

Abstract
Background
Poor prognosis of glioblastoma patients and the extensive heterogeneity of glioblastoma at both the molecular and cellular level necessitates developing novel individualized treatment modalities via genomics-driven approaches.
Methods
This study leverages numerous pharmacogenomic and tissue databases to examine drug repositioning for glioblastoma. RNA-seq of glioblastoma tumor samples from The Cancer Genome Atlas (TCGA, n = 117) were compared to “normal” frontal lobe samples from Genotype-Tissue Expression Portal (GTEX, n = 120) to find differentially expressed genes (DEGs). Using compound gene expression data and drug activity data from the Library of Integrated Network-Based Cellular Signatures (LINCS, n = 66,512 compounds) CCLE (71 glioma cell lines), and Chemical European Molecular Biology Laboratory (ChEMBL) platforms, we employed a summarized reversal gene expression metric (sRGES) to “reverse” the resultant disease signature for GBM and its subtypes. A multiparametric strategy was employed to stratify compounds capable of blood-brain barrier penetrance with a favorable pharmacokinetic profile (CNS-MPO).
Results
Significant correlations were identified between sRGES and drug efficacy in GBM cell lines in both ChEMBL(r = 0.37, P < .001) and Cancer Therapeutic Response Portal (CTRP) databases (r = 0.35, P < 0.001). Our multiparametric algorithm identified two classes of drugs with highest sRGES and CNS-MPO: HDAC inhibitors (vorinostat and entinostat) and topoisomerase inhibitors suitable for drug repurposing.
Conclusions
Our studies suggest that reversal of glioblastoma disease signature correlates with drug potency for various GBM subtypes. This multiparametric approach may set the foundation for an early-phase personalized -omics clinical trial for glioblastoma by effectively identifying drugs that are capable of reversing the disease signature and have favorable pharmacokinetic and safety profiles.
Keywords: bioinformatics, drug repositioning, glioblastoma, LINCS
Key Points.
We leverage numerous pharmacogenetic databases to discover brain penetrant drugs that have the potential to reverse the glioblastoma disease signature.
This multiparametric approach may lay the foundation for an early-phase personalized -omics clinical trial.
Importance of the Study.
Given the relative resistance and significant molecular heterogeneity of glioblastoma, personalized treatments that reverse disease phenotypes must be explored. This manuscript leverages numerous pharmacogenetic and tissue databases to discover brain penetrant compounds that reverse the glioblastoma transcriptomic signature and selectively target glioblastoma cells. Our multiparametric approach also uniquely integrates pharmacokinetic profiles of drugs that stratifies compounds on their safety, neuro-penetrance, and treatment efficacy. This multiparametric approach may lay the foundation for an early-phase personalized -omics clinical trial for glioblastoma.
Glioblastoma (GBM) is the most common primary malignant brain tumor, accounting for nearly half of all central nervous system neoplasms.1–3 Although there have been significant advances in molecular and clinical phenotyping of GBM, median survival of patients has remained unchanged in the last 25 years with a 5-year survival rate of 13.4%.4–6 Due to the prognosis and the extensive heterogeneity of glioblastoma, at both a molecular and cellular level, attention has shifted to individualized treatment modalities based on transcriptional assessment of tumors.
The goal of genomics-driven oncology is to replace failed chemotherapeutic strategies with promising tailored treatments using profiling to find druggable targets. This form of precision medicine correlates individual phenotypic signatures to cellular responses via high-throughput drug screening. Over the last few years, large-scale bioinformatics databases have been established to catalog disease signatures and transcriptomic responses to environmental and genetic perturbagens. Of these, the Library of Integrated Network-Based Cellular Signatures (LINCS), Cancer Cell Line Encyclopedia (CCLE), and Chemical European Molecular Biology Laboratory (ChEMBL) have offered new computational approaches for drug repositioning by aligning disease signatures, drug-induced gene expression changes, and cellular responses.7–10 Using these systems, the Reversal of Gene Expression Score (RGES) was recently established as a measure of a compound's ability to reverse the disease-specific transcriptional signature.11 Because RGES positively correlates to half-maximal inhibitory concentrations (IC50), RGES may be used as a surrogate to identify promising new therapeutics for certain diseases.
Here, we leverage extensive clinical and genomic databases (GTEX and TCGA) to link genomic signatures of GBM subtypes (classical, mesenchymal, proneural) to drug-induced gene expression changes. In this study, we employ the previously described RGES system to stratify drug candidates capable of treating GBM subtypes by integrating GBM TCGA and CCLE gene expression profiles and cellular drug response data. To date, this approach has not been utilized in glioblastoma for several reasons: 1) cellular/genetic heterogeneity in tissue samples, 2) poor CNS penetration, and 3) failure to find a druggable target. To address these issues, our multiparametric algorithm incorporates the CNS-Multiparameter optimization (CNS-MPO) score to facilitate the discovery of compounds with low toxicity, neuro-penetrance, and optimal pharmacokinetic profiles.12
This manuscript focuses on individualized treatments and genomics-driven oncology for glioblastoma treatment by repositioning drugs with optimal pharmacokinetic profiles and blood brain penetrance that may reverse the disease signature of glioblastoma in a subtype specific manner as GBM has been classified into mesenchymal, classical, and proneural subtypes. Since differential gene expression with The Cancer Genome Atlas (TCGA) tumor data is limited in GBM due to lack of corresponding same tissue controls, we utilized normal frontal lobe tissue from Genotype-Tissue Expression project (GTEx) as normal controls for calculating disease signatures. We then utilized a previously described summarized reversal gene expression metric (sRGES) and the CNS-Multiparameter optimization (CNS-MPO) score to stratify drugs that may reverse the disease signature with low toxicity, neuro-penetrance, and optimal pharmacokinetic profiles. This multiparametric approach may set the foundation for an early-phase, highly personalized -omics clinical trial for the treatment of glioblastoma.
Methods
Gene Expression Data Sets
We downloaded preprocessed raw read counts from GBM samples in The Cancer Genome Atlas (TCGA) using TCGAbiolinks.13,14 Forty GBM cell lines were identified from the Cancer Cell Line Encyclopedia (CCLE), and their gene expression profiles were downloaded from the CCLE Portal (http://www.broadinstitute.org/ccle). Tumor samples from TCGA were matched to GBM cancer cell lines from CCLE, and their gene expression profiles were correlated using previous described methods.11,15 Briefly, the top 5000 genes ranked by interquartile range from CCLE were correlated to tumor sample gene expression using Spearman rank correlation. Additionally, for normal brain controls, we collected preprocessed RNA-seq data from human frontal lobe from the Genome Tissue Expression Project (GTEX) portal (http://www.gtexportal.org).
For drug gene expression profiles, we accessed the level 4 data from the LINCS L1000 assay using the LINCS Data Portal (http://lincsportal.ccs.miami.edu/datasets/). The transcriptomic responses of 978 landmark genes after exposure to over 25,000 perturbagens, together with the corresponding metadata from the L1000 dataset, were retrieved including drug concentration, cell line, and treatment duration.16,17 Only compound-induced gene expressions that demonstrated reproducibility among replicates were included in our analysis, as defined as gold signatures in the array. While the L1000 dataset does not specifically report on the responses within GBM cell lines, we have previously shown that using these signatures to reverse a GBM disease signature is predictive of the actual pharmacologic cellular response in GBM in vitro and in vivo.18
Compound Activity Profiles
All GBM cell lines from CCLE were manually matched to cell lines in the ChEMBL Version 27 (https://www.ebi.ac.uk/chembl/) database (Supplementary Table S1). Half-maximal inhibitory concentrations (IC50) were extracted from ChEMBL for all GBM cell lines; studies with genetically/environmentally manipulated cell lines were excluded. For duplicate assays using the same compound and cell line, the median IC50 was calculated (Supplementary Table S2). For external validation of the ChEMBL data, compound efficacy was accessed from the Cancer therapeutic Response Portal (CTRPv2, https://portals.broadinstitute.org/ctrp.v2.1/) for GBM cell lines.19,20 Using previously described methods, the median area under the curve (AUC) using percent-viability scores was computed with <10 μM as an activity threshold for each compound.21
Disease Signatures
Differential gene expression (DGE) between The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression Study (GTEx) RNA-seq data were calculated using the ReCount2 (Bioconductor.ord/packages/recount).22,23 We queried the TCGA/GTEx data using the TCGAquery_recount2 function of TCGAbiolinks (v2.8) for glioblastoma. Individual patient samples were parsed into their respective subtypes (Mesenchymal, Classical, Proneural). Given similarities in the transcriptomic profile, glioma CpG island methylator phenotype (G-CIMP) was included into the proneural/neural phenotype.24,25Supplementary Fig. S1. Given the molecular distinction between IDH-mutant and IDH wild-type high-grade gliomas, a separate analysis was also conducted for the proneural subgroup after excluding IDH-mutant tumors. The GTEx data from the frontal lobe was used as a control set for normal brain for our analysis. There were 49 mesenchymal samples, 39 classical samples, and 29 proneural samples used in our dataset, for a glioblastoma aggregate of 117 samples. These were compared against 120 GTEx frontal lobe samples. By using the limma-voom function, we carried out DGE analysis using a single-pipeline to avoid batch effects between heterogeneous datasets.26
Summarized Reversal of Gene expression Score
Originally adapted from the connectivity score, the reversal of gene expression score (RGES) was computed as described previously by Chen et al. Briefly, the RGES was defined as esup−esdown, where the enrichment score (es), is defined as a compilation of upregulated (esup) and downregulated disease genes (esdown) based on a ranked gene expression list from the LINCS L1000 dataset. The RGES recapitulates the potential of a certain compound/drug to reverse the disease signature at a given concentration and exposure period. The summarized reversal of gene expression score (sRGES) was adapted from Chen et al. to merge the multiple drug expression profiles (varying concentrations and incubation periods) into a unified normalized score using a reference condition (10 μM, 24 h treatment duration). We correlated the disease expression profile of the GBM subtype to the tested cell lines, and incorporated it into the sRGES score. The code for derivation of the sRGES score is provided on the following link: github.com/Bin-Chen-Lab/RGES.11 As described in previous studies, we performed a Spearman rank correlation test to assess the nonparametric correlation between sRGES and cell viability (IC50) from ChEMBL and AUC from CTRP.11,21
Reversed Genes
Our analysis also identified several reversed genes for each subtype of glioblastoma and glioblastoma aggregate samples that were specifically reversed by effective compounds (IC50 < 10 μM). If a compound had multiple IC50 values in the ChEMBL database, only the median of the values was used. We sorted each gene expression profile with upregulated and downregulated genes ranked high (on the top) and low (on the bottom) respectively. Reversal of upregulated gene designation was given to those genes that were ranked lower eg downregulated in the effective group (<10 μM) than the ineffective group (>10 μM). We applied the same principle for downregulated genes. Further, to obtain a rigorous list of reversed genes, a leave-one-compound-out cross-validation approach was employed with P < .25 as previously described. Finally, only genes present in all trials were kept.11 The genes quantified by the L1000 assay represent transcriptomic network nodes, the expression of which have been reported to adequately reflect the activities and state of the cell as a whole as a reduced version of the transcriptome. 16 A disease signature is derived by comparing tumor tissue to a nontransformed tissue reference (ie GTex). Using a nonlinear correlation to quantify RGES, a strong negative correlation of a compound signature with the disease signature would reflect a reversal of a signature specific to tumor tissue (ie target genes). A more positive correlation coefficient would reflect a larger effect on off-target genes, specific to untransformed reference tissue (GTex).
Compound CNS Multiparametric Optimization Scoring
Based on previously published methods, each tested compound in the LINCS library was attributed to a corresponding CNS Multiparametric Optimization (CNS-MPO) score to identify safe CNS compounds with optimal pharmacokinetic properties and blood-brain barrier penetration. Each compound is aligned based on the InChI key (the IUPAC International Chemical Identifier), and physicochemical parameters were obtained including molecular weight, calculated partition coefficient (ClogP), calculated distribution coefficient (ClogD), topological polar surface area (TPSA), hydrogen bond donors (HBD), and most basic pKa (Supplementary Table S3). Using a previously described algorithm, a CNS-MPO index was derived, and compounds stratified with an CNS-MPO index >4 (a threshold that correlated well to clinical success of marketed CNS drugs).12,27
An overview of our methodology is illustrated in Figure 1. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. IRB approval was not required for this study, as all databases used were public and did not include any identifiable patient information.
Figure 1.

Pathway of methodology. Created with BioRender.com.
Results
Generation of Differential Gene Expression Profiles
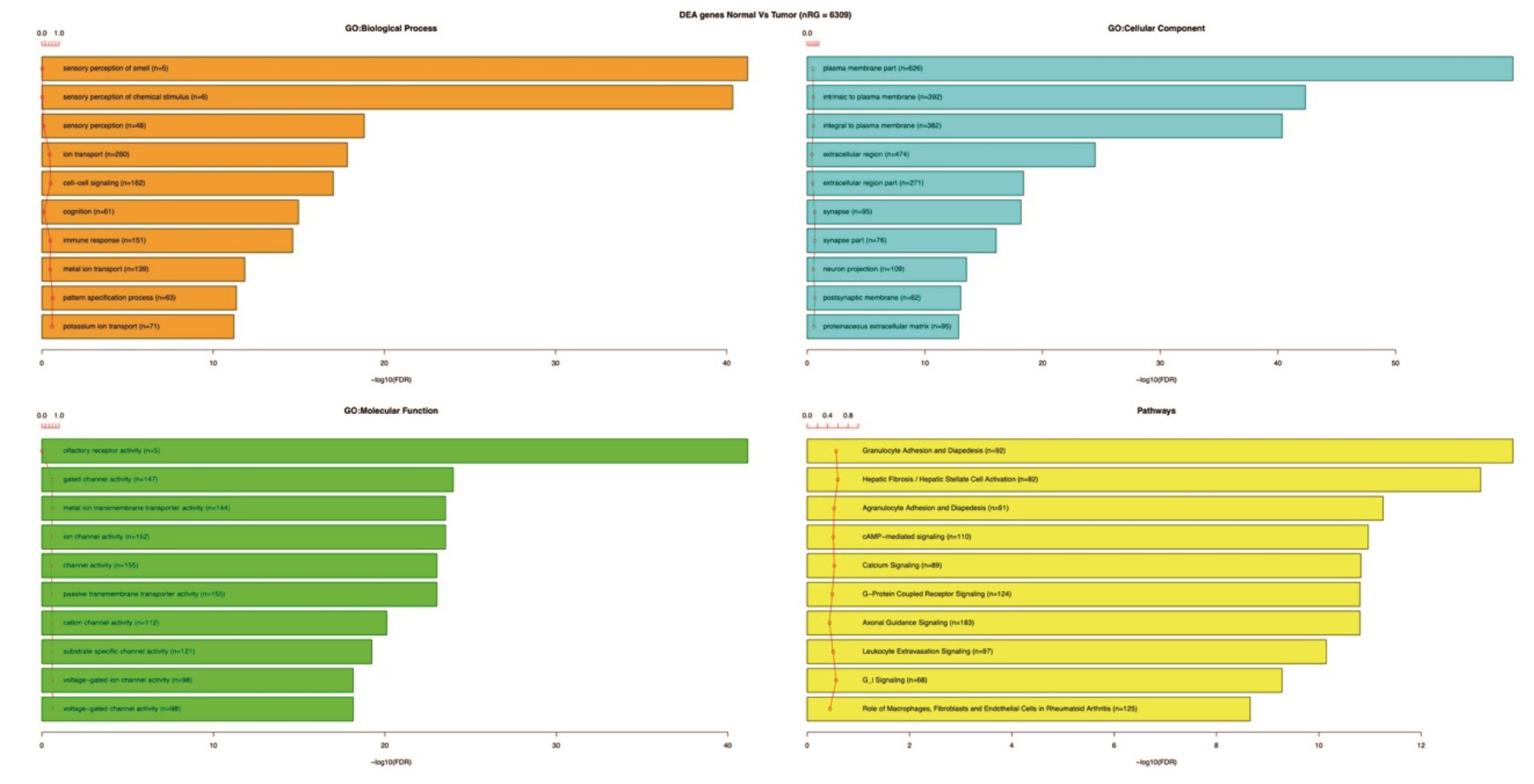
Using Recount2, we generated disease expression signatures by comparing RNA-Seq data from 117 glioblastoma and 120 normal frontal lobe samples from TCGA and GTEX, respectively. To compare the disease expression profiles between the samples, we generated principal component analysis (PCA) plots between the GBM TCGA samples and GTEx control samples for the entire brain and frontal lobe samples Figure 2. PCA analysis revealed differential clustering between the tumor samples (TCGA) and control brain samples (GTEx frontal lobe). Of note, GTEx total brain samples (Figure 2B) demonstrate significant locoregional heterogeneity depending on the sampled area, thereby justifying the choice of frontal lobe as a control. A functional pathway annotation of DGE between GTEX and TCGA was performed, which demonstrated significant disruption of granulocyte adhesion, cAMP-mediated signaling, and G-protein coupled receptor signaling (Supplementary Fig. S2).
Figure 2.

Dimensional principal component analysis (PCA) using ReCount2 of TCGA GBM subtypes (A), GTEx locoregional normal brain samples (B), TCGA GBM subtypes and GTEx total normal brain samples (C), and TCGA GBM subtypes and GTEx frontal lobe samples (D). Plots revealed locoregional separation of normal brain samples between cerebellum and supratentorial cortical tissue (B), and separation of control samples (frontal lobe) and TCGA GBM subtypes (C + D).
Summarized RGES Performance
In order to investigate the efficacy and validity of the summarized RGES method, the sRGES values of the compounds were compared with the compounds' IC50 (half-minimal inhibitory concentration) values through Spearman correlation tests. The sRGES values were significantly correlated with median IC50 value for the glioblastoma aggregate sample (rho = 0.45 and p = 3.07e-6) and each of its subtypes (CLA: rho = 0.47 and p = 6.92e-07, MES: rho = 0.32 and p = 1.28e-3, PRO: rho = 0.51 and p = 4.87e-8) as in Figure 3. Similar correlations were found for IDH wild-stype proneural glioblastoma samples (rho = 0.45, p = 1.5e-4) Supplementary Fig. S1.
Figure 3.

Drug efficacy data correlates with sRGES. Spearman correlations between sRGES (summarized reversal gene expression score) and IC50/drug efficacy data (ChEMBL dataset) for the cancer groups. The correlation coefficient for (A) GBM is rho = 0.45 p = 3.07e-6, (B) CLA is rho = 0.38 p = 6.92e-7, (C) MES is rho = 0.32 p = 1.28e-3, (D) PRO is rho = 0.51 p = 4.87e-8.
To further investigate the efficacy and validity of the summarized RGES method, Spearman's correlations were computed between sRGES values and the compound's Area under Concentration–Response (AUC) curve. As with the IC50 values, the sRGES and AUC scores significantly correlated for the glioblastoma aggregate sample (rho = 0.41 and p = 4.42e-10) and each of its three subtypes (CLA: rho = 0.4 and p = 1.05e-9, MES: rho = 0.36 and p = 8.39e-8, PRO: rho = 0.36 and p = 4.63e-08) as in Figure 4. To determine tumor type specificity for our model, we compared sRGES for other cancers (Breast adenocarcinoma, Liver Hepatocellular Carcinoma, and Colon Adenocarcinoma) to IC50 for GBM. In this analysis, the correlation sRGES and IC50 demonstrated a lower correlation coefficient (mean: 0.45 vs. 0.41), which is similar to findings described by Chen et al. Based on the comparisons with IC50 and AUC scores, the sRGES model may correlate with drug potency for the given data.
Figure 4.

AUC data correlates with sRGES. Spearman correlations between sRGES (summarized reversal gene expression score) and AUC (areas under the concentration–response curve) data for the cancer groups. The correlation coefficient for (A) GBM is rho = 0.41 p = 4.42e-10, (B) CLA is rho = 0.4 p = 1.05e-9, (C) MES is rho = 0.36 p = 8.39e-8, (D) PRO is rho = 0.36 p = 4.63e-8. Data for AUC was retrieved from Cancer Therapeutic Response Portal (CTRP v2), and the median AUC value was used for AUC data available across multiple cell lines.
Identified Reversed Genes
Based on a leave-one-compound-out validation approach, reversed genes were identified that were significantly reversed by effective compounds for each of the tumor groups for P < .25. The program identified 9 genes in glioblastoma aggregate (GBM) including CDK1, MELK, and PCNA; 4 genes in classical subtype (CLA) including MCM3, PPIC, and BIRC5; 14 genes in mesenchymal subtype (MES) including CDK5R1, NOTCH1, and TERT; and 23 genes in proneural subtype (PRO) including ABHD4, ETS1, and TIMP2. The complete reversal gene heatmap for GBM is in Figure 5, and heatmaps for CLA, MES, and PRO are in Figure 6.
Figure 5.

Reversed genes heatmap for GBM. Using a leave-one out compound validation approach, only genes with reversal designation present in every trial with P < .25 were kept to create a list of reversed genes. Green means upregulated and red means downregulated regarding the compound's effect on the gene. Effective compounds have IC50 < 10 μM, and ineffective have IC50 >= 10 μM. For GBM, genes of interest were found to be CDK1, MELK, PCNA, POLE2, CCNA2, CDCA4, IQGAP1, CHEK2, and BACE2.
Figure 6.

Using a leave-one out compound validation approach, only genes with reversal designation present in every trial with P < .25 were kept to create a list of reversed genes for (A) CLA, (B) MES, (C) PRO. Green means upregulated and red means downregulated regarding the compound's effect on the gene. Effective compounds have IC50 < 10 μM, and ineffective have IC50 >= 10 μM.
CNS-MPO Scoring
The CNS-MPO Scoring metric could only be computed on drugs that had each of the necessary 6 components (ClogP, ClogD, MW, TPSA, HBD, and most basic pKa). For the glioblastoma aggregate group and each of the subtypes, a list of sRGES values for 11,951 compounds was generated. The characteristics of each compound were then retrieved from LINCS small molecules records and, if the necessary information was present, a CNS-MPO score was calculated. Then a new list of compounds was created based on filtering for sRGES < 0 and CNS-MPO >= 4 (Table 1). For the glioblastoma aggregate group (Table 2), mesenchymal subtype, classical subtype, and proneural subtype, this yielded 3069, 3036, 2999, and 2988 compounds, respectively.
Table 1.
Top 5 FDA-Approved Drugs for Each Group Ranked by Most Negative sRGES and CNS-MPO > 4
| pert_id | pert_iname | sRGES | CNS.MPO |
|---|---|---|---|
| BRD-K81418486 | Vorinostat | −0.2186586 | 5.16666667 |
| BRD-A30437061 | Camptothecin | −0.1817918 | 4.82333333 |
| BRD-K15108141 | Gemcitabine | −0.1562845 | 4.554 |
| BRD-K77908580 | Entinostat | −0.1478504 | 4.46978571 |
| BRD-K33106058 | Cytarabine | −0.1117964 | 4 |
Table 2.
Effective Compounds (S2) With CNS-MPO > 4 Ranked by Most Negative sRGES for GBM
| Drug Rank | GBM | MES | CLA | PRO |
|---|---|---|---|---|
| 1 | Nadifloxacin | Levalbuterol | Panobinostat | Nadifloxacin |
| 2 | Panobinostat | Enalaprilat | Trichostatin-a | Enalaprilat |
| 3 | Enalaprilat | Panobinostat | Nadifloxacin | Panobinostat |
| 4 | Ranolazine | Nadifloxacin | Dacinostat | Levisoprenaline |
| 5 | Trichostatin-a | Ranolazine | Ranolazine | Ranolazine |
Discussion
We have adapted a multiparametric pharmacogenetic strategy for drug repurposing for glioblastoma to “reverse” the disease transcriptomic signature based on the LINCS1000 dataset. Here we demonstrate that our strategy may serve as an in silico tool to identify compounds that 1) target aberrant genetic pathways in glioblastoma subtypes, 2) induce cell GBM cell death, and 3) align with the pharmacokinetic profile of marketed CNS drugs. By integrating data from publicly available pharmacogenomic databases (LINCS, TCGA, GTEx, CheMBL, CTRP), we demonstrate a correlation between drug activity and reversal of differentially expressed genes within each GBM subtype.
The tools for the sRGES computation have been previously described by Chen et al., who effectively demonstrated a correlation between sRGES and cell viability for several cancer types. However, application of this algorithm was not translated to GBM since astrocytic tumors do not have adjacent normal counterparts in TCGA databases. However, with new data to suggest that the GTEx repository can serve as a surrogate for a normal control, differential gene expression in TCGA could now be investigated.28 Given locoregional transcriptional heterogeneity in the brain, we utilized normal frontal lobe (one of the primary sites for GBM) as a relevant control dataset. Here, we use the GTEx dataset as a reference normal set using the ReCount2 R-package to assess differential gene expression. In parallel, we extracted data from the LINCS pharmacogenomic database, which categorizes drug-induced cellular responses for many cancer cell lines through transcriptional analysis postexposure to over 41,000 small molecules (perturbagens). The vector of transcriptional changes from each perturbagen is then utilized to calculate a sRGES score that recapitulates the compounds' ability to reverse aberrant cancer gene expression. Validating this strategy, the sRGES score for each compound significantly correlated with in vitro cell toxicity and viability in two separate public databases (CHEMbL and CTRP) for each GBM subtype respectively.
Over the last decade, three main subtypes of glioblastoma have been identified with corresponding mutations: proneural (IDH1, PDGFR), classical (EGFRVIII), and mesenchymal (NF1); the neural subtype was removed because it was nontumor specific.24,25,29 Given the significant transcriptional heterogeneity between these subtypes, responses to traditional chemotherapeutics may vary, ultimately affecting treatment response and prognosis. Our study demonstrates that sRGES inversely correlates to IC50 concentrations/AUC not only for glioblastoma aggregate samples but also for each subtype of glioblastoma (proneural, mesenchymal, classical). Additionally, this correlation existed for drug activity within the CTRP database, suggesting that sRGES is a useful measure for predicting drug efficacy for glioblastoma.
Various synthetic lethality (SL) based computational approaches have been proposed to target tumor-specific susceptibilities thereby facilitating drug repositioning.30–32 SL approaches leverage loss-of-function mutations to identify vulnerable cellular targets like targeting an intact DNA repair gene in a cancer cell with intrinsic cellular repair deficiency (BRCA mutation). Although SL uniquely offers a potential mechanism to overcome cancer resistance by exploiting a susceptible cellular pathway, our sRGES approach targets multiple aberrant genes in the cancer cell to maximize reversal of the oncogenic phenotype towards a normal cell. Although less specific for individual cellular pathways, the sRGES approach offers a broader perspective on drug repositioning by identifying drugs that may restore regulation of the most aberrant genes for each cancer cell.
Importantly, we employed a previously described multiparametric strategy to identify compounds with ideal pharmacokinetic properties for drug repurposing in neuro-oncology. The CNS-MPO score can be utilized to isolate compounds that optimally align with marketed and clinically-proven CNS therapeutics; therefore, this tool is well positioned to identify drugs with blood brain penetration and an ideal safety profile.12 Recently, Rankovic et. al validated the CNS-MPO scoring system by demonstrating that increasing CNS-MPO scores correlated to increased unbound drug concentrations in the brain for over 600 compounds.33 The CNS-MPO score has been previously used to evaluate druggability of novel CNS compounds for seizures, depression, and substance abuse disorders, but this scoring system has yet to be investigated in neuro-oncology.34,35 Several factors principally determine the biopharmaceutic success of neurological drugs including low molecular weight, polar surface area, and hydrogen bond donors/acceptors. With a threshold value of 4 or greater, the CNS-MPO score incorporates these factors to identify promising marketed or newly developed neuro-pharmaceuticals.
Clinical Safety and Efficacy of Repositioned Drugs
The aforementioned analyses have identified several therapeutic classes which may be repurposed for -omics driven chemotherapy. Histone Deacetylase inhibitors (HDACi) [vorinostat, panobinostat, Trichostatin-A] remained one of the top therapeutic classes with a high sRGES and CNS-MPO score (>4). HDACi have demonstrated marked in vitro efficacy in reducing invasion, vascular mimicry, and proliferation of glioblastoma stem cells via epigenetic and metabolic reprogramming.36,37 Promising preclinical results have facilitated early phase I clinical trials investigating HDACi in recurrent GBM patients with a tolerable safety profile, but without significant improvement in PFS.38 Although no significant dose-limiting toxicities (DLT were noted in early phase trials, HDACi may have minor toxicities including thromocytopenia and lymphopenia.39 Other repositioned drugs for GBM include gemcitabine and cytarabine which are currently approved for other solid tumors. Cytarabine (Ara-C) is used widely for CNS metastatic disease and has a demonstrated safety profile when administered intrathecally. Small retrospective studies have been conducted to evaluate its utility in treating subventricular spread of glioma, and a few studies demonstrated modest disease control (~6 months) in treatment refractory patients with ventricular spread of GBM.40 However, these limited prospective studies using cytarabine in combination with other chemotherapeutics (cisplatin and hydroxyurea) did not demonstrate improved overall survival for recurrent high-grade gliomas.41 Other preclinical studies using captothecin have demonstrated efficacy in cytoreduction in patient-derived cell lines and reducing tumor recurrence in vivo.42,43 Future individualized -omics approaches for GBM drug repositioning may be warranted to stratify patients who would benefit from targeted treatment using a sRGES-guided algorithm.
Limitations
Our approach for drug repurposing for GBM subtypes may have a few inherent limitations prior to clinical translation. Primarily, the data from our analysis is extrapolated from the LINCS dataset of other cancer cell lines from a separate lineage since the LINCS data does not specifically assay GBM cell lines. Nevertheless, it has been previously demonstrated that sRGES from other nonlineage cell lines significantly correlate with pharmacologic cellular response.11,21 Moreover, we have previously demonstrated that response signatures generated from the data of non-GBM cell lines within the L1000 dataset are predictive of pharmacological response in GBM cells in vitro and in vivo when integrated with GBM expression data.18 Lastly, we must consider any batch effects associated with analyzing RNA-seq from multiple sources such as GTEx and TCGA. However, since all samples were processed using a uniform pipeline (ReCount2), the confounding results due to batch effects were substantially mitigated.
Additionally, the differential gene expression from TCGA and GTEx does not account for individual intratumoral heterogeneity since our methods globally assay transcriptomic changes across dozens of patients. By incorporating single-cell RNA-seq, we may identify several cellular subpopulations within each sample with varying sRGES values. New developments in scRNA-seq techniques have been suggested that intratumoral heterogeneity may identify treatment-resistant cellular subpopulations that are not readily identified by bulk-RNA sequencing techniques.44 Additionally, the cellular plasticity of malignant glioma cells also complicates drug repositioning by theoretically shifting the gene expression signature; we could aim more accurately at this “moving target” by expanding our multiparametric protocol for scRNA-seq platforms.45,46 Such scRNA-seq-driven drug repositioning programs could identify therapeutic options for treatment-resistant cellular niches within the tumor microenvironment.
Conclusion
Our approach helps validate that reversal of glioblastoma disease signature may serve as a surrogate for drug potency for various GBM subtypes. This computational method helps identify aberrant gene pathways and targets that may play a role in GBM pathophysiology. Since we may collect individual patient tumor tissue, our multiparametric strategy may facilitate the clinical translation of personalized treatment paradigms that may be validated by in vitro cytotoxicity assays. The combination of in vitro drug screening of matched patient-derived glioma cell lines and in silico transcriptomic profiling may envisage the clinical response of the tumor to certain FDA-approved drugs. Other -omics based approaches (metabolome, epigenome, proteome) have also recently been utilized for glioblastoma; together, these have helped validate novel classification systems, improved prognostication, and developed precision-based therapeutic approaches. Of these, multi-omic approaches that incorporate metabolic and neurodevelopmental pathways have facilitated discovery of novel subtypes of GBM (ie mitochondrial subtype), which may portend therapeutic susceptibility.47 By incorporating single-cell and bulk tumor RNA-seq, computational approaches have improved their sensitivity to detect distinct disease signatures, and uncover intratumoral heterogeneity, tumor infiltration, and diverse regulatory mechanisms.48–50 Single-cell transcriptomic techniques have also been employed to identify potential therapeutic avenues to target invasive tumor cells, glioma stem-cell niches and the tumor microenvironment.50–52 In this manner, we may identify personalized treatment options for most patients using this novel computational screening method that can be validated in both patient-specific cell lines and in vivo models. This multiparametric approach may set the foundation for an early-phase personalized -omics clinical trial for glioblastoma by effectively identifying drugs that are capable of not only reversing the disease signature, but also reside in an optimal neuro-pharmaceutical niche.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank all members of the University of Miami Brain Tumor Initiative for helpful discussions.
Funding
Funding from the Sylvester Comprehensive Cancer Center, FCBTR, and FACCA to NGA and National Institute of Neurological Disorders and Stroke (R25NS108937-01) to AS. This research was funded (in part) by the intramural research division of the National Institutes of Health (NIH), National Institute of Neurological Disorders and Stroke (NINDS).
Conflict of interest statement. NGA is a consultant for Epigenetix, Inc., and is a founder of Decode Therapeutics. All other authors declare no potential conflicts of interest.
Authorship statement. AHS, PG, RS, VG all contributed to data collection, analysis, design and preparation of manuscript. VS manuscript review, data analysis. IC data analysis, statistics, MLF project overview, AM, VL, MF, MI, SS manuscript review, TDO, RJK, DGE, EL critically reviewed paper. NA and MG provided senior mentorship and design of research proposal.
References
- 1. Frankel SA, German WJ. Glioblastoma multiforme; review of 219 cases with regard to natural history, pathology, diagnostic methods, and treatment. J Neurosurg. 1958; 15(5):489–503. [DOI] [PubMed] [Google Scholar]
- 2. Kelly KA, Kirkwood JM, Kapp DS. Glioblastoma multiforme: pathology, natural history and treatment. Cancer Treat Rev. 1984; 11(1):1–26. [DOI] [PubMed] [Google Scholar]
- 3. Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005; 109(1):93–108. [DOI] [PubMed] [Google Scholar]
- 4. Rock K, McArdle O, Forde P, et al. A clinical review of treatment outcomes in glioblastoma multiforme - the validation in a non-trial population of the results of a randomised phase III clinical trial: has a more radical approach improved survival? Br J Radiol. 2012; 85(1017):e729––.33. doi: 10.1259/bjr/83796755. PMID: 22215883; PMCID: PMC3487092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Birol Sarica F, Tufan K, Cekinmez M, et al. Effectiveness of temozolomide treatment used at the same time with radiotherapy and adjuvant temozolomide; concomitant therapy of glioblastoma multiforme: multivariate analysis and other prognostic factors. J Neurosurg Sci. 2010; 54(1):7–19. [PubMed] [Google Scholar]
- 6. Chaudhry NS, Shah AH, Ferraro N, et al. Predictors of long-term survival in patients with glioblastoma multiforme: advancements from the last quarter century. Cancer Invest. 2013; 31(5):287–308. [DOI] [PubMed] [Google Scholar]
- 7. Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012; 483(7391):603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koleti A, Terryn R, Stathias V, et al. Data portal for the Library of Integrated Network-based Cellular Signatures (LINCS) program: integrated access to diverse large-scale cellular perturbation response data. Nucleic Acids Res. 2018; 46(D1):D558–D566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Keenan AB, Jenkins SL, Jagodnik KM, et al. The Library of Integrated Network-Based Cellular Signatures NIH Program: system-level cataloging of human cells response to perturbations. Cell Syst. 2018; 6(1):13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davies M, Nowotka M, Papadatos G, et al. ChEMBL web services: streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015; 43(W1):W612–W620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen B, Ma L, Paik H, et al. Reversal of cancer gene expression correlates with drug efficacy and reveals therapeutic targets. Nat Commun. 2017; 8:16022. doi: 10.1038/ncomms16022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wager TT, Hou X, Verhoest PR, Villalobos A. Central nervous system multiparameter optimization desirability: application in drug discovery. ACS Chem Neurosci. 2016; 7(6):767–775. [DOI] [PubMed] [Google Scholar]
- 13. Colaprico A, Silva TC, Olsen C, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016; 44(8):e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mounir M, Lucchetta M, Silva TC, et al. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput Biol. 2019; 15(3):e1006701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen B, Sirota M, Fan-Minogue H, Hadley D, Butte AJ. Relating hepatocellular carcinoma tumor samples and cell lines using gene expression data in translational research. BMC Med Genomics. 2015; 8(Suppl 2):S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Subramanian A, Narayan R, Corsello SM, et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017; 171(6):1437–1452 e1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stathias V, Turner J, Koleti A, et al. LINCS Data Portal 2.0: next generation access point for perturbation-response signatures. Nucleic Acids Res. 2020; 48(D1):D431–D439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stathias V, Jermakowicz AM, Maloof ME, et al. Drug and disease signature integration identifies synergistic combinations in glioblastoma. Nat Commun. 2018; 9(1):5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Seashore-Ludlow B, Rees MG, Cheah JH, et al. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. 2015; 5(11):1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rees MG, Seashore-Ludlow B, Cheah JH, et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat Chem Biol. 2016; 12(2):109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim IW, Jang H, Kim JH, et al. Computational drug repositioning for gastric cancer using reversal gene expression profiles. Sci Rep. 2019; 9(1):2660. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22. Collado-Torres L, Nellore A, Jaffe AE. Recount workflow: accessing over 70,000 human RNA-seq samples with Bioconductor. F1000Res. 2017; 6:1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Collado-Torres L, Nellore A, Kammers K, et al. Reproducible RNA-seq analysis using recount2. Nat Biotechnol. 2017; 35(4):319–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Q, Hu B, Hu X, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2017; 32(1):42–56 e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013; 155(2):462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Law CW, Chen Y, Shi W, Smyth GK. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014; 15(2):R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wager TT, Hou X, Verhoest PR, Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem Neurosci. 2010; 1(6):435–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zeng WZD, Glicksberg BS, Li Y, Chen B. Selecting precise reference normal tissue samples for cancer research using a deep learning approach. BMC Med Genomics. 2019; 12(Suppl 1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010; 17(1):98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yar MS, Haider K, Gohel V, Siddiqui NA, Kamal A. Synthetic lethality on drug discovery: an update on cancer therapy. Expert Opin Drug Discov. 2020; 15(7):823–832. [DOI] [PubMed] [Google Scholar]
- 31. Zhang B, Tang C, Yao Y, et al. The tumor therapy landscape of synthetic lethality. Nat Commun. 2021; 12(1):1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Apaolaza I, San Jose-Eneriz E, Tobalina L, et al. An in-silico approach to predict and exploit synthetic lethality in cancer metabolism. Nat Commun. 2017; 8(1):459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rankovic Z. CNS drug design: balancing physicochemical properties for optimal brain exposure. J Med Chem. 2015; 58(6):2584–2608. [DOI] [PubMed] [Google Scholar]
- 34. Odi R, Bibi D, Wager T, Bialer M. A perspective on the physicochemical and biopharmaceutic properties of marketed antiseizure drugs—from phenobarbital to cenobamate and beyond. Epilepsia. 2020; 61(8):1543–1552. doi: 10.1111/epi.16597. PMID: 32614073. [DOI] [PubMed] [Google Scholar]
- 35. Sniecikowska J, Gluch-Lutwin M, Bucki A, et al. Novel aryloxyethyl derivatives of 1-(1-benzoylpiperidin-4-yl)methanamine as the extracellular regulated kinases ½ (ERK1/2) phosphorylation-preferring serotonin 5-HT1A receptor-biased agonists with robust antidepressant-like activity. J Med Chem. 2019; 62(5):2750–2771. [DOI] [PubMed] [Google Scholar]
- 36. Nguyen TTT, Zhang Y, Shang E, et al. HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models. J Clin Invest. 2020; 130(7):3699–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pastorino O, Gentile MT, Mancini A, et al. Histone deacetylase inhibitors impair vasculogenic mimicry from glioblastoma cells. Cancers (Basel). 2019; 11(6):747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Peters KB, Lipp ES, Miller E, et al. Phase I/II trial of vorinostat, bevacizumab, and daily temozolomide for recurrent malignant gliomas. J Neurooncol. 2018; 137(2):349–356. [DOI] [PubMed] [Google Scholar]
- 39. Lee EQ, Puduvalli VK, Reid JM, et al. Phase I study of vorinostat in combination with temozolomide in patients with high-grade gliomas: North American Brain Tumor Consortium Study 04-03. Clin Cancer Res. 2012; 18(21):6032–6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Scott BJ, van Vugt VA, Rush T, et al. Concurrent intrathecal methotrexate and liposomal cytarabine for leptomeningeal metastasis from solid tumors: a retrospective cohort study. J Neurooncol. 2014; 119(2):361–368. [DOI] [PubMed] [Google Scholar]
- 41. Swinnen LJ, Rankin C, Carraway H, et al. A phase II study of cisplatin preceded by a 12-h continuous infusion of concurrent hydroxyurea and cytosine arabinoside (Ara-C) for adult patients with malignant gliomas (Southwest Oncology Group S9149). J Neurooncol. 2008; 86(3):353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lu YJ, Lan YH, Chuang CC, et al. Injectable hermos-sensitive chitosan hydrogel containing CPT-11-loaded EGFR-targeted graphene oxide and SLP2 shRNA for localized drug/gene delivery in glioblastoma therapy. Int J Mol Sci. 2020; 21(19):7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Checa-Chavarria E, Rivero-Buceta E, Sanchez Martos MA, et al. Development of a Prodrug of Camptothecin for Enhanced Treatment of Glioblastoma Multiforme. Mol Pharm. 2021; 18(4):1558–1572. doi: 10.1021/acs.molpharmaceut.0c00968. PMID: 33645231; PMCID: PMC8482753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tirosh I, Suva ML. Dissecting human gliomas by single-cell RNA sequencing. Neuro Oncol. 2018; 20(1):37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Al Mahi N, Zhang EY, Sherman S, Yu JJ, Medvedovic M. Connectivity map analysis of a single-cell RNA-sequencing-derived transcriptional signature of mTOR signaling. Int J Mol Sci. 2021; 22(9):4371. doi: 10.3390/ijms22094371. PMID: 33922083; PMCID: PMC8122562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Neftel C, Laffy J, Filbin MG, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. 2019; 178(4):835–849 e821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Garofano L, Migliozzi S, Oh YT, et al. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat Cancer. 2021; 2(2):141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014;344(6190):1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Darmanis S, Sloan SA, Croote D, et al. Single-cell RNA-seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep. 2017; 21(5):1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Couturier CP, Ayyadhury S, Le PU, et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat Commun. 2020; 11(1):3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang J, Guan M, Wang Q, Zhang J, Zhou T, Sun X. Single-cell transcriptome-based multilayer network biomarker for predicting prognosis and therapeutic response of gliomas. Brief Bioinform. 2020; 21(3):1080–1097. [DOI] [PubMed] [Google Scholar]
- 52. Lopes MB, Vinga S. Tracking intratumoral heterogeneity in glioblastoma via regularized classification of single-cell RNA-Seq data. BMC Bioinf. 2020; 21(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.