

Abstract
Periodontitis is characterized by alveolar bone loss leading to tooth loss. A small proportion of patients develop severe periodontitis at the juvenile or adolescent age without exposure to the main risk factors of the disease. It is considered that these cases carry rare variants with large causal effects, but the specific variants are largely unknown. In this study, we performed exome sequencing of 5 families with children who developed stage IV, grade C, periodontitis between 3 and 18 y of age. In 1 family, we found compound heterozygous variants in the gene CTSC (p.R272H, p.G139R), 1 of which was previously identified in a family with prepubertal periodontitis. Subsequent targeted resequencing of the CTSC gene in 24 patients <25 y of age (stage IV, grade C) identified the known mutation p.I453V (odds ratio = 4.06, 95% CI = 1.6 to 10.3, P = 0.001), which was previously reported to increase the risk for adolescent periodontitis. An affected sibling of another family carried a homozygous deleterious mutation in the gene TUT7 (p.R560Q, CADD score >30 [Combined Annotation Dependent Depletion]), which is implicated in regulation of interleukin 6 expression. Two other affected siblings shared heterozygous deleterious mutations in the interacting genes PADI1 and FLG (both CADD = 36), which contribute to the integrity of the environment–tissue barrier interface. Additionally, we found predicted deleterious mutations in the periodontitis risk genes ABCA1, GLT6D1, and SIGLEC5. We conclude that the CTSC variants p.R272H and p.I453V have different expressivity and diagnostic relevance for prepubertal and adolescent periodontitis, respectively. We propose additional causal variants for early-onset periodontitis, which also locate within genes that carry known susceptibility variants for common forms. However, the genetic architecture of juvenile periodontitis is complex and differs among the affected siblings of the sequenced families.
Keywords: prepubertal periodontitis, juvenile periodontitis, mutation, CTSC, SIGLEC5, GLT6D1
Introduction
Periodontitis (PD) is a common complex inflammatory disease of the oral cavity. It is characterized by inflammation of oral keratinized mucosa and alveolar bone loss. PD has a range of manifestations that differ in severity and progression of tissue destruction and age of disease onset. The basis of phenotypic variation is genetic variability among individuals (Timpson et al. 2018). Specifically, the number of variants, the magnitude of their effects on the disease phenotype, and their interactions with one another and factors in the environment influence and shape disease phenotypes (Wray et al. 2018). Genetic variants are mostly single-nucleotide polymorphisms but also small DNA sequence insertions and deletions. Many of them frequently occur in the general population, but a small proportion are rare and found in few individuals (1000 Genomes Project Consortium et al. 2015). A small proportion of PD cases, with a frequency <0.01%, are characterized by an early age of disease onset and often show familial aggregation (Susin et al. 2014). These disease phenotypes are mostly characterized by the absence of common risk factors of PD, such as long-term smoking, diabetes mellitus, or advanced age, and commonly show absence of bacterial plaque. Sometimes, they correlate with monogenetic syndromes, such as Papillon-Lefèvre (Machado et al. 2019) and Ehlers-Danlos (Kapferer-Seebacher et al. 2016).
Various genome-wide association studies (GWASs) have searched the genome for common single-nucleotide polymorphisms that contribute to the increased disease susceptibility and identified several loci that were associated with PD at a genome-wide significance level, such as GLT6D1 (Schaefer et al. 2010), DEFA1A3 (Munz et al. 2017), SIGLEC5 (Munz et al. 2017; Shungin et al. 2019), and ATP6V1C1 (Munz et al. 2019). Common susceptibility variants have small to moderate effects, with odds ratios generally <1.5, because large effects mostly result in more severe and/or early-onset diagnoses and are thus inconsistent with common diseases (Wray et al. 2018). This is why GWAS but also large exome or genome sequencing studies of common diseases largely failed to confirm the presence of large effect size variants, which influence susceptibility to common diseases (Fuchsberger et al. 2016; Genovese et al. 2016). Instead, large effect sizes, caused by rare variants or mutations, were identified in rare disorders, which are distinct from common forms by age of onset, familial clusters, or segregation and progression rate. However, risk genes detected by GWAS may harbor rare, high-effect susceptibility variants that cause severe early-onset disease manifestations and are independent of the variants associated with common diseases (Flannick et al. 2014; Do et al. 2015; Fuchsberger et al. 2016; Luo et al. 2017). Therefore, not only can rare disorders identify rare variants with large effect sizes, but they can also serve as tools to identify candidate susceptibility genes of common disease phenotypes. These can subsequently be tested for associations in available GWAS data sets (Antonarakis and Beckmann 2006).
In the current study, we performed whole exome sequencing (WES) in 5 families with children showing juvenile or adolescent generalized stage III or IV, grade C, PD to identify rare deleterious variants that may contribute to the etiology of PD.
Material and Methods
Study Samples
Five families were selected for WES with stage III or IV, grade B or C, PD diagnosed in their underage siblings. The family members are described in Figure 1.
Figure 1.

Genogram of the families. The arrow indicates the proband in each family, who was first diagnosed with periodontitis by a specialized periodontologist. Family 1 was recruited at the Department of Conservative Dentistry, Periodontology and Preventive Dentistry, Hannover Medical School, Hannover, Germany. The hands and feet of the siblings showed no palmoplantar hyperkeratosis. Family 2 was recruited at the Department of Operative and Conservative Dentistry and Periodontology, University Hospital Tübingen, Tübingen, Germany. Families 3 and 4 were recruited at the Department of Periodontology, Operative and Preventive Dentistry, University of Bonn, Bonn, Germany. Family 5 was recruited at the Institute of Periodontology, Department of Dentistry, Faculty of Medicine, University of Coimbra, Coimbra, Portugal. Square, male; circle, female; black fill, periodontitis diagnosed; white fill, no periodontitis diagnosed. PD, periodontal disease; PPP, prepubertal periodontitis.
Whole Exome and Targeted Resequencing
Sequencing was performed at the Competence Centre for Genomic Analysis, Kiel, Germany.
FASTQ files were analyzed with the FastQC software tool. Alignment to the human reference genome (GRCh37; hs37d5.fa) was performed with the Burrows Wheeler Aligner (version 0.7.17), and deviations from the reference sequence were detected with GATK HaplotypeCaller (version 3.7) and reported in a VCF file. We determined the allele frequencies of the identified variants in the general population with gnomAD exomes database (Karczewski et al. 2020) and VarFish (Holtgrewe et al. 2020).
We filtered for variants with a minor allele frequency (MAF) ≤0.01. (To exclude technical errors, a mutation was ignored if it was present in >20 exomes of our in-house database of ~3,000 exome samples.) The functional effect of each variant was determined via comparisons with ClinVar, a database on pathogenic changes in the genome (Landrum et al. 2018) and with software tools that estimate the influence on the encoded protein based on predicted structural changes, conservation, and other parameters. These tools were SIFT (Ng and Henikoff 2003), Polyphen2 (Adzhubei et al. 2013), MutationTaster (Schwarz et al. 2010), and the CADD score (version 1.4; Combined Annotation-Dependent Depletion), a method that integrates the information from various functional annotations and condenses this information into a single score (Kircher et al. 2014). A scaled CADD score of 20 assigned a variant among the top 1% of deleterious variants in the human genome; a scaled CADD score of 30 assigned a variant among the top 0.1%; and so on. In addition to the analysis of autosomal dominant inheritance patterns, accumulation of homozygous or compound heterozygous changes in genes was investigated to detect recessive inheritance patterns. To be reported, a mutation had to change an amino acid or splice site and had to show a CADD score ≥20. The quality criterion cutoffs for all analyses were a read depth ≥10 for heterozygous variants and ≥5 for homozygous variants, an allelic balance ≥0.2 for heterozygous variants, a genotype quality ≥10, and an allelic depth ≥0.3.
For the parents of family 1, no WES data were available. In this family, putative compound heterozygous variants were required to be present in both siblings. We filtered mutations with a MAF < 0.002 and a CADD score ≥ 20 from the WES data of the siblings. Putative de novo variants (rare allele present in only 1 sibling) with a CADD score ≥ 30 were validated in the parents with Sanger sequencing.
The filtering approach is illustrated in Figure 2. In a first step, we screened for homozygous, compound heterozygous, and de novo mutations. In a second step that included all families, we searched for heterozygous variants other than compound heterozygous mutations. These were also partly carried by healthy family members. Here, we applied 4 inclusion criteria: 1) variants with a CADD score ≥ 30, 2) at least 2 variants with a CADD score ≥ 20 in the same gene, 3) variants with a CADD score ≥ 20 in genes with described autosomal dominant disease mechanisms (as designated by the Human Phenotype Ontology Project; Kohler et al. 2019), or 4) variants with a CADD score ≥ 20 in genes with genome-wide significant association with PD (ATP6V1C1, DEFA1A3, GLT6D1, and SIGLEC5). The MAF cutoff was 5% and a maximum of 50 carriers in our in-house database of ~3,000 individual exomes.
Figure 2.
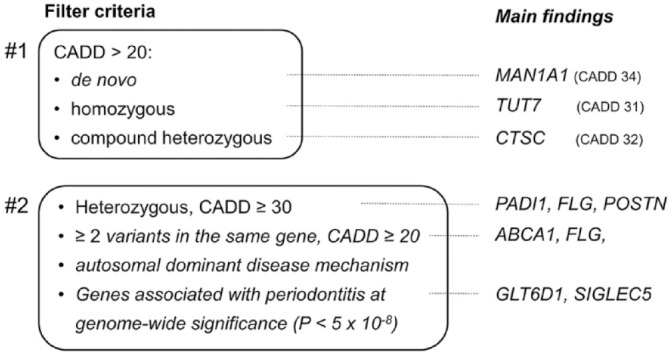
Screening approach to identify nonsynonymous variants in the affected children for nonsynonymous mutations and main findings. The minor allele frequency cutoff to find putative causal nonsynonymous variants was ≤0.01 in step 1 and ≤0.05 in step 2. However, all nonsynonymous exon variants with a CADD score >20 were rare. CADD, Combined Annotation Dependent Depletion.
The genomic regions of the genes CTSC, FLG, PADI1, POSTB, PRB3, and TUT7 ±200 kilobase pairs were searched for common variant associations in a sample of 896 patients with PD stage III or IV, grade C, who were ≤35 years of age, and 7,104 healthy controls of German and Dutch descent (described by Munz et al. 2017). These genes were identified as candidate genes in the 2 filtering steps. The association tests were conducted under the allelic genetic model with the Student’s t test and were adjusted for the covariates smoking and sex by logistic regression.
Case Samples for Targeted Resequencing and Candidate Gene Analysis
The gene CTSC was sequenced in 24 additional cases with PD stage IV, grade C, at ≥3 teeth with ≤25 y of age (age, 12 to 25 y; mean age of first diagnosis, 19 y; SD, 3.5 years), as first described by Schaefer et al. (2010). The study was approved by the local ethical boards.
Results
Nonsynonymous De Novo, Homozygous, and Compound Heterozygous Variants in the Affected Children
Homozygous and compound heterozygous mutations have a high penetrance because both gene copies are affected and, unlike heterozygous mutations, the second gene copy cannot compensate the loss of function. Additionally, de novo mutations can have dominant effects that can have phenotypic expression, although the second gene copy is functional (e.g., by gene dosage effects). To identify such variants, we first screened the exomes for nonsynonymous de novo, homozygous, and compound heterozygous variants in the affected children.
In family 1, both siblings carried a compound heterozygous mutation in the gene CTSC. p.G139R (CADD score = 32, MAF = 2 × 10-5; gnomAD exome database) was inherited from the mother and p.R272H (CADD score = 26.7) from the father (Tables 1 and 2, Fig. 2). p.R272H was reported in heterozygous state in a child who had prepubertal PD (PPP; Hewitt et al. 2004), a disease characterized by PD similar to that in Papillon-Lefèvre syndrome.
Table 1.
De Novo, Homozygous Recessive, and Compound Heterozygous Mutations Selected by Frequency of ≤0.01 and CADD Score ≥20.
| Genotypes | ||||||||
|---|---|---|---|---|---|---|---|---|
| Gene Symbol | Variant | CADD Score | Mode of Inheritance | Son (PD IV, C) | Daughter a | Mother (Unaffected) | Father (Unaffected) | |
| Family 1 b | ||||||||
| PRB3 | p.P146Tfs*109 | 21.6 | RH | 0/1 | 1/1 | 0/1 | 0/1 | |
| CTSC | p.G139R | 32.0 | CH | 0/1 | 0/1 | 0/1 | 0/0 | |
| CTSC | p.R272H | 26.7 | CH | 0/1 | 0/1 | 0/0 | 0/1 | |
| MAN1A1 | c.1061 + 1G > A | 34.0 | De novo | 0/1 | 0/0 | 0/0 | 0/0 | |
| Daughter 1 (PD III, C) | Daughter 2 (PD III, C) | Mother (PD IV, C) | Father (PD III, C) | |||||
| Family 2 | ||||||||
| TUT7 | p.R560Q | 31.0 | RH | 0/1 | 1/1 | 0/1 | 0/1 | |
| DNAH5 | p.D1462Y | 25.9 | CH | 0/1 | 0/1 | 0/0 | 0/1 | |
| DNAH5 | p.E3450K | 21.6 | CH | 0/1 | 0/1 | 0/1 | 0/0 | |
| USP18 | p.C320S | 24.3 | De novo | 0/1 | 0/0 | 0/0 | 0/0 | |
| Daughter (PD III, B) | Son (Unaffected) | Mother (PD III, B) | Father (PD III, B) | |||||
| Family 3 | ||||||||
| GRAMD4 | p.R207W | 21.2 | De novo | 0/1 | 0/0 | 0/0 | 0/0 | |
| Daughter 1 (PD III, C) | Daughter 2 (PD III, C) | Son (Unaffected) | Mother (PD III, B) | Father (PD IV, B) | ||||
| Family 4 | ||||||||
| OBSCN | p.G3058E | 25.7 | CH | 0/1 | 0/1 | 0/1 | 0/0 | 0/1 |
| OBSCN | p.E1235A | 23.0 | CH | 0/1 | 0/1 | 0/0 | 0/1 | 0/0 |
| KMT5C | p.R76C | 25.4 | De novo | 0/1 | 0/0 | 0/0 | 0/0 | 0/0 |
| ABCA1 | p.R666W | 25.3 | De novo | 0/1 | 0/0 | 0/0 | 0/0 | 0/0 |
| U2AF2 | p.R81H | 22.9 | De novo | 0/0 | 0/1 | 0/0 | 0/0 | 0/0 |
| POFUT1 | p.Q74P | 22.6 | De novo | 0/0 | 0/1 | 0/0 | 0/0 | 0/0 |
| CTC1 | p.R624Q | 21.7 | De novo | 0/0 | 0/1 | 0/0 | 0/0 | 0/0 |
| EFNA3 | c.128 + 7C > T | 21.2 | De novo | 0/1 | 0/0 | 0/0 | 0/0 | 0/0 |
Mutations of the affected sibling of family 5 are not listed, as no germline DNA was available from the mother due to a bone marrow transplantation.
CADD, Combined Annotation Dependent Depletion; CH, compound heterozygous; PD, periodontitis (stage, grade); RH, recessive homozygous.
Prepubertal PD, age 5 y; no PD since.
Genotypes for parents from family 1 were inferred from Sanger sequencing.
Table 2.
Detailed Information on De Novo, Homozygous Recessive, and Compound Heterozygous Mutations From Filtering Step 1.
| Chromosomal Position (hg19) | Gene Symbol | Variant | Reference Sequence ID | Common / Rare Allele | Mutation | CADD Score | MAF gnomAD Exomes |
|---|---|---|---|---|---|---|---|
| chr12:11,420,754 | PRB3 | p.P146Tfs*109 | NA | T/187-bp deletion | Frameshift truncation | 21.6 | 0.0004 |
| chr11:88,045,626 | CTSC | p.G139R | rs749103588 | C/T | Missense | 32.0 | 0.00002 |
| chr11:88,029,375 | CTSC | p.R272H | rs587777534 | C/T | Missense | 26.7 | 0 |
| chr6:119,569,424 | MAN1A1 | c.1061 + 1G > A | NA | C/T | Splice site changed (intronic) | 34.0 | 0.00000 |
| chr9:88,940,359 | TUT7 | p.R560Q | rs41310053 | C/T | Missense | 31.0 | 0.00355 |
| chr5:13,864,718 | DNAH5 | p.D1462Y | rs1189846120 | C/A | Missense | 25.9 | 0 |
| chr5:13,759,026 | DNAH5 | p.E3450K | rs758739748 | C/T | Missense | 21.6 | 0.00001 |
| chr22:18,655,984 | USP18 | p.C320S | NA | G/C | Missense | 24.3 | 0 |
| chr22:47,059,754 | GRAMD4 | p.R207W | rs1282915359 | C/T | Missense | 21.2 | 0 |
| chr1:228,473,947 | OBSCN | p.G3058E | NA | G/A | Missense | 25.7 | 0 |
| chr1:228,433,336 | OBSCN | p.E1235A | rs758725573 | A/C | Missense | 23.0 | 0.0001 |
| chr19:55,853,698 | KMT5C | p.R76C | rs866476245 | C/T | Missense | 25.4 | 0.00001 |
| chr9:107,591,316 | ABCA1 | p.R666W | rs201599169 | G/A | Missense | 25.3 | 0.00006 |
| chr19:56,171,893 | U2AF2 | p.R81H | rs779075792 | G/A | Missense | 22.9 | 0.00001 |
| chr20:30,797,970 | POFUT1 | p.Q74P | NA | A/C | Missense | 22.6 | 0 |
| chr17:8,136,298 | CTC1 | p.R624Q | rs377423237 | C/T | Missense | 21.7 | 0.00002 |
| chr1:155,051,552 | EFNA3 | c.128 + 7C > T | rs1254227036 | C/T | Missense | 21.2 | 0.00003 |
bp, base pairs; CADD, Combined Annotation Dependent Depletion; MAF, minor allele frequency; NA, not available.
In addition, the affected daughter was homozygous for a 180–base pair deletion (p.P146Tfs*109, CADD score = 22.6) in the gene PRB3, encoding a major protein component of saliva. The son was carrier of a heterozygous de novo variant in the gene MAN1A1 (CADD score = 34), which is involved in glycosylation of immune and other cells.
In family 2, 1 affected sibling was homozygous for a mutation in the gene zinc-finger CCHC domain-containing protein 6 (TUT7, p.R560Q, CADD score = 31). Although for this variant a MAF of 0.003 was reported (gnomAD), we did not identify homozygous genotypes for this variant in current exome sequencing projects.
The affected daughter of family 3 (age of onset, 16 y) carried a heterozygous de novo mutation in the gene GRAM domain containing 4 (GRAMD4; p.R207W, CADD score = 21.2).
For family 4, the affected siblings showed a compound heterozygous variant in the gene obscurin (OBSCN; p.E1235A, CADD score = 23; p.G3058E, CADD score = 25.7). Additional de novo mutations with CADD score ≥ 25 were carried by the affected older daughter in the genes lysine methyltransferase 5C (KMT5C; p.R76C, CADD score = 25.4) and the suggestive PD risk gene ATP binding cassette subfamily A member 1 (ABCA1; p.R666W, CADD score = 25; Teumer et al. 2013).
Because in family 5, the mother was genetically chimeric due to bone marrow transplantations, de novo and compound heterozygous mutations could not be distinguished, because the DNA sampled from the mother stemmed from the bone marrow donor. No homozygous variants with CADD score ≥20 were observed in the affected son.
Putative Causal Nonsynonymous Heterozygous Variants
Unaffected or mildly affected parents might carry genetic variants with moderate expressivity. However, recombination of gene × gene interacting variants in the siblings may result in high expressivity (Wray et al. 2018). To identify such putative interacting causal variants, we searched the exomes for heterozygous nonsynonymous variants with CADD scores ≥30, the presence of >1 variant with a CADD score ≥20 in the same gene, and a described autosomal dominant disease mechanism of the gene carrying the mutation. Additionally we screened the coding regions of genes that were previously reported to be associated with PD at a genome-wide significant level. The results are listed in Appendix Tables 1 to 4. We observed that 4 genes with putative pathogenic variants were shared among families: TTN (p.E20484K, CADD score = 25, family 3; p.R14131Q, CADD score = 22, family 4; and p.R28682K, p.A25959T, and p.E10001K, CADD score = 23, family 5), FLG (p.S805F, CADD score = 22, family 1; p.R2613, CADD score = 36, family 4), the PD risk gene ABCA1 (Teumer et al. 2013; p.S1181F, CADD score = 24, family 3; p.R666W, CADD score = 25.3, family 4), and ABCA7 (p.V1599M in family 1 and p.R1812H in family 2). Notably, in family 4, the affected daughters who carried the FLG mutation p.R2613 also carried a heterozygous mutation in the gene PADI1 (p.R240*, CADD score = 36). The PADI1 protein biologically interacts with FLG (Hsu et al. 2011). The affected daughters inherited the variants in FLG and PADI from the affected father, which were not carried by the unaffected mother and brother.
In addition, we observed that the mother and both siblings of family 3 carried heterozygous variants in the PD risk gene SIGLEC5 (Munz et al. 2018; Shungin et al. 2019; SIGLEC5; p.A277Cfs*71, CADD score = 23). Furthermore, both sisters of family 4 were heterozygous for a premature stop codon in the PD stage III, grade C, risk gene GLT6D1 (Schaefer et al. 2010; p.R108*, CADD score = 35). In this family, p.R108 showed the highest CADD score next to PADI1, FLG, and ADIPOQ. We observed other mutations that implied a role in the etiology of PD according to their gene functions in the affected son of family 5, who carried heterozygous mutations in the genes periostin (POSTN; p.T73M, CADD score = 33) and MMP8 (c.347 + 2T > C, CADD score = 32) that were not found in his unaffected sister.
Targeted Resequencing of CTSC
We investigated whether variants in CTSC were enriched in adolescent stage IV, grade C, PD cases. Resequencing of CTSC in an independent sample of 24 unrelated cases with an age of disease <25 y (first described by Schaefer et al. 2010) revealed that 6 cases were heterozygous for rs3888798 (p.I453V, MAF gnomAD exome 0.06, CADD score = 24.2). We performed an association test with the genotypes of our 24 sequenced cases and 4,299 controls of the NHLBI Exome Sequencing Project. In this test, the rare allele of rs3888798 showed an odds ratio of 4.06 (95% CI = 1.6 to 10.3) with P = 0.001. The effect allele of rs3888798 was also carried by the affected child and the unaffected mother of another PPP family (Hewitt et al. 2004) and was significantly enriched in a sample of 110 German cases and 50 controls (odds ratio = 3.35, 95% CI = 1.2 to 9.7; Noack et al. 2008). We found no other predicted pathogenic CTSC mutations in our targeted resequenced cases.
Candidate Gene Analysis for the Search of Common Variant Associations
We analyzed imputed genotypes ±200 kilobase pairs at the genes CTSC, FLG, PADI1, POSTN, PRB3, and TUT7/ZCCHC6 for associations of common single-nucleotide polymorphisms (MAF ≥ 5%) in 896 PD cases with stage III or IV, grade C (18 to 35 y of age), and 7,104 controls from our recent GWAS (Munz et al. 2017). For these genes, we found no evidence of association of common variants with this disease phenotype (Appendix Figs. 1–6).
Discussion
In this study, we analyzed the exome of 5 families with siblings who had early-onset severe generalized PD. We identified compound heterozygous variants in the gene CTSC. Defects in the encoded protein were shown to be a cause of Papillon-Lefèvre syndrome, but a few studies reported CTSC variants to be associated with nonsyndromic PPP: Tyr347Cys (Hart et al. 2000), p.R272H/p.Y412C/p.I453 (Hewitt et al. 2004), and p.W101S (Molitor et al. 2019). In our study, the compound heterozygous variants of both siblings of family 1 consisted of the known PPP variant p.R272H and the hitherto in nonsyndromic PD unobserved variant p.G139R. In agreement with the observation that p.R272H was found in nonsyndromic PPP, 1 of the affected siblings was treated for PPP. Additionally, studies that investigated PD stage IV, grade C, cases revealed the nonsynonymous CTSC variants p.I453V (Noack et al. 2008) and p.W101S (Molitor et al. 2019). By targeted resequencing of CTSC in PD stage IV, grade C, cases <25 y of age, we identified significant enrichment of the pathogenic variant p.I453V in these cases, indicating that CTSC is a susceptibility locus for early-onset PD. Our findings also imply that the variants p.R272H and p.I453V have a different expressivity and contribute to susceptibility of prepubertal and adolescent PD, respectively.
In family 2, we found a homozygous deleterious variant (CADD score = 31) in only 1 of the 2 affected siblings, located in the gene TUT7. The gene product enhances IL-6 expression in chondrocytes (Ansari et al. 2019). Additionally, both siblings carried a heterozygous variant in the gene “monocyte to macrophage differentiation associated” (MMD; CADD score = 41) inherited from the father, who had adult PD. This gene belongs to the progestin and adipoQ receptor family and is associated with heel bone mineral density (Morris et al. 2019). Interestingly, a variant within another adipoQ receptor family gene, ADIPOQ, exhibited the third-highest CADD score in family 4.
Notably, in family 3, the mother (stage III, grade B, PD as a young adult), the daughter (stage III, grade B, PD at 16 y), and the 14-y-old son with no diagnosed PD carried heterozygous variants in the PD risk genes SIGLEC5 (Munz et al. 2018; Shungin et al. 2019) and ABCA1 (Teumer et al. 2013). Interestingly, we also identified a predicted pathogenic heterozygous de novo variant in the PD risk gene ABCA1 (CADD score = 25) in 1 of the affected daughters of family 4. We find it remarkable that we found mutations in SIGLEC5 and that the siblings of families 3 and 4 had 2 predicted deleterious variants in ABCA1.
In family 4, both affected siblings shared heterozygous deleterious variants in the genes PADI1, FLG, and ADIPOQ, which all showed the second-highest CADD score (36) in this family and were inherited from the affected father but not present in the healthy brother. PADI1 and FLG have direct functional interaction. PADI1 deaminases FLG, which contributes to the integrity of the environmental–tissue barrier interface function (Hsu et al. 2011). ADIPOQ has anti-inflammatory activities (Yamauchi et al. 2001). Interestingly, a variant with the fifth-highest CADD score (35) in this family was located in the PD risk gene GLT6D1 (Schaefer et al. 2010). This variant was heterozygous in the affected siblings and not present in the unaffected sibling. However, the expressivity of this heterozygous variant is currently unclear.
Shared putative genetic susceptibility loci among families were found in TTN (potentially deleterious variants in 3 families), FLG, ABCA1, and ABCA7. TTN exhibits the largest number of exons in the human genome (Bang et al. 2001), encoding around 35,000 amino acids. The occurrence of TTN variants in several families therefore may be due to chance effects. In contrast, the occurrence of FLG variants in families 1 and 3 emphasizes a potential role of FLG in the etiology of PD.
This study has limitations. The chimeric DNA of the mother of family 5 did not allow us to follow the transmission of mutated alleles from the mother to the son and, thus, differentiation of de novo mutations from heterozygous mutations. Following the established design of WES projects, the current study proposed candidate genes but did not give experimental evidence for the causality of the identified mutations. Future studies are needed to follow up the molecular consequences of the nonsynonymous mutations and their etiologic implications. We consider CTSC, FLG, PADI1, POSTN, ABCA1, and GLT6D1 promising candidates.
We conclude that the genetic architecture of early-onset PD is complex. However, we found pathogenic variants in the PD risk genes CTSC, SIGLEC5, ABCA1, and GLT6D1. This shows that rare, high-effect susceptibility variants that cause severe early-onset disease manifestations also locate to genes that harbor more frequent susceptibility variants that increase the risk for common diseases. This confirms that rare disorders can be used as tools to identify candidate susceptibility genes of common disease phenotypes, which can be tested for associations in large GWAS data sets.
Author Contributions
G.M. Richter, A.S. Schaefer, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; G. Wagner, K. Reichenmiller, I. Staufenbiel, O. Martins, S. Jepsen, H. Dommisch, contributed to data acquisition, critically revised the manuscript; B.S. Löscher, M. Holtgrewe, contributed to data analysis, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
Supplemental material, sj-docx-1-jdr-10.1177_00220345211029266 for Exome Sequencing of 5 Families with Severe Early-Onset Periodontitis by G.M. Richter, G. Wagner, K. Reichenmiller, I. Staufenbiel, O. Martins, B.S. Löscher, M. Holtgrewe, S. Jepsen, H. Dommisch and A.S. Schaefer in Journal of Dental Research
Footnotes
A supplemental appendix to this article is available online.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant of the German Society of Periodontology to K. Reichenmiller, I. Staufenbiel, and A.S. Schaefer and a performance-related bonus of the medical faculty of the Charité–University Medicine Berlin, Germany, to G.M. Richter and A.S. Schaefer.
ORCID iD: H. Dommisch  https://orcid.org/0000-0003-1043-2651
https://orcid.org/0000-0003-1043-2651
References
- 1000 Genomes Project Consortium; Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. 2015. A global reference for human genetic variation. Nature. 526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei I, Jordan DM, Sunyaev SR. 2013. Predicting functional effect of human missense mutations using polyphen-2. Curr Protoc Hum Genet. Chapter 7:Unit7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari MY, Khan NM, Ahmad N, Green J, Novak K, Haqqi TM. 2019. Genetic inactivation of ZCCHC6 suppresses interleukin-6 expression and reduces the severity of experimental osteoarthritis in mice. Arthritis Rheumatol. 71(4):583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis SE, Beckmann JS. 2006. Mendelian disorders deserve more attention. Nat Rev Genet. 7(4):277–282. [DOI] [PubMed] [Google Scholar]
- Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, Witt CC, Labeit D, Gregorio CC, Granzier H, et al. 2001. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res. 89(11):1065–1072. [DOI] [PubMed] [Google Scholar]
- Do R, Stitziel NO, Won HH, Jorgensen AB, Duga S, Merlini PA, Kiezun A, Farrall M, Goel A, Zuk O, et al. 2015. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 518(7537):102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannick J, Thorleifsson G, Beer NL, Jacobs SB, Grarup N, Burtt NP, Mahajan A, Fuchsberger C, Atzmon G, Benediktsson R, et al. 2014. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet. 46(4):357–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchsberger C, Flannick J, Teslovich TM, Mahajan A, Agarwala V, Gaulton KJ, Ma C, Fontanillas P, Moutsianas L, McCarthy DJ, et al. 2016. The genetic architecture of type 2 diabetes. Nature. 536(7614):41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Fromer M, Stahl EA, Ruderfer DM, Chambert K, Landen M, Moran JL, Purcell SM, Sklar P, Sullivan PF, et al. 2016. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat Neurosci. 19(11):1433–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart TC, Hart PS, Michalec MD, Zhang Y, Marazita ML, Cooper M, Yassin OM, Nusier M, Walker S. 2000. Localisation of a gene for prepubertal periodontitis to chromosome 11q14 and identification of a cathepsin C gene mutation. J Med Genet. 37(2):95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt C, McCormick D, Linden G, Turk D, Stern I, Wallace I, Southern L, Zhang L, Howard R, Bullon P, et al. 2004. The role of cathepsin C in Papillon-Lefevre syndrome, prepubertal periodontitis, and aggressive periodontitis. Hum Mutat. 23(3):222–228. [DOI] [PubMed] [Google Scholar]
- Holtgrewe M, Stolpe O, Nieminen M, Mundlos S, Knaus A, Kornak U, Seelow D, Segebrecht L, Spielmann M, Fischer-Zirnsak B, et al. 2020. VarFish: comprehensive DNA variant analysis for diagnostics and research. Nucleic Acids Res. 48(W1):W162–W169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CY, Henry J, Raymond AA, Mechin MC, Pendaries V, Nassar D, Hansmann B, Balica S, Burlet-Schiltz O, Schmitt AM, et al. 2011. Deimination of human filaggrin-2 promotes its proteolysis by calpain 1.J Biol Chem. 286(26):23222–23233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapferer-Seebacher I, Pepin M, Werner R, Aitman TJ, Nordgren A, Stoiber H, Thielens N, Gaboriaud C, Amberger A, Schossig A, et al. 2016. Periodontal Ehlers-Danlos syndrome is caused by mutations in C1R and C1S, which encode subcomponents C1R and C1S of complement. Am J Hum Genet. 99(5):1005–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. 2020. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 581(7809):434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 46(3):310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler S, Carmody L, Vasilevsky N, Jacobsen JOB, Danis D, Gourdine JP, Gargano M, Harris NL, Matentzoglu N, McMurry JA, et al. 2019. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res. 47(D1):D1018–D1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al. 2018. Clinvar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46(D1):D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, De Lange KM, Jostins L, Moutsianas L, Randall J, Kennedy NA, Lamb CA, McCarthy S, Ahmad T, Edwards C, et al. 2017. Exploring the genetic architecture of inflammatory bowel disease by whole-genome sequencing identifies association at ADCY7. Nat Genet. 49(2):186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado RA, Cuadra-Zelaya FJM, Martelli-Junior H, Miranda RT, Casarin RCV, Correa MG, Nociti F, Coletta RD. 2019. Clinical and molecular analysis in Papillon-Lefevre syndrome. Am J Med Genet A. 179(10):2124–2131. [DOI] [PubMed] [Google Scholar]
- Molitor A, Prud’homme T, Miao Z, Conrad S, Bloch-Zupan A, Pichot A, Hanauer A, Isidor B, Bahram S, Carapito R. 2019. Exome sequencing identifies a novel missense variant in CTSC causing nonsyndromic aggressive periodontitis. J Hum Genet. 64(7):689–694. [DOI] [PubMed] [Google Scholar]
- Morris JA, Kemp JP, Youlten SE, Laurent L, Logan JG, Chai RC, Vulpescu NA, Forgetta V, Kleinman A, Mohanty ST, et al. 2019. An atlas of genetic influences on osteoporosis in humans and mice. Nat Genet. 51(2):258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munz M, Richter GM, Loos BG, Jepsen S, Divaris K, Offenbacher S, Teumer A, Holtfreter B, Kocher T, Bruckmann C, et al. 2018. Genome-wide association meta-analysis of coronary artery disease and periodontitis reveals a novel shared risk locus. Sci Rep. 8(1):13678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munz M, Richter GM, Loos BG, Jepsen S, Divaris K, Offenbacher S, Teumer A, Holtfreter B, Kocher T, Bruckmann C, et al. 2019. Meta-analysis of genome-wide association studies of aggressive and chronic periodontitis identifies two novel risk loci. Eur J Hum Genet. 27(1):102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munz M, Willenborg C, Richter GM, Jockel-Schneider Y, Graetz C, Staufenbiel I, Wellmann J, Berger K, Krone B, Hoffmann P, et al. 2017. A genome-wide association study identifies nucleotide variants at SIGLEC5 and DEFA1A3 as risk loci for periodontitis. Hum Mol Genet. 26(13):2577–2588. [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. 2003. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31(13):3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noack B, Gorgens H, Hempel U, Fanghanel J, Hoffmann T, Ziegler A, Schackert HK. 2008. Cathepsin C gene variants in aggressive periodontitis. J Dent Res. 87(10):958–963. [DOI] [PubMed] [Google Scholar]
- Schaefer AS, Richter GM, Nothnagel M, Manke T, Dommisch H, Jacobs G, Arlt A, Rosenstiel P, Noack B, Groessner-Schreiber B, et al. 2010. A genome-wide association study identifies GLT6D1 as a susceptibility locus for periodontitis. Hum Mol Genet. 19(3):553–562. [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. 2010. Mutationtaster evaluates disease-causing potential of sequence alterations. Nat Methods. 7(8):575–576. [DOI] [PubMed] [Google Scholar]
- Shungin D, Haworth S, Divaris K, Agler CS, Kamatani Y, Lee MK, Grinde K, Hindy G, Alaraudanjoki V, Pesonen P, et al. 2019. Genome-wide analysis of dental caries and periodontitis combining clinical and self-reported data. Nat Commun. 10(1):2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin C, Haas AN, Albandar JM. 2014. Epidemiology and demographics of aggressive periodontitis. Periodontol 2000. 65(1):27–45. [DOI] [PubMed] [Google Scholar]
- Teumer A, Holtfreter B, Volker U, Petersmann A, Nauck M, Biffar R, Volzke H, Kroemer HK, Meisel P, Homuth G, et al. 2013. Genome-wide association study of chronic periodontitis in a general German population. J Clin Periodontol. 40(11):977–985. [DOI] [PubMed] [Google Scholar]
- Timpson NJ, Greenwood CMT, Soranzo N, Lawson DJ, Richards JB. 2018. Genetic architecture: the shape of the genetic contribution to human traits and disease. Nat Rev Genet. 19(2):110–124. [DOI] [PubMed] [Google Scholar]
- Wray NR, Wijmenga C, Sullivan PF, Yang J, Visscher PM. 2018. Common disease is more complex than implied by the core gene omnigenic model. Cell. 173(7):1573–1580. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, et al. 2001. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 7(8):941–946. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-docx-1-jdr-10.1177_00220345211029266 for Exome Sequencing of 5 Families with Severe Early-Onset Periodontitis by G.M. Richter, G. Wagner, K. Reichenmiller, I. Staufenbiel, O. Martins, B.S. Löscher, M. Holtgrewe, S. Jepsen, H. Dommisch and A.S. Schaefer in Journal of Dental Research