

Summary
Specific, potent ligands drive precision medicine and fundamental biology. Proteins, peptides, and small molecules constitute effective ligand classes. Yet greater molecular diversity would aid the pursuit of ligands to elicit precise biological activity against challenging targets. We demonstrate a platform to discover protein-small molecule (PriSM) hybrids to combine unique pharmacophore activities and shapes with constrained, efficiently engineerable proteins. A fibronectin protein library is yeast displayed with a single cysteine coupled to acetazolamide via a maleimide-poly(ethylene glycol) linker. Magnetic and flow cytometric sorts enrich specific binders to carbonic anhydrase isoforms. Isolated PriSMs exhibit potent, specific inhibition of carbonic anhydrase isoforms with superior efficacy to acetazolamide or protein alone including an 80-fold specificity increase and 9-fold potency gain. PriSMs are engineered with multiple linker lengths, protein conjugation sites, and sequences against two different isoforms, which reveals platform flexibility and impacts of molecular designs. PriSMs advance the molecular diversity of efficiently engineerable ligands.
Graphical Abstract
eTOC
Lewis and Harthorn et al. demonstrate a platform to discover protein-small molecule hybrids that merge pharmacophore activity with engineerable proteins to achieve potency and specificity superior to either component alone. Hybrids with multiple linker lengths, conjugation sites, and protein sequences against two enzymes reveal platform flexibility and design impacts.
Introduction
Molecular targeting ligands empower precision medicine and fundamental biological research (Carter and Lazar, 2018; Carter et al., 2019). A multitude of platforms have effectively empowered ligand discovery including computational design (Chevalier et al., 2017; Yin et al., 2007) and combinatorial library screening via phenotype-genotype linked display methods (Chao et al., 2006; Winter et al., 1994) for proteins as well as structure-guided screening (Renaud et al., 2016) and fragment-based drug discovery (Erlanson et al., 2016; Murray and Rees, 2009) for small molecules. Proteins have large surface area to drive affinity (Chen et al., 2013) and specificity (Nadalin and Carbone, 2018), structural organization to limit entropic cost upon binding (Searle and Williams, 1992), and are ribosomally encoded to permit high-throughput genotype-phenotype selections. Small molecules provide smaller size that is more amenable to target concave epitopes, broader chemical diversity, and different opportunities for shape complementarity. These complementary properties, as well as different advances in discovery platforms, have led both molecular classes to yield successful ligands for an array of applications.
Nevertheless, the ability to target select epitopes and specifically elicit desired biological activities remains challenging in some systems, including enzymes and highly homologous targets, which has inspired an array of advanced molecular designs and engineering strategies. Extended antibody complementarity-determining regions (de los Rios et al., 2015) and alternative scaffold topologies (Gilbreth et al., 2011) have facilitated cavity targeting. Hybrid molecules with small molecule pharmacophores conjugated to peptides have been engineered to increase specificity on kinases (Gower et al., 2015; Meyer et al., 2007; Schneider et al., 2005) and to increase affinity or potency against lectins (Ng et al., 2015), enzymes (Andersson et al., 2005; Chen et al., 2020), and microbes (Li and Roberts, 2003).
A key challenge in these latter systems is to efficiently engineer the peptide and conjugated small molecule to form a synergistic binding interface. Peptide-pharmacophore hybrids have been identified via design (Hosseinzadeh et al., 2021; Parang et al., 2001) and discovered from combinatorial libraries via phage (Chen et al., 2020; Derda and Ng, 2019; Meyer et al., 2007; Ng et al., 2015; Wu et al., 2021) and mRNA (Derda and Ng, 2019; Horiya et al., 2014) display. In the current work, we evaluated the ability to merge the benefits of small molecule pharmacophores with folded protein scaffolds, which provide greater surface area, more diverse topology, and reduced entropic costs relative to previous peptide-based hybrids. We evaluated the implementation of protein-pharmacophore hybrid engineering within the context of yeast surface display, which enables quantitative flow cytometric selection for affinity (VanAntwerp and Wittrup, 2000) and specificity (Puri et al., 2013); multivalent display for avid selection (Chao et al., 2006) of moderate affinities to begin the discovery and evolutionary process, and eukaryotic processing for effective folding. Previous work by Van Deventer and colleagues has demonstrated the ability to conjugate small molecules to non-canonical amino acids in yeast-displayed proteins (Deventer et al., 2016; Islam et al., 2021). In the current work, we used the free thiol residue of a single cysteine moiety to enable focused conjugation of a small molecule pharmacophore, which builds on work conjugating small molecules to antibody fragment cysteines in phage display (Jespers et al., 2004, 1994).
The approach can be considered as an analog of fragment-based drug discovery (Derda and Ng, 2019; Jespers et al., 1994; Murray and Rees, 2009) in which the pharmacophore extends the binding capabilities of an engineered protein in line with the aforementioned peptide-pharmacophore hybrids (Andersson et al., 2005; Chen et al., 2020; Derda and Ng, 2019; Gower et al., 2015; Horiya et al., 2014; Hosseinzadeh et al., 2021; Li and Roberts, 2003; Meyer et al., 2007; Ng et al., 2015; Parang et al., 2001; Schneider et al., 2005; Wu et al., 2021). As another perspective, an integrated binding site comprising protein-target and small molecule-target interfaces builds on the success of heterobivalent AND gates (Vauquelin and Charlton, 2013; Xu et al., 2012), which avidly couple two interactions of moderate intrinsic affinity to achieve high specificity and strong macroscopic affinity. In a third perspective, the conjugated small molecule can be viewed as a non-canonical amino acid to expand the natural repertoire (Link et al., 2003) without the need to engineer translational machinery.
As an initial test system for the protein-small molecule (PriSM) hybrid engineering platform, we aimed to selectively inhibit enzymes of both fundamental and clinical importance with off-target homologs. We chose carbonic anhydrases (CAs), a family of zinc metalloenzymes that catalyze interconversion of carbon dioxide and bicarbonate. CA inhibitors have been pursued for cancer, infectious disease, and ocular indications (Pastorekova et al., 2004; Potter and Harris, 2003; Supuran, 2008, 2012, 2017). CA-IX exhibits aberrant expression in a multitude of tumors (Masereel et al., 2006; Mcdonald et al., 2012; Pastorekova and Gillies, 2019; Supuran et al., 2018), is associated with tumor hypoxia (Pastorek and Pastorekova, 2015; Pastorekova and Gillies, 2019; Rafajov et al., 2004), and drives tumor invasiveness (Robertson et al., 2004; Svastova et al., 2012; Ward et al., 2015). CA-IX inhibition has exhibited preclinical and clinical efficacy in multiple cancers (Mcdonald et al., 2012; Supuran et al., 2018; Ward et al., 2015). Also, while largely cytosolic, extracellular CA-II activity is problematic in proliferative diabetic retinopathy (Gao et al., 2007). Numerous small molecule inhibitors demonstrate activity but modest specificity (Supuran, 2008, 2012), because of high isoform homology (Figure S1), which hinders fundamental study and causes detrimental side effects (Supuran, 2008). A frequently studied small molecule, acetazolamide (AAZ), exhibits an inhibition constant (Ki) in the tens of nM for several CA isoforms (Supuran, 2008) including 12 nM for CA-II and 25 nM for CA-IX. Extensive structural modifications have been explored to identify enhanced potency and/or improved selectivity, albeit with mixed results and substantial effort (Supuran, 2008). Thus, we chose to pursue specific inhibitors of CA-IX and, separately, CA-II as useful proofs of concept that also have potential biomedical utility. A set of recently developed triazinyl-substituted benzenesulfonamides inhibit CA-II with ~10-fold selectivity (Ki ~ 13 nM) and CA-IX with ~700-fold selectivity (Ki ~ 0.15 nM), respectively (Carta et al., 2011; Garaj et al., 2004). These molecules were re-engineered to incorporate symmetric amino acid pairs into their tail regions and yield altered selectivity, producing a set of sulfonamides that are selective to CA-I, CA-IV, or CA-IX (Mikulová et al., 2020; Mikuš et al., 2018). We integrate AAZ into a binding loop of the tenth type III domain of human fibronectin (Fn, also termed monobody or adnectin) (Lipovsek, 2011) as a cysteine-free antibody mimetic scaffold. Evolution of the amino acid sequence in its solvent-exposed loops has enabled development of specific, high affinity binders to a multitude of targets (Koide et al., 1998; Lipovsek, 2011). Genetic incorporation of a single cysteine enables site-specific conjugation to maleimide pharmacophores.
DNA-encoded library strategies have also been used to identify CA ligands. Peptides were selected from phage display libraries for binding to CA-IX extracellular domain (Hebling et al., 2010) or proteoglycan region (Rana et al., 2013). Million-member DNA-encoded libraries were screened to identify small molecules that bind (Oehler et al., 2021) and inhibit (Buller et al., 2011) CA-IX.
The current study validates a platform for PriSM engineering via sorting a combinatorial library of 108 Fn-AAZ hybrids to discover inhibitors of CA-II and CA-IX with superior potency and selectivity relative to small molecule or protein alone. Protein-small molecule synergy can be achieved by engineering various combinations of conjugation sites, linker lengths, protein sequences, and target isoforms. Beyond discovery of selective CA inhibitors, this ligand engineering platform provides broad opportunity for targeting epitopes to selectively elicit biological activity.
Results
PriSM library design and construction
We sought to engineer PriSMs with strong potency and isoform selectivity to CA-II or CA-IX via modulation of the protein interface, AAZ conjugation site, and linker length. We designed combinatorial Fn libraries conserving a sole cysteine at a central, solvent-exposed site in either the central loop or the most protruding loop (sites 28 and 80 in the BC and FG loops, respectively; Figure 1A) and biased diversity within the adjacent three loops via guidance from previous protein evolution studies (Woldring et al., 2015). Eight sites were diversified with all 20 amino acids, biased toward tyrosine, serine, and other amino acids prevalent in antibody complementarity-determining regions and evolved Fn domains (Figure 1B, Figure S2); 11 sites had reduced diversity to balance inter- and intramolecular constraints. Loop lengths were diversified to provide shape modulation: BC loop: −1, 0, +1 relative to wild-type; DE loop: −1, 0; FG loop: −2, −1, 0. Diverse genes, assembled from degenerate oligonucleotides, were homologously recombined into a Saccharomyces cerevisiae yeast display system as genetic fusions to N-terminal Aga2p with a semi-flexible polypeptide linker (Figure 1A). Both Cys28 and Cys80 libraries had 200 million unique transformants. Sequence analysis confirms that library construction generally matches design (median deviation from design (|fobserved – fdesign|) of 0.1% for both Cys28 and Cys80 libraries) (Figure S2).
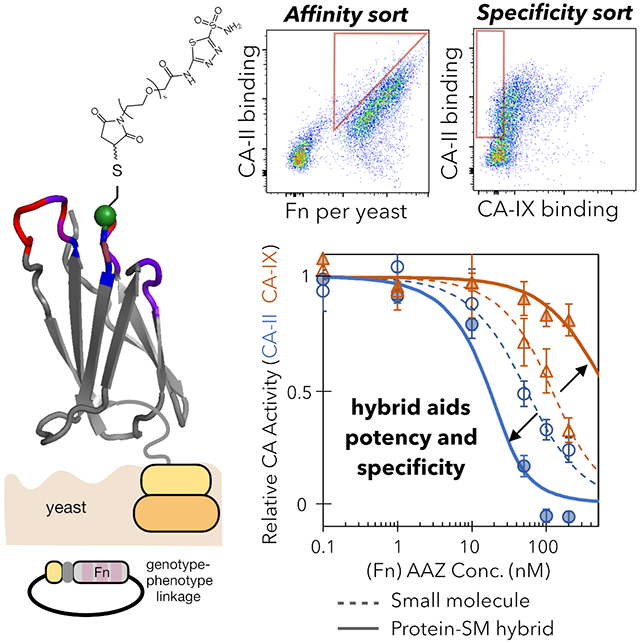
Figure 1. PriSM design integrates a pharmacophore with combinatorial protein diversity.

(A) The Fn domain is genetically diversified at up to 19 sites in three solvent-exposed loops surrounding a conserved cysteine (green). Cys28 is shown explicitly with thiol; the semi-transparent green circle indicates site 80, which was alternatively conserved in a second library. Fn is tethered to the yeast cell wall via protein fusion to a semi-flexible peptide linker to Aga2p mating protein, which is bound to Aga1p anchored in the cell wall. Treatment with maleimide-PEG-AAZ yields the protein-small molecule conjugate PriSM (as well as conjugation to other available thiols). (B) Left: top view of Fn. Right: amino acid diversity at each site with wild-type residues indicated below (gray: deleted in shortened loop lengths; arrow: insertion in lengthened loops). The Cys28 library is shown; the Cys80 library is equivalent except the diversities at sites 28 and 80 are swapped.
Engineered PriSMs exhibit potent, isoform-specific inhibition
Yeast were grown to replicate variants, induced to display Fn, and treated with maleimide-PEGx-AAZ (x = 2, 3, 5, or 7). We first describe the development of Fn-PEG-AAZ PriSMs that target CA-II and are conjugated at site 28 with a PEG7 linker. In later sections, we will show that this technology is also effective with an FG loop conjugation site, linker lengths between 2 and 7 PEG units, and targeting to inhibit CA-IX with isoform specificity. The library was sorted twice for binding to biotinylated CA-II, and binders were pulled down with streptavidin-coated magnetic beads. Analysis of the sorted population revealed numerous binders of at least low affinity but also a majority of clones without detectable binding (Figure 2A). The presence of a multitude of non-binders highlights the need to precisely engineer the combination of protein sequence, conjugation site, linker, and pharmacophore for any target. Moreover, it demonstrates that conjugation of PEG-AAZ to the multitude of non-fibronectin cysteines (or alternative maleimide-reactive sites) on the yeast surface does not result in problematic background binding at the concentrations used (10 nM). The presence of numerous binders indicates the potential for the PriSM concept to function in the context of a Fn protein scaffold, AAZ pharmacophore, and CA-II target.
Figure 2. Selective, high-affinity PriSM discovery.

(A) The magnetic bead-sorted population of yeast-displayed Fn was incubated with maleimide-PEG7-AAZ, washed, and incubated with 10 nM biotin-CA-II followed by streptavidin-AlexaFluor647. Fn display was quantified with mouse anti-Myc and anti-mouse-AlexaFluor488. Flow cytometric analysis of 10,000 random variants is shown. (B) The enriched population was labeled with DL650-CA-IX, biotin-CA-II+streptavidin-AF488, and mouse anti-Myc+anti-mouse-PE. Flow cytometric sorts enriched CA-II specific binders (left) with high affinity (right). (C) Final enriched population was labeled with CA-II or CA-IX at 0.1 or 10 nM each; binding was evaluated by flow cytometry. The distribution of 10,000 variants is presented. * indicates significant difference between binding CA-II and CA-IX (p < 0.001).
To promote increased diversity in subsequent sorts, Fn variants enriched from sorts with PEG2, PEG3, PEG5, and PEG7 versions of maleimide-PEGx-AAZ were pooled after each bead sort. The pooled population was sorted twice via stringent flow cytometric sorting – the first for affinity, the second for affinity and specificity. Yeast-displayed conjugates were incubated with both DyLight650-labeled CA-II and biotinylated CA-IX (detected by streptavidin-AF488). Conjugates that displayed high CA-II-DL650 labeling and low CA-IX-AF488 labeling were gated for sorting (Figure 2B). A second sort was performed with greater stringency to further enrich binders with high affinity and selectivity for CA-II. The resulting population was expressed on yeast, conjugated, and confirmed to selectively bind CA-II compared to CA-IX (Figure 2C).
Fn-encoding genes from the population enriched for strong, selective binders were shuttled to an E. coli expression vector. Clones were selected from this population at random, and Fn was expressed in the soluble fraction, purified via metal affinity chromatography, and conjugated with maleimide-PEG7-AAZ. Purity and conjugation were verified by SDS-PAGE and matrix-assisted laser desorption-ionization (MALDI) mass spectrometry (Figure S3). Conjugation yields averaged 98 ± 2%.
Potency and specificity in inhibiting CA activity were evaluated via spectrophotometric monitoring of the conversion of 4-nitrophenyl acetate to 4-nitrophenol and acetic acid by carbonic anhydrase (Figure 3A).(lnnocenti et al., 2008) FnII.28.7-PEG7-AAZ exhibits potent inhibition of CA-II (KI = 4.8 nM) that is highly specific as CA-IX inhibition was 120-fold weaker (KI = 580 nM; p = 0.01; Figure 3BC; confidence intervals are indicated in figures).
Figure 3. Engineered PriSMs exhibit potent, isoform-specific inhibition.

(A) 4-nitrophenyl acetate is converted to 4-nitrophenol and acetic acid by carbonic anhydrase. The ability of PriSMs to inhibit this activity is titrated by spectrophotometrically measuring 4-nitrophenol production over time at different PriSM concentrations. (B) Titration curves of FnII-28.7-PEG7-AAZ (solid lines) and PEG7-AAZ (dashed lines) with CA-II and CA-IX. (C) Inhibition constants. Data are presented as the mean ± 68% confidence interval for three replicates. Inhibition was not detected for FnII-28.7.
The PriSM dramatically enhances specificity, while maintaining potency, compared to AAZ alone (KI,CA-II = 3.3 nM; KI,CA-IX = 6.6 nM). Moreover, consistent with prior work modifying CA inhibitors(Krishnamurthy et al., 2008), the PEG linker hinders AAZ activity; thus it is also relevant to compare performance of the PriSM to unreacted PEG7-AAZ. The PriSM is 9-fold more potent (KI,CA-II = 42 nM for PEG7-AAZ) and 80-fold more specific (KI,CA-IX = 6 nM for PEG7-AAZ) than unreacted PEG7-AAZ. Notably, the evolved FnII.28.7 protein alone, without PEG7-AAZ, does not inhibit CA-II activity. Thus, the engineered protein component of the hybrid provides beneficial interaction that compensates for the inhibitory detriment of the linker and provides substantial specificity.
Potent, specific PriSMs can be engineered with multiple linker lengths, conjugation sites, and targets
To assess discovery of PriSMs with alternative linker length, analogous selection campaigns were performed with maleimide-PEGx-AAZ linkers of length x = 2, 3, and 5 (along with the previous x = 7, these provide a range of lengths while being experimentally efficient). Random colony picking of the enriched populations yielded clones with significant specificity for FnII.28.5-PEG5-AAZ and FnII.28.3-PEG3-AAZ, albeit not for the clone with PEG2 (Figure 4: Cys28). FnII.28.5-PEG5-AAZ increases affinity 3.6-fold and specificity 9-fold relative to PEG5-AAZ. FnII.28.3-PEG3-AAZ exhibits 11-fold specificity for CA-II relative to CA-IX by essentially eliminating observable inhibition of CA-IX but with an appreciable reduction in potency for CA-II. Thus, functional PriSMs can be engineered with a range of linker lengths. While it could be tempting to attribute reduced efficacy with shorter linker length, this is not the general case as shown by deeper analysis of linker length trends (see Figures 5C and S5) and demonstration of specificity and potency gains from short linkers in the context of other conjugation sites and isoform targets (Figure 4: Cys80 and CA-IX, detailed next).
Figure 4. PriSMs can be engineered with multiple linker lengths.

Enzymatic inhibition constants – measured via the assay detailed in Figure 3 – for AAZ, PEG-AAZ, and Fn-PEG-AAZ PriSMs. Clones were isolated from each of the four linker campaigns from the Cys28 and Cys80 libraries for CA-II binding and the Cys28 library for CA-IX binding. Data are presented as the mean ± 68% confidence interval for three replicates.
Figure 5. The PriSM-enzyme interface is finely tuned to linker length.

(A,B) Modifying the linker from the length of original discovery hinders specificity. Enzyme inhibition constants for FnII.28.7-Lx-AAZ (A) and FnII.80.2-Lx-AAZ (B) (and both Fn proteins alone) are presented as the mean ± 68% confidence interval of triplicate measurements. Inhibition was not detected for Fn without conjugation. (C) Relative frequency across different linker length campaigns for CA-II binders from Cys80 library. Data exist at lengths 2, 3, 5, and 7; connecting lines are drawn for visual guidance.
To assess the ability to engineer PriSMs with an alternate protein conjugation site, binder selections were performed using the library conserving cysteine at site 80, conjugated to each of the four lengths of maleimide-PEG-AAZ, in parallel. Site 80 was chosen for its central location in the extended FG loop (Figure 1). A broad panel of specific, high-affinity binding variants was enriched (Figure S4A,B). A selected clone at each linker length was evaluated for enzymatic inhibition. In all four cases, Fn conjugation engendered isoform specificity, with ratios of KI values for CA-II to CA-IX of 11-42 (Figure 4: Cys80). Relative to PEG-AAZ alone, potency to CA-II was maintained (9 – 88 nM) while the engineered Fn drove the desired diminished activity against CA-IX. Conjugation yield on the yeast surface was >99% at sites 28 and site 80 (standard errors: 6% and 4%) (Figures S4C). Thus, the Fn-PEG-AAZ concept is amenable to multiple Fn conjugation sites.
All conjugates to this point have been selected for specificity to CA-II compared to CA-IX. To assess the generality of the PriSM concept, we aimed to identify conjugates with the converse specificity. An analogous library selection scheme using magnetic bead and flow cytometric selections was used to isolate CA-IX binders. The selected conjugates achieved high potency (3-48 nM KI values) and enhanced selectivity by 46-112 fold relative to small molecule alone (Figure 4: CA-IX-specific). Notably, FnIX.28.5-PEG5-AAZ moderately enhanced potency to CA-IX relative to AAZ (to 3.4 nM from 6.6 nM) and drastically enhanced potency relative to PEG5-AAZ (170 nM), clearly demonstrating the addition of beneficial interactions from the Fn component. The performance of the collection of conjugates shows the applicability of the PriSM discovery platform to multiple targets.
The PriSM-enzyme interface is finely tuned to linker length
Each aforementioned construct is associated with a particular PEG linker length with which it was discovered in the process of ligand selection. The intention of our design is that the AAZ pharmacophore inserts into the enzyme catalytic site, while the PEG linker creates space for the Fn component to find a low-energy, bound state. Specificity is the result of Fn finding energetically favorable interactions with its designed target, while generating unfavorable interactions with other non-targets. We hypothesize that an effective linker connects the pharmacophore and protein in their optimal interfacial configurations, is constrained to thereby minimize entropic cost upon binding (Krishnamurthy et al., 2006; Mammen et al., 1998), and does not itself hinder the binding interface. Notably, inclusion of the PEG linker, especially the longer two lengths tested, alters potency of AAZ (Figure 4); yet potent, selective PriSMs were engineered in all four lengths tested (Figure 4). To determine the extent to which the PEG linker length drives performance as a design parameter of each clone, we varied the linker length and assayed enzyme inhibition for FnII.28.7-Lx-AAZ and FnII.80.2-Lx-AAZ.
For FnII.28.7-PEG7-AAZ, shortening the linker to 5 units results in a 40-fold specificity loss via dramatically elevating off-target CA-IX potency – either via removal of originally detrimental interactions or creation of beneficial interactions – while only nominally aiding CA-II potency (Figure 5A). Further reduction in linker length to 3 or 2 units drastically hinders CA-II potency by ~100-fold and also hinders CA-IX potency but less thereby resulting in a specificity reversal. The resulting potency toward both isoforms is substantially worse than the small molecule, with or without the linker, indicating that the Fn component, which was selected for enhancing potency with a longer linker, introduces detrimental interaction when constrained by a shorter linker. It is also notable, that the Fn protein alone does not provide measurable inhibition of either isoform.
Starting with a variant discovered to be effective with the shortest linker, FnII.80.2-PEG2-AAZ, specificity reduction is again observed with linker modification. CA-II potency steadily worsens with longer linkers while CA-IX potency improves with extension to PEG3 and then PEG5 but is drastically hindered by further extension to PEG7 (Figure 5B). Also, as with the previous clone, the Fn protein alone is not inhibitory.
To broaden the analysis of linker length impact, we used the power of flow cytometry and deep sequencing. Each of the populations enriched during selection of high affinity, specific binders via flow cytometry (as in Figure 2B) were deep sequenced. The relative frequency of each variant reports on its efficacy; thus, comparison of frequencies across different length campaigns informs on linker length impact. An array of outcomes is observed (Figures 5C and S5). As a few examples from the CA-II binders with Cys80, one clone (orange in Figure 5C) performs better with longer PEG but tolerates all four lengths; another (blue) has substantially better performance with PEG5 but tolerates two units shorter or longer; another (red) is only effective with PEG7; conversely another (green) is broadly tolerant at all four lengths tested. Thus, the ability of several lengths, separately, to yield specific, high-affinity binders supports an efficient approach in which a single length is used in future campaigns. Yet the improved performance achieved by different lengths for different protein sequences indicates that a broader diversity of PriSM sequences will be discovered, including improvement of many clones, if multiple linker lengths are used in parallel during discovery.
Analysis of the sequence landscape of functional PriSMs
Deep sequencing of selected populations also empowers analysis of the sequence landscape of functional PriSMs. Populations of engineered PriSMs are diverse, as evidenced by median Hamming distances (numbers of sites with differing amino acids) of 9-14 within a campaign (Figure 6A). Most campaigns yielded moderate preferential enrichment of more functional variants with a consistent slope of 10-fold reduction in frequency per order of magnitude of diversity (Figure 6B). Notably, the clones randomly chosen from the final selection populations for inhibitory analysis were relatively broadly distributed. For example, the clones for PEG5 and PEG7 in Cys28 and PEG7 in Cys80 were the 3rd, 6th, and 4th most frequent in their populations. Yet, other analyzed clones were 51st, 64th, 91st, and 240th. All were specific and potent except FnII.28.2-PEG2-AAZ (which ranked 240th thereby explaining its lack of efficacy on unfortunate stochastic selection). The achievement of specific potency from these stochastically-chosen clones that did not dominate enrichment indicates that a broad array of functional PriSMs were selected. This diversity is beneficial so that subsequent steps could be used to enrich for alternative properties, such as developability(Jain et al., 2017), or increase stringency for potency or specificity.
Figure 6. PriSM sequence landscape.

(A) The Hamming distance between all pairs of variants in each population. Black represents the unsorted libraries. (B) The relative frequency of each variant in each population. (C) The relative frequency of each loop length for each campaign (Cys28 or Cys80 conjugation sites; naïve library or sorted for binding in the presence of conjugates with linkers of PEG2, PEG3, PEG5, or PEG7. (D) Sitewise amino acid enrichment, from the naïve library to the sorted binding population, is presented for each target (CA-II or CA-IX) and conjugation site (Cys28 or Cys80) combination, with results from all linker lengths aggregated. Sites with possible gaps for loop length diversity are underlined.
The functionally enriched populations were also evaluated for Fn loop length to guide future library design (Figure 6C). When containing the Cys28 conjugation site, BC loop length trends counter the PEG linker length, perhaps to provide sufficient cavity penetration for short linkers. The analog is not observed for FG loops with Cys80 conjugation, which is consistent with the already extended structure of the FG loop (Figure 1A). When AAZ is conjugated via short linkers to Cys28, short lengths are enriched in the adjacent FG loop, perhaps to avoid steric hindrance of the extended structure. Conversely, in Cys28 campaigns, longer DE loops are enriched, which could enable better target engagement of this otherwise smaller loop.
The PriSM sequence-function landscape is elucidated by analysis of amino acid enrichment at each site during selection of specific, high-affinity binders (Figure 6D). Cys is not enriched at other sites, which is consistent with the effective utility of the proposed conjugation sites C28 and C80. Multiple amino acid preferences at sites proximal to the AAZ conjugation site are revealed. Within Cys28 conjugation campaigns, site 27 is heavily enriched in W. Interestingly, this is counter to two other aromatics, the homologous F and Y, which are consistently depleted at the four sites flanking Cys28. At site 26, I, Q, and D are heavily enriched, which curiously represents a diverse chemical repertoire albeit with similar size; which may represent the importance of spatial packing at this location or diversity of engineered interfaces. Two sites from Cys28 AAZ conjugation on the other side, P30 is heavily enriched while numerous amino acids are substantially depleted. When the BC loop is at its longest, making p26 a non-gap amino acid, it is strongly enriched in G and P suggesting importance of loop conformation. In campaigns with Cys80 conjugation, the adjacent site 79 is enriched in G (from 17% to 39%), which may provide conformational flexibility. Conversely, the adjacent site on the other side, 81, is most heavily enriched in the conformationally constraining P. Lastly, the sites with initial diversity constraint (24, 25, 29, 31, 53-55, 76, 77, 79) generally do not exhibit substantial enrichment or depletion, which is consistent with effective library design; mild exceptions are enrichment of Y at sites 53 and 55 and S at 55. Collectively, analysis of the diverse PriSMs contributes to elucidation of sequence-function relationships that will be further enlightened by future campaigns and structural analysis.
Discussion
The functional specificity and potency achieved herein highlight the power of hybridizing proteins and small molecule pharmacophores as well as the PriSM discovery platform. The systematic evaluation of hybrid performance across molecular design parameters demonstrates the generality of the approach as PriSM discovery was successful with two distinct conjugation sites in two different Fn loops, using four different PEG linker lengths, a broad diversity of protein sequences, and targeting two different enzyme isoforms. The distinctiveness of solutions across the different molecular designs reveals the impact of PriSM elements and underscores opportunities for further advancement via expansion and tuning of these design elements.
Activity-based selection with high throughput is a noted challenge of combinatorial discovery and directed evolution (Lutz and Patrick, 2004; Packer and Liu, 2015). A key strength of the PriSM concept, and the chemical biology discovery platform, may prove to be the ability of the pharmacophore to focus the search of molecular space on functionally active molecules (i.e. inhibitors in the current context, but could be agonists in other PriSM campaigns). Although high-throughput yeast display sorts were performed based on target binding not inhibitory potency, presence of the AAZ pre-conditioned hybrids for activity. Indeed, potent inhibition was achieved across numerous campaigns in the current work. Moreover, despite providing potency and selectivity enhancement, the engineered Fn domains without conjugated pharmacophore did not exhibit appreciable inhibition. The PriSM platform empowered efficient discovery of functional inhibitors. It is also striking that the current PriSMs were engineered without iterative evolution; that is, they were identified via direct discovery from a substantially undersampled set of sequence space (108 experimentally tested genetic variants within a focused library design of 1017 possible protein variants with unbiased potential diversity of 1026).
In the 12 combinations of conjugation site, target, and linker length, specificity was always enhanced via hindrance of off-target potency. In 50% of these cases, on-target potency was also enhanced (Figure S6). The potency of the pharmacophore likely plays a role in this balance (reducing off-target potency versus enhancing on-target potency) when selecting for potent, selective PriSMs. Mutations more readily hinder interactions than aid (Tokuriki et al., 2007), thus a diverse population will tend to contain more variants that reduce off-target potency than enhance on-target potency. With a relatively strong pharmacophore, reduced off-target potency may be sufficient leading to enrichment of such variants. With a weaker pharmacophore, on-target potency enhancement becomes more requisite thereby increasing enrichment of such variants. Future efforts could explore the impact of pharmacophore affinity as well as other aforementioned design elements including additional linkers, protein conjugation sites, and protein scaffolds. While we elucidated the impact of linker length in the current work, modulation of linker stiffness, hydrophilicity, and bulk could also play key roles. Linkers could be engineered, for example via peptide linkers, to beneficially engage the active site, provide preferential interactions relative to non-target site, and reduce entropic cost upon binding (Krishnamurthy et al., 2006; Mammen et al., 1998). The yeast display bioconjugation PriSM engineering platform demonstrated here can efficiently empower such studies.
Alternative conjugation chemistries could also be explored. While maleimide-cysteine chemistry has been widely used with significant success (Fredy et al., 2018; Ravasco et al., 2019), instability (Ross and Wolfe, 2016) and the production of diastereomers could motivate pursuit of other options such as chloroacetamide-cysteine (Lindley, 1960), non-canonical amino acid options (Islam et al., 2021; Link et al., 2003), or others (DeGruyter et al., 2017). Future studies would be needed to determine the relative activity of each diastereomer in the current hybrids. CA binding to AAZ conjugated at alternative (non-Fn) sites on the yeast surface was not sufficiently strong to cause problematic background enrichment at the experimental conditions for conjugation and target labeling (Fig. 2A). Nevertheless, more site selective chemistry could reduce this potentiality. For example, phage lacking cysteine in gene 3 protein have been engineered (Kather et al., 2005) to enable display-selective thiol conjugation (Heinis et al., 2009), non-canonical amino acids have empowered site-specific conjugation in yeast display(lslam et al., 2021), bacterial display (Navaratna et al., 2020), mRNA display (Horiya et al., 2014), and phage display (Tian et al., 2004).
Significance
Diverse molecular architectures are needed to elicit precise biological function via active binding at select epitopes. While proteins, peptides, and small molecules have all exhibited efficacy in numerous cases, each format balances advantages and shortcomings. Previous work on peptide-pharmacophore hybrids has exemplified the utility of a complementary merger of formats and – along with preliminary work on antibody-pharmacophore systems – motivates further expansion of the molecular repertoire. Herein, we demonstrate the effectiveness of protein-small molecule hybrids or PriSMs within the context of fibronectin domain proteins and acetazolamide pharmacophore. Moreover, we develop an effective platform for hybrid discovery via yeast display library selections. The PriSM platform efficiently advances specificity and potency in selection from a single library without evolution. The success of the PriSM approach can be viewed from several lenses of drug discovery, thereby building on the strong precedent for each: a spatially integrated version of heterobivalent ligand design to boost avidity and specificity via dual binding interfaces; a hybrid approach to fragment-based drug discovery; or a synthetic chemistry approach to non-canonical amino acid incorporation. Demonstration of hybrid functionality with multiple linker lengths, conjugation sites, and protein sequences against a pair of isoforms highlights the potential to further tune molecular design from each of these perspectives. Also, opportunities exist to modify conjugation chemistry for selectivity and stability. This study adds to the strong body of work on chemical biology strategies to engineer integrated hybrid interfaces as well as to apply the technological utility to a broad array of targets and biological functions.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be addressed to and will be fulfilled by the Lead Contact, Benjamin Hackel (hackel@umn.edu).
Materials Availability
Key plasmids created in this study are available upon request.
Data and Code Availability
Raw source data are available upon reasonable request.
All original code is publicly available at https://github.com/HackelLab-UMN_and https://doi.org/10.5281/zenodo.5093631.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial Strains
Escherichia coli (E. coli) DH5α strains were used for plasmid amplification. E. coli DH5α cells were grown in LB medium at 37 °C. E. coli T7 strains were used for protein purification for inhibition assays. Cells were grown at 37 °C until OD600 of 0.5-1.0, followed by induction with 0.5 mM isopropyl-β-d-thiogalactoside (IPTG) at 30 °C for 4 hours.
Cell Lines
Saccharomyces cerevisiae (S. cerevisiae) EBY100 cells were obtained from the American Type Culture Collection (ATCC). S. cerevisiae EBY100 cells were routinely cultured at 30 °C in rich YPD media. Once transformed with the yeast display pCT40-FnLoopHP plasmid (or library variants thereof), cells were grown in glucose-rich yeast minimal media minus tryptophan (SD-CAA) at 30 °C until OD600 of 6.0, followed by induction of yeast display with change into galactose-rich SG-CAA (0.1M sodium phosphate, pH 6.0, 6.7 g/L yeast nitrogen base, 5 g/L casamino acids, 19 g/L galactose, 1 g/L glucose).
METHOD DETAILS
Materials and general methods
Synthetic DNA oligonucleotides (Table S1) were purchased from Integrated DNA Technologies (Coralville, IA). PCR and restriction digest products were purified by gel electrophoresis and extracted using Zymoclean Gel DNA Recovery kit. Plasmid DNA was purified using GenCatch Plasmid DNA Miniprep kit. Sanger DNA sequencing was conducted by Eurofins Genomics (Louisville, KY).
Library Construction
A genetic library was constructed based on the human tenth domain of fibronectin type III (Fn), in which the sequence encoding for the three loops was systematically diversified using degenerate oligonucleotides encoding for an amino acid distribution (Table S2). Degenerate nucleotides, at diversified positions 26-28, 78, and 80-83, had 20% A, 19% C, 27% G, and 34% T in the first position; 44% A, 21% C, 12% G, and 23% T in the second position; and 23% G, and 77% T in the third position as motivated by earlier library engineering (Woldring et al., 2015). The design also included loop length diversity with an insertion, deletion, or neither in the BC loop, a deletion or wild-type in the DE loop, and zero, one, or two deletions in the FG loop. Overlap extension PCR reactions were carried out to construct the full-length Fn genes. Gene reactions were transformed into EBY100 yeast using homologous recombination with linearized yeast surface display pCT40-FnLoopHP vector (Woldring et al., 2015).
Labeling of CA-II and CA-IX for FACS Detection
CA-II was first buffer exchanged with Zeba Spin Desalting Columns 7K MWCO to phosphate-buffered saline (PBS) and then incubated for four hours at 4 °C with 1:10 NHS-LC-biotin. CA-IX was reconstituted according to manufacturer recommendation and incubated for four hours at 4 °C with 1:100 with DyLight 650 NHS ester. Both conjugated CA-II and CA-IX were desalting by running twice through separate Zeba columns. Concentration was quantified via gel electrophoresis.
Maleimide-PEGx-AAZ on-yeast conjugation
Appropriate yeast populations were washed three times with PBS. No reduction step was included because of the disulfide-bonded connection of Aga1p (anchored in the yeast cell wall) to Aga2p (fused to Fn via a polypeptide linker to achieve yeast display). Yeast were then incubated for two hours at room temperature with 8 μM maleimide-PEGx-AAZ in 100 μL per 10 million cells. Conjugated yeast were then washed with PBSA (containing 0.1% BSA) to quench and clear any unbound maleimide-PEGx-AAZ. Functional yield was calculated by incubating conjugated yeast with 10 nM CA-II and measuring binding via flow cytometry. Binding signal of unconjugated yeast was subtracted as a background, and the difference was normalized relative to binding signal from yeast conjugated at saturating concentrations of maleimide-PEGx-AAZ (no change observed across 3, 8, 10, 30 μM).
Binder and specificity selection
Yeasts displaying the Fn Cys28 library were washed and conjugated in four campaigns to maleimide-PEGx-AAZ (x= 2, 3, 5, or 7) as described above. Conjugated yeast underwent magnetic activated cell sorting, where yeasts were incubated with control avidin-coated magnetic beads for one hour at room temperature, followed by another incubation of control avidin-coated beads to remove any non-specific binding interactions. Yeasts were then exposed to magnetic beads with immobilized CA-II biotinylated target protein and bound yeasts were selected. Magnetic sorts on the initial library were performed at room temperature and washed twice. Yeasts from each of the four linker length campaigns were pooled, grown, induced, and again conjugated, separately, to each of the four linker lengths. Non-naïve populations underwent a more stringent secondary magnetic sort. Conjugated yeasts were incubated twice with control avidin-coated beads, as before, incubated with 10 nM biotin-conjugated CA-II for 30 minutes, and then followed with incubation of avidin-coated beads for an additional 30 minutes for selection. Non-naïve populations were sorted at room temperature and washed four times. Two rounds of flow cytometry selections — the first with 10 nM biotinylated CA-II target, the second with 10 nM CA-II target and 10 nM DL650-labeled CA-IX non-target, and both with mouse anti-c-MYC and anti-mouse-PE — were used to isolate full-length (c-MYC positive) that bind selectively and with high affinity toward CA-II. The sort scheme was repeated for the Cys80 library.
Additionally, yeasts displaying the Fn Cys28 library were selected for CA-IX binders at each of the four PEG linker lengths. The naïve population first underwent the more stringent magnetic sort, instead incubated with 36 nM CA-IX biotinylated target, and selected with avidin-coated magnetic beads, as before. Yeasts were pooled, grown, induced, and reconjugated at each of the four lengths. Yeasts were sorted thrice via three-color FACS at 0.75 nM CA-IX and 50 nM CA-II, 0.5 nM CA-IX and 100 nM CA-IX, and again at 0.5 nM CA-IX and 100 nM CA-II.
Cloning, Protein Expression, and Purification
Fn-encoding regions in DNA recovered from the final CA-II/CA-IX flow cytometry sort were amplified by polymerase chain reaction (see Table S1), digested with NheI-HF and BamHI-HF reaction enzymes (New England Biolabs), and ligated with T4 DNA ligase into pET-24b vector2 containing a C-terminal hexa-histidine tag. Plasmids were transformed into T7 Express Competent E. coli and plated on lysogeny broth (LB) plates containing 50 mg/L kanamycin. Transformants were Sanger sequenced for full-length gene (Table S3), and proper transformants were grown in 5 mL liquid LB with kanamycin (50 mg/L) at 37 °C at 250 rpm for 12-16 hours. Saturated cultures were added to 100 mL LB, grown, and induced. Cells were pelleted and resuspended in lysis buffer (50 mM sodium phosphate (pH 8.0), 0.5 M sodium chloride, 5% glycerol, 5 mM 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate, and 25 mM imidazole), frozen and thawed five times, centrifuged for 10 minutes at 4 °C, and 0.25 μm filtered. The resulting cell lysate were run through 0.25 mL Cobalt HisPur resin volume spin columns and conjugated to maleimide-PEGx-AAZ on-column (x = 2, 3, 5, or 7). The lysate-resin was washed with 30 mM imidazole, incubated with 1 mM TCEP for 15 minutes, washed with PBS, incubated for 30 minutes with 400 μM maleimide-PEGx-AAZ, washed with 30 imidazole, and then eluted with 300 mM imidazole. PriSMs were further purified via dialysis with three 1:1000 PBS buffer exchanges. Concentrations were quantified via NanoDrop One C (ThermoFisher Scientific, Madison, VA). PriSM purity was analyzed via gel electrophoresis, and small molecule conjugation was analyzed using AB-Sciex 5800 MALDI/TOF-MS.
Carbonic anhydrase inhibition assay
Carbonic anhydrase esterase activity was assayed by following the conversion of 4-nitrophenyl acetate (4-NPA) hydrolyzed to 4-nitrophenol and acetic acid in the presence of the enzyme via absorbance at 400 nm with a BioTek Synergy H1 microplate reader. CA-II or CA-IX was pre-incubated with inhibitor at various concentrations for 30 minutes prior to substrate addition, and the reaction was initiated upon addition of the substrate. The absorbance was measured every minute for 30 minutes at room temperature. The final concentration of the reaction components were 17.5 mM Tris, 105 mM NaCl buffer, pH 7.4, 25 nM CA-II or 100 nM CA-IX, 0, 1, 10, 50, 100, or 200 nM inhibitor, and 2 mM 4-NPA substrate in a final enzymatic assay volume of 100 μL containing 2% acetone. A higher concentration of CA-IX was used as CA-IX was less active than CA-II and therefore produced a much weaker signal. The absorbance of the 4-NPA with various inhibitor concentrations 0-200 nM in buffer, in the absence of enzyme, was subtracted from the enzymatic measurements. Each inhibitor was assayed with either CA-II or CA-IX with various concentrations at least three times.
Illumina MiSeq analysis of functional PriSMs
Plasmid DNA for the naive Fn Cys28 and Fn Cys80 libraries as well as the final sorted populations was isolated from yeast using Zymolyase and extracted via silica spin column. The full Fn gene was amplified via PCR using full gene amplification primer (Table S1). Illumina adapters were added in a second PCR to differentiate between populations, and DNA was quantified by Nanodrop and mixed in equal molar ratios for a final concentration of 5 nM. Samples were submitted to the University of Minnesota Genomics Center for quality control analysis and sequenced using Illumina MiSeq (2x300, v3). Sequences were processed using USearch (Edgar, 2010) and filtered for a maximum of 1 total expected error for all bases in the read. Fn scaffold analysis was completed via homemade Python scripts to evaluate the frequency of unique variants, Hamming distance, loop length diversity, and site enrichment of final sorted populations.
QUANTIFICATION AND STATISTICAL ANALYSIS
Determination of apparent KI
The absorbance values over time at different inhibitor concentrations were linearly fit to determine the slope. The fitted slope for various inhibitor concentrations in the absence of enzyme were used as a background subtraction. Inhibitor concentration did not affect substrate hydrolysis. Apparent Ki analyses were carried out in Matlab R2020a, fitting v0 and to Equation 1 using nlinfit. Default options were used for the solver except TolX and TolFun were both set to 1e-15. ET was set as 25 nM for CA-II or 100 nM for CA-IX. Enzyme activity was then normalized to one using the fitted v0, the enzyme activity in the absence of inhibitor. The function nlparci was used to determine the 68% confidence interval for , where alpha equaled 0.32.
| [Eq. 1] |
Statistical analyses
Fluorescence single-cell data of final Fn Cys28-PEG7-AAZ population (Figure 2C) was exported, and a t-test was used to calculate p. Statistically significant conditions are marked with an asterisk (*) in the figure. Standard error of three independent experiments are shown in FnII.28.7-PEG7-AAZ spectrophotometer data (Figure 3A) and titration curves (Figure 3B). A t-test was used to calculate p and is found the Results section.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse c-MYC Monoclonal Antibody (9E10) | Invitrogen | Cat# MA1-980, RRID AB_558470 |
| Streptavidin, Alexa Fluor™ 488 conjugate | Invitrogen | Cat# S11223 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary antibody, PE | Invitrogen | Cat# P-852, RRID AB_2539848 |
| Bacterial and Viral Strains | ||
| Escherichia coli: DH5α | New England Biolabs | Cat# C2987C |
| Escherichia coli: T7 Express | New England Biolabs | Cat# C2566H |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 4-Nitrophenyl acetate | Sigma-Aldrich | Cat# N8130-5G |
| DyLight™ 650 NHS Ester, 5 x 50 ug | Thermo Scientific | Cat# 62266 |
| EZ-Link™ Sulfo-NHS-LC-Biotin | Thermo Scientific | Cat# 21335 |
| HisPur™ Cobalt Resin | Thermo Scientific | Cat# 89964 |
| Human Carbonic Anhydrase II Protein (Recombinant) | LifeSpan BioSciences, Inc | Cat# LS-G551-100 |
| Human Carbonic Anhydrase IX / CA9 (38-414) Protein, His Tag | ACRO Biosystems | Cat# CA9-H5226-1 mg |
| Carbonic Anhydrase IX Protein, Human, Recombinant (His Tag) | Sino Biological Inc. | Cat# 10107-H08H |
| Maleimide-PEG2-acetazolamide | Peptech | N/A |
| Maleimide-PEG3-acetazolamide | Peptech | N/A |
| Maleimide-PEG5-acetazolamide | Peptech | N/A |
| Maleimide-PEG7-acetazolamide | Peptech | N/A |
| Critical commercial assays | ||
| Zymoclean Gel DNA Recovery Kit | Zymo Research | Cat# D4002 |
| Zeba Spin Desalting column, 7 kDa | ThermoFisher | Cat# 89882 |
| LongLife Zymolyase | G-Biosciences | Cat#: 786-036 |
| Experimental models: cell lines | ||
| Saccharomyces cerevisiae: EBY100 | ATCC | MYA-4941 |
| Oligonucleotides | ||
| See Table S1 for PCR primers used in this study | ||
| Recombinant DNA | ||
| pCT40-FnLoopHP | Woldring et al., 2015 | N/A |
| pET-24b | Lipovšek et al., 2007 (Novagen) | N/A |
| Software and Algorithms | ||
| BioTek Gen5 Software Version 2.09 | Biotek | https://www.biotek.com/products/software-robotics/ |
| Matlab R2020a | MathWorks | https://www.mathworks.com/ |
| PyMol 2.4.0 Open-Source | PyMol | https://www.pymol.org/ |
| Python 3.8.8 | Python | https://www.python.org/ |
| Code / Scripts | This manuscript | https://github.com/HackelLab-UMN |
Highlights.
Protein-small molecule hybrids engineered via yeast display platform
Hybrids are superior to individual components in inhibitory potency and specificity
Multiple linker lengths, conjugation sites, sequences, and targets show flexibility
Hybrid engineering platform advances engineerable ligand diversity
Acknowledgements
We thank Alex Golinski for guidance on sequence analysis and Jim Van Deventer for useful discussions. We thank the University of Minnesota Flow Cytometry Resource for cell sorting, the University of Minnesota Genomics Center for assistance with deep sequencing, and the Minnesota Supercomputing Institute for computing resources. Research was supported by the National Institutes of Health (R01 EB023339 and R01 EB028274) and Itara Biotherapeutics.
Footnotes
Declaration of Interests
R.R.L. was an employee of Itara Biotherapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersson T, Lundquist M, Dolphin GT, Enander K, Jonsson BH, Nilsson JW, and Baltzer L (2005). The binding of human carbonic anhydrase II by functionalized folded polypeptide receptors. Chem. Biol 12, 1245–1252. [DOI] [PubMed] [Google Scholar]
- Buller F, Steiner M, Frey K, Mircsof D, Kalisch M, Peter B, Supuran CT, and Neri D (2011). Selection of Carbonic Anhydrase IX Inhibitors from One Million DNA-Encoded Compounds. ACS Chem. Biol 6, 336–344. [DOI] [PubMed] [Google Scholar]
- Carta F, Garaj V, Maresca A, Wagner J, Avvaru BS, Robbins AH, Scozzafava A, McKenna R, and Supuran CT (2011). Sulfonamides incorporating 1,3,5-triazine moieties selectively and potently inhibit carbonic anhydrase transmembrane isoforms IX, XII and XIV over cytosolic isoforms i and II: Solution and X-ray crystallographic studies. Bioorganic Med. Chem 19, 3105–3119. [DOI] [PubMed] [Google Scholar]
- Carter PJ, and Lazar GA (2018). Next generation antibody drugs: pursuit of the ‘high-hanging fruit.’ Nat. Rev. Drug Discov 17, 197–223. [DOI] [PubMed] [Google Scholar]
- Carter AJ, Kraemer O, Zwick M, Mueller-Fahrnow A, Arrowsmith CH, and Edwards AM (2019). Target 2035: probing the human proteome. Drug Discov. Today 2513, 1–5. [DOI] [PubMed] [Google Scholar]
- Chao G, Lau WLWL, Hackel BJBJ, Sazinsky SLSL, Lippow SMSM, and Wittrup KDD (2006). Isolating and engineering human antibodies using yeast surface display. Nat. Protoc 1, 755–768. [DOI] [PubMed] [Google Scholar]
- Chen J, Sawyer N, and Regan L (2013). Protein-protein interactions: general trends in the relationship between binding affinity and interfacial buried surface area. Protein Sci. 22, 510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Lovell S, Lee S, Fellner M, Mace PD, and Bogyo M (2020). Identification of highly selective covalent inhibitors by phage display. Nat. Biotechnol 39, 490–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier A, Silva D, Rocklin GJ, Hicks DR, Vergara R, Murapa P, Bernard SM, Zhang L, Lam K, Yao G, et al. (2017). Massively parallel de novo protein design for targeted therapeutics. Nature 550, 74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGruyter JN, Malins LR, and Baran PS (2017). Residue-Specific Peptide Modification: A Chemist’s Guide. Biochemistry 56, 3863–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derda R, and Ng S (2019). Genetically encoded fragment-based discovery. Curr. Opin. Chem. Biol 50, 128–137. [DOI] [PubMed] [Google Scholar]
- Deventer J.A. Van, Le DN, Zhao J, Kehoe HP, and Kelly RL (2016). A platform for constructing , evaluating , and screening bioconjugates on the yeast surface. Protein Eng. Des. Sel 29, 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. [DOI] [PubMed] [Google Scholar]
- Erlanson DA, Fesik SW, Hubbard RE, Jahnke W, and Jhoti H (2016). Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov 15, 605–619. [DOI] [PubMed] [Google Scholar]
- Fredy JW, Renard P, and Sabot C (2018). Covalent Modification of Biomolecules through Maleimide-Based Labeling Strategies. Bioconjug. Chem 29, 2497–2513. [DOI] [PubMed] [Google Scholar]
- Gao BB, Clermont A, Rook S, Fonda SJ, Srinivasan VJ, Wojtkowski M, Fujimoto JG, Avery RL, Arrigg PG, Bursell SE, et al. (2007). Extracellular carbonic anhydrase mediates hemorrhagic retinal and cerebral vascular permeability through prekallikrein activation. Nat. Med 13, 181–188. [DOI] [PubMed] [Google Scholar]
- Garaj V, Puccetti L, Fasolis G, Winum JY, Montero JL, Scozzafava A, Vullo D, Innocenti A, and Supuran CT (2004). Carbonic anhydrase inhibitors: Synthesis and inhibition of cytosolic/tumor-associated carbonic anhydrase isozymes I, II, and IX with sulfonamides incorporating 1,2,4-triazine moieties. Bioorganic Med. Chem. Lett 14, 5427–5433. [DOI] [PubMed] [Google Scholar]
- Gilbreth RN, Truong K, Madu I, Koide A, Wojcik JB, Li N-S, Piccirilli JA, Chen Y, and Koide S (2011). Isoform-specific monobody inhibitors of small ubiquitin-related modifiers engineered using structure-guided library design. Proc. Natl. Acad. Sci 108, 7751–7756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gower CM, Thomas JR, Harrington E, Murphy J, Chang MEK, Cornella-Taracido I, Jain RK, Schirle M, and Maly DJ (2015). Conversion of a Single Polypharmacological Agent into Selective Bivalent Inhibitors of Intracellular Kinase Activity. ACS Chem. Biol 11, 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebling U, Askoxylakis V, Garcia-Boy R, Rana S, Kra S, and Haberkorn U (2010). A New Peptide Ligand for Targeting Human Carbonic Anhydrase IX, Identified through the Phage Display Technology. PLoS One 5, e15962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinis C, Rutherford T, Freund S, and Winter G (2009). Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol 5, 502–507. [DOI] [PubMed] [Google Scholar]
- Horiya S, Bailey JK, Temme JS, Guillen Schlippe YV, and Krauss IJ (2014). Directed evolution of multivalent glycopeptides tightly recognized by HIV antibody 2G12. J. Am. Chem. Soc 136, 5407–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseinzadeh P, Watson PR, Craven TW, Li X, Rettie S, Pardo-avila F, Bera K, Mulligan VK, Lu P, Ford AS, et al. (2021). Anchor extension: a structure-guided approach to design cyclic peptides targeting enzyme active sites. Nat. Commun 12, 3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocenti A, Scozzafava A, Parkkila S, Puccetti L, De Simone G, and Supuran CT (2008). Investigations of the esterase, phosphatase, and sulfatase activities of the cytosolic mammalian carbonic anhydrase isoforms I, II, and XIII with 4-nitrophenyl esters as substrates. Bioorganic Med. Chem. Lett 18, 2267–2271. [DOI] [PubMed] [Google Scholar]
- Islam M, Kehoe HP, Lissoos JB, Huang M, Ghadban CE, Lane HZ, and Deventer J.A. Van (2021). Chemical Diversification of Simple Synthetic Antibodies. ACS Chem. Biol 16, 344–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz I, Caffry I, Yu Y, et al. (2017). Biophysical properties of the clinical-stage antibody landscape. Proc. Natl. Acad. Sci 114, 944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jespers L, Bonnert TP, and Winter G (2004). Selection of optical biosensors from chemisynthetic antibody libraries. Protein Eng. Des. Sel 17, 709–713. [DOI] [PubMed] [Google Scholar]
- Jespers LSAT, Winter GP, Bonnert TP, and Simon TM (1994). Bacteriophage library displaying immunoglobulin repertoires with a chemical moiety covalently bound within the binding site: production and selection thereof. United States Patent 6,017,732.
- Kather I, Bippes CA, and Schmid FX (2005). A Stable Disulfide-free Gene-3-protein of Phage fd Generated by In vitro Evolution. J. Mol. Biol 354, 666–678. [DOI] [PubMed] [Google Scholar]
- Koide A, Bailey CW, Huang X, and Koide S (1998). The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol 284, 1141–1151. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy VM, Estroff LA, and Whitesides GM (2006). Multivalency in Ligand Design. In Fragment-Based Approaches in Drug Discovery, Mannhold R, Kubinyi H, and Folkers G, eds. pp. 11–53. [Google Scholar]
- Krishnamurthy VM, Kaufman GK, Urbach AR, Gitlin I, Gudiksen KL, Weibel DB, and Whitesides GM (2008). Carbonic anhydrase as a model for biophysical and physical-organic studies of proteins and protein-ligand binding. Chem. Rev 108, 946–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, and Roberts RW (2003). A Novel Strategy for In Vitro Selection of Peptide-Drug Conjugates. Chem. Biol 10, 233–239. [DOI] [PubMed] [Google Scholar]
- Lindley H (1960). A Study of the Kinetics of the Reaction between Thiol Compounds and Chloroacetamide. Biochemistry 74, 577–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link AJ, Mock ML, and Tirrell DA (2003). Non-canonical amino acids in protein engineering. Curr. Opin. Biotechnol 14, 603–609. [DOI] [PubMed] [Google Scholar]
- Lipovsek D (2011). Adnectins: engineered target-binding protein therapeutics. Protein Eng. Des. Sel 24, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipovsek D, Lippow SM, Hackel BJ, Gregson MW, Cheng P, Kapila A, and Wittrup KD (2007). Evolution of an Interloop Disulfide Bond in High-Affinity Antibody Mimics Based on Fibronectin Type III Domain and Selected by Yeast Surface Display: Molecular Convergence with Single-Domain Camelid and Shark Antibodies. J. Mol. Biol 368, 1024–1041. [DOI] [PubMed] [Google Scholar]
- de los Rios M, Criscitiello MF, and Smider VV (2015). Structural and genetic diversity in antibody repertoires from diverse species. Curr. Opin. Struct. Biol 33, 27–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz S, and Patrick WM (2004). Novel methods for directed evolution of enzymes: quality, not quantity. Curr. Opin. Biotechnol 15, 291–297. [DOI] [PubMed] [Google Scholar]
- Mammen M, Choi S, and Whitesides GM (1998). Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors. Angew Chem Int Ed Engl 37, 2754–2794. [DOI] [PubMed] [Google Scholar]
- Masereel B, Supuran CT, Thiry A, and Dogne J (2006). Targeting tumor-associated carbonic anhydrase IX in cancer therapy. Trends Pharmacol. Sci 27, 566–573. [DOI] [PubMed] [Google Scholar]
- Mcdonald PC, Winum J, Supuran CT, and Dedhar S (2012). Recent Developments in Targeting Carbonic Anhydrase IX for Cancer Therapeutics. Oncotarget 3, 84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer SC, Shomin CD, Gaj T, and Ghosh I (2007). Tethering small molecules to a phage display library: discovery of a selective bivalent inhibitor of protein kinase A. J Am Chem Soc 129, 13812–13813. [DOI] [PubMed] [Google Scholar]
- Mikulová MB, Kružlicová D, Pecher D, Supuran CT, and Mikuš P (2020). Synthetic Strategies and Computational Inhibition Activity Study for Triazinyl-Substituted Benzenesulfonamide Conjugates with Polar and Hydrophobic Amino Acids as Inhibitors of Carbonic Anhydrases. Int. J. Mol. Sci 21, 3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikuš P, Krajčiová D, Mikulová M, Horváth B, Pecher D, Garaj V, Bua S, Angeli A, and Supuran CT (2018). Novel sulfonamides incorporating 1,3,5-triazine and amino acid structural motifs as inhibitors of the physiological carbonic anhydrase isozymes I, II and IV and tumor-associated isozyme IX. Bioorg. Chem 81, 241–252. [DOI] [PubMed] [Google Scholar]
- Murray CW, and Rees DC (2009). The rise of fragment-based drug discovery. Nat. Chem 1, 187–192. [DOI] [PubMed] [Google Scholar]
- Nadalin F, and Carbone A (2018). Protein – protein interaction specificity is captured by contact preferences and interface composition. Bioinformatics 34, 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navaratna T, Atangcho L, Mahajan M, Subramanian V, Case M, Min A, Tresnak D, and Thurber GM (2020). Directed Evolution Using Stabilized Bacterial Peptide Display. JACS 142, 1882–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng S, Lin E, Kitov PI, Tjhung KF, Gerlits OO, Deng L, Kasper B, Sood A, Paschal BM, Zhang P, et al. (2015). Genetically encoded fragment-based discovery of glycopeptide ligands for carbohydrate-binding proteins. J Am Chem Soc 137, 5248–5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehler S, Catalano M, Scapozza I, Bigatti M, Bassi G, Favalli N, Mortensen MR, Samain F, and Scheuermann J (2021). Affinity Selections of DNA-Encoded Chemical Libraries on Carbonic Anhydrase IX-Expressing Tumor Cells Reveal a Dependence on Ligand Valence. Chemistry (Easton). 27, 1–10. [DOI] [PubMed] [Google Scholar]
- Packer MS, and Liu DR (2015). Methods for the directed evolution of proteins. Nat. Rev. Genet 16, 379–394. [DOI] [PubMed] [Google Scholar]
- Parang K, Till JH, Ablooglu AJ, Kohanski RA, Hubbard SR, and Cole PA (2001). Mechanism-based design of a protein kinase inhibitor. Nat. Struct. Biol 8, 37–41. [DOI] [PubMed] [Google Scholar]
- Pastorek J, and Pastorekova S (2015). Hypoxia-induced carbonic anhydrase IX as a target for cancer therapy: From biology to clinical use. Semin. Cancer Biol 31, 52–64. [DOI] [PubMed] [Google Scholar]
- Pastorekova S, and Gillies RJ (2019). The role of carbonic anhydrase IX in cancer development: links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev. 38, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT, Pastorekova S, Parkkila S, Pastorek J, and Supuran CT (2004). Carbonic Anhydrases: Current State of the Art, Therapeutic Applications and Future Prospects. J. Enzyme Inhib. Med. Chem 19, 199–229. [DOI] [PubMed] [Google Scholar]
- Potter CPS, and Harris AL (2003). Diagnostic , prognostic and therapeutic implications of carbonic anhydrases in cancer. Br. J. Cancer 89, 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri V, Streaker E, Prabakaran P, Zhu Z, and Dimiter S (2013). Highly efficient selection of epitope specific antibody through competitive yeast display library sorting. MAbs 5, 533–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafajov M, Zat M, Gibadulinov A, Casini A, Cecchi A, Scozzafava A, Pastorekov S, Supuran CT, and Fiorentino S (2004). Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett. 577, 439–445. [DOI] [PubMed] [Google Scholar]
- Rana S, Nissen F, Lindner T, Altmann A, Mier W, Debus J, Haberkorn U, and Askoxylakis V (2013). Screening of a Novel Peptide Targeting the Proteoglycan-Like Region of Human Carbonic Anhydrase IX. Mol. Imaging 12, 1–12. [DOI] [PubMed] [Google Scholar]
- Ravasco JMJM, Faustino H, Trindade A, and Gois PMP (2019). Bioconjugation with Maleimides: A Useful Tool for Chemical Biology. Chem. Eur. J 25, 43–59. [DOI] [PubMed] [Google Scholar]
- Renaud J, Chung C, Danielson UH, Egner U, Hennig M, Hubbard RE, and Nar H (2016). Biophysics in drug discovery: impact, challenges and opportunities. Nat. Rev. Drug Discov 15, 679–698. [DOI] [PubMed] [Google Scholar]
- Robertson N, Potter C, and Harris AL (2004). Role of Carbonic Anhydrase IX in Human Tumor Cell Growth, Survival, and Invasion. Cancer Res. 64, 6160–6165. [DOI] [PubMed] [Google Scholar]
- Ross PL, and Wolfe JL (2016). Physical and Chemical Stability of Antibody Drug Conjugates: Current Status. J. Pharm. Sci 105, 391–397. [DOI] [PubMed] [Google Scholar]
- Schneider TL, Mathew RS, Rice KP, Tamaki K, Wood JL, and Schepartz A (2005). Increasing the kinase specificity of K252a by protein surface recognition. Org. Lett 7, 1695–1698. [DOI] [PubMed] [Google Scholar]
- Searle MS, and Williams DH (1992). The cost of conformational order: entropy changes in molecular associations. J. Am. Chem. Soc 114, 10690–10697. [Google Scholar]
- Supuran CT (2008). Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Struct. Mol. Biol 7, 168–181. [DOI] [PubMed] [Google Scholar]
- Supuran CT (2012). Structure-based drug discovery of carbonic anhydrase inhibitors Structure-based drug discovery of carbonic anhydrase inhibitors. J. Enzyme Inhib. Med. Chem 27, 759–772. [DOI] [PubMed] [Google Scholar]
- Supuran CT (2017). Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin. Drug Discov 12, 61–88. [DOI] [PubMed] [Google Scholar]
- Supuran CT, Alterio V, Fiore A. Di, Ambrosio KD, Carta F, Monti SM, and Simone G. De (2018). Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: Three for the price of one. Med Res Rev 38, 1799–1836. [DOI] [PubMed] [Google Scholar]
- Svastova E, Witarski W, Csaderova L, Kosik I, Skvarkova L, Hulikova A, Zatovicova M, Barathova M, Kopacek J, Pastorek J, et al. (2012). Carbonic Anhydrase IX Interacts with Bicarbonate Transporters in Lamellipodia and Increases Cell Migration via Its Catalytic Domain. J. Biol. Chem 287, 3392–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian F, Tsao M-L, and Schultz PG (2004). A phage display system with unnatural amino acids. J. Am. Chem. Soc 126, 15962–15963. [DOI] [PubMed] [Google Scholar]
- Tokuriki N, Stricher F, Schymkowitz J, Serrano L, and Tawfik DS (2007). The Stability Effects of Protein Mutations Appear to be Universally Distributed. J. Mol. Biol 369, 1318–1332. [DOI] [PubMed] [Google Scholar]
- VanAntwerp JJ, and Wittrup KD (2000). Fine affinity discrimination by yeast surface display and flow cytometry. Biotechnol. Prog 16, 31–37. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, and Charlton SJ (2013). Exploring avidity: understanding the potential gains in functional affinity and target residence time of bivalent and heterobivalent ligands. Br. J. Pharmacol 168, 1771–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward C, Meehan J, Mullen P, Supuran C, Dixon JM, Thomas JS, Winum J-Y, Lambin P, Dubois L, Pavathaneni N-K, et al. (2015). Evaluation of carbonic anhydrase IX as a therapeutic target for inhibition of breast cancer invasion and metastasis using a series of in vitro breast cancer models. Oncotarget 6, 24856–24870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G, Griffiths AD, Hawkins RE, and Hoogenboom HR (1994). Making antibodies by phage display technology. Annu. Rev. Immunol 12, 433–455. [DOI] [PubMed] [Google Scholar]
- Woldring DR, Holec PV, Zhou H, and Hackel BJ (2015). High-Throughput Ligand Discovery Reveals a Sitewise Gradient of Diversity in Broadly Evolved Hydrophilic Fibronectin Domains. PLoS One 10, e0138956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Williams J, Calder EDD, and Walport LJ (2021). Strategies to expand peptide functionality through hybridisation with a small molecule component. RSC Chem. Biol 2, 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Josan JS, Vagner J, Caplan MR, Hruby VJ, Mash EA, Lynch RM, Morse DL, and Gillies RJ (2012). Heterobivalent ligands target cell-surface receptor combinations in vivo. Proc. Natl. Acad. Sci 109, 21295–21300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Slusky JS, Berger BW, Walters RS, Vilaire G, Litvinov RI, Lear JD, Caputo GA, Bennett JS, and Degrado WF (2007). Computational Design of Peptides That Target Transmembrane Helices. Science. 315, 1817–1823. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw source data are available upon reasonable request.
All original code is publicly available at https://github.com/HackelLab-UMN_and https://doi.org/10.5281/zenodo.5093631.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.