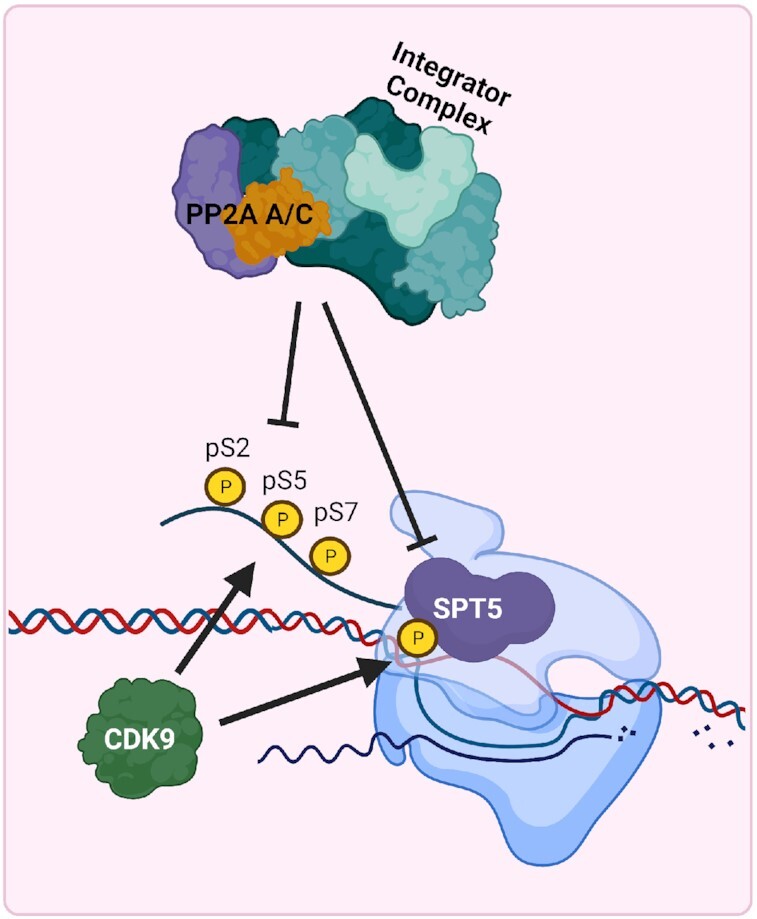
PP2A contributes to regulation of transcription as part of the Integrator-PP2A (INTAC) complex. The PP2A A/C subunits bind to the Integrator complex and promote dephosphorylation of S2/5/7 residues on the tail of RNA Polymerase II in addition to SPT5. These actions prevent efficient transcriptional initiation and elongation of RNA transcripts. INTAC dephosphorylation of RNA Polymerase II and SPT5 is counteracted by CDK9 phosphorylation.
Cancer cells commonly maintain high levels of transcription to accommodate increased proliferation rates and oncogene-induced gene expression (136). This high transcriptional output may represent a cancer-specific vulnerability that can be leveraged through activation of INTAC complex activity. Cyclin Dependent Kinase 9 (CDK9) promotes transcriptional elongation and increases mRNA transcription through RNA Pol II phosphorylation at Serine 2 and 5 (137). Multiple types of solid and hematopoietic malignancies are addicted to elevated transcription rates and are, therefore, sensitive to CDK9 inhibitors (138–140). It was recently discovered that the INTAC complex directly opposes CDK9 phosphorylation and that treatment with a small molecule activator of PP2A (SMAP) synergizes with CDK9 inhibition (132). In addition, SPT5, an essential positive regulator of transcription and a target of CDK9 phosphorylation, has also been observed to be negatively regulated by the INTAC complex, further supporting CDK9 inhibitor/PP2A activator synergy (Figure 3) (133,141). While CDK9 inhibitors alone have demonstrated antitumoral activity in preclinical research, CDK9 inhibitors have been largely unsuccessful in the clinic due to limited drug efficacy and adverse side effects (142). Therefore, combination treatment with CDK9 inhibitors and SMAPs may increase anti-tumor responses while reducing adverse side effects in patients.
THERAPEUTIC TARGETING OF PP2A IN CANCER
As PP2A regulates a wide range of targets, its activity and holoenzyme composition are tightly controlled through expression, localization, phosphorylation, methylation and association with cellular inhibitors (24). There are three major endogenous cellular inhibitors that regulate PP2A activity: Inhibitor 2 of PP2A (I2PP2A or SET), CIP2A and PME1, all of which have been heavily implicated in tumor promotion and are overexpressed in many cancer types (30). Kauko et al. demonstrated that genetic knockdown of SET, CIP2A or PME1 globally increases the sensitivity of cancer cells to a panel of kinase inhibitors, while knockdown of the PP2A A subunit drives global therapeutic resistance (41). These studies also show that CIP2A is predominately cytoplasmic, whereas PME1 and SET are nuclear. Together, these studies implicate PP2A as an important rheostat for therapeutic response and suggest that the spatial regulation of PP2A activity may lead to distinct phenotypes and unique therapeutic combinations depending on cellular context. In an effort to modify PP2A activity in tumors, three main therapeutic strategies have been explored: preventing cellular inhibitors from binding to PP2A, directly nucleating the PP2A holoenzyme or global inhibition of PP2A (Table 2) (143,144).
Inhibiting the inhibitors of PP2A
SET is a global inhibitor of PP2A that binds to the C subunit of PP2A and inhibits catalytic activity of the PP2A holoenzyme (145,146). High SET expression has been implicated in poor survival outcomes in a diverse set of cancers, including colorectal cancer, breast cancer and hepatocellular carcinoma (147–149). Suppression of SET leads to decreased phosphorylation of proteins involved in histone methylation and chromatin organization, consistent with SET nuclear localization (41). FTY720 (Fingolimod) is a ceramide derivative and sphingosine analog that inhibits the binding of SET to the PP2A holoenzyme, thereby indirectly promoting PP2A phosphatase activity (150,151). FTY720-mediated inhibition of SET has shown promising anti-tumor effects through the activation of PP2A in lung cancer, colorectal cancer and various hematologic malignancies (152–155). In colorectal cancer, FTY720 was found to promote PP2A-mediated degradation of Polycomb Group RING Finger Protein 4, BMI1, thereby preventing Polycomb Repressive Complex 1 (PRC1)-based epigenetic modifications (156). Given that PRC1 is implicated in promoting stem-like states and metastasis in multiple cancers, particularly through BMI1-containing PRC1 complexes, the re-activation of PP2A in this context may help to target relevant cell sub-populations within tumors (157–159). Other studies have indicated that FTY720 can suppress transcription by altering histone modifications, including H3K27 trimethylation, although the direct prevention of PRC1-mediated oncogenesis by PP2A has not been explored (160,161). Despite the anti-tumor effects of FTY720, this compound elicits severe cardiac toxicities among other side effects. An analog compound, SH-BC-893, potentially addresses these issues and is currently in preclinical phases (162,163). While there are ongoing efforts in the cancer field to develop direct small molecule inhibitors of PME1 and CIP2A, these strategies are lagging compared to those that target SET.
Direct activation of PP2A phosphatase activity
While cellular PP2A inhibitors play a significant role in PP2A function, many have PP2A-independent functions, making the therapeutic inhibition of these factors problematic (164,165). Therefore, a considerable amount of effort has been spent developing direct activators of PP2A. Early in the development of PP2A activators, the phenothiazine class of antipsychotics were found to have antiproliferative effects on cancer cells (166) by indirect activation of PP2A through inhibition of calmodulin (167). Due to off-target effects of D2 dopamine receptor antagonism, phenothiazines could not be used therapeutically in cancer but provided the structural basis for the development of more specific PP2A activators: improved Heterocyclic Activators of PP2A (iHAPs) and Small Molecule Activators of PP2A (SMAPs) (168). These allosteric activators directly increase PP2A activity by nucleating and stabilizing various PP2A holoenzymes (169,170). Interestingly, both iHAPs and SMAPs promote the incorporation of B56 family members (B56ϵ and B56α, respectively) into the complex (169,170). However, studies have suggested that SMAPs, such as DT061 can promote incorporation of other B subunits, including B55α (169). Differences in PP2A complex stabilization have been observed between cancers such as lung adenocarcinoma (170) or in various hematologic malignancies (169). These conflicting results may be due to variances in expression of PP2A complexes or through differences in post-translational modification of PP2A within each unique malignancy (74). SMAPs have shown significant pre-clinical efficacy in vivo, both as a single agent and in combination with other compounds but has yet to be assessed in the context of epigenetic-based therapeutics (168).
Direct inhibition of PP2A phosphatase activity
While most cancers have been shown to be sensitive to PP2A re-activation, there are some contexts in which tumors display increased sensitivity to PP2A inhibition. Lung cancer, pancreatic cancer and leukemias have been reported to be sensitive to therapeutic re-activation of PP2A activity (169,171–173). On the other hand, cancers such as glioblastoma show therapeutic efficacy with PP2A catalytic inhibitors, such as LB100, particularly when used in combination with DNA damaging therapeutics (122,174–177). The combination of PP2A and epigenetic inhibitors are currently being explored and may prove effective in malignancies of the nervous system (122,144). In either context (activation or inhibition), PP2A holds great promise as a therapeutic target as more specific compounds are developed (Table 2). However, the complex regulation of PP2A demands a greater understanding of the context-specific functions of the PP2A holoenzyme for the rational design of new therapeutic compounds and the use of combination therapies.
DISCUSSION
PP2A therapeutic targeting challenges
One of the most exciting aspects of PP2A-centric therapeutics has been the low toxicity observed in vivo (170). While the exact mechanism underlying the potential cancer cell selectivity of PP2A targeting agents is still being explored, it is likely that as tumors progress, cancer cells become reliant on low PP2A activity for survival. Therefore, when PP2A is therapeutically activated, cancer cells are unable to compensate for the loss in oncogenic signaling and undergo cell death. In contrast, normal cells, which are predominately quiescent, have a much higher threshold for the amount of PP2A activity that can be tolerated before losing critical survival signals (Figure 4). Intriguingly, HDAC inhibitors seem to function similarly, with cancer cells displaying a unique vulnerability to epigenetic-based therapeutics (178). Ultimately, these findings indicate that cancer cells develop unique regulatory mechanisms that differ from normal cell populations.
Figure 4.
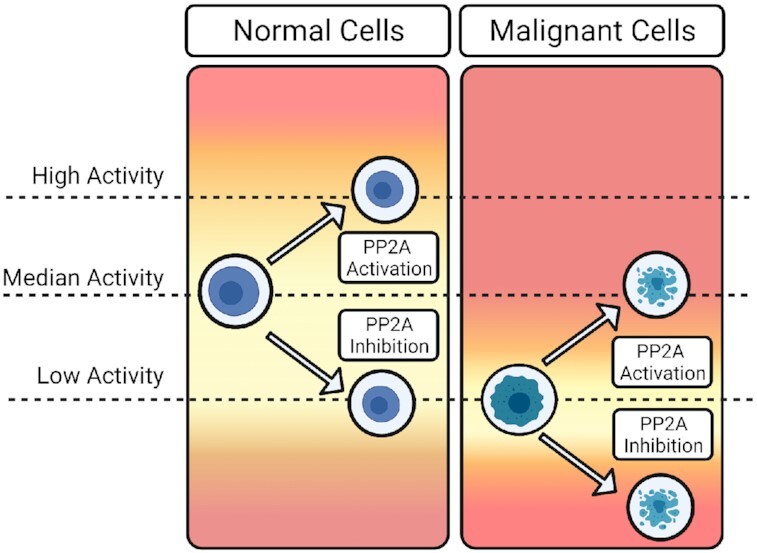
Cancer cells have different thresholds for PP2A activity than normal cells Normal cells (left) maintain median PP2A activity levels and have a larger tolerance for fluctuations in PP2A phosphatase activity. In contrast, malignant cells (right) become addicted to low levels of PP2A activity and, therefore, become more susceptible to fluctuations in activity. Upon therapeutic activation of PP2A, malignant cells cannot withstand PP2A activation returning to baseline levels and the subsequent loss of oncogenic signaling. Similarly, given that malignant cells already function on low levels of PP2A activity, inhibition of PP2A activity results in a loss of survival signals and cell apoptosis. Since PP2A activators and inhibitors display little to no toxicity, this would suggest that normal cells are able to maintain the signals necessary for survival in both contexts.
Several studies demonstrate that PP2A contributes to the regulation of various epigenetic and transcriptional processes, but most of these studies inappropriately treat PP2A as a singular entity. For example, many studies utilize the phosphatase inhibitor, OA, as proof that PP2A regulates specific signaling pathways. However, OA inhibits both PP1 and PP2A, which confounds experimental conclusions (179). Similarly, very few research studies tease apart the contribution of distinct PP2A holoenzymes from global PP2A activity. As a result of these oversimplifications, several reports indicate potentially contradictory roles for PP2A in cancer epigenetics. Additionally, it is likely that some PP2A subunits can in part compensate for the loss of other subunits. Although variants of B subunits within each of the B subunit families (B55, B56, PR72 and Striatins) have high degrees of structural homology, the targets and function of these B subunits can be distinct. These gaps in knowledge severely limit our understanding of PP2A-dependent phenotypes and complicate the clinical use of PP2A therapeutics.
Just as PP2A is not ‘one’ protein, the localization and function of PP2A complexes may not be restricted to one cellular compartment or be uniform in response. The functional consequence of higher-order chromatin architecture, including TAD domains, phase separation and chromosome territories are all being interrogated in the context of cancer phenotypes (180,181). The presence of these complex nuclear microdomains impacts not only inter- and intratumoral heterogeneity but potentially drives differential regulation of the epigenetic processes by phosphatases in distinct locations. Delineating the spatial and kinetic activities of PP2A will potentially uncover the critical cancer-specific mechanisms verses those that are dispensable.
Leveraging the PP2A interactome for personalized medicine
Extensive research efforts have been spent determining the consequence of specific phosphorylation sites on protein function. Interestingly, the number of kinase-regulated proteins that have been identified significantly overshadows the number of proteins known to be direct targets of phosphatases, suggesting that the cancer-associated PP2A interactome is far from complete (182). The transient nature of the protein-protein interactions between phosphatases and their substrates has severely limited our ability to identify substrates by traditional methods of detection (e.g. co-immunoprecipitation and mass spectrometry-based assays). To overcome this barrier and make significant strides in PP2A substrate identification, studies are now incorporating more advanced techniques, such as proximity labeling and crosslinking to identify PP2A complexes in live cells. For example, TurboID (a modified version of BioID) has been performed to create an interactome map of specific PP2A B subunits utilizing a unique biotin ligase that covalently label proteins within close proximity (183). This unbiased approach generates a large number of potential targets but lacks spatial information about these interactions and generates a high rate of false positives due to the promiscuous nature of the ligase. Recently, a conditional version of this assay, SplitID, has been developed, in which two proteins each get one half of the biotin ligase and only form a functional ligase when complexed together (184). This strategy has been used to identify targets of specific PP1 complexes but has not yet been used specifically with individual PP2A B subunits.
Proximity ligation assays (PLA) has been used as a targeted approach to identify not only if two proteins interact (phosphatase-substrate) but the location of the complex within the cell. This additional spatial information is invaluable for dissecting potential subcellular domains of phosphatase function (nuclear, perinuclear, cytoplasmic, etc.) and dynamic shifts in protein localization or PP2A holoenzyme composition in response to perturbations such as oncogenic mutation. To decipher transcriptional regulatory complexes, chromatin immunoprecipitation with selective isolation of chromatin-associated proteins (ChIP-SICAP) is now being used to isolate both protein complexes and their associated DNA in the same experiment. This unique approach uses crosslinking to stabilize transient protein–protein interactions and could be applied to identify specific PP2A-regulated genes and associated binding partners. Ultimately, the toolbox available to interrogate PP2A interactions is growing rapidly and will no doubt increase our understanding of this complex family of regulators.
As we begin to unravel the context-specific roles of PP2A in the regulation of epigenetic and transcriptional mechanisms, new compounds could be developed to capitalize on the unique functions of individual PP2A B subunits. Already, compounds are emerging that preferentially activate unique PP2A complexes (SMAPs and iHAP1), suggesting that eventually we may be able to tailor therapeutics to specific PP2A-dependent cancer phenotypes (170). Since the dynamic regulation of PP2A holoenzyme composition can greatly differ between tissues and cancer types, this strategy would allow for a more personalized therapeutic approach.
ACKNOWLEDGEMENTS
Figures 1-4 and graphical abstract were created using BioRender.com. We would like to thank the Purdue University Center for Cancer Research and the Indiana University Pancreatic Challenges and Solutions Working Group for their continued support and members of the Allen-Petersen lab for thoughtful comments during the editing process.
Contributor Information
Samantha L Tinsley, Department of Biological Sciences, Purdue University, West Lafayette, IN 47907, USA; Purdue University Interdisciplinary Life Sciences, Purdue University, West Lafayette, IN 47907, USA.
Brittany L Allen-Petersen, Department of Biological Sciences, Purdue University, West Lafayette, IN 47907, USA; Purdue University Center for Cancer Research, Purdue University, West Lafayette, IN 47907, USA.
FUNDING
National Cancer Institute [K22 CA237620-01A1 to B.A.P.]; Purdue University Ross-Lynn Research Scholar Fund; Ralph W. and Grace M. Showalter Research Trust; Purdue Center for Cancer Research SIRG Graduate Research Assistantship (to S.L.T.).
Conflict of interest statement. None declared.
REFERENCES
- 1. Ciriello G., Magnani L. The many faces of cancer evolution. Iscience. 2021; 24:102403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Flavahan W.A., Gaskell E., Bernstein B.E. Epigenetic plasticity and the hallmarks of cancer. Science. 2017; 357:eaal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marine J.-C., Dawson S.-J., Dawson M.A. Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer. 2020; 20:743–756. [DOI] [PubMed] [Google Scholar]
- 4. Baylin S.B., Jones P.A. A decade of exploring the cancer epigenome — biological and translational implications. Nat. Rev. Cancer. 2011; 11:726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Audia J.E., Campbell R.M. Histone modifications and cancer. Csh Perspect Biol. 2016; 8:a019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Topper M.J., Vaz M., Marrone K.A., Brahmer J.R., Baylin S.B. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 2020; 17:75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Furtado C.L.M., Luciano M.C.D.S., Santos R.D.S., Furtado G.P., Moraes M.O., Pessoa C. Epidrugs: targeting epigenetic marks in cancer treatment. Epigenetics. 2019; 14:1164–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hyun K., Jeon J., Park K., Kim J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017; 49:e324–e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vicente-Dueñas C., Hauer J., Cobaleda C., Borkhardt A., Sánchez-García I. Epigenetic priming in cancer initiation. Trends Cancer. 2018; 4:408–417. [DOI] [PubMed] [Google Scholar]
- 10. Liu F., Wang L., Perna F., Nimer S.D. Beyond transcription factors: how oncogenic signalling reshapes the epigenetic landscape. Nat. Rev. Cancer. 2016; 16:359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bahl S., Seto E. Regulation of histone deacetylase activities and functions by phosphorylation and its physiological relevance. Cell Mol. Life Sci. 2021; 78:427–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang Y., Li G. Post-translational modifications of PRC2: signals directing its activity. Epigenet Chromatin. 2020; 13:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gil R.S., Vagnarelli P. Protein phosphatases in chromatin structure and function. Biochim. Biophys. Acta Mol. Cell Res. 2018; 1866:90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dolgin E. The most popular genes in the human genome. Nature. 2017; 551:427–431. [DOI] [PubMed] [Google Scholar]
- 15. Vainonen J.P., Momeny M., Westermarck J. Druggable cancer phosphatases. Sci. Transl. Med. 2021; 13:eabe2967. [DOI] [PubMed] [Google Scholar]
- 16. Westermarck J. Targeted therapies don’t work for a reason; the neglected tumor suppressor phosphatase PP2A strikes back. FEBS J. 2018; 285:4139–4145. [DOI] [PubMed] [Google Scholar]
- 17. Mao Z., Liu C., Lin X., Sun B., Su C. PPP2R5A: a multirole protein phosphatase subunit in regulating cancer development. Cancer Lett. 2018; 414:222–229. [DOI] [PubMed] [Google Scholar]
- 18. Meeusen B., Janssens V. Tumor suppressive protein phosphatases in human cancer: emerging targets for therapeutic intervention and tumor stratification. Int. J. Biochem. Cell Biol. 2018; 96:98–134. [DOI] [PubMed] [Google Scholar]
- 19. Ruvolo P.P., Qui Y.H., Coombes K.R., Zhang N., Ruvolo V.R., Borthakur G., Konopleva M., Andreeff M., Kornblau S.M. Low expression of PP2A regulatory subunit B55α is associated with T308 phosphorylation of AKT and shorter complete remission duration in acute myeloid leukemia patients. Leukemia. 2011; 25:1711–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arnold H.K., Sears R.C. Protein phosphatase 2A regulatory subunit B56α associates with c-Myc and negatively regulates c-Myc accumulation†. Mol. Cell Biol. 2006; 26:2832–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lin S.S., Bassik M.C., Suh H., Nishino M., Arroyo J.D., Hahn W.C., Korsmeyer S.J., Roberts T.M. PP2A regulates BCL-2 phosphorylation and proteasome-mediated degradation at the endoplasmic reticulum*. J. Biol. Chem. 2006; 281:23003–23012. [DOI] [PubMed] [Google Scholar]
- 22. Letourneux C., Rocher G., Porteu F. B56-containing PP2A dephosphorylate ERK and their activity is controlled by the early gene IEX-1 and ERK. Embo J. 2006; 25:727–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ruvolo P.P. The broken “Off” switch in cancer signaling: PP2A as a regulator of tumorigenesis, drug resistance, and immune surveillance. BBA Clin. 2016; 6:87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sangodkar J., Farrington C.C., McClinch K., Galsky M.D., Kastrinsky D.B., Narla G. All roads lead to PP2A: exploiting the therapeutic potential of this phosphatase. FEBS J. 2016; 283:1004–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Seshacharyulu P., Pandey P., Datta K., Batra S.K. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett. 2013; 335:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen W., Arroyo J.D., Timmons J.C., Possemato R., Hahn W.C. Cancer-associated PP2A Aα subunits induce functional haploinsufficiency and tumorigenicity. Cancer Res. 2005; 65:8183–8192. [DOI] [PubMed] [Google Scholar]
- 27. Haesen D., Asbagh L.A., Derua R., Hubert A., Schrauwen S., Hoorne Y., Amant F., Waelkens E., Sablina A., Janssens V. Recurrent PPP2R1A mutations in uterine cancer act through a dominant-negative mechanism to promote malignant cell growth. Cancer Res. 2016; 76:5719–5731. [DOI] [PubMed] [Google Scholar]
- 28. Muggerud A.A., Rønneberg J.A., Wärnberg F., Botling J., Busato F., Jovanovic J., Solvang H., Bukholm I., Børresen-Dale A.-L., Kristensen V.N. et al. Frequent aberrant DNA methylation of ABCB1, FOXC1, PPP2R2B and PTEN in ductal carcinoma in situ and early invasive breast cancer. Breast Cancer Res. Bcr. 2010; 12:R3–R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tan J., Lee P.L., Li Z., Jiang X., Lim Y.C., Hooi S.C., Yu Q. B55β-associated PP2A complex controls PDK1-directed Myc signaling and modulates rapamycin sensitivity in colorectal cancer. Cancer Cell. 2010; 18:459–471. [DOI] [PubMed] [Google Scholar]
- 30. Kauko O., Westermarck J. Non-genomic mechanisms of protein phosphatase 2A (PP2A) regulation in cancer. Int. J. Biochem. Cell Biol. 2018; 96:157–164. [DOI] [PubMed] [Google Scholar]
- 31. Bitman-Lotan E., Orian A. Nuclear organization and regulation of the differentiated state. Cell Mol. Life Sci. 2021; 78:3141–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Becker J.S., Nicetto D., Zaret K.S. H3K9me3-dependent heterochromatin: barrier to cell fate changes. Trends Genet. 2016; 32:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Poleshko A., Smith C.L., Nguyen S.C., Sivaramakrishnan P., Wong K.G., Murray J.I., Lakadamyali M., Joyce E.F., Jain R., Epstein J.A. H3K9me2 orchestrates inheritance of spatial positioning of peripheral heterochromatin through mitosis. Elife. 2019; 8:e49278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Solovei I., Wang A.S., Thanisch K., Schmidt C.S., Krebs S., Zwerger M., Cohen T.V., Devys D., Foisner R., Peichl L. et al. LBR and Lamin A/C Sequentially Tether Peripheral Heterochromatin and Inversely Regulate Differentiation. Cell. 2013; 152:584–598. [DOI] [PubMed] [Google Scholar]
- 35. Akhtar W., de Jong J., Pindyurin A.V., Pagie L., Meuleman W., de Ridder J., Berns A., Wessels L.F.A., van Lohuizen M., van Steensel B. Chromatin Position Effects Assayed by Thousands of Reporters Integrated in Parallel. Cell. 2013; 154:914–927. [DOI] [PubMed] [Google Scholar]
- 36. Poleshko A., Mansfield K.M., Burlingame C.C., Andrake M.D., Shah N.R., Katz R.A. The human protein PRR14 tethers heterochromatin to the nuclear lamina during interphase and mitotic exit. Cell Rep. 2013; 5:292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hertz E.P.T., Kruse T., Davey N.E., López-Méndez B., Sigurðsson J.O., Montoya G., Olsen J.V., Nilsson J. A conserved motif provides binding specificity to the PP2A-B56 phosphatase. Mol. Cell. 2016; 63:686–695. [DOI] [PubMed] [Google Scholar]
- 38. Wang X., Bajaj R., Bollen M., Peti W., Page R. Expanding the PP2A interactome by defining a B56-specific SLiM. Structure. 2016; 24:2174–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dunlevy K.L., Medvedeva V., Wilson J.E., Hoque M., Pellegrin T., Maynard A., Kremp M.M., Wasserman J.S., Poleshko A., Katz R.A. The PRR14 heterochromatin tether encodes modular domains that mediate and regulate nuclear lamina targeting. J. Cell Sci. 2020; 133:jcs240416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murray-Nerger L.A., Cristea I.M. Lamin post-translational modifications: emerging toggles of nuclear organization and function. Trends. Biochem. Sci. 2021; 46:832–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kauko O., Imanishi S.Y., Kulesskiy E., Yetukuri L., Laajala T.D., Sharma M., Pavic K., Aakula A., Rupp C., Jumppanen M. et al. Phosphoproteome and drug-response effects mediated by the three protein phosphatase 2A inhibitor proteins CIP2A, SET, and PME-1. J. Biol. Chem. 2020; 295:4194–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ikegami K., Secchia S., Almakki O., Lieb J.D., Moskowitz I.P. Phosphorylated Lamin A/C in the nuclear interior binds active enhancers associated with abnormal transcription in progeria. Dev. Cell. 2020; 52:699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kochin V., Shimi T., Torvaldson E., Adam S.A., Goldman A., Pack C.-G., Melo-Cardenas J., Imanishi S.Y., Goldman R.D., Eriksson J.E. Interphase phosphorylation of lamin A. J. Cell Sci. 2014; 127:2683–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Worman H.J., Bonne G. Laminopathies”: A wide spectrum of human diseases. Exp. Cell Res. 2007; 313:2121–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Arib G., Akhtar A. Multiple facets of nuclear periphery in gene expression control. Curr. Opin. Cell Biol. 2011; 23:346–353. [DOI] [PubMed] [Google Scholar]
- 46. Kadota S., Ou J., Shi Y., Lee J.T., Sun J., Yildirim E. Nucleoporin 153 links nuclear pore complex to chromatin architecture by mediating CTCF and cohesin binding. Nat. Commun. 2020; 11:2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Myant K., Qiao X., Halonen T., Come C., Laine A., Janghorban M., Partanen J.I., Cassidy J., Ogg E.-L., Cammareri P. et al. Serine 62-phosphorylated MYC associates with nuclear lamins and its regulation by CIP2A is essential for regenerative proliferation. Cell Rep. 2015; 12:1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Su Y., Pelz C., Huang T., Torkenczy K., Wang X., Cherry A., Daniel C.J., Liang J., Nan X., Dai M.-S. et al. Post-translational modification localizes MYC to the nuclear pore basket to regulate a subset of target genes involved in cellular responses to environmental signals. Gene Dev. 2018; 32:1398–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McAnulty J., DiFeo A. The molecular ‘Myc-anisms’ behind Myc-driven tumorigenesis and the relevant Myc-directed therapeutics. Int. J. Mol. Sci. 2020; 21:9486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Farrell A.S., Allen-Petersen B., Daniel C.J., Wang X., Wang Z., Rodriguez S., Impey S., Oddo J., Vitek M.P., Lopez C. et al. Targeting inhibitors of the tumor suppressor PP2A for the treatment of pancreatic cancer. Mol. Cancer Res. 2014; 12:924–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang L., Zhou H., Li X., Vartuli R.L., Rowse M., Xing Y., Rudra P., Ghosh D., Zhao R., Ford H.L. Eya3 partners with PP2A to induce c-Myc stabilization and tumor progression. Nat. Commun. 2018; 9:1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang J., Okkeri J., Pavic K., Wang Z., Kauko O., Halonen T., Sarek G., Ojala P.M., Rao Z., Xu W. et al. Oncoprotein CIP2A is stabilized via interaction with tumor suppressor PP2A/B56. Emb. Rep. 2017; 18:437–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Thomson S., Clayton A.L., Hazzalin C.A., Rose S., Barratt M.J., Mahadevan L.C. The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. Emb. J. 1999; 18:4779–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhu X., Li D., Zhang Z., Zhu W., Li W., Zhao J., Xing X., He Z., Wang S., Wang F. et al. Persistent phosphorylation at specific H3 serine residues involved in chemical carcinogen-induced cell transformation. Mol. Carcinogen. 2017; 56:1449–1460. [DOI] [PubMed] [Google Scholar]
- 55. Choi H.S., Choi B.Y., Cho Y.-Y., Mizuno H., Kang B.S., Bode A.M., Dong Z. Phosphorylation of Histone H3 at Serine 10 is indispensable for neoplastic cell transformation. Cancer Res. 2005; 65:5818–5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Castellano-Pozo M., Santos-Pereira J.M., Rondón A.G., Barroso S., Andújar E., Pérez-Alegre M., García-Muse T., Aguilera A. R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol. Cell. 2013; 52:583–590. [DOI] [PubMed] [Google Scholar]
- 57. Komar D., Juszczynski P. Rebelled epigenome: histone H3S10 phosphorylation and H3S10 kinases in cancer biology and therapy. Clin. Epigenetics. 2020; 12:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zippo A., Robertis A.D., Serafini R., Oliviero S. PIM1-dependent phosphorylation of histone H3 at serine 10 is required for MYC-dependent transcriptional activation and oncogenic transformation. Nat. Cell Biol. 2007; 9:932–944. [DOI] [PubMed] [Google Scholar]
- 59. Panchal N.K., Sabina E.P. A serine/threonine protein PIM kinase as a biomarker of cancer and a target for anti-tumor therapy. Life Sci. 2020; 255:117866. [DOI] [PubMed] [Google Scholar]
- 60. Ma J., Arnold H.K., Lilly M.B., Sears R.C., Kraft A.S. Negative regulation of Pim-1 protein kinase levels by the B56β subunit of PP2A. Oncogene. 2007; 26:5145–5153. [DOI] [PubMed] [Google Scholar]
- 61. Wang J., Anderson P.D., Luo W., Gius D., Roh M., Abdulkadir S.A. Pim1 kinase is required to maintain tumorigenicity in MYC-expressing prostate cancer cells. Oncogene. 2012; 31:1794–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Harms K.L., Chen X. Histone deacetylase 2 modulates p53 transcriptional activities through regulation of p53-DNA binding activity. Cancer Res. 2007; 67:3145–3152. [DOI] [PubMed] [Google Scholar]
- 63. Stojanovic N., Hassan Z., Wirth M., Wenzel P., Beyer M., Schäfer C., Brand P., Kroemer A., Stauber R.H., Schmid R.M. et al. HDAC1 and HDAC2 integrate the expression of p53 mutants in pancreatic cancer. Oncogene. 2017; 36:1804–1815. [DOI] [PubMed] [Google Scholar]
- 64. Mamdani H., Jalal S.I. Histone deacetylase inhibition in non-small cell lung cancer: hype or hope. Front. Cell Dev. Biol. 2020; 8:582370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Donati B., Lorenzini E., Ciarrocchi A. BRD4 and cancer: going beyond transcriptional regulation. Mol. Cancer. 2018; 17:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zullo J.M., Demarco I.A., Piqué-Regi R., Gaffney D.J., Epstein C.B., Spooner C.J., Luperchio T.R., Bernstein B.E., Pritchard J.K., Reddy K.L. et al. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell. 2012; 149:1474–1487. [DOI] [PubMed] [Google Scholar]
- 67. Mathias R.A., Guise A.J., Cristea I.M. Post-translational modifications regulate Class IIa histone deacetylase (HDAC) function in health and disease* [S]. Mol. Cell Proteomics. 2015; 14:456–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wu S.-Y., Lee A.-Y., Lai H.-T., Zhang H., Chiang C.-M. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol. Cell. 2013; 49:843–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sanz-Álvarez M., Cristóbal I., Luque M., Santos A., Zazo S., Madoz-Gúrpide J., Caramés C., Chiang C.-M., García-Foncillas J., Eroles P. et al. Expression of phosphorylated BRD4 is markedly associated with the activation status of the PP2A pathway and shows a strong prognostic value in triple negative breast cancer patients. Cancers. 2021; 13:1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang R., Cao X.-J., Kulej K., Liu W., Ma T., MacDonald M., Chiang C.-M., Garcia B.A., You J. Uncovering BRD4 hyperphosphorylation associated with cellular transformation in NUT midline carcinoma. Proc. Natl Acad. Sci. 2017; 114:E5352–E5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lovén J., Hoke H.A., Lin C.Y., Lau A., Orlando D.A., Vakoc C.R., Bradner J.E., Lee T.I., Young R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013; 153:320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Truica M.I., Burns M.C., Han H., Abdulkadir S.A. Turning up the heat on MYC: progress in small-molecule inhibitors. Cancer Res. 2021; 81:248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shu S., Lin C.Y., He H.H., Witwicki R.M., Tabassum D.P., Roberts J.M., Janiszewska M., Huh S.J., Liang Y., Ryan J. et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016; 529:413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Westermarck J., Neel B.G. Piecing together a broken tumor suppressor phosphatase for cancer therapy. Cell. 2020; 181:514–517. [DOI] [PubMed] [Google Scholar]
- 75. Martin M., Geudens I., Bruyr J., Potente M., Bleuart A., Lebrun M., Simonis N., Deroanne C., Twizere J., Soubeyran P. et al. PP2A regulatory subunit Bα controls endothelial contractility and vessel lumen integrity via regulation of HDAC7. Embo J. 2013; 32:2491–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Henriksson E., Säll J., Gormand A., Wasserstrom S., Morrice N.A., Fritzen A.M., Foretz M., Campbell D.G., Sakamoto K., Ekelund M. et al. SIK2 regulates CRTCs, HDAC4 and glucose uptake in adipocytes. J. Cell Sci. 2014; 128:472–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cadot B., Brunetti M., Coppari S., Fedeli S., de Rinaldis E., Russo C.D., Gallinari P., Francesco R.D., Steinkühler C., Filocamo G. Loss of Histone Deacetylase 4 Causes Segregation Defects during Mitosis of p53-Deficient Human Tumor Cells. Cancer Res. 2009; 69:6074–6082. [DOI] [PubMed] [Google Scholar]
- 78. Weeks K.L., Ranieri A., Karaś A., Bernardo B.C., Ashcroft A.S., Molenaar C., McMullen J.R., Avkiran M. β-Adrenergic Stimulation Induces Histone Deacetylase 5 (HDAC5) Nuclear Accumulation in Cardiomyocytes by B55α-PP2A-Mediated Dephosphorylation. J. Am. Heart Assoc. 2017; 6:e004861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Paroni G., Fontanini A., Cernotta N., Foti C., Gupta M.P., Yang X.-J., Fasino D., Brancolini C. Dephosphorylation and Caspase Processing Generate Distinct Nuclear Pools of Histone Deacetylase 4▿ †. Mol. Cell Biol. 2007; 27:6718–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Martin M., Potente M., Janssens V., Vertommen D., Twizere J.-C., Rider M.H., Goris J., Dimmeler S., Kettmann R., Dequiedt F. Protein phosphatase 2A controls the activity of histone deacetylase 7 during T cell apoptosis and angiogenesis. Proc. Natl Acad. Sci. 2008; 105:4727–4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sucharov C.C., Langer S., Bristow M., Leinwand L. Shuttling of HDAC5 in H9C2 cells regulates YY1 function through CaMKIV/PKD and PP2A. Am. J. Physiol. 2006; 291:C1029–C1037. [DOI] [PubMed] [Google Scholar]
- 82. Hein A.L., Seshacharyulu P., Rachagani S., Sheinin Y.M., Ouellette M.M., Ponnusamy M.P., Mumby M.C., Batra S.K., Yan Y. PR55α subunit of protein phosphatase 2A supports the tumorigenic and metastatic potential of pancreatic cancer cells by sustaining hyperactive oncogenic signaling. Cancer Res. 2016; 76:2243–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bajpai R., Makhijani K., Rao P.R., Shashidhara L.S. Drosophila Twins regulates Armadillo levels in response to Wg/Wnt signal. Development. 2004; 131:1007–1016. [DOI] [PubMed] [Google Scholar]
- 84. Conza G.D., Cafarello S.T., Loroch S., Mennerich D., Deschoemaeker S., Matteo M.D., Ehling M., Gevaert K., Prenen H., Zahedi R.P. et al. The mTOR and PP2A pathways regulate PHD2 phosphorylation to fine-tune HIF1α levels and colorectal cancer cell survival under hypoxia. Cell Rep. 2017; 18:1699–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhang W., Yang J., Liu Y., Chen X., Yu T., Jia J., Liu C. PR55α, a regulatory subunit of PP2A, specifically regulates PP2A-mediated β-catenin dephosphorylation. J. Biol. Chem. 2009; 284:22649–22656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ory S., Zhou M., Conrads T.P., Veenstra T.D., Morrison D.K. Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr. Biol. 2003; 13:1356–1364. [DOI] [PubMed] [Google Scholar]
- 87. Jiang X., Hu J., Wu Z., Cafarello S.T., Matteo M.D., Shen Y., Dong X., Adler H., Mazzone M., de Almodovar C.R. et al. Protein phosphatase 2A mediates YAP activation in endothelial cells upon VEGF stimulation and matrix stiffness. Front. Cell Dev. Biol. 2021; 9:675562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bai X.-L., Zhang Q., Ye L.-Y., Hu Q.-D., Fu Q.-H., Zhi X., Su W., Su R.-G., Ma T., Chen W. et al. Inhibition of protein phosphatase 2A enhances cytotoxicity and accessibility of chemotherapeutic drugs to hepatocellular carcinomas. Mol. Cancer Ther. 2014; 13:2062–2072. [DOI] [PubMed] [Google Scholar]
- 89. Bai X., Zhi X., Zhang Q., Liang F., Chen W., Liang C., Hu Q., Sun X., Zhuang Z., Liang T. Inhibition of protein phosphatase 2A sensitizes pancreatic cancer to chemotherapy by increasing drug perfusion via HIF-1α-VEGF mediated angiogenesis. Cancer Lett. 2014; 355:281–287. [DOI] [PubMed] [Google Scholar]
- 90. Hagelkruys A., Sawicka A., Rennmayr M., Seiser C. Histone deacetylases: the biology and clinical implication. Handb. Exp. Pharmacol. 2011; 206:13–37. [DOI] [PubMed] [Google Scholar]
- 91. Varadkar P., Abbasi F., Takeda K., Dyson J.J., McCright B. PP2A-B56γ is required for an efficient spindle assembly checkpoint. Cell Cycle. 2017; 16:1210–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Vallardi G., Allan L.A., Crozier L., Saurin A.T. Division of labour between PP2A-B56 isoforms at the centromere and kinetochore. Elife. 2019; 8:e42619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kruse T., Zhang G., Larsen M.S.Y., Lischetti T., Streicher W., Nielsen T.K., Bjørn S.P., Nilsson J. Direct binding between BubR1 and B56–PP2A phosphatase complexes regulate mitotic progression. J. Cell Sci. 2013; 126:1086–1092. [DOI] [PubMed] [Google Scholar]
- 94. Hayward D., Bancroft J., Mangat D., Alfonso-Pérez T., Dugdale S., McCarthy J., Barr F.A., Gruneberg U. Checkpoint signaling and error correction require regulation of the MPS1 T-loop by PP2A-B56PP2A-B56 dephosphorylates the MPS1 T-loop. J. Cell Biol. 2019; 218:3188–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Li Y., Tang C.B., Kilian K.A. Matrix mechanics influence fibroblast–myofibroblast transition by directing the localization of histone deacetylase 4. Cell Mol. Bioeng. 2017; 10:405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Butti R., Nimma R., Kundu G., Bulbule A., Kumar T.V.S., Gunasekaran V.P., Tomar D., Kumar D., Mane A., Gill S.S. et al. Tumor-derived osteopontin drives the resident fibroblast to myofibroblast differentiation through Twist1 to promote breast cancer progression. Oncogene. 2021; 40:2002–2017. [DOI] [PubMed] [Google Scholar]
- 97. Underwood T.J., Hayden A.L., Derouet M., Garcia E., Noble F., White M.J., Thirdborough S., Mead A., Clemons N., Mellone M. et al. Cancer-associated fibroblasts predict poor outcome and promote periostin-dependent invasion in oesophageal adenocarcinoma. J. Pathol. 2015; 235:466–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Steele N.G., Biffi G., Kemp S.B., Zhang Y., Drouillard D., Syu L., Hao Y., Oni T.E., Brosnan E., Elyada E. et al. Inhibition of hedgehog signaling alters fibroblast composition in pancreatic cancer. Clin. Cancer Res. 2021; 27:2023–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tsujino T., Seshimo I., Yamamoto H., Ngan C.Y., Ezumi K., Takemasa I., Ikeda M., Sekimoto M., Matsuura N., Monden M. Stromal myofibroblasts predict disease recurrence for colorectal cancer. Clin. Cancer Res. 2007; 13:2082–2090. [DOI] [PubMed] [Google Scholar]
- 100. Glenisson W., Castronovo V., Waltregny D. Histone deacetylase 4 is required for TGFβ1-induced myofibroblastic differentiation. Biochim. Biophys. Acta Mol Cell Res. 2007; 1773:1572–1582. [DOI] [PubMed] [Google Scholar]
- 101. Paroni G., Cernotta N., Russo C.D., Gallinari P., Pallaoro M., Foti C., Talamo F., Orsatti L., Steinkühler C., Brancolini C. PP2A Regulates HDAC4 Nuclear Import. Mol. Biol. Cell. 2008; 19:655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Husmann D., Gozani O. Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 2019; 26:880–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yang Y., Bedford M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer. 2013; 13:37–50. [DOI] [PubMed] [Google Scholar]
- 104. Lei Y., Han P., Tian D. Protein arginine methyltransferases and hepatocellular carcinoma: A review. Transl. Oncol. 2021; 14:101194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Seth-Vollenweider T., Joshi S., Dhawan P., Sif S., Christakos S. Novel Mechanism of Negative Regulation of 1,25-Dihydroxyvitamin D3-induced 25-Hydroxyvitamin D3 24-Hydroxylase (Cyp24a1) Transcription EPIGENETIC MODIFICATION INVOLVING CROSS-TALK BETWEEN PROTEIN-ARGININE METHYLTRANSFERASE 5 AND THE SWI/SNF COMPLEX*. J. Biol. Chem. 2014; 289:33958–33970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Tang J., Frankel A., Cook R.J., Kim S., Paik W.K., Williams K.R., Clarke S., Herschman H.R. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells*. J. Biol. Chem. 2000; 275:7723–7730. [DOI] [PubMed] [Google Scholar]
- 107. Wang H., Huang Z.-Q., Xia L., Feng Q., Erdjument-Bromage H., Strahl B.D., Briggs S.D., Allis C.D., Wong J., Tempst P. et al. Methylation of histone H4 at Arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science. 2001; 293:853–857. [DOI] [PubMed] [Google Scholar]
- 108. Strahl B.D., Briggs S.D., Brame C.J., Caldwell J.A., Koh S.S., Ma H., Cook R.G., Shabanowitz J., Hunt D.F., Stallcup M.R. et al. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr. Biol. 2001; 11:996–1000. [DOI] [PubMed] [Google Scholar]
- 109. Seligson D.B., Horvath S., Shi T., Yu H., Tze S., Grunstein M., Kurdistani S.K. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005; 435:1262–1266. [DOI] [PubMed] [Google Scholar]
- 110. Giuliani V., Miller M.A., Liu C.-Y., Hartono S.R., Class C.A., Bristow C.A., Suzuki E., Sanz L.A., Gao G., Gay J.P. et al. PRMT1-dependent regulation of RNA metabolism and DNA damage response sustains pancreatic ductal adenocarcinoma. Nat. Commun. 2021; 12:4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yao B., Gui T., Zeng X., Deng Y., Wang Z., Wang Y., Yang D., Li Q., Xu P., Hu R. et al. PRMT1-mediated H4R3me2a recruits SMARCA4 to promote colorectal cancer progression by enhancing EGFR signaling. Genome Med. 2021; 13:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Liu L.-M., Tang Q., Hu X., Zhao J.-J., Zhang Y., Ying G.-G., Zhang F. Arginine methyltransferase PRMT1 regulates p53 activity in breast cancer. Life. 2021; 11:789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Zhao J., Adams A., Weinman S.A., Tikhanovich I. Hepatocyte PRMT1 protects from alcohol induced liver injury by modulating oxidative stress responses. Sci. Rep-uk. 2019; 9:9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Duong F.H.T., Christen V., Berke J.M., Penna S.H., Moradpour D., Heim M.H. Upregulation of protein phosphatase 2Ac by Hepatitis C Virus modulates NS3 helicase activity through inhibition of protein arginine methyltransferase 1. J. Virol. 2005; 79:15342–15350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Duong F.H.T., Christen V., Lin S., Heim M.H. Hepatitis C virus–induced up-regulation of protein phosphatase 2A inhibits histone modification and DNA damage repair. Hepatology. 2010; 51:741–751. [DOI] [PubMed] [Google Scholar]
- 116. Chowdhury D., Keogh M.-C., Ishii H., Peterson C.L., Buratowski S., Lieberman J. γ-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol. Cell. 2005; 20:801–809. [DOI] [PubMed] [Google Scholar]
- 117. Beketova E., Owens J.L., Asberry A.M., Hu C.-D. PRMT5: a putative oncogene and therapeutic target in prostate cancer. Cancer Gene. Ther. 2021; 10.1038/s41417-021-00327-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Shifteh D., Sapir T., Pahmer M., Haimowitz A., Goel S., Maitra R. Protein arginine methyltransferase 5 as a therapeutic target for KRAS mutated colorectal cancer. Cancers. 2020; 12:2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Lee M.K.C., Grimmond S.M., McArthur G.A., Sheppard K.E. PRMT5: an emerging target for pancreatic adenocarcinoma. Cancers. 2021; 13:5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Ichikawa T., Shanab O., Nakahata S., Shimosaki S., Manachai N., Ono M., Iha H., Shimoda K., Morishita K. Novel PRMT5-mediated arginine methylations of HSP90A are essential for maintenance of HSP90A function in NDRG2low ATL and various cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2020; 1867:118615. [DOI] [PubMed] [Google Scholar]
- 121. Morishita K., Nakahata S., Ichikawa T. Pathophysiological significance of NDRG2 in cancer development through PP2A phosphorylation regulation. Cancer Sci. 2020; 112:22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Otani Y., Sur H., Rachaiah G., Namagiri S., Chowdhury A., Lewis C.T., Shimizu T., Gangaplara A., Wang X., Vézina A. et al. Inhibiting protein phosphatase 2A increases the antitumor effect of protein arginine methyltransferase 5 inhibition in models of glioblastoma. Neuro-oncology. 2021; 23:1481–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Tang T., Wang H., Han Y., Huang H., Niu W., Fei M., Zhu Y. The role of N-myc downstream-regulated gene family in glioma based on bioinformatics analysis. DNA Cell Biol. 2021; 40:949–968. [DOI] [PubMed] [Google Scholar]
- 124. Weber A.R., Krawczyk C., Robertson A.B., Kuśnierczyk A., Vågbø C.B., Schuermann D., Klungland A., Schär P. Biochemical reconstitution of TET1–TDG–BER-dependent active DNA demethylation reveals a highly coordinated mechanism. Nat. Commun. 2016; 7:10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Wu X., Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet. 2017; 18:517–534. [DOI] [PubMed] [Google Scholar]
- 126. Wu D., Hu D., Chen H., Shi G., Fetahu I.S., Wu F., Rabidou K., Fang R., Tan L., Xu S. et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature. 2018; 559:637–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Hausser A., Link G., Hoene M., Russo C., Selchow O., Pfizenmaier K. Phospho-specific binding of 14-3-3 proteins to phosphatidylinositol 4-kinase III β protects from dephosphorylation and stabilizes lipid kinase activity. J. Cell Sci. 2006; 119:3613–3621. [DOI] [PubMed] [Google Scholar]
- 128. Kundu A., Shelar S., Ghosh A.P., Ballestas M., Kirkman R., Nam H., Brinkley G.J., Karki S., Mobley J.A., Bae S. et al. 14-3-3 proteins protect AMPK-phosphorylated ten-eleven translocation-2 (TET2) from PP2A-mediated dephosphorylation. J. Biol. Chem. 2020; 295:1754–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Chen H., Yu D., Fang R., Rabidou K., Wu D., Hu D., Jia P., Zhao Z., Wu Z., Peng J. et al. TET2 stabilization by 14-3-3 binding to the phosphorylated Serine 99 is deregulated by mutations in cancer. Cell Res. 2019; 29:248–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Baillat D., Hakimi M.-A., Näär A.M., Shilatifard A., Cooch N., Shiekhattar R. Integrator, a multiprotein mediator of small nuclear RNA processing, associates with the C-terminal repeat of RNA polymerase II. Cell. 2005; 123:265–276. [DOI] [PubMed] [Google Scholar]
- 131. Zheng H., Qi Y., Hu S., Cao X., Xu C., Yin Z., Chen X., Li Y., Liu W., Li J. et al. Identification of integrator-PP2A complex (INTAC), an RNA polymerase II phosphatase. Science. 2020; 370:eabb5872. [DOI] [PubMed] [Google Scholar]
- 132. Vervoort S.J., Welsh S.A., Devlin J.R., Barbieri E., Knight D.A., Offley S., Bjelosevic S., Costacurta M., Todorovski I., Kearney C.J. et al. The PP2A-integrator-CDK9 axis fine-tunes transcription and can be targeted therapeutically in cancer. Cell. 2021; 184:3143–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Huang K.-L., Jee D., Stein C.B., Elrod N.D., Henriques T., Mascibroda L.G., Baillat D., Russell W.K., Adelman K., Wagner E.J. Integrator recruits protein phosphatase 2A to prevent pause release and facilitate transcription termination. Mol. Cell. 2020; 80:345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Egloff S., O’Reilly D., Chapman R.D., Taylor A., Tanzhaus K., Pitts L., Eick D., Murphy S. Serine-7 of the RNA polymerase II CTD is specifically required for snRNA gene expression. Science. 2007; 318:1777–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Cossa G., Parua P.K., Eilers M., Fisher R.P. Protein phosphatases in the RNAPII transcription cycle: erasers, sculptors, gatekeepers, and potential drug targets. Gene Dev. 2021; 35:658–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ferreira R., Schneekloth J.S., Panov K.I., Hannan K.M., Hannan R.D. Targeting the RNA Polymerase I transcription for cancer therapy comes of age. Cells. 2020; 9:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Parua P.K., Booth G.T., Sansó M., Benjamin B., Tanny J.C., Lis J.T., Fisher R.P. A Cdk9–PP1 switch regulates the elongation–termination transition of RNA polymerase II. Nature. 2018; 558:460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Wu T., Qin Z., Tian Y., Wang J., Xu C., Li Z., Bian J. Recent developments in the biology and medicinal chemistry of CDK9 inhibitors: an update. J. Med. Chem. 2020; 63:13228–13257. [DOI] [PubMed] [Google Scholar]
- 139. Chao S.-H., Price D.H. Flavopiridol inactivates P-TEFb and blocks most RNA Polymerase II transcription in vivo *. J. Biol. Chem. 2001; 276:31793–31799. [DOI] [PubMed] [Google Scholar]
- 140. Martin R.D., Hébert T.E., Tanny J.C. Therapeutic targeting of the general RNA Polymerase II transcription machinery. Int. J. Mol. Sci. 2020; 21:3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Hu S., Peng L., Xu C., Wang Z., Song A., Chen F.X. SPT5 stabilizes RNA polymerase II, orchestrates transcription cycles, and maintains the enhancer landscape. Mol. Cell. 2021; 81:4425–4439. [DOI] [PubMed] [Google Scholar]
- 142. Morales F., Giordano A. Overview of CDK9 as a target in cancer research. Cell Cycle. 2016; 15:519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. O’Connor C.M., Perl A., Leonard D., Sangodkar J., Narla G. Therapeutic targeting of PP2A. Int. J. Biochem. Cell Biol. 2018; 96:182–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Bryant J.-P., Levy A., Heiss J., Banasavadi-Siddegowda Y.K. Review of PP2A tumor biology and antitumor effects of PP2A inhibitor LB100 in the nervous system. Cancers. 2021; 13:3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Li M., Makkinje A., Damuni Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A (∗). J. Biol. Chem. 1996; 271:11059–11062. [DOI] [PubMed] [Google Scholar]
- 146. Adachi Y., Pavlakis G.N., Copeland T.D. Identification and characterization of SET, a nuclear phosphoprotein encoded by the translocation break point in acute undifferentiated leukemia. J. Biol. Chem. 1994; 269:2258–2262. [PubMed] [Google Scholar]
- 147. Cristóbal I., Torrejón B., Rubio J., Santos A., Pedregal M., Caramés C., Zazo S., Luque M., Sanz-Alvarez M., Madoz-Gúrpide J. et al. Deregulation of SET is associated with tumor progression and predicts adverse outcome in patients with early-stage colorectal cancer. J. Clin. Med. 2019; 8:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Huang Y.-H., Chu P.-Y., Chen J.-L., Huang C.-T., Lee C.-H., Lau K.-Y., Wang W.-L., Wang Y.-L., Lien P.-J., Tseng L.-M. et al. SET overexpression is associated with worse recurrence-free survival in patients with primary breast cancer receiving adjuvant tamoxifen treatment. J. Clin. Med. 2018; 7:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Hung M.-H., Chen Y.-L., Chu P.-Y., Shih C.-T., Yu H.-C., Tai W.-T., Shiau C.-W., Chen K.-F. Upregulation of the oncoprotein SET determines poor clinical outcomes in hepatocellular carcinoma and shows therapeutic potential. Oncogene. 2016; 35:4891–4902. [DOI] [PubMed] [Google Scholar]
- 150. Dobrowsky R.T., Kamibayashi C., Mumby M.C., Hannun Y.A. Ceramide activates heterotrimeric protein phosphatase 2A. J. Biol. Chem. 1993; 268:15523–15530. [PubMed] [Google Scholar]
- 151. Mukhopadhyay A., Saddoughi S.A., Song P., Sultan I., Ponnusamy S., Senkal C.E., Snook C.F., Arnold H.K., Sears R.C., Hanniui Y.A. et al. Direct interaction between the inhibitor 2 and ceramide via sphingolipid-protein binding is involved in the regulation of protein phosphatase 2A activity and signaling. FASEB J. 2009; 23:751–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Yang Y., Huang Q., Lu Y., Li X., Huang S. Reactivating PP2A by FTY720 as a Novel therapy for AML with C-KIT tyrosine kinase domain mutation. J. Cell Biochem. 2012; 113:1314–1322. [DOI] [PubMed] [Google Scholar]
- 153. Saddoughi S.A., Gencer S., Peterson Y.K., Ward K.E., Mukhopadhyay A., Oaks J., Bielawski J., Szulc Z.M., Thomas R.J., Selvam S.P. et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. Embo. Mol. Med. 2013; 5:105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Oaks J.J., Santhanam R., Walker C.J., Roof S., Harb J.G., Ferenchak G., Eisfeld A.-K., Brocklyn J.R.V., Briesewitz R., Saddoughi S.A. et al. Antagonistic activities of the immunomodulator and PP2A-activating drug FTY720 (Fingolimod, Gilenya) in Jak2-driven hematologic malignancies. Blood. 2013; 122:1923–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Cristóbal I., Manso R., Rincón R., Caramés C., Senin C., Borrero A., Martínez-Useros J., Rodriguez M., Zazo S., Aguilera O. et al. PP2A inhibition is a common event in colorectal cancer and its restoration using FTY720 shows promising therapeutic potential. Mol. Cancer Ther. 2014; 13:938–947. [DOI] [PubMed] [Google Scholar]
- 156. Sardoiwala M.N., Kushwaha A.C., Dev A., Shrimali N., Guchhait P., Karmakar S., Choudhury S.R. Hypericin-loaded transferrin nanoparticles induce PP2A-regulated BMI1 degradation in colorectal cancer-specific chemo-photodynamic therapy. ACS Biomater. Sci. Eng. 2020; 6:3139–3153. [DOI] [PubMed] [Google Scholar]
- 157. Jia L., Zhang W., Wang C.-Y. BMI1 inhibition eliminates residual cancer stem cells after PD1 blockade and activates antitumor immunity to prevent metastasis and relapse. Cell Stem Cell. 2020; 27:238–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Chen D., Wu M., Li Y., Chang I., Yuan Q., Ekimyan-Salvo M., Deng P., Yu B., Yu Y., Dong J. et al. Targeting BMI1+ cancer stem cells overcomes chemoresistance and inhibits metastases in squamous cell carcinoma. Cell Stem Cell. 2017; 20:621–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Su W., Han H.H., Wang Y., Zhang B., Zhou B., Cheng Y., Rumandla A., Gurrapu S., Chakraborty G., Su J. et al. The Polycomb Repressor Complex 1 drives double-negative prostate cancer metastasis by coordinating stemness and immune suppression. Cancer Cell. 2019; 36:139–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Hait N.C., Avni D., Yamada A., Nagahashi M., Aoyagi T., Aoki H., Dumur C.I., Zelenko Z., Gallagher E.J., Leroith D. et al. The phosphorylated prodrug FTY720 is a histone deacetylase inhibitor that reactivates ERα expression and enhances hormonal therapy for breast cancer. Oncogenesis. 2015; 4:e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Mazzola M.A., Raheja R., Murugaiyan G., Rajabi H., Kumar D., Pertel T., Regev K., Griffin R., Aly L., Kivisakk P. et al. Identification of a novel mechanism of action of fingolimod (FTY720) on human effector T cell function through TCF-1 upregulation. J. Neuroinflamm. 2015; 12:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Kim S.M., Roy S.G., Chen B., Nguyen T.M., McMonigle R.J., McCracken A.N., Zhang Y., Kofuji S., Hou J., Selwan E. et al. Targeting cancer metabolism by simultaneously disrupting parallel nutrient access pathways. J. Clin. Invest. 2016; 126:4088–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Camm J., Hla T., Bakshi R., Brinkmann V. Cardiac and vascular effects of fingolimod: mechanistic basis and clinical implications. Am. Heart J. 2014; 168:632–644. [DOI] [PubMed] [Google Scholar]
- 164. Kim J.-Y., Kim K.-B., Son H.-J., Chae Y.-C., Oh S.-T., Kim D.-W., Pak J.H., Seo S.-B. H3K27 methylation and H3S28 phosphorylation-dependent transcriptional regulation by INHAT subunit SET/TAF-Iβ. Febs. Lett. 2012; 586:3159–3165. [DOI] [PubMed] [Google Scholar]
- 165. Khanna A., Pimanda J.E., Westermarck J. Cancerous inhibitor of protein phosphatase 2A, an emerging human oncoprotein and a potential cancer therapy target. Cancer Res. 2013; 73:6548–6553. [DOI] [PubMed] [Google Scholar]
- 166. Hait W.N., Lazo J.S. Calmodulin: a potential target for cancer chemotherapeutic agents. J. Clin. Oncol. 1986; 4:994–1012. [DOI] [PubMed] [Google Scholar]
- 167. Takahashi K., Suzuki K. Regulation of protein phosphatase 2A-mediated recruitment of IQGAP1 to β1 integrin by EGF through activation of Ca2+/calmodulin-dependent protein kinase II. J. Cell Physiol. 2006; 208:213–219. [DOI] [PubMed] [Google Scholar]
- 168. Brautigan D.L., Farrington C., Narla G. Targeting protein phosphatase PP2A for cancer therapy: development of allosteric pharmaceutical agents. Clin. Sci. 2021; 135:1545–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Morita K., He S., Nowak R.P., Wang J., Zimmerman M.W., Fu C., Durbin A.D., Martel M.W., Prutsch N., Gray N.S. et al. Allosteric activators of protein phosphatase 2A display broad antitumor activity mediated by dephosphorylation of MYBL2. Cell. 2020; 10.1016/j.cell.2020.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 170. Leonard D., Huang W., Izadmehr S., O’Connor C.M., Wiredja D.D., Wang Z., Zaware N., Chen Y., Schlatzer D.M., Kiselar J. et al. Selective PP2A enhancement through biased heterotrimer stabilization. Cell. 2020; 181:688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Tohmé R., Izadmehr S., Gandhe S., Tabaro G., Vallabhaneni S., Thomas A., Vasireddi N., Dhawan N.S., Ma’ayan A., Sharma N. et al. Direct activation of PP2A for the treatment of tyrosine kinase inhibitor–resistant lung adenocarcinoma. JCI Insight. 2019; 4:e125693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Allen-Petersen B.L., Risom T., Feng Z., Wang Z., Jenny Z.P., Thoma M.C., Pelz K.R., Morton J.P., Sansom O.J., Lopez C.D. et al. Activation of PP2A and inhibition of mTOR synergistically reduce MYC signaling and decrease tumor growth in pancreatic ductal adenocarcinoma. Cancer Res. 2018; 79:209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Sangodkar J., Perl A., Tohme R., Kiselar J., Kastrinsky D.B., Zaware N., Izadmehr S., Mazhar S., Wiredja D.D., O’Connor C.M. et al. Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J. Clin. Invest. 2017; 127:2081–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Hong C.S., Ho W., Zhang C., Yang C., Elder J.B., Zhuang Z. LB100, a small molecule inhibitor of PP2A with potent chemo- and radio-sensitizing potential. Cancer Biol. Ther. 2015; 16:821–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Hao S., Song H., Zhang W., Seldomridge A., Jung J., Giles A.J., Hutchinson M.-K., Cao X., Colwell N., Lita A. et al. Protein phosphatase 2A inhibition enhances radiation sensitivity and reduces tumor growth in chordoma. Neuro-oncology. 2017; 20:799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Mirzapoiazova T., Xiao G., Mambetsariev B., Nasser M.W., Miaou E., Singhal S.S., Srivastava S., Mambetsariev I., Nelson M.S., Nam A. et al. Protein phosphatase 2A as a therapeutic target in small cell lung cancer. Mol. Cancer Ther. 2021; 20:1820–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Gordon I.K., Lu J., Graves C.A., Huntoon K., Frerich J.M., Hanson R.H., Wang X., Hong C.S., Ho W., Feldman M.J. et al. Protein phosphatase 2A inhibition with LB100 enhances radiation-induced mitotic catastrophe and tumor growth delay in glioblastoma. Mol. Cancer Ther. 2015; 14:1540–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Eckschlager T., Plch J., Stiborova M., Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017; 18:1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Takai A., Mieskes G. Inhibitory effect of okadaic acid on the p-nitrophenyl phosphate phosphatase activity of protein phosphatases. Biochem. J. 1991; 275:233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Song S.-H., Kim T.-Y. CTCF, cohesin, and chromatin in human cancer. Genom Inform. 2017; 15:114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Fritz A.J., Gillis N.E., Gerrard D.L., Rodriguez P.D., Hong D., Rose J.T., Ghule P.N., Bolf E.L., Gordon J.A., Tye C.E. et al. Higher order genomic organization and epigenetic control maintain cellular identity and prevent breast cancer. Genes Chromosomes Cancer. 2019; 58:484–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Fahs S., Lujan P., Köhn M. Approaches to study phosphatases. ACS Chem. Biol. 2016; 11:2944–2961. [DOI] [PubMed] [Google Scholar]
- 183. Cho K.F., Branon T.C., Udeshi N.D., Myers S.A., Carr S.A., Ting A.Y. Proximity labeling in mammalian cells with TurboID and split-TurboID. Nat. Protoc. 2020; 15:3971–3999. [DOI] [PubMed] [Google Scholar]
- 184. Schopp I.M., Ramirez C.C.A., Debeljak J., Kreibich E., Skribbe M., Wild K., Béthune J. Split-BioID a conditional proteomics approach to monitor the composition of spatiotemporally defined protein complexes. Nat. Commun. 2017; 8:15690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Huang W., Batra S., Atkins B.A., Mishra V., Mehta K.D. Increases in intracellular calcium dephosphorylate histone H3 at serine 10 in human hepatoma cells: Potential role of protein phosphatase 2A–protein kinase CβII complex. J. Cell Physiol. 2005; 205:37–46. [DOI] [PubMed] [Google Scholar]
- 186. Sif S., Stukenberg P.T., Kirschner M.W., Kingston R.E. Mitotic inactivation of a human SWI/SNF chromatin remodeling complex. Gene. Dev. 1998; 12:2842–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Asencio C., Davidson I.F., Santarella-Mellwig R., Ly-Hartig T.B.N., Mall M., Wallenfang M.R., Mattaj I.W., Gorjánácz M. Coordination of kinase and phosphatase activities by lem4 enables nuclear envelope reassembly during mitosis. Cell. 2012; 150:122–135. [DOI] [PubMed] [Google Scholar]
- 188. Yoon S., Kook T., Min H.-K., Kwon D.-H., Cho Y.K., Kim M., Shin S., Joung H., Jeong S.H., Lee S. et al. PP2A negatively regulates the hypertrophic response by dephosphorylating HDAC2 S394 in the heart. Exp. Mol. Med. 2018; 50:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Weeks K.L., Avkiran M. Roles and post-translational regulation of cardiac class IIa histone deacetylase isoforms. J. Physiol. 2015; 593:1785–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Wang A.H., Kruhlak M.J., Wu J., Bertos N.R., Vezmar M., Posner B.I., Bazett-Jones D.P., Yang X.-J. Regulation of histone deacetylase 4 by binding of 14-3-3 proteins. Mol. Cell Biol. 2000; 20:6904–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Lin T., Hsieh M., Lai C., Cheng J., Wang H., Chau Y., Chen G., Peng H. Melatonin relieves neuropathic allodynia through spinal MT2-enhanced PP2Ac and downstream HDAC4 shuttling-dependent epigenetic modification of hmgb1 transcription. J. Pineal. Res. 2016; 60:263–276. [DOI] [PubMed] [Google Scholar]
- 192. Wu Q., Yang X., Zhang L., Zhang Y., Feng L. Nuclear accumulation of histone deacetylase 4 (HDAC4) exerts neurotoxicity in models of Parkinson’s disease. Mol. Neurobiol. 2017; 54:6970–6983. [DOI] [PubMed] [Google Scholar]
- 193. Illi B., Russo C.D., Colussi C., Rosati J., Pallaoro M., Spallotta F., Rotili D., Valente S., Ragone G., Martelli F. et al. Nitric oxide modulates chromatin folding in human endothelial cells via protein phosphatase 2A activation and class II histone deacetylases nuclear shuttling. Circ. Res. 2008; 102:51–58. [DOI] [PubMed] [Google Scholar]
- 194. Darling N.J., Cohen P. Nuts and bolts of the salt-inducible kinases (SIKs). Biochem. J. 2021; 478:1377–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. He T., Huang J., Chen L., Han G., Stanmore D., Krebs-Haupenthal J., Avkiran M., Hagenmüller M., Backs J. Cyclic AMP represses pathological MEF2 activation by myocyte-specific hypo-phosphorylation of HDAC5. J. Mol. Cell Cardiol. 2020; 145:88–98. [DOI] [PubMed] [Google Scholar]
- 196. Harrison B.C., Huynh K., Lundgaard G.L., Helmke S.M., Perryman M.B., McKinsey T.A. Protein kinase C-related kinase targets nuclear localization signals in a subset of class IIa histone deacetylases. Febs Lett. 2010; 584:1103–1110. [DOI] [PubMed] [Google Scholar]
- 197. Christen V., Duong F., Bernsmeier C., Sun D., Nassal M., Heim M.H. Inhibition of Alpha Interferon Signaling by Hepatitis B Virus▿. J. Virol. 2007; 81:159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Li X., Nan A., Xiao Y., Chen Y., Lai Y. PP2A–B56ε complex is involved in dephosphorylation of γ-H2AX in the repair process of CPT-induced DNA double-strand breaks. Toxicology. 2015; 331:57–65. [DOI] [PubMed] [Google Scholar]
- 199. Kopp B., Khoury L., Audebert M. Validation of the γH2AX biomarker for genotoxicity assessment: a review. Arch Toxicol. 2019; 93:2103–2114. [DOI] [PubMed] [Google Scholar]
- 200. Kim S.W., Kim H.J., Chun Y.J., Kim M.Y. Ceramide produces apoptosis through induction of p27kip1 by protein phosphatase 2A-dependent Akt dephosphorylation in PC-3 prostate cancer cells. J. Toxicol. Environ. Heal. Part. 2010; 73:1465–1476. [DOI] [PubMed] [Google Scholar]
- 201. Neviani P., Perrotti D. SETting OP449 into the PP2A-activating drug family. Clin. Cancer Res. 2014; 20:2026–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Matsuoka Y., Nagahara Y., Ikekita M., Shinomiya T. A novel immunosuppressive agent FTY720 induced Akt dephosphorylation in leukemia cells. Brit. J. Pharmacol. 2003; 138:1303–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Liu C.-Y., Shiau C.-W., Kuo H.-Y., Huang H.-P., Chen M.-H., Tzeng C.-H., Chen K.-F. Cancerous inhibitor of protein phosphatase 2A determines bortezomib-induced apoptosis in leukemia cells. Haematologica. 2013; 98:729–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Tseng L.-M., Liu C.-Y., Chang K.-C., Chu P.-Y., Shiau C.-W., Chen K.-F. CIP2A is a target of bortezomib in human triple negative breast cancer cells. Breast. Cancer Res. 2012; 14:R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Chen K.-F., Yeh P.-Y., Hsu C., Hsu C.-H., Lu Y.-S., Hsieh H.-P., Chen P.-J., Cheng A.-L. Bortezomib overcomes tumor necrosis factor-related apoptosis-inducing ligand resistance in hepatocellular carcinoma cells in part through the inhibition of the phosphatidylinositol 3-Kinase/Akt Pathway*. J. Biol. Chem. 2009; 284:11121–11133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Lv P., Wang Y., Ma J., Wang Z., Li J.-L., Hong C.S., Zhuang Z., Zeng Y.-X. Inhibition of protein phosphatase 2A with a small molecule LB100 radiosensitizes nasopharyngeal carcinoma xenografts by inducing mitotic catastrophe and blocking DNA damage repair. Oncotarget. 2014; 5:7512–7524. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 207. Agarwal A., MacKenzie R.J., Pippa R., Eide C.A., Oddo J., Tyner J.W., Sears R., Vitek M.P., Odero M.D., Christensen D.J. et al. Antagonism of SET Using OP449 Enhances the Efficacy of Tyrosine Kinase Inhibitors and Overcomes Drug Resistance in Myeloid Leukemia. Clin. Cancer Res. 2014; 20:2092–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Farrington C.C., Yuan E., Mazhar S., Izadmehr S., Hurst L., Allen-Petersen B.L., Janghorban M., Chung E., Wolczanski G., Galsky M. et al. Protein phosphatase 2A activation as a therapeutic strategy for managing MYC-driven cancers. J. Biol. Chem. 2020; 295:757–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. McClinch K., Avelar R.A., Callejas D., Izadmehr S., Wiredja D., Perl A., Sangodkar J., Kastrinsky D.B., Schlatzer D., Cooper M. et al. Small-molecule activators of protein phosphatase 2A for the treatment of castration-resistant prostate cancer. Cancer Res. 2018; 78:2065–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]