

Key Points
Question
Does treatment with a subcutaneous combination of casirivimab and imdevimab prevent progression to symptomatic COVID-19 when given to recently infected, asymptomatic individuals?
Findings
In this randomized clinical trial that included 314 SARS-CoV-2 reverse transcriptase–quantitative polymerase chain reaction–positive individuals living with an infected household contact, 29.0% of asymptomatic seronegative participants treated with subcutaneous casirivimab and imdevimab, 1200 mg (600 mg of each antibody), developed symptomatic COVID-19 over 28 days vs 42.3% of those treated with placebo. This difference was statistically significant.
Meaning
Treatment with subcutaneous casirivimab and imdevimab antibody combination compared with placebo significantly reduced the incidence of symptomatic COVID-19 among recently exposed, asymptomatic individuals.
Abstract
Importance
Easy-to-administer anti–SARS-CoV-2 treatments may be used to prevent progression from asymptomatic infection to symptomatic disease and to reduce viral carriage.
Objective
To evaluate the effect of combination subcutaneous casirivimab and imdevimab on progression from early asymptomatic SARS-CoV-2 infection to symptomatic COVID-19.
Design, Setting, and Participants
Randomized, double-blind, placebo-controlled, phase 3 trial of close household contacts of a SARS-CoV-2–infected index case at 112 sites in the US, Romania, and Moldova enrolled July 13, 2020–January 28, 2021; follow-up ended March 11, 2021. Asymptomatic individuals (aged ≥12 years) were eligible if identified within 96 hours of index case positive test collection. Results from 314 individuals positive on SARS-CoV-2 reverse transcriptase–quantitative polymerase chain reaction (RT-qPCR) testing are reported.
Interventions
Individuals were randomized 1:1 to receive 1 dose of subcutaneous casirivimab and imdevimab, 1200 mg (600 mg of each; n = 158), or placebo (n = 156).
Main Outcomes and Measures
The primary end point was the proportion of seronegative participants who developed symptomatic COVID-19 during the 28-day efficacy assessment period. The key secondary efficacy end points were the number of weeks of symptomatic SARS-CoV-2 infection and the number of weeks of high viral load (>4 log10 copies/mL).
Results
Among 314 randomized participants (mean age, 41.0 years; 51.6% women), 310 (99.7%) completed the efficacy assessment period; 204 were asymptomatic and seronegative at baseline and included in the primary efficacy analysis. Subcutaneous casirivimab and imdevimab, 1200 mg, significantly prevented progression to symptomatic disease (29/100 [29.0%] vs 44/104 [42.3%] with placebo; odds ratio, 0.54 [95% CI, 0.30-0.97]; P = .04; absolute risk difference, −13.3% [95% CI, −26.3% to −0.3%]). Casirivimab and imdevimab reduced the number of symptomatic weeks per 1000 participants (895.7 weeks vs 1637.4 weeks with placebo; P = .03), an approximately 5.6-day reduction in symptom duration per symptomatic participant. Treatment with casirivimab and imdevimab also reduced the number of high viral load weeks per 1000 participants (489.8 weeks vs 811.9 weeks with placebo; P = .001). The proportion of participants receiving casirivimab and imdevimab who had 1 or more treatment-emergent adverse event was 33.5% vs 48.1% for placebo, including events related (25.8% vs 39.7%) or not related (11.0% vs 16.0%) to COVID-19.
Conclusions and Relevance
Among asymptomatic SARS-CoV-2 RT-qPCR–positive individuals living with an infected household contact, treatment with subcutaneous casirivimab and imdevimab antibody combination vs placebo significantly reduced the incidence of symptomatic COVID-19 over 28 days.
Trial Registration
ClinicalTrials.gov Identifier: NCT04452318
This randomized trial assesses the effect of a single dose of subcutaneous casirivimab and imdevimab antibody combination vs placebo on 28-day progression to symptomatic COVID-19 among asymptomatic SARS-CoV-2 reverse transcriptase–quantitative polymerase chain reaction–positive individuals living with an infected household contact.
Introduction
Control of SARS-CoV-2 and the COVID-19 pandemic has been challenging, due in part to a highly variable incubation period (range, 2-14 days), high rates of transmission from asymptomatic individuals, and the emergence of numerous SARS-CoV-2 variants of concern/variants of interest (VOCs/VOIs), including B.1.617.2 (Delta).1,2,3 Even with the arrival of effective SARS-CoV-2 vaccines,4 many individuals remain at risk of infection and potential progression to severe COVID-19 or longer-term complications due to limited vaccine uptake in certain geographic areas, inadequate immune responses to vaccination in certain risk groups, or immune-evading VOCs.5,6,7 For individuals not protected by vaccination, complementary approaches such as anti-SARS-CoV-2 monoclonal antibodies are needed.8,9
Casirivimab and imdevimab are 2 neutralizing, human sequence monoclonal antibodies that bind nonoverlapping epitopes on the SARS-CoV-2 spike protein receptor–binding domain and block virus entry.10 The 2-antibody combination reduces the risk of emergence of treatment-induced SARS-CoV-2 variants and retains neutralization potency in vitro against already circulating VOCs/VOIs, including B.1.1.7 (Alpha), B.1.351 (Beta), P.1 (Gamma), and B.1.617.2 (Delta).11,12 Casirivimab and imdevimab combination treatment has proven effective in treating COVID-19 outpatients13 and in preventing infection in close-contact settings14 and is currently authorized in the US under an Emergency Use Authorization (EUA) for treatment of mild to moderate COVID-19 and for postexposure prophylaxis in certain individuals.15
A 2-part, phase 3 randomized clinical trial among asymptomatic household contacts of SARS-CoV-2–infected individuals was conducted to assess whether subcutaneous casirivimab and imdevimab, 1200 mg (600 mg of each antibody), would prevent infection and/or COVID-19 in this high-risk setting. In Part A of the trial, casirivimab and imdevimab significantly prevented symptomatic SARS-CoV-2 infection compared with placebo (81.4% risk reduction) in uninfected close contacts.14 The current report describes results from Part B, wherein asymptomatic, infected close contacts were treated with subcutaneous casirivimab and imdevimab, 1200 mg.
Methods
Trial Design and Oversight
This randomized, double-blind, placebo-controlled, phase 3 trial assessed the efficacy of casirivimab and imdevimab in preventing progression to symptomatic SARS-CoV-2 infection among asymptomatic, infected household contacts of infected individuals. The trial enrolled participants at 112 study sites in the United States, Romania, and Moldova and was managed jointly by Regeneron, the COVID-19 Prevention Network, and the National Institute of Allergy and Infectious Diseases.
The trial was conducted in accordance with the principles of the Declaration of Helsinki,16 Good Clinical Practices/International Council for Harmonisation E-9 guidelines, and all applicable laws and regulations. The trial protocol (Supplement 1) and all amendments were reviewed and approved by an independent institutional review board/ethics committee. All participants provided written informed consent. Additional details are provided in eAppendix 1 in Supplement 2.
Nasopharyngeal swab samples were collected at the screening/baseline visit for central laboratory reverse transcriptase–quantitative polymerase chain reaction (RT-qPCR) testing and were used to assign participants into the 2 analysis cohorts: Part A analyses included those who at baseline were asymptomatic and RT-qPCR negative; Part B analyses included those who at baseline were asymptomatic and RT-qPCR positive (eFigure 1 in Supplement 2). The primary results for Part B are described in this report.
The trial consisted of a 1-day screening/baseline period, a 28-day efficacy assessment period, and a 7-month follow-up period (eFigure 1 in Supplement 2). Protocols (original and final for this analysis/amendment 6) and a detailed summary of all protocol amendments with rationales for these changes are included in Supplement 1. Briefly, the primary purpose for each of the 6 protocol amendments was to (1) change the collection of respiratory samples from nasal swabs and saliva samples to nasopharyngeal swabs; (2) remove the requirement to have at least 48 hours of sustained exposure to the index case; (3) include participants aged at least 12 to less than 18 years; (4) increase the sample size, modify study end points, and include participants younger than 12 years and those who were pregnant; (5) add an administrative assessment for assumption verification and sample size estimation; and (6) revise primary objectives and end points based on the administrative assessment.
Trial Participants
The trial included adults (aged ≥18 years) and adolescents (aged ≥12 to <18 years) who were household contacts of the first known household member with SARS-CoV-2 infection (index case) and who were asymptomatic (having no active respiratory or nonrespiratory symptoms consistent with COVID-19). Part B participants were SARS-CoV-2 positive by RT-qPCR central laboratory testing. COVID-19 vaccination was prohibited prior to enrollment but was allowed after completing the 28-day efficacy assessment period. The full list of inclusion and exclusion criteria are provided in eAppendix 1 in Supplement 2. Because COVID-19 outcomes have been observed to differ by race and ethnicity,17 data on participants’ self-reported race and ethnicity (both based on fixed categories) were collected to describe the demographics of the trial population and to assess the homogeneity of the treatment effect across various subgroups.
Randomization
Trial participants were randomized (1:1) to receive casirivimab and imdevimab or placebo within 96 hours of collection of the index case’s positive SARS-CoV-2 test sample. Participants were stratified by age and SARS-CoV-2 local diagnostic results, when available (see eAppendix 1 in Supplement 2). Participants were randomized according to a central randomization scheme provided by an interactive web response system. A permuted block randomization scheme with a fixed block length of 2 was used. Randomization was performed at the individual study participant level, not by households.
Intervention and Assessments
At baseline (day 1), participants received a single dose of casirivimab and imdevimab, 1200 mg (120 mg/mL), or placebo (matching saline solution), via subcutaneous injection (4 injections of 2.5-mL solution).
Serum samples were collected at the screening/baseline visit for serology testing for anti–SARS-CoV-2 antibodies (antispike [S1] IgA, antispike [S1] IgG, and antinucleocapsid IgG) to determine whether participants had evidence of immunity to SARS-CoV-2 (ie, seropositive as opposed to seronegative).
Signs and symptoms of COVID-19 were collected by weekly investigator- (or designee-) led interviews. At each visit/contact, investigators interviewed participants about adverse events they were experiencing or could have experienced since the last visit/contact. If a participant developed signs and/or symptoms, these data were collected weekly until resolved, even if the participant had completed the efficacy assessment period.
Serial nasopharyngeal swabs were collected at baseline, prior to study drug administration, and weekly during the efficacy assessment period or the follow-up period to determine SARS-CoV-2 viral load by RT-qPCR until participants tested negative on 2 consecutive swabs.
Serious adverse events, treatment-emergent adverse events (TEAEs), and adverse events of special interest, defined as grade 3 or higher injection site reactions or hypersensitivity reactions, were assessed.
Details on the intervention, assessments, and analytical methods are provided in eAppendix 1 of Supplement 2 or have been previously described.18
End Points
The primary efficacy end point was the proportion of participants who had a positive RT-qPCR result at baseline or during the 28-day efficacy assessment period and who developed signs and symptoms of COVID-19 within 14 days of the positive RT-qPCR result. A broad definition (broad term) of what constituted symptomatic COVID-19 was used for the primary analysis (eAppendix 1 in Supplement 2).
There were 2 key secondary efficacy end points: (1) the number of weeks of symptomatic SARS-CoV-2 infection (broad term) and (2) the number of weeks of high viral load (>4 log10 copies/mL) in nasopharyngeal samples over 28 days. There were an additional 19 prespecified secondary efficacy end points; analyses for 9 of these are reported herein (eTable 1 in Supplement 2).
Sample Size Calculation
The sample size calculation assumed that approximately 200 seronegative participants would be enrolled and 50% of placebo participants would develop symptoms, providing greater than 90% power to detect a relative risk of 0.5 at a 2-sided α = .05. The relative risk assumption of 0.5 was based on the virologic and clinical treatment effect observed in the interim analysis from the outpatient treatment study with casirivimab and imdevimab.18
Statistical Analysis
The statistical analysis plan for the presented analysis was finalized prior to database lock and treatment unblinding. The primary analysis population used for efficacy analyses was the seronegative modified full analysis set, which included all randomized participants who were asymptomatic, negative for SARS-CoV-2 antibodies (ie, seronegative), and SARS-CoV-2 positive by central laboratory RT-qPCR at baseline (ie, Part B in this 2-part trial). Hierarchical testing was used for the primary and key secondary end points to control for type I error at a 2-sided α = .05 (eAppendix 1 in Supplement 2). The primary end point was analyzed using a logistic regression model; additional analyses of the primary end point were performed in prespecified subgroups of interest (see Supplement 1 for details). Key secondary end points were analyzed by Van Elteren tests stratified by region and age. Analysis of other end points was performed using logistic regression (binary end points), Fisher exact test (medically attended visit end points), and analysis of covariance (virologic end points). Findings for analyses of end points other than the primary and key secondary end points should be interpreted as exploratory due to the potential for type I error from multiple comparisons. Efficacy analyses are reported through the 28-day efficacy assessment period unless otherwise noted. Adverse events occurring in all participants who received study drug (active or placebo) were assessed. Efficacy analyses and the number and frequency of adverse events are reported for participants randomized through January 28, 2021, until the data cutoff date of March 11, 2021.
Participants with COVID-19 symptoms who were missing central laboratory RT-qPCR test results were considered as having a symptomatic infection if any symptoms occurred within 14 days of a positive SARS-CoV-2 local test result.
All analyses were performed using SAS version 9.4 or higher (SAS Institute Inc). All statistical methods are described in detail in Supplement 1.
Post Hoc and Exploratory Analyses
Post hoc analyses included examinations of symptomatic infections and viral load trajectories in placebo-treated patients only (ie, natural history); only symptomatic infections that began 3 days or longer after treatment; symptomatic infections in participants with risk factors for progression to severe COVID-19; and the number of participants with detectable virus at each week during the efficacy assessment period. Casirivimab and imdevimab efficacy according to participants’ baseline serologic status (seronegative, seropositive, or undetermined) was also explored.
Results
Trial Population
Between July 13, 2020, and January 28, 2021, a total of 314 household contacts were confirmed to be SARS-CoV-2 RT-qPCR positive at baseline based on central laboratory nasopharyngeal RT-qPCR; 156 study participants received placebo and 155 received subcutaneous casirivimab and imdevimab, 1200 mg (3 randomized participants did not receive any study drug) (Figure 1).
Figure 1. Flow of Participants Through the Trial.

aIndividuals who were randomized and were negative for SARS-CoV-2 by reverse transcriptase–quantitative polymerase chain reaction were included in Part A (n = 2621); individuals whose SARS-CoV-2 status at baseline was undetermined (ie, inconclusive or missing results) were not included in either Part A or Part B (n = 94).
bOne randomized participant in the CAS/IMD group and 2 randomized participants in the placebo group were determined after randomization to have been symptomatic at baseline and were excluded from the primary analysis. This is the seronegative modified full analysis set.
The primary, key secondary, and other secondary analyses were performed in participants without evidence of immunity (ie, seronegative). Of the 314 RT-qPCR–positive participants randomized, 207 (66%) were seronegative, 84 (27%) were seropositive, and 23 (7%) had undetermined status. Three seronegative participants were excluded from efficacy analyses because they were determined after randomization to have been symptomatic at baseline. A post hoc analysis evaluated the effect of preexisting immunity (seropositive vs seronegative) on virologic and clinical outcomes in placebo-treated patients (eAppendix 2 in Supplement 2).
Demographics and baseline characteristics were balanced across treatment groups (Table 1). For the primary analysis population, the mean age was 40.9 years, 45.4% were male, 5.3% identified as Black or African American, and 34.8% identified as Hispanic or Latino. Based on the scientific understanding of COVID-19 at the beginning of the trial, 32% of participants had 1 or more risk factor for severe COVID-19. According to the current understanding (August 2021),19 the proportion of participants with 1 or more risk factor for severe COVID-19 was 71%, including 62.3% who were overweight (body mass index >25, calculated as weight in kilograms divided by height in meters squared), 18.6% who had cardiovascular disease or hypertension, and 10.3% who were aged 65 years or older (Table 1). Demographics and baseline characteristics for participants who were seropositive at baseline are provided in eTable 2 in Supplement 2.
Table 1. Demographics and Baseline Characteristicsa.
| Characteristics | Casirivimab and imdevimab, 1200 mg (n = 101) | Placebo (n = 106) |
|---|---|---|
| Age, y | ||
| Mean (SD) [range] | 39.2 (17.7) [12-87] | 42.5 (18.3) [12-87] |
| No. (%) | ||
| ≥12 to <18 | 15 (14.9) | 11 (10.4) |
| ≥50 | 31 (30.7) | 39 (36.8) |
| Sex, No. (%) | ||
| Female | 50 (49.5) | 63 (59.4) |
| Male | 51 (50.5) | 43 (40.6) |
| Race, No. (%) | ||
| American Indian or Alaska Native | 1 (1.0) | 0 |
| Asian | 9 (8.9) | 3 (2.8) |
| Black or African American | 7 (6.9) | 4 (3.8) |
| White | 79 (78.2) | 96 (90.6) |
| Otherb | 5 (5.0) | 3 (2.8) |
| Hispanic or Latino ethnicity, No./total (%)c | 34/100 (34.0) | 38/105 (36.2) |
| Weight, mean (SD), kg | 81.7 (22.86) | 78.5 (19.04) |
| BMI, mean (SD)d | 28.3 (6.68) | 27.8 (6.46) |
| Participants with any high risk factor for COVID-19, No./total (%)e | 72/100 (72.0) | 73/104 (70.2) |
| BMI >25 (adults) or ≥85th percentile (adolescents)d | 64/100 (64.0) | 63/104 (60.6) |
| BMI ≥35 (adults)d | 16/100 (16.0) | 11/104 (10.6) |
| Cardiovascular disease or hypertension | 20/100 (20.0) | 18/104 (17.3) |
| Aged ≥65 y | 8/100 (8.0) | 13/104 (12.5) |
| Diabetes | 5/100 (5.0) | 11/104 (10.6) |
| Immunosuppressive disease or treatment | 4/100 (4.0) | 1/104 (1.0) |
| Chronic kidney disease | 2/100 (2.0) | 3/104 (2.9) |
| Chronic lung disease | 1/100 (1.0) | 10/104 (9.6) |
| Total No. of households | 97 | 99 |
| Households by No. of participants in seronegative modified full analysis set, No./total (%)f | ||
| 1 | 86/97 (88.7) | 88/99 (88.9) |
| 2 | 9/97 (9.3) | 9/99 (9.1) |
| 3 | 2/97 (2.1) | 2/99 (2.0) |
Abbreviation: BMI, body mass index.
Demographics and baseline characteristics for the seronegative population.
Includes unknown and not reported.
In participants with available ethnicity data.
Calculated as weight in kilograms divided by the square of the height in meters.
Percentages are based on the seronegative modified full analysis set.
Percentages are based on the total number of households. Household size is calculated by counting only participants in the seronegative modified full analysis set without considering the index case or household members not participating in Part B of this study. A household with more than 1 seronegative participant could consist of participants in both the casirivimab and imdevimab group and the placebo group and is summarized in both treatment groups.
Primary Outcome
In asymptomatic, infected participants who were seronegative at baseline, subcutaneous casirivimab and imdevimab, 1200 mg, significantly reduced the risk of developing a symptomatic infection (broad-term definition) compared with placebo (29/100 [29.0%] vs 44/104 [42.3%]; odds ratio, 0.54 [95% CI, 0.30-0.97]; P = .04; absolute risk difference, −13.3% [95% CI, −26.3% to −0.3%]) (Table 2 and Figure 2A). The effect of treatment with casirivimab and imdevimab on the primary outcome was consistent across prespecified subgroups of interest (eTable 3 in Supplement 2) and in a sensitivity analysis using multiple imputation for missing data (eTable 4 in Supplement 2).
Table 2. Study Outcomesa.
| Outcomes | Casirivimab and imdevimab, 1200 mg (n = 100) | Placebo (n = 104) | Difference (95% CI) | Adjusted odds ratio (95% CI)b | P value |
|---|---|---|---|---|---|
| Primary outcome | |||||
| Participants who subsequently developed signs and symptoms (broad term) within 14 d of a positive RT-qPCR result at baseline or during the efficacy assessment period, No. (%) | 29 (29.0) | 44 (42.3) | −13.3 (−26.3 to −0.3) | 0.54 (0.30-0.97) | .04b |
| Key secondary outcomesc | |||||
| No. of weeks of symptomatic SARS-CoV-2 infection (broad term) within 14 d of a positive RT-qPCR result at baseline or during the efficacy assessment period | |||||
| Total per 1000 participants | 895.7 | 1637.4 | −741.7 | .03d | |
| Per symptomatic participant, mean (SD) | 3.1 (4.1) | 3.9 (4.5) | −0.8 | ||
| Per participant, mean (SD) | 0.9 (2.6) | 1.6 (3.5) | −0.7 | ||
| No. of weeks of high viral load (>4 log10 copies/mL) in nasopharyngeal swab samples during the efficacy assessment period | |||||
| Total per 1000 participants | 489.8 | 811.9 | −322.1 | .001d | |
| Per participant, mean (SD) | 0.5 (0.7) | 0.8 (0.8) | −0.3 | ||
Abbreviation: RT-qPCR, reverse transcriptase–quantitative polymerase chain reaction.
Analyses of the primary and key secondary end points were conducted in the seronegative modified full analysis set. Three seronegative participants (1 in the casirivimab and imdevimab group and 2 in placebo group) were excluded from efficacy analyses because they were determined after randomization to have been symptomatic at baseline.
Based on logistic regression model adjusted by region (United States or outside of United States) and age group (aged 12 to less than 50 years vs aged 50 years or older).
Key secondary end points are presented in order of the hierarchical testing sequence.
Based on stratified Wilcoxon rank sum test (van Elteren test) with region (United States or outside of United States) and age group (aged 12 to less than 50 years vs aged 50 years or older) as strata.
Figure 2. Prevention of Progression From Asymptomatic SARS-CoV-2 Infection to Symptomatic COVID-19 With Subcutaneous Casirivimab and Imdevimab.
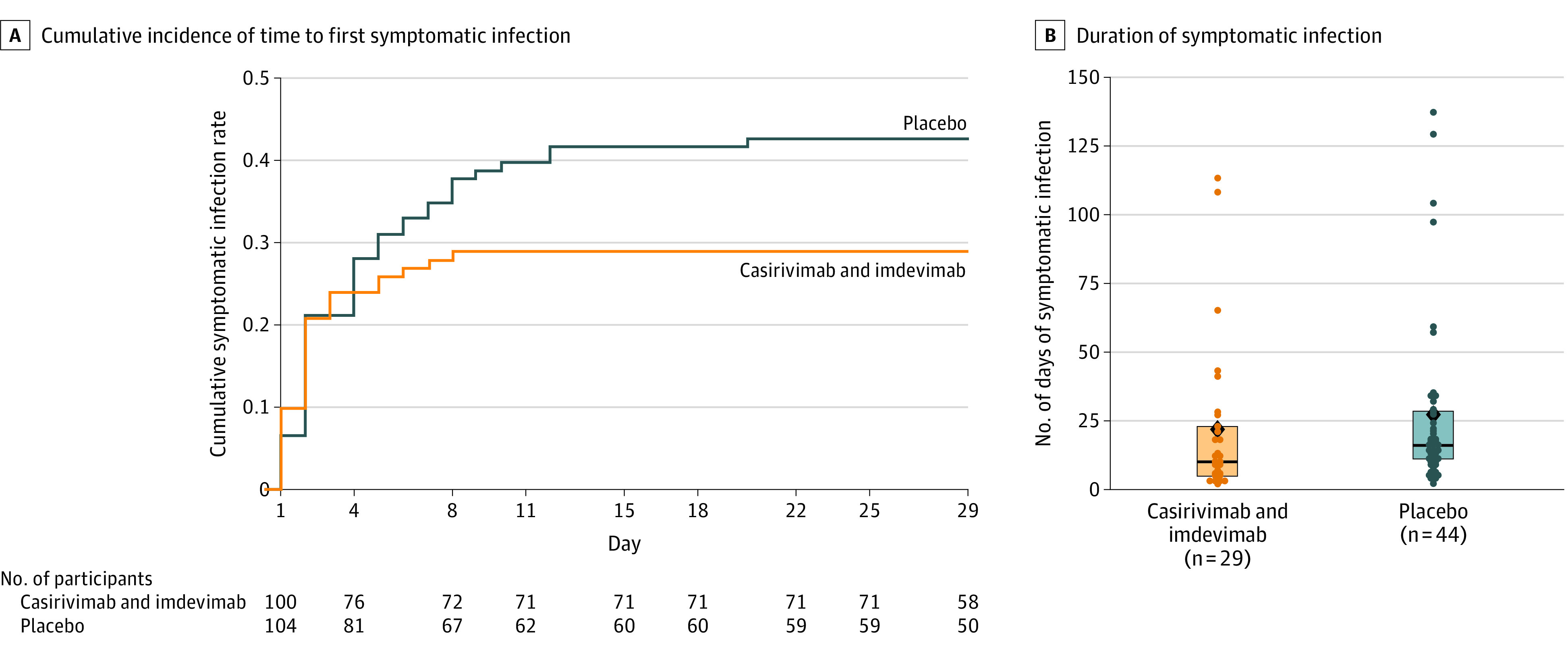
Analyses were conducted using the seronegative modified full analysis set. A, Cumulative incidence of time to first symptomatic infection within 14 days of a positive reverse transcriptase–quantitative polymerase chain reaction result at baseline or during the efficacy assessment period. B, Duration of symptomatic infection. The box tops and bottoms represent the interquartile range; horizontal bars within boxes, median; and diamonds, mean. Individual participant data are represented by circles.
Key Secondary Outcomes
Casirivimab and imdevimab treatment significantly reduced the number of weeks with symptoms vs placebo (895.7 weeks vs 1637.4 weeks per 1000 participants; P = .03) (Table 2). This corresponded to a 5.6-day reduction in the mean duration of symptoms per symptomatic participant treated with casirivimab and imdevimab (21.7 days; 3.1 weeks) vs placebo (27.3 days; 3.9 weeks) (Table 2 and Figure 2B).
The total number of weeks of high viral load (>4 log10 copies/mL) in nasopharyngeal swab samples was significantly reduced with casirivimab and imdevimab vs placebo (489.8 weeks vs 811.9 weeks per 1000 participants; P = .001) (Table 2 and Figure 3A). This corresponded to a 0.3-week reduction per participant in weeks with high viral load from 0.8 weeks with placebo to 0.5 weeks with casirivimab and imdevimab (Table 2).
Figure 3. Reduction in SARS-CoV-2 Viral Load With Subcutaneous Casirivimab and Imdevimab.
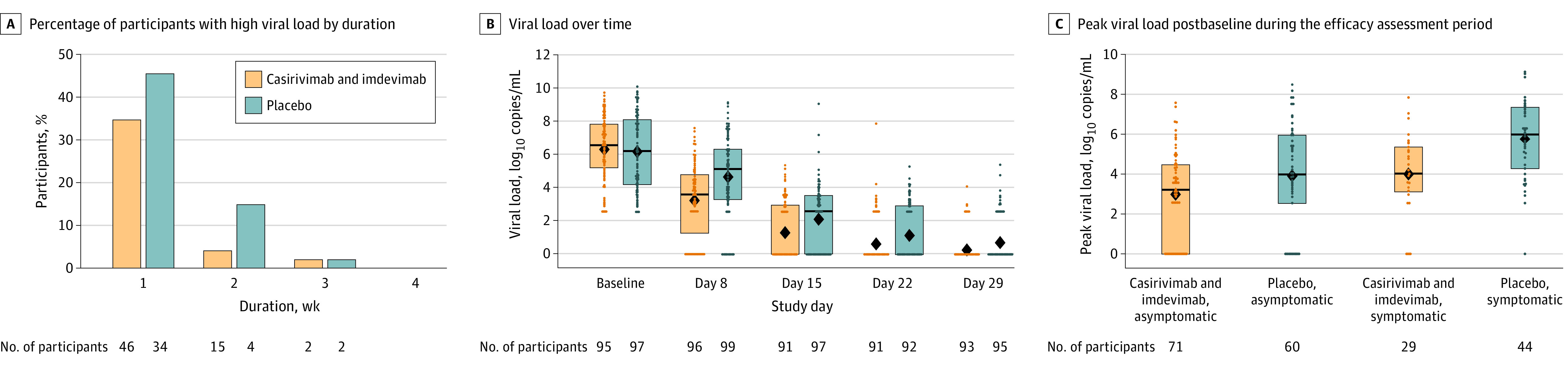
Analyses were conducted using the seronegative modified full analysis set. Only nonmissing available nasopharyngeal swab viral load data were used for the analysis of viral load end points. Only participants with at least 1 postbaseline viral load data point (in nasopharyngeal swab samples) were included in the analyses. A, Percentage of participants with high nasopharyngeal viral load (>4 log10 copies/mL) by duration. B, Viral load over time, and C, peak viral load postbaseline during the efficacy assessment period. The box tops and bottoms represent the interquartile range; horizontal bars within boxes, median; and diamonds, mean. Individual participant data are represented by circles.
Other Secondary Outcomes
Symptomatic Infection by Definition
Similar to findings observed for the primary outcome analysis, which was based on a broad-term definition, subcutaneous casirivimab and imdevimab, 1200 mg, reduced the risk of developing symptomatic infection vs placebo when using the study-defined strict-term (10.0% vs 19.2%) or Centers for Disease Control and Prevention (CDC)–defined (27.0% vs 39.4%) definitions of COVID-19 signs and symptoms (eTable 5 in Supplement 2).
Virologic Outcomes
There was a more rapid decline in nasopharyngeal SARS-CoV-2 viral load in participants treated with subcutaneous casirivimab and imdevimab, 1200 mg, compared with those treated with placebo, with an adjusted least-square mean difference of −1.5 log10 copies/mL in favor of the antibody combination at day 8 (Figure 3B; eTable 6 in Supplement 2). The reduction in viral load with subcutaneous casirivimab and imdevimab, 1200 mg, observed in the current study and the reduction observed with intravenous casirivimab and imdevimab, 1200 mg, in a separate trial in symptomatic outpatients with COVID-19 are shown in eFigure 2 in Supplement 2.13
Compared with placebo, casirivimab and imdevimab reduced peak viral load by approximately 1 to 2 log10 copies/mL in participants who became symptomatic and in those who remained asymptomatic throughout the efficacy assessment period (Figure 3C; eTable 6 in Supplement 2).
Hospitalization Outcomes
Subcutaneous casirivimab and imdevimab, 1200 mg, reduced the proportion of participants who had a COVID-19–related hospitalization or emergency department visit vs placebo (0/100 vs 6/104, respectively) (eTable 7 in Supplement 2). Of the 6 participants in the placebo group, 3 had emergency department visits, 1 was hospitalized, and 2 had emergency department visits and were subsequently hospitalized. In contrast, no participants receiving casirivimab and imdevimab had emergency department visits or hospitalizations. Other hospitalization outcomes are provided in eTable 8 in Supplement 2.
Post Hoc and Exploratory Analyses
Symptomatic Infection Post Hoc Analyses
Casirivimab and imdevimab reduced the risk of developing symptomatic infection (the primary end point) vs placebo when assessing only infections that began 3 days or longer after treatment (day 4 to end of efficacy assessment period; 5.0% vs 21.2%; eTable 9 in Supplement 2). In a subgroup analysis including only participants who were more likely to progress to severe COVID-19 based on the protocol definition and updated current understanding of risk factors for severe disease, casirivimab and imdevimab reduced the incidence of symptomatic infections vs placebo (eTable 10 in Supplement 2).
Proportion of Participants With No Detectable Virus
Casirivimab and imdevimab treatment also led to a higher proportion of participants with no RT-qPCR–detectable virus at each week during the efficacy assessment period (eTable 11 in Supplement 2).
Efficacy in Participants According to Baseline Serology Status
Although the primary analysis population focused on participants without evidence of prior infection (seronegative), prespecified exploratory analyses conducted in all participants combined (seronegative, seropositive, and undetermined status) and in the seropositive population analyzed separately showed that casirivimab and imdevimab reduced the risk of developing symptomatic infection vs placebo by 35.4% and 33.9%, respectively (eTable 12 in Supplement 2). In all participants combined, casirivimab and imdevimab reduced the duration of symptoms in those who became symptomatic, reduced the duration of weeks of detectable viral load and high viral load, and reduced peak viral load, with similar numerical trends in the seropositive-only population in most analyses (eTables 13-16 in Supplement 2).
Adverse Events
Casirivimab and imdevimab treatment was generally well tolerated. The proportions of participants in the casirivimab and imdevimab group who experienced 1 or more TEAE, 1 or more COVID-19–related TEAE, or 1 or more non–COVID-19–related TEAE were 33.5%, 25.8%, and 11.0%, respectively; the proportions of participants in the placebo group who experienced these events were 48.1%, 39.7%, and 16.0%, respectively (eTables 17-18 in Supplement 2). Serious TEAEs were reported in no participants (0%) in the casirivimab and imdevimab group and 4 participants (2.6%) in the placebo group; 1 of the 4 participants in the placebo group had a non–COVID-19–related serious TEAE (eTable 19 in Supplement 2). The most frequent TEAEs and serious TEAEs were COVID-19 related (eTables 18-19 in Supplement 2). There were no grade 3 or higher injection site reactions or grade 3 or higher hypersensitivity reactions in either group (eTable 17 in Supplement 2). Injection site reactions (grade 1-2) occurred in 6 participants (4%) in the casirivimab and imdevimab group and 1 participant (1%) in the placebo group (eTable 19 in Supplement 2). No deaths were reported up to the data cutoff date.
Pharmacokinetics
Following subcutaneous administration of a single 1200-mg dose to study participants (in either sentinel or safety groups; for description, see eAppendix 1 in Supplement 2), casirivimab and imdevimab were rapidly absorbed, with mean concentrations in serum 1 day after dosing of 23.3 (SD, 15.0) mg/L and 22.7 (SD, 14.8) mg/L; at this time point, concentrations were greater than 100 times the 90% inhibitory concentration for the Delta variant (eFigures 3-4 in Supplement 2). Both antibodies reached maximal concentrations in serum at a median of 7.5 days. Casirivimab and imdevimab exhibited linear elimination, with a mean half-life of 30.2 (SD, 5.31) days and 26.5 (SD, 5.31) days, respectively. Mean concentrations in serum 28 days after dosing were 33.5 (SD, 12.3) mg/L for casirivimab and 26.9 (SD, 9.12) mg/L for imdevimab. A summary of pharmacokinetic parameters after a single 1200-mg subcutaneous dose is shown in eTable 20 in Supplement 2. A comparison of antibody concentrations in serum over 28 days following a single subcutaneous dose (this trial) or intravenous doses (2 outpatient trials: NCT04425629 and NCT04666441) of casirivimab and imdevimab is shown in eFigure 5 in Supplement 2.
Discussion
It was previously reported in Part A of this phase 3 clinical trial that subcutaneous casirivimab and imdevimab prevented SARS-CoV-2 infection (asymptomatic and symptomatic) in uninfected individuals living with an infected household contact.14 In Part B of the trial, which evaluated the effect of casirivimab and imdevimab in early asymptomatic infection in the same household contact setting, the antibody combination reduced the incidence of symptomatic COVID-19 over 28 days vs placebo. Secondary outcomes demonstrated differences in favor of casirivimab and imdevimab compared with placebo in the duration of symptomatic infection, duration of RT-qPCR–detectable SARS-CoV-2 in the nasopharynx, and proportion of patients with COVID-19–related hospitalizations or emergency department visits. Reductions in progression to symptomatic infection and in other outcomes as observed herein would be of potential clinical relevance for use of monoclonal antibody therapies in early treatment of COVID-19.
A post hoc analysis that evaluated the progression from asymptomatic infection to symptomatic COVID-19 (the primary end point) when symptoms began 3 days or longer after treatment suggested benefit of casirivimab and imdevimab in reducing the occurrence of symptomatic disease, but this finding should be considered hypothesis generating.
A prespecified exploratory analysis assessed the primary end point in all participants regardless of baseline serostatus (in comparison with the primary end-point analysis, which included the seronegative population only). This analysis demonstrated that casirivimab and imdevimab reduced the progression from asymptomatic infection to symptomatic COVID-19 in the overall population, regardless of serostatus. Point-of-care serology tests may therefore have limited utility in guiding decisions in the clinic to prevent COVID-19 in exposed individuals.
Casirivimab and imdevimab has received an EUA from the US Food and Drug Administration for treatment of high-risk outpatients with mild to moderate COVID-19.15,20 Data from a phase 3 clinical trial involving outpatients with COVID-19 showed that casirivimab and imdevimab, 1200 mg administered intravenously, reduced the risk of COVID-19–related hospitalization or death, reduced the time to resolution of symptoms, and reduced viral load faster than placebo.13 The EUA allows use of 1200-mg subcutaneous administration as an alternative route of administration when intravenous infusion is not feasible and would lead to a delay in treatment. This alternative route of administration under the EUA is supported by virologic outcome data21 and is further supported by viral load reduction data presented in the current report. In the current study, analysis of the secondary end point of COVID-19–related medically attended visits during the 28-day efficacy assessment period demonstrated that 6 placebo-treated participants had an emergency department visit or hospitalization while no participants receiving subcutaneous casirivimab and imdevimab had these events, in support of the use of 1200-mg subcutaneous administration as an alternative route of administration for outpatient treatment of COVID-19, as authorized under the current EUA.15
As has been shown consistently in casirivimab and imdevimab clinical studies, a larger proportion of participants who received placebo experienced 1 or more TEAE, with the difference attributed to the higher number of COVID-19–related events observed in that group.13
Following subcutaneous dosing, concentrations of each antibody in serum were above the predicted neutralization target concentration, based on in vitro and preclinical data,10,12,22 on the first day following dosing and throughout the 28-day efficacy assessment period. A comparison of pharmacokinetic profiles following intravenous or subcutaneous single doses of casirivimab and imdevimab (eFigure 5 in Supplement 2) demonstrated that although intravenous administration of casirivimab, 600 mg, and imdevimab, 600 mg, achieved higher drug concentrations at early time points, subcutaneous administration of casirivimab, 600 mg, and imdevimab, 600 mg, achieved mean concentrations in serum 1 day after administration of 22.1 mg/L and 25.8 mg/L, respectively; these day 1 levels were above the estimated target dose for neutralization of SARS-CoV-2 (20 mg/L).
Limitations
This study has several limitations. First, although this study was conducted at multiple sites, all were located in the United States, Romania, or Moldova, with the majority of sites in the United States. Second, this study was conducted prior to both widespread vaccination and circulation of the Delta (B.1.617.2) and Omicron (B.1.1.529) variants in these locales. Third, despite efforts by sites to recruit non-White participants, there were relatively few non-White participants enrolled; also, few adolescents were enrolled. Fourth, although the study was adequately powered, the sample size was relatively small due to a study design in which the infection status of asymptomatic participants was not confirmed at inclusion. Fifth, the trial was ongoing at the time of this report, so while all participants completed the 28-day efficacy assessment period, some were early in the follow-up period. Therefore, for any participant experiencing symptom(s) at the time of the analysis, symptom duration was calculated from the start date to the data cutoff date.
Conclusions
Among asymptomatic SARS-CoV-2 RT-qPCR–positive individuals living with an infected household contact, treatment with subcutaneous casirivimab and imdevimab antibody combination vs placebo significantly reduced the incidence of symptomatic COVID-19 over 28 days.
Trial Protocol
COVID-19 Phase 3 Prevention Trial Team (REGN 2069/CoVPN 3502)
eAppendix 1. Supplementary Methods
eAppendix 2. Supplementary Results
eFigure 1. Schematic Overview of the Study Design
eFigure 2. Mean Viral Load in Symptomatic and Asymptomatic SARS-CoV-2 PCR+ Individuals
eFigure 3. Mean (+SD) Concentrations of Casirivimab and Imdevimab in Serum Over Time (Part B Sentinel Group)
eFigure 4. Mean (+SD) Concentrations of Casirivimab and Imdevimab in Serum Over Time (Part B Sentinel + Safety Group)
eFigure 5. Mean (±SD) Concentrations of Casirivimab and Imdevimab in Serum Over Time (Study 2069 [SC] vs Study 2067 [IV] or 20145 [IV])
eTable 1. Listing of Primary and Secondary End Points
eTable 2. Demographics and Baseline Characteristics
eTable 3. Subgroup Analyses of the Primary Endpoint (Seronegative)
eTable 4. Sensitivity Analysis of the Primary Endpoint to Address Missing Data (Seronegative)
eTable 5. Proportion of Participants Who Had a Symptomatic RT-qPCR-Confirmed SARS-CoV-2 Infection by Definition (Seronegative)
eTable 6. Summary of Viral Load Endpoints (Seronegative)
eTable 7. Proportion of Participants Who Had a COVID-19-Related Hospitalization or ER Visit (Seronegative)
eTable 8. Hospitalization and Hospitalization Outcomes (Seronegative)
eTable 9. Proportion of Participants Who Had a Symptomatic RT-qPCR-Confirmed SARS-CoV-2 Infection (Broad-Term) by Period (Seronegative)
eTable 10. Proportion of Participants Who Had a Symptomatic RT-qPCR-Confirmed SARS-CoV-2 Infection (Broad-Term) in Those Who Have ≥1 Risk Factor for Severe COVID-19 (Seronegative)
eTable 11. Proportion of Participants With a Confirmed Negative SARS-CoV-2 RT-qPCR During the EAP (Seronegative)
eTable 12. Proportion of Participants Who Had a Symptomatic RT-qPCR-Confirmed SARS-CoV-2 Infection (Broad-Term) by Baseline Serology Status
eTable 13. Number of Weeks of Symptomatic SARS-CoV-2 Infection (Broad-Term) by Baseline Serology Status
eTable 14. Number of Weeks of High Viral Load (>4 Log10 Copies/mL) in NP Swab Samples by Baseline Serology Status
eTable 15. Maximum SARS-CoV-2 RT-qPCR Viral Load (Log10 Copies/mL) in NP Swab Samples by Baseline Serology Status
eTable 16. Number of Weeks of RT-qPCR Confirmed SARS-CoV-2 Infection (Regardless of Symptoms) by Baseline Serology Status
eTable 17. Overview of Treatment-Emergent Adverse Events
eTable 18. Treatment-Emergent Adverse Events Occurring in ≥2% of Participants
eTable 19. Serious Treatment-Emergent Adverse Events
eTable 20. Summary of PK Parameters for Casirivimab and Imdevimab After a Single 1200 mg Subcutaneous Dose in Part B Participants
Nonauthor Collaborators. COVID-19 Phase 3 Prevention Trial Team
Data Sharing Statement
References
- 1.Buitrago-Garcia D, Egli-Gany D, Counotte MJ, et al. Occurrence and transmission potential of asymptomatic and presymptomatic SARS-CoV-2 infections: a living systematic review and meta-analysis. PLoS Med. 2020;17(9):e1003346. doi: 10.1371/journal.pmed.1003346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention . Clinical Questions About COVID-19: Questions and Answers. Accessed April 23, 2021. https://www.cdc.gov/coronavirus/2019-ncov/hcp/faq.html#Transmission
- 3.GISAID . hCov19 Variants. Accessed July 20, 2021. https://www.gisaid.org/hcov19-variants/
- 4.Higdon MM, Wahl B, Jones CB, et al. . A systematic review of COVID-19 vaccine efficacy and effectiveness against SARS-CoV-2 infection and disease. medRxiv. Preprint posted September 25, 2021. doi: 10.1101/2021.09.17.21263549 [DOI]
- 5.World Health Organization . Coronavirus (COVID-19) dashboard. Accessed November 2, 2021. https://covid19.who.int/table
- 6.Garcia-Beltran WF, Lam EC, St Denis K, et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell. 2021;184(9):2372-2383.e9. doi: 10.1016/j.cell.2021.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boyarsky BJ, Werbel WA, Avery RK, et al. Antibody response to 2-dose SARS-CoV-2 mRNA vaccine series in solid organ transplant recipients. JAMA. 2021;325(21):2204-2206. doi: 10.1001/jama.2021.7489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kreuzberger N, Hirsch C, Chai KL, et al. SARS-CoV-2-neutralising monoclonal antibodies for treatment of COVID-19. Cochrane Database Syst Rev. 2021;9:CD013825. doi: 10.1002/14651858.CD013825.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boutron I, Chaimani A, Devane D, et al. Interventions for the prevention and treatment of COVID-19: a living mapping of research and living network meta-analysis. Published online November 3, 2020. Cochrane Database Syst Rev. doi: 10.1002/14651858.CD013769 [DOI] [Google Scholar]
- 10.Hansen J, Baum A, Pascal KE, et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science. 2020;369(6506):1010-1014. doi: 10.1126/science.abd0827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Copin R, Baum A, Wloga E, et al. The monoclonal antibody combination REGEN-COV protects against SARS-CoV-2 mutational escape in preclinical and human studies. Cell. 2021;184(15):3949-3961. doi: 10.1016/j.cell.2021.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baum A, Fulton BO, Wloga E, et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020;369(6506):1014-1018. doi: 10.1126/science.abd0831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinreich DM, Sivapalasingam S, Norton T, et al. ; Trial Investigators . REGEN-COV antibody combination and outcomes in outpatients with Covid-19. N Engl J Med. 2021;385(23):e81. doi: 10.1056/NEJMoa2108163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Brien MP, Forleo-Neto E, Musser BJ, et al. ; Covid-19 Phase 3 Prevention Trial Team . Subcutaneous REGEN-COV antibody combination to prevent Covid-19. N Engl J Med. 2021;385(13):1184-1195. doi: 10.1056/NEJMoa2109682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.US Food and Drug Administration . Fact sheet for health care providers—Emergency Use Authorization (EUA) of REGEN-COV (casirivimab and imdevimab). Revised December 2021. Accessed January 6, 2022. https://www.fda.gov/media/145611/download
- 16.World Medical Association . World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191-2194. doi: 10.1001/jama.2013.281053 [DOI] [PubMed] [Google Scholar]
- 17.Magesh S, John D, Li WT, et al. Disparities in COVID-19 outcomes by race, ethnicity, and socioeconomic status: a systematic-review and meta-analysis. JAMA Netw Open. 2021;4(11):e2134147. doi: 10.1001/jamanetworkopen.2021.34147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weinreich DM, Sivapalasingam S, Norton T, et al. ; Trial Investigators . REGN-COV2, a neutralizing antibody cocktail, in outpatients with Covid-19. N Engl J Med. 2021;384(3):238-251. doi: 10.1056/NEJMoa2035002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Centers for Disease Control and Prevention . COVID-19: people with certain medical conditions. Accessed September 1, 2021. https://www.cdc.gov/coronavirus/2019-ncov/need-extra-precautions/people-with-medical-conditions.html
- 20.US Food and Drug Administration . Letter of authorization for emergency use of REGEN-COV (casirivimab and imdevimab). November 17, 2021. Accessed January 6, 2022. https://www.fda.gov/media/145610/download
- 21.Portal-Celhay C, Forleo-Neto E, Eagan W, et al. Dose-ranging virology study of the combination COVID-19 antibodies casirivimab and imdevimab in the outpatient setting—poster 04769. Presented at: 31st European Congress of Clinical Microbiology and Infectious Diseases [virtual meeting]; July 9-12, 2021. [Google Scholar]
- 22.Baum A, Ajithdoss D, Copin R, et al. REGN-COV2 antibodies prevent and treat SARS-CoV-2 infection in rhesus macaques and hamsters. Science. 2020;370(6520):1110-1115. doi: 10.1126/science.abe2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial Protocol
COVID-19 Phase 3 Prevention Trial Team (REGN 2069/CoVPN 3502)
eAppendix 1. Supplementary Methods
eAppendix 2. Supplementary Results
eFigure 1. Schematic Overview of the Study Design
eFigure 2. Mean Viral Load in Symptomatic and Asymptomatic SARS-CoV-2 PCR+ Individuals
eFigure 3. Mean (+SD) Concentrations of Casirivimab and Imdevimab in Serum Over Time (Part B Sentinel Group)
eFigure 4. Mean (+SD) Concentrations of Casirivimab and Imdevimab in Serum Over Time (Part B Sentinel + Safety Group)
eFigure 5. Mean (±SD) Concentrations of Casirivimab and Imdevimab in Serum Over Time (Study 2069 [SC] vs Study 2067 [IV] or 20145 [IV])
eTable 1. Listing of Primary and Secondary End Points
eTable 2. Demographics and Baseline Characteristics
eTable 3. Subgroup Analyses of the Primary Endpoint (Seronegative)
eTable 4. Sensitivity Analysis of the Primary Endpoint to Address Missing Data (Seronegative)
eTable 5. Proportion of Participants Who Had a Symptomatic RT-qPCR-Confirmed SARS-CoV-2 Infection by Definition (Seronegative)
eTable 6. Summary of Viral Load Endpoints (Seronegative)
eTable 7. Proportion of Participants Who Had a COVID-19-Related Hospitalization or ER Visit (Seronegative)
eTable 8. Hospitalization and Hospitalization Outcomes (Seronegative)
eTable 9. Proportion of Participants Who Had a Symptomatic RT-qPCR-Confirmed SARS-CoV-2 Infection (Broad-Term) by Period (Seronegative)
eTable 10. Proportion of Participants Who Had a Symptomatic RT-qPCR-Confirmed SARS-CoV-2 Infection (Broad-Term) in Those Who Have ≥1 Risk Factor for Severe COVID-19 (Seronegative)
eTable 11. Proportion of Participants With a Confirmed Negative SARS-CoV-2 RT-qPCR During the EAP (Seronegative)
eTable 12. Proportion of Participants Who Had a Symptomatic RT-qPCR-Confirmed SARS-CoV-2 Infection (Broad-Term) by Baseline Serology Status
eTable 13. Number of Weeks of Symptomatic SARS-CoV-2 Infection (Broad-Term) by Baseline Serology Status
eTable 14. Number of Weeks of High Viral Load (>4 Log10 Copies/mL) in NP Swab Samples by Baseline Serology Status
eTable 15. Maximum SARS-CoV-2 RT-qPCR Viral Load (Log10 Copies/mL) in NP Swab Samples by Baseline Serology Status
eTable 16. Number of Weeks of RT-qPCR Confirmed SARS-CoV-2 Infection (Regardless of Symptoms) by Baseline Serology Status
eTable 17. Overview of Treatment-Emergent Adverse Events
eTable 18. Treatment-Emergent Adverse Events Occurring in ≥2% of Participants
eTable 19. Serious Treatment-Emergent Adverse Events
eTable 20. Summary of PK Parameters for Casirivimab and Imdevimab After a Single 1200 mg Subcutaneous Dose in Part B Participants
Nonauthor Collaborators. COVID-19 Phase 3 Prevention Trial Team
Data Sharing Statement