

Abstract
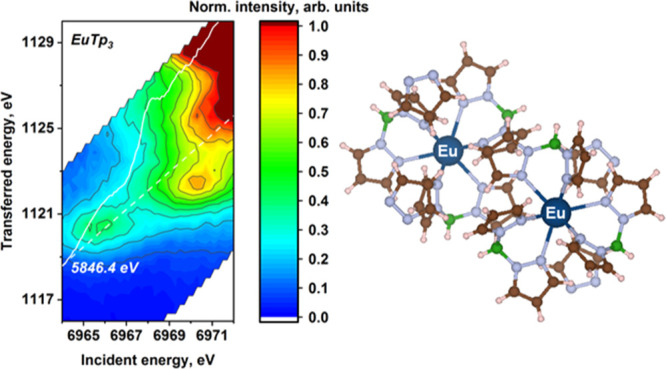
We performed a systematic study of the complexes of trivalent lanthanide cations with the hydridotris(1-pyrazolyl)borato (Tp) ligand (LnTp3; Ln = La, Ce, Pr, Nd, Sm, Eu, Tb, Dy, Ho, Er, Tm, Yb, and Lu) using both high-energy-resolution fluorescence-detected X-ray absorption near-edge structure (HERFD-XANES) and resonant inelastic X-ray scattering (RIXS) at the lanthanide L3 absorption edge. Here, we report the results obtained and we discuss them against calculations performed using density functional theory (DFT) and atomic multiplet theory. The spectral shape and the elemental trends observed in the experimental HERFD-XANES spectra are well reproduced by DFT calculations, while the pre-edge energy interval is better described by atomic multiplet theory. The RIXS data show a generally rather complex pattern that originates from the intra-atomic electron–electron interactions in the intermediate and final states, as demonstrated by the good agreement obtained with calculations using an atomic-only model of the absorber. Guided by theoretical predictions, we discuss the possible origins of the observed spectral features and the trends in energy splitting across the series. The insight into the electronic structure of trivalent lanthanide compounds demonstrated here and obtained with advanced X-ray spectroscopies coupled with theoretical calculations can be applied to any lanthanide-bearing compound and be of great interest for all research fields involving lanthanides.
Short abstract
This work represents a systematic investigation of the complexes of trivalent lanthanide cations with the hydridotris(1-pyrazolyl)borato (Tp) ligand (LnTp3; Ln = La, Ce, Pr, Nd, Sm, Eu, Tb, Dy, Ho, Er, Tm, Yb, and Lu) using both high-energy-resolution fluorescence-detected X-ray absorption near-edge structure and resonant inelastic X-ray scattering at the lanthanide L3 absorption edge. The experimental results are discussed against the calculations performed with the density functional theory and atomic multiplet theory approaches.
1. Introduction
The 15 chemical elements with atomic numbers from 57, that is, lanthanum (La), to 71, that is, lutetium (Lu), constitute the lanthanide series. These elements are of tremendous relevance in industrial, technological, and scientific domains. Lanthanides and their compounds are widely used in various fields, such as catalysis, metallurgy, ceramics and glass production, electronic, renewable energy, and biomedical devices.1−3 A distinctive feature of the lanthanide series is the progressive filling of the 4f electronic shell, which confers many remarkable physical and chemical properties to lanthanide compounds.1 The presence of a partially filled 4f shell, which is weakly involved in chemical bonding but determines many of the physicochemical properties of lanthanide compounds, stimulates considerable scientific interest in the fundamental understanding of its electronic structure. In these respects, systematic studies comparing isostructural complexes having different lanthanides as a metal center are particularly useful to correlate specific trends in the electronic structure to chemical properties.
X-ray spectroscopies are powerful element-selective methods to investigate the electronic structure of matter.1,4,5 Different techniques belonging to this family are widely applied to investigate lanthanide-containing materials, for example, X-ray absorption near-edge structure (XANES), resonant inelastic X-ray scattering (RIXS), X-ray emission spectroscopy (XES), X-ray photoelectron spectroscopy (XPS), and X-ray magnetic circular dichroism (XMCD).1,4−21 Nowadays, the possibility of performing XANES in the high-energy-resolution fluorescence-detected (HERFD) mode is of particular interest for the lanthanide series. The conventional L3-edge XANES of lanthanides is broadened by the large 2p core-hole lifetime. The HERFD mode of detection allows a great improvement of energy resolution and sensitivity.5,8,22 In this mode, the X-ray absorption spectrum is measured by selecting only a small bandwidth around the maximum of a characteristic fluorescence line and the resulting spectral broadening is a combination of the core-hole lifetimes of the intermediate and final states. For the lanthanide series, the gain in resolution at the L3 edge is remarkable. In particular, the pre-edge features originating from quadrupolar transitions to the 4f states, invisible in conventional XANES, are well resolved. As a result, HERFD-XANES on lanthanides can probe at once the 4f shell (at the pre-edge) and the 5d shell (at and beyond the main edge), where the first determines the oxidation state and the second is involved in bonding. However, one should be very careful when interpreting a HERFD-XANES spectrum because additional features may arise due to the presence of a shallower core hole in the final state.5 RIXS spectroscopy, which records the scattering cross section along both the incident and emitted energy axes, considerably helps in the correct interpretation of experimental spectra.1,4,5,8,11−17 The combined use of HERFD-XANES and RIXS was shown to be a rather powerful approach to investigate transition metal,23 actinide,24 and lanthanide8 compounds.
Systematic X-ray spectroscopy studies, where a set of isostructural samples hosting different metal centers are investigated, are particularly helpful to understand which information can be extracted from the spectra. As for lanthanides, these studies are very rare. One first challenge is represented by the synthesis of the series of isostructural samples. If, on the one hand, the whole lanthanide series share a common stable oxidation state, that is, Ln3+,1 making it easy to fulfill valence requirements, the ionic radius markedly decreases when going from La to Lu, making it hard to maintain the same structure throughout the series. To the best of our knowledge, there are only a few reports on the synthesis of lanthanide-containing isostructural compounds at present.25−31 In almost all cases, the isostructural complexes are formed with multidentate ligands that demonstrate enough flexibility to accommodate ions of different sizes while encapsulating them within a fixed coordination environment.30 Hydridotris(1-pyrazolyl)borato (Tp) is a widely applied tridentate ligand with chemical formula of [HB(C3N2H3)3]−, which is known to form stable complexes with lanthanides.32,33 Using this ligand, we succeeded in overcoming the highlighted chemical challenge and synthesized the stable Tp complexes with a series of trivalent lanthanide compounds (Ln3+) that are isostructural over almost the entire series.
In this work, we present experimental L3-edge HERFD-XANES and 2p3d RIXS on the series of LnTp3 complexes (Ln = La, Ce, Pr, Nd, Sm, Eu, Tb, Dy, Ho, Er, Tm, Yb, and Lu) and analyze the experimental results with theoretical calculations based on the density functional theory (DFT) and the atomic multiplet theory for XANES and RIXS, respectively. The isostructural LnTp3 (Ln = La, Pr, Nd, Sm, and Eu) complexes were chosen as the model series for DFT calculations of the lanthanide XANES spectra. The whole lanthanide series was computed using the atomic multiplet theory because, as explained later, the pre-edge transitions measured by RIXS spectroscopy are mainly affected by atomic electronic interactions and the structure of the complex has limited effect. We show the sensitivity of specific postedge features to the local environment, bond distances and cation type by the analysis of edge and postedge XANES regions. On the other hand, we demonstrate that atomic multiplet calculations nicely reproduce the complexity observed in RIXS measurements of the pre-edge region. We additionally discuss RIXS and pre-edge HERFD-XANES, which are simple RIXS cuts, to highlight the importance of collecting the full RIXS plane for a correct interpretation of the observed features.
2. Experimental and Theoretical Details
2.1. Sample Synthesis
The complexes have been synthesized by the reaction of the lanthanide trichlorides with K[HB(N2C3H3)3] (KTp), following the procedure described in the literature.34 The LnTp3 compounds from La to Tb are nine-coordinated, while the compounds of the heavier ions (Dy to Lu) are eight-coordinated. Detailed structural and spectroscopic investigations have been carried out on LnTp3 (Ln = La, Ce, Pr, Nd, Sm, Eu, Tb, and Yb).35−42
2.2. Experimental Techniques
The experiment was performed at the ID26 beamline of the European Synchrotron Radiation Facility (ESRF) in Grenoble43 using LnTp3-pressed powder pellets. The incident energy was selected using the ⟨111⟩ reflection from a double Si crystal monochromator. Rejection of higher harmonics was achieved by two Si mirrors working under total reflection at an angle of 2.5 mrad. The emission energy for measuring XANES in the HERFD mode and 2p3d RIXS spectra was selected using spherically bent crystal analyzers (bending radius R = 1 m) aligned at the Bragg angle, as detailed in Table 1.44 The Ln HERFD spectra at the L3 edge were obtained by recording the intensity of the Ln Lα1 emission line as a function of the incident energy.
Table 1. Experimental Parameters and Crystal Analyzer Reflections Set to Perform HERFD-XANES and RIXS Measurements of the LnTp3 Compounds.
| lanthanide (Ln) | L3 edge (eV) | Lα1 line (eV) | crystal analyzer | Bragg angle (deg) | estimated energy resolution (eV) |
|---|---|---|---|---|---|
| La | 5483 | 4647.0 | Si(440) | 79.3 | 1.2 |
| Ce | 5723 | 4839.2 | Ge(331) | 80.7 | 1.3 |
| Pr | 5964 | 5035.2 | Si(331) | 81.2 | 1.3 |
| Nd | 6208 | 5227.6 | Si(331) | 72.2 | 1.3 |
| Sm | 6716 | 5632.6 | Si(422) | 83.2 | 1.4 |
| Eu | 6977 | 5849.5 | Ge(333) | 76.7 | 1.4 |
| Gd | 7243 | 6053.4 | Si(333) | 78.5 | 1.4 |
| Tb | 7514 | 6272.9 | Ge(440) | 81.2 | 1.4 |
| Dy | 7790 | 6498.0 | Si(440) | 83.5 | 1.5 |
| Ho | 8071 | 6720.0 | Si(440) | 74.0 | 1.5 |
| Er | 8358 | 6949.0 | Ge(620) | 85.7 | 1.5 |
| Tm | 8648 | 7180.0 | Ge(620) | 74.8 | 1.6 |
| Yb | 8944 | 7416.0 | Si(620) | 76.7 | 1.6 |
| Lu | 9244 | 7416.0 | Ge(444) | 82.6 | 1.6 |
2.3. Theoretical Calculations
XANES spectroscopy at the L3 edge of lanthanides probes both the 5d and 4f orbitals. While 5d orbitals are involved in chemical bonding and therefore strongly affected by the local environment, 4f orbitals are highly localized and are only weakly affected by the surrounding atoms. Consequently, the intra-atomic electron–electron interactions will be dominating for transitions to the 4f orbitals, while they can be approximated for transitions to the 5d orbitals.5 Thus, we used two theoretical methods to analyze the LnTp3 spectra: first-principles calculations based on DFT to reproduce transitions to the 5d orbitals, that is, edge and postedge regions of L3 edge spectra, and atomic multiplet theory to calculate the transitions to the more localized 4f orbitals, probed in the pre-edge region of L3-edge spectra with 2p3d RIXS.
The XANES spectra of Ln3+ L3-edge (Ln = La, Pr, Nd, Sm, and Eu) were calculated with the FDMNES code that is based on DFT with local density approximation.45 The calculations are relativistic following Wood and Boring46 with a full treatment of spin–orbit interaction, and the finite difference method is used to solve the XANES equation. Both dipolar and quadrupolar transitions were included, as well as scalar relativistic and spin–orbit effects. The atomic potential around the absorber was calculated self-consistently within a radius of 6 Å, and the absorber was set to be excited. The spectra were calculated in the energy range from −15 to +65 eV relative to the Fermi level. The atomic coordinates of LnTp3 molecules were obtained by X-ray diffraction measurements on single crystals but were available only for a subset of lanthanides (La, Pr, Nd, Sm, and Eu); therefore, only a subset of experimental data could be calculated with this approach that requires the coordinates of a cluster of atoms around the absorber as input. The computed XANES spectra were convoluted after calculation.45
 |
Here, Γf(ω) is the energy-dependent full width at half-maximum (fwhm) of the Lorentzian function which accounts for the core-hole broadening (Γhole) and to the spectral width γ(ω) of the final state. The convolution was limited to the unoccupied states which lie above the Fermi level (EF).
The core-hole broadening (Γhole), which for lanthanids has values between 3.41 and 4.68 eV, was reduced to reproduce the high resolution of HERFD-XANES spectra. The energy-dependent part [γ(ω)] was calculated using an empirical arctangent model.45
The best agreement between theoretical spectra and experimental data was found for Γhole, Γmax (maximum width of the final state), Ectr – EFermi (the center of the arctangent function), and El (width of the arctangent function) set to 1.2, 15, 30, and 30 eV, respectively. A subsequent convolution was done using a Gaussian function with a 1 eV fwhm. The smaller value compared to the experiment was used to better distinguish features from the edge region.
The 2p3d RIXS planes corresponding to the 2p → 4f absorption and 3d → 2p emission transitions were calculated with the multiplet theory as implemented in the Quanty library47−50 using the input files generated by the Crispy graphical user interface.51 We have used an atomic representation to describe each system, that is, we did not include the effect of the coordinating atoms. The multiplet calculations are semiempirical, and, therefore, parameters are required for the terms included in the Hamiltonian. We have used the suite of codes developed by Robert Cowan52 to calculate the parameters required for the present Hamiltonian, which included the electron–electron term and spin–orbit coupling term (calculations details for the parameters can be found in the Supporting Information). The calculated values for the Slater integrals, which parameterize the electron–electron interaction, and the spin–orbit coupling integrals are tabulated in Table 2. In the spectroscopy calculations, the former values have been scaled using a 0.8 factor to account for the intra-atomic configuration interaction neglected in the Hartree–Fock approximation (see pages 464–465 of ref (52) for a detailed discussion). It is common practice to further tune these scaled values to increase the agreement with the experimental spectra. However, as detailed later in the text, this was not required in the present study. The spectra are calculated using Green’s function approach, as detailed in the Quanty publication,47 and are shifted in energy to match the experimental data. The Lorentzian broadening parameters for the incident energy and energy transfer directions, corresponding to the lifetime broadenings of the intermediate and final-state core holes involved in the RIXS process, are applied as parameters in the Green’s function approach calculations (Γ in eq 3, ref (47)). The two-lifetime broadenings are set to the previously published values (see Table S1)53,54 which were reasoned by a generally satisfactory agreement with the fwhm values derived from the fitting of the experimental RIXS data cuts. To better distinguish the features in the theoretical RIXS data, we decided not to perform additional convolution of the natural spectra by a Gaussian function which is commonly applied to account for the experimental broadening. The energy step was set to be 0.3 eV in the incident energy direction and 0.03 eV in the energy-transfer one.
Table 2. Slater Integrals and Spin–Orbit Coupling Constants for the Lanthanide Trivalent Cations (Ln3+)a.
| Ln | configuration | F2 (4f, 4f) | F4 (4f, 4f) | F6 (4f, 4f) | F2 (2p, 4f) | G2 (4f, 4f) | G4 (2p, 4f) | F2 (3d, 4f) | F4 (3d, 4f) | G1 (3d, 4f) | G3 (3d, 4f) | G5 (3d, 4f) | ξ (4f) | ξ (2p) | ξ (3d) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| La | 4f0 | 0.000 | 0.000 | 0.000 | 0.000 | ||||||||||
| 2p54f1 | 0.000 | 0.000 | 0.000 | 1.305 | 0.116 | 0.075 | 0.091 | 281.477 | |||||||
| 3d94f1 | 0.000 | 0.000 | 0.000 | 7.063 | 3.167 | 4.723 | 2.761 | 1.905 | 0.092 | 6.799 | |||||
| Ce | 4f1 | 0.000 | 0.000 | 0.000 | 0.087 | ||||||||||
| 2p54f2 | 12.436 | 7.808 | 5.619 | 1.408 | 0.130 | 0.083 | 0.106 | 304.694 | |||||||
| 3d94f2 | 12.628 | 7.940 | 5.717 | 7.486 | 3.384 | 5.073 | 2.968 | 2.048 | 0.107 | 7.446 | |||||
| Pr | 4f2 | 12.227 | 7.670 | 5.518 | 0.103 | ||||||||||
| 2p54f3 | 12.920 | 8.114 | 5.839 | 1.508 | 0.143 | 0.092 | 0.123 | 329.429 | |||||||
| 3d94f3 | 13.096 | 8.235 | 5.929 | 7.889 | 3.591 | 5.410 | 3.166 | 2.186 | 0.123 | 8.139 | |||||
| Nd | 4f3 | 12.725 | 7.985 | 5.744 | 0.119 | ||||||||||
| 2p54f4 | 13.381 | 8.404 | 6.048 | 1.604 | 0.156 | 0.100 | 0.140 | 355.758 | |||||||
| 3d94f4 | 13.543 | 8.516 | 6.132 | 8.275 | 3.790 | 5.734 | 3.357 | 2.318 | 0.141 | 8.879 | |||||
| Pm | 4f4 | 13.197 | 8.282 | 5.959 | 0.136 | ||||||||||
| 2p54f5 | 13.823 | 8.682 | 6.248 | 1.698 | 0.169 | 0.109 | 0.159 | 383.763 | |||||||
| 3d94f5 | 13.975 | 8.787 | 6.326 | 8.650 | 3.983 | 6.049 | 3.543 | 2.447 | 0.160 | 9.668 | |||||
| Sm | 4f5 | 13.648 | 8.566 | 6.163 | 0.155 | ||||||||||
| 2p54f6 | 14.251 | 8.950 | 6.441 | 1.790 | 0.182 | 0.117 | 0.180 | 413.526 | |||||||
| 3d94f6 | 14.393 | 9.049 | 6.515 | 9.014 | 4.171 | 6.357 | 3.724 | 2.572 | 0.180 | 10.510 | |||||
| Eu | 4f6 | 14.083 | 8.839 | 6.360 | 0.175 | ||||||||||
| 2p54f7 | 14.665 | 9.209 | 6.628 | 1.881 | 0.195 | 0.126 | 0.202 | 445.137 | |||||||
| 3d94f7 | 14.800 | 9.303 | 6.698 | 9.368 | 4.354 | 6.657 | 3.901 | 2.695 | 0.202 | 11.405 | |||||
| Gd | 4f7 | 14.505 | 9.103 | 6.550 | 0.197 | ||||||||||
| 2p54f8 | 15.070 | 9.462 | 6.809 | 1.970 | 0.208 | 0.134 | 0.226 | 478.688 | |||||||
| 3d94f8 | 15.197 | 9.551 | 6.876 | 9.715 | 4.533 | 6.951 | 4.075 | 2.815 | 0.225 | 12.357 | |||||
| Tb | 4f8 | 14.915 | 9.360 | 6.734 | 0.221 | ||||||||||
| 2p54f9 | 15.465 | 9.709 | 6.987 | 2.057 | 0.221 | 0.142 | 0.251 | 514.272 | |||||||
| 3d94f9 | 15.586 | 9.794 | 7.050 | 10.055 | 4.709 | 7.240 | 4.245 | 2.933 | 0.251 | 13.368 | |||||
| Dy | 4f9 | 15.315 | 9.610 | 6.914 | 0.246 | ||||||||||
| 2p54f10 | 15.852 | 9.951 | 7.160 | 2.143 | 0.234 | 0.151 | 0.279 | 551.991 | |||||||
| 3d94f10 | 15.968 | 10.032 | 7.221 | 10.389 | 4.882 | 7.524 | 4.412 | 3.049 | 0.278 | 14.439 | |||||
| Ho | 4f10 | 15.707 | 9.854 | 7.089 | 0.273 | ||||||||||
| 2p54f11 | 16.233 | 10.188 | 7.330 | 2.228 | 0.247 | 0.159 | 0.308 | 591.947 | |||||||
| 3d94f11 | 16.343 | 10.265 | 7.389 | 10.718 | 5.052 | 7.803 | 4.577 | 3.163 | 0.307 | 15.575 | |||||
| Et | 4f11 | 16.091 | 10.094 | 7.261 | 0.302 | ||||||||||
| 2p54f12 | 16.607 | 10.421 | 7.497 | 2.312 | 0.260 | 0.167 | 0.339 | 634.250 | |||||||
| 3d94f12 | 16.713 | 10.496 | 7.554 | 11.042 | 5.219 | 8.080 | 4.740 | 3.276 | 0.337 | 16.777 | |||||
| Tm | 4f12 | 16.469 | 10.330 | 7.431 | 0.333 | ||||||||||
| 2p54f13 | 16.975 | 10.650 | 7.662 | 2.395 | 0.273 | 0.176 | 0.372 | 679.013 | |||||||
| 3d94f13 | 17.077 | 10.722 | 7.716 | 11.362 | 5.385 | 8.352 | 4.901 | 3.387 | 0.371 | 18.048 | |||||
| Yb | 4f13 | 16.841 | 10.561 | 7.597 | 0.366 | ||||||||||
| 2p54f14 | 17.339 | 10.876 | 7.824 | 2.477 | 0.285 | 0.184 | 0.408 | 726.353 | |||||||
| 3d94f14 | 17.437 | 10.946 | 7.877 | 11.677 | 5.548 | 8.622 | 5.060 | 3.497 | 0.406 | 19.391 |
All values are in electronvolts. In the spectroscopy calculations, the Slater integrals were scaled by 0.8 to correct for the Hartree–Fock approximations (see the text for details). Only the unfilled orbitals are shown in the configuration column.
3. Results and Discussion
3.1. HERFD-XANES Studies of the LnTp3 Systems
The experimental L3-edge HERFD-XANES of the LnTp3 compounds (Ln = La, Ce, Pr, Nd, Sm, Eu, Tb, Dy, Ho, Er, Tm, Yb, and Lu) are provided in Figure 1. To compare the results on a common energy scale, the maximum of the absorption edge has been set to zero. The spectra present four main spectral features: the main absorption peak (so-called white line), some pre-edge structures, and two postedge peaks. These spectral features are marked in Figure 1 as W, A, B, and C, respectively. The white line (W) and the postedge features originate from 2p65d0 → 2p55d1 dipolar transitions.55 The pre-edge features (A) originate from quadrupolar transitions to the Ln3+ 4f orbitals, 2p64fN → 2p54fN+1. The reason why quadrupole transitions appear at lower energies than dipole ones is that the intermediate state with an extra 4f electron (2p54fN+1) is more strongly bound than the intermediate state with an extra 5d electron (2p54fN5d1).10,11 Looking at the relative energy positions of spectral features over the lanthanide series, it can be noticed that the energy separation between the pre-edge (A) and the white line and between the white line and the second postedge peak (C) increases, while the energy separation of the first postedge peak (B) and the white line decreases (see Figure 1). Possible origins of the observed trends will be discussed in the following section.
Figure 1.
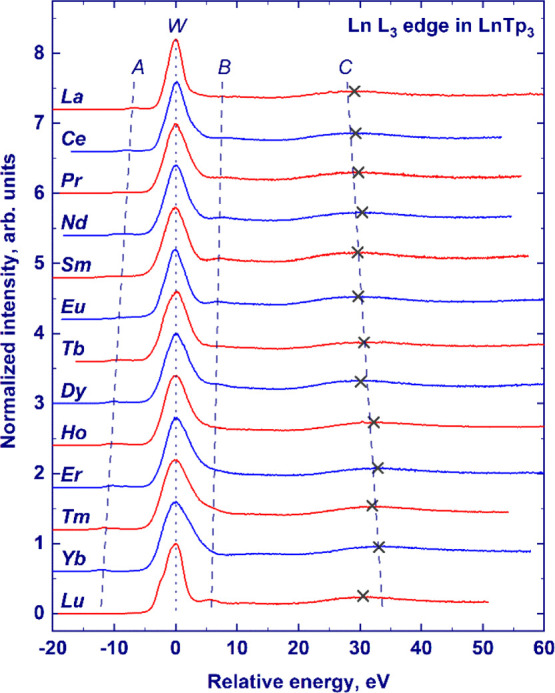
Experimentally measured XANES spectra of the Ln3+ L3-edge absorption in the LnTp3 compounds (Ln = La, Ce, Pr, Nd, Sm, Eu, Tb, Dy, Ho, Er, Tm, Yb, and Lu). For the sake of clarity, the spectra have been scaled to the same maximum height and offset along the Y-axis. The curves are shown in a unified energy scale, with zero corresponding to the position of the white line (W). Other spectral features (pre-edge structure and two postedge peaks) are marked as A, B, and C, respectively. Maximal positions of feature C are indicated with crosses. Dashed and dotted lines are guides to the eye.
Figure 2 shows the comparison of experimental HERFD-XANES spectra and finite-difference calculations on the selected samples (LnTp3 with Ln = La, Pr, Nd, Sm, and Eu) for which experimental crystal structures were available. Theoretical spectra are in very good agreement with experimental data and the shift of the postedge features B and C is reproduced, as indicated by the vertical lines interpolating the maximum of features B and C along the series. A closer look at the white line region reveals that the calculated spectra for all examined elements present a fine structure of the white line (W), specifically a shoulder on the left side of the main peak that is not distinguished in the experimental data. The effect can be observed in Figure 2 and in Figure 3, which reports the detail of the edge region for the calculated spectra and the density of states (DOS) of Ln and Eu cases (Figures S1 and S2 shows La, Pr, Nd, Sm, and Eu cases). Inspection of the projected DOS reveals that the fine structure arises from the crystal field splitting acting on the 5d-DOS. The local environment of the lanthanide atom consists of nine nitrogen atoms in the tricapped trigonal prism geometry (D3h symmetry, see Figure 2). In this local symmetry, d orbitals split into three groups as follows: dz2 (a1′), dx2–y2 and dxy (e′), and dxz and dyz (e‴) (the d orbitals are provided in the order of increasing energy as resulting from the finite-difference calculations). The discrepancy may originate from an overestimation of the attractive force of the core-hole potential on d orbitals of different symmetries. At the same time, the limited experimental resolution makes it difficult to establish if the experimental spectral features are split, especially by an amount smaller than that provided by calculations. Increasing the Gaussian convolution of theoretical spectra results in the vanishing of the fine structure of the white line, but for Eu, the overall white line width after additional convolution exceeds the experimental one (as shown in Figure S3, EuTp3 exhibits the highest crystal field splitting of the white line across the series). Therefore, we conclude that the crystal field splitting is overestimated by calculations, and it is not distinguished in experimental data due to limited energy resolution.
Figure 2.

XANES spectra of the Ln3+ L3-edge absorption in the LnTp3 compounds (Ln = La, Pr, Nd, Sm, and Eu): experimentally measured (a) and calculated with the finite-difference calculations (b). For the sake of clarity, the spectra have been scaled to the same maximum height and offset along the Y-axis. The curves are shown in a unified energy scale, with zero corresponding to the position of the white line (W). Other spectral features (pre-edge structure and two postedge peaks) are marked in the left panel as A, B, and C, respectively. Dashed and dotted lines are guides to the eye. The fragment of the NdTp3 structure (Nd and the first coordination shell of D3h symmetry) is provided in the right panel. Bond lengths are in angstroms.
Figure 3.

Finite-difference-computed ligand DOS (Ln3+ cation d- and f-orbitals and N atom s- and p-orbitals) and normalized XANES spectra (left panel); dz2, dx2–y2 and dxy, dxz, and dyz orbitals of the lanthanide atom (right panel) in the LaTp3 and EuTp3 compounds. The experimental spectra are provided with the dashed–dotted lines. The spectral intensities have been scaled to the same maximum height, while the ligand DOSs are presented in arbitrary units (note that the ligand DOSs are scaled by the factors indicated in the legend). The theoretical curves are shown in a unified energy scale, with zero corresponding to the position of the white line. The Fermi-level positions are marked with vertical dashed lines. The experimental spectra are aligned along the X-axis to match the theoretical data.
Another difference between experiments and calculations is the presence of low-intensity spectral features at ca. 2 eV above the Fermi level in the theoretical spectra (see Figures 3 and S2) which are not visible in the experiment. The projected DOS shown in Figure 3 demonstrates that these features stem from transitions to relatively low-lying states of d symmetry, hybridized with p orbitals of ligand atoms (Figures 3 and S2 show, as an example, the calculations for nitrogen atoms), as indicated by the superposition of the outlined DOS. The presence of the low-intensity features in the theoretical spectra may again be related to an overestimation of the attracting effect of the core-hole potential, which pulls down the projected DOS (the effect of a core hole on the calculated spectra of LaTp3 and projected DOS is shown in Figure S4).
The first postedge peak observed in the experiment, feature B, is reproduced by calculations and originates from d states as it could be concluded from the inspection of the projected DOS of the lanthanide atom (see Figures 3 and S2). According to the finite-difference calculations, a noticeable contribution of the ligand p orbitals occurs at the position of feature B, suggesting possible hybridization of the ligand p and metal d orbitals of the absorbing atom in correspondence with this feature. However, finite-difference-calculated XANES of the LaTp3 molecule with different cluster radii shows that feature B appears only when including atoms beyond 3 Å, that is, second nearest neighbors. Furthermore, the energy separation between feature B and the white line increases with the enlargement of the cluster radius (see Figure S5). This result implies that distant coordination shells contribute to the intensity and the relative position of feature B, which therefore cannot be ascribed to hybridization with only nearest neighbors. It can be concluded that this feature originates from the promotion of a photoelectron to the continuum states of d symmetry (as suggested by Bartolomé et al.11). Furthermore, it is worth noticing that feature B, observed in the L3-edge XANES spectra of several Ln2O3 substances, was also interpreted in terms of continuum resonance caused by more distant coordination shells,7 which supports the present assignment.
As could be seen in the experimental spectra in Figure 1, the energy gap between the white line and feature B gradually decreases from La to Lu. The decrease of the separation presumably results from the increased screening of the core hole by the f electrons, leading to a weaker attraction of the 5d states and, therefore, to a higher energy position of the white line. Because the d states originating feature B are in the continuum, they are less affected by the core hole and the position of feature B remains unaffected by the additional screening. The experimentally and finite-difference-calculated energy gaps between feature B and the white line for La, Pr, Nd, Sm, and Eu are provided in Figure 4. We may notice that the relative positions of the maximum of feature B, as calculated by the FDMNES code, are in good agreement with the measured ones within the experimental error. To investigate the dependence of feature B on bond distances and the lanthanide species, we performed finite-difference calculations in which La was placed in different Tp3 structures (see Figure S5). Interestingly, the relative position of feature B is approximately constant in these calculations, in contrast to feature C that follows the same trend shown in the experimental spectra despite the change of the lanthanide. This result indicates that weak structural variations have little impact on feature B, which appears to be more affected by the lanthanide species and confirm the fact that screening of the core hole by f electrons might be the reason for the observed changes.
Figure 4.
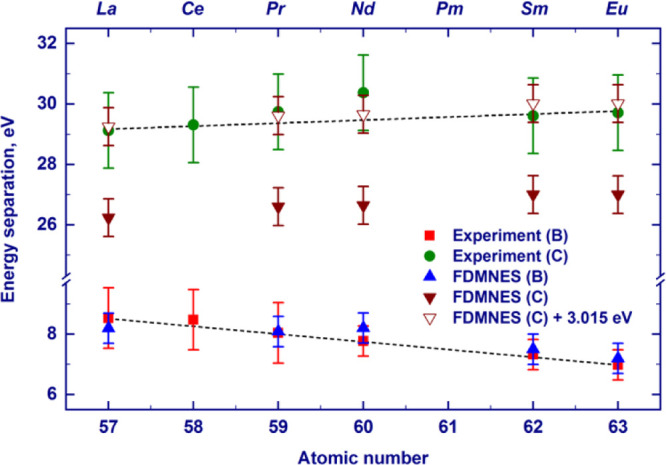
Dependence of the energy separation between the white-line position and the maximum of the feature B (feature C) on the atomic number of the absorbing lanthanide atom obtained by the analysis of the experimental L3-absorption-edge XANES spectra of the LnTp3 compounds (B, squares; C, circles) and the corresponding spectra calculated by the FDMNES code (B, apex-up triangles; C, solid apex-down triangles). To fit the experimental data for feature C, the offset of 3.015 eV was added to the finite-difference calculated values (empty apex-down triangles). The offset value was obtained by the least-square fitting procedure. Dashed lines are guides to the eye.
As shown in Figure 2, finite-difference calculations also reproduce the second postedge peak observed experimentally (labeled C in Figure 1). The experimental energy separation between the maximum of feature C and the white line increases from La to Eu (Figure 1). Theoretical calculations well reproduce this elemental trend. However, an offset of ca. 3 eV to the theoretical values is needed to achieve agreement with the absolute position (see Figure 4). A similar disagreement has already been observed at L3 edges of heavy elements and may be due to the underestimation of the attracting effect of the core-hole potential on the white line or to other factors affecting the position of the spectral features involved. The shift to higher energies of feature C when going from La to Eu recalls the behavior of a postedge feature corresponding to a bond that shortens across the series. Indeed, analyzing the structure of LnTp3 (Ln = La, Pr, Nd, Sm, and Eu) used for calculations, one can see that the R(Ln–N) distance decreases from La to Eu (Eu 2.53 Å < Sm 2.54 Å < Nd 2.60 Å < Pr 2.61 Å < La 2.65 Å). A similar decrease of nearest-neighbor distances across the series, previously observed for other isostructural complexes of lanthanides, was ascribed to lanthanide contraction.25−31 Further confirmation of the structural nature of feature C comes from the finite-difference calculations where lanthanum was placed inside the Tp3 structures of Nd and Eu (see Figure S5). The relative position of feature C increases with the decreasing R(Ln–N) distance and is not significantly affected by the nature of the lanthanide.
It is worth noting that feature C appears already for clusters including only the nearest nitrogen atoms, indicating that multiple scattering from the closest nitrogen atoms is the main contributor to this feature (see Figure S5). Calculations with smaller convolution parameters show that feature C consists of two peaks that are unresolved experimentally, and this probably reflects the splitting of the first-shell bond lengths. The first shell of the lanthanide atom consists of six nitrogen atoms at the 2.5–2.7 Å distance from Ln and three more nitrogen atoms at 2.7–2.8 Å (Figure 2 shows the example of the Nd first shell).
The pre-edge region marked A in Figure 1 presents features corresponding to quadrupole transitions to the 4f orbitals. From the experimental HERFD-XANES spectra in Figure 1, it can be noticed that the energy separation between the white line and the pre-edge structures A increases while progressing along the lanthanide series, in reasonable agreement with the literature data10 (additionally, see Figure S6). This trend can be understood by considering the change in the energy of the involved transitions when the number of 4f electrons increases. The first effect is the increased screening of the 2p core-hole potential which weakens the attraction of the 5d orbitals. A second effect that goes in the same direction is the repulsion between 4f and the excited 5d electron, which becomes stronger and leads to the increase of the 2p → 5d transition energy. In contrast, the stronger repulsion of 4f electrons along the series increases the 2p → 4f transition energy. The tendency observed in Figure 1 indicates that the sum of the first two effects prevails over the latter. The absolute values of energy separation of the white line and the pre-edge A obtained in this work are a few electronvolts larger than those observed by Bartolomé et al. for the Ln2Fe14B intermetallic series.10 The difference can be easily understood as an effect of the chemical environment, which significantly affects the energy of the 2p → 5d excitation. As an example, the maximum of the L3 edge of ionic TbF3 lies at higher incident energy (+3.3 eV) than that of the intermetallic TbCo2.12 In intermetallic TbCo2, Tb 5d states are found in correspondence to the Fermi level due to the strong overlap with Co 3d8 states. In the more ionic TbF3, 5d orbitals lie higher in energy because of the insulating gap. In contrast, the position of the 2p → 4f excitation remains unchanged because of the localized nature of 4f orbitals, less sensitive to the chemical environment. As a result, the separation between the white line and the pre-edge is larger for more insulating systems, such as LnTp3, compared to the intermetallic compounds of refs (10) and (11).
A closer look at the pre-edge region along the series shows a rich structure (Figure S6), arising from the complex intra-atomic interactions among the 4f electrons and the core holes involved in the process. Former RIXS studies on lanthanide compounds interpreted the pre-edge structures as made by two groups of features whose origin may be simplistically explained by the following scheme:10,11 a low-energy feature (features) originating from the promotion of a core electron in the partially filled “spin-up” 4f subshell and a high-energy feature (features) originating from the promotion of a core electron in the full empty “spin-down” 4f subshell. Owing to the first Hund rule, the former configuration is less energetic than the latter one. For La3+, with no electrons in the 4f shell, a single transition to 4f states is observed. From Figure S6, showing the HERFD-XANES pre-edge of the entire series, it is clear that such a distinction is hard to make because even for lanthanides with a filled “spin-up” 4f subshell (Tb–Yb), the pre-edge region has a complex structure with multiplet peaks. As mentioned in the Introduction, the complex interelectronic interactions in the intermediate and final states of the process may induce additional features in the HERFD-XANES spectra. More precisely, the intra-4f subshell interaction and that between 4f electrons and the core holes (2p in the intermediate and 3d in the final state) are both relevant. These interactions cause sizable energy splitting of the multiplet states and have received special attention in the attempts to provide a theoretical interpretation of X-ray spectroscopies results.56−58 As a consequence, HERFD-XANES spectra are not sufficient to properly visualize the pre-edge region in 4f systems and multiple scans varying both the incident and the emitted energy are required. Previously published systematic examinations of lanthanide compounds by RIXS10,11,14 provide stacks of emission energy spectra recorded at fixed incident energy. This way of visualizing RIXS data makes it complex to follow the dispersion of peaks along the incident and transferred energy axes. Organizing the same scans of the pre-edge region as a 2D map, referred to as the RIXS plane, allows one to readily visualize all electronic transitions constituting the pre-edge and helps to extract the information on the observed trends.23 We recorded the RIXS planes for the pre-edge region of all investigated LnTp3 and calculated them with atomic multiplet theory to verify that the rich structures observed are due to intra-atomic interactions. In this regard, the FDMNES code is not appropriate since it is based on the single-electron approximation and neglects intrashell electron correlations, which are the main contributors to the pre-edge region. In the following section, we discuss the experimental results against atomic multiplet theory calculations.
3.2. RIXS Planes of the LnTp3 Series
The experimental 2p3d RIXS planes for the investigated lanthanide compounds are provided in Figure 5 together with the results of the calculations (experimental data on Gd and Lu are provided in Figure S7). As expected, the pre-edge structure starts quite simple, increases in complexity up to Eu3+, and then gradually regains a relative simplicity going toward Yb3+, which presents a single and unstructured peak. Lu3+ has a filled 4f shell and has no pre-edge (see Figures S7 and S8), which further confirms the assignment of pre-edge features to 2p → 4f transitions. The increasing complexity of the pre-edge structures obviously correlates with the increasing number of 4f electrons. A closer inspection shows that starting from Ce, two groups of structures can be identified, the first at a lower incident and emitted energy and the second closer to the white line. These groups become more structured and separated along the series up to Eu, where almost three groups may be distinguished. The energy splitting up to ca. 5 and 6 eV in the incident and transferred energy directions, respectively, demonstrates the magnitude of interelectronic interactions in the intermediate and final states of the RIXS process. A decrease in the number of features is then observed from the Gd3+ ion, most probably because we are left with only “spin-down” available transitions. However, also “spin-down” 4f transitions in the heavy lanthanide trivalent cations result in a rich structure of peaks that is generally less spread in the incident energy direction compared to the first half of the series.
Figure 5.


Atomic multiplet-calculated (first and the third columns) and experimentally measured (second and fourth columns) 2p3d RIXS planes for the LnTp3 (Ln = La, Ce, Pr, Nd, Sm, Eu, Tb, Dy, Er, Ho, Tm, and Yb). Positions of the HERFD cut are marked with dashed lines. The emission energy used to record the HERFD cut is given in the top-left corner.
All the described trends are very well reproduced by the calculated RIXS planes, as shown in Figure 5 (theoretical results on Pm and Gd are provided in Figure S7). The calculated results have been shifted in energy to have the best correspondence with the experimental data. The scales for the incident and emitted energy are the same. We recall that the calculations were performed considering only atomic interactions, that is, neglecting the effect of ligands that could be included as crystal field and charge-transfer effects. The good agreement confirms that they play the main role in shaping the pre-edge region of 4f systems. It has been previously shown8 that the energy separation of the RIXS features in the incident energy direction is influenced by 4f–4f interactions. For La3+, Ce4+, and Yb3+ systems, where the 4f ground- or intermediate-state configuration is either full or empty, no splitting in the horizontal direction has been detected. Concerning the vertical direction, the energy separation of the RIXS features is mainly influenced by the 4f–3d interactions. These interactions are present along the whole series and explain the richness of structures found also in the second half of the series.
Some discrepancies between atomic calculations and experimental data may be observed for Er3+, where calculations predict a much higher intensity of the feature at higher energy transfer. Additionally, we notice that the experimental pre-edge of Yb3+ is more asymmetric than the calculated one. Overall, the main trends are very well reproduced and minor discrepancies may be because the Slater integrals and the spin–orbit coupling parameter have been kept to the uniformly scaled (80 per cent) values calculated using Hartree–Fock. Another explanation for the minor discrepancies is the absence in our calculations of weaker interactions such as the crystal field splitting. The experimental results suggest that further investigation in this direction may start from Yb3+, which is the simplest case showing sizable discrepancies with atomic calculations.
To compare our results with previous calculations by Nakazawa et al.,56 who calculated the pre-edges of Ln ions for conventional XANES, we integrated our calculated RIXS planes along the energy transfer axis, that is, the Y-axis. The resulting curves, shown in Figure 6, are the pre-edges of conventional XANES, which in real measurements are not visible due to the large core-hole lifetime broadening of the white line. The richness of structures observed in the RIXS plane of Figure 5 is reduced to two main features, whose energy separation is in excellent agreement with previous calculations, also shown in Figure 6. The good agreement of our calculated RIXS with both Nakasawa’s results and experimental RIXS confirms the goodness of atomic multiplet calculations in predicting 2p → 4f transitions. Additionally, the separation between these features gradually increases with the increasing atomic number, which was previously observed experimentally.10,11 The larger splitting is due to the increase of the exchange energy with the progressive addition of electrons with parallel spins. The double structure was interpreted to be due to “spin-up” and “spin-down” transitions. However, our 2D RIXS plane shows that several features, spread along the vertical axis, contribute to the same peak and a clear-cut separation between “spin-up” and “spin-down” transitions is hard to make.
Figure 6.

Pre-edge regions of the Ln3+ L3-edge absorptions (Ln = La, Ce, Pr, Nd, Pm, Sm, and Eu) were obtained by the integration of the calculated 2p3d RIXS planes along the Y-axis. The curves are shown in a unified energy scale, with zero corresponding to the position of the white line. For Pm, the energy of the pre-edge feature was set to fit the data for the other lanthanides. A1 and A2 labels represent low- and high-energy quadrupolar features, respectively (left panel). The energy separation between the A1 and A2 features across the series was derived by the integration of the calculated 2p3d RIXS plane along the energy-transfer axis. Theoretical data from ref (56) are shown by open circles. Dashed lines are guides to the eye.
The good agreement between the experimental and theoretical results can be further checked on HERFD cuts of the calculated RIXS planes, as shown in Figure S8. We did not subtract the white line intensity from the experimental data; therefore, the intensity of pre-edge features closer to the white line is different between data and the calculated cuts. Since we carefully aligned the calculations to the experimental data, we use the same emitted energy used in the experiments to cut the calculated RIXS planes. It is worth highlighting that the shape of the HERFD-XANES pre-edge depends strongly on where we cut the RIXS plane, and a demonstration is provided in Figure 7. Our overview on the pre-edge RIXS planes of the LnTp3 series nicely shows that the interpretation of the pre-edge into a “spin-up” and “spin-down” group of transitions is far too simple and that it is not straightforward to ascribe features to one group, especially if only a single cut of the RIXS plane is measured, such as in HERFD-XANES. At the same time, it is very encouraging to observe that the rich pre-edge structure is very well reproduced by the atomic multiplet calculations along the whole series. Our recommendation is therefore to always record the full RIXS planes of the pre-edge region when investigating 4f systems.
Figure 7.
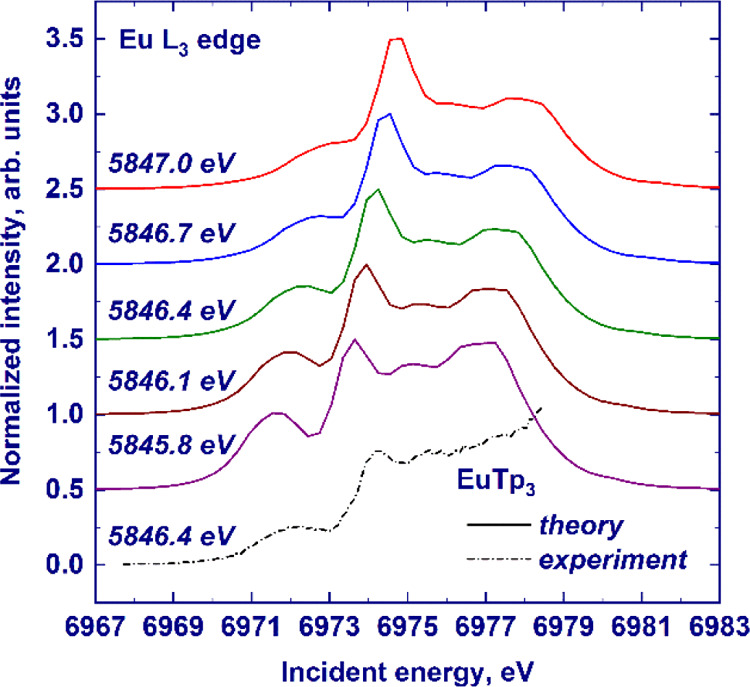
HERFD-XANES spectra (solid lines) of the Eu3+ L3 absorption edge in EuTp3 calculated in the pre-edge region by atomic multiplet theory. The experimental spectrum is shown by the dashed–dotted line. For the sake of clarity, the spectra have been scaled to the same maximum height and offset along the Y-axis. The emitted energy at which the HERFD cut was recorded is provided on the left-hand side.
4. Summary and Conclusions
In the present work, we systematically studied the LnTp3 complexes (Ln = La, Ce, Pr, Nd, Sm, Eu, Tb, Dy, Ho, Er, Tm, Yb, and Lu) via a combination of L3-edge HERFD-XANES and 2p3d RIXS spectroscopies. We described and discussed the trends observed along the series in detail. In particular, the energy separation between the main features constituting the spectra (pre-edge, white line, and postedge features) and the nature of postedge features with the help of finite-difference calculations. The postedge features showed a high sensitivity to the local structure around the absorber, while crystal field effects at the white line were not resolved with the present experimental resolution. The effects of the core-hole potential and the 4f screening on the observed trends were also discussed and found to be important. The XANES spectra calculated by the FDMNES code were found to reproduce the main absorption edge and two postedge features detected in the experiment, giving overall reasonable agreement. However, finite-difference calculations failed to correctly calculate the f-DOS and corresponding pre-edge features originating from the quadrupolar transition, which is better described in the framework of atomic multiplet theory.
We showed that the pre-edge region of the considered complexes is very rich and complex, and it is better visualized by 2p3d RIXS maps. Atomic multiplet calculations of the 2p3d RIXS maps show very good agreement with the experimental data and demonstrate that the complex pre-edge pattern arises from intra-atomic electron–electron interactions in the intermediate and final states of the RIXS process. The comparison with previous RIXS experimental and theoretical investigation is reported in detail, and our results are found to be in good agreement. Overall, RIXS maps greatly facilitate the visualization and interpretation of the pre-edge region. It confirms that the L3 pre-edge structure of Ln is due to the 2p–4f quadrupole transitions. We found excellent agreement between experimental data and theoretical calculations where we did not take into account the 5d electron configuration and the interaction between 5d-, core-, and valence-electron levels. This is an indication of the strong localized character of the 4f states in the investigated Ln compounds. In conclusion, our work constitutes the first systematic collection of experimental and calculated HERFD-XANES and 2p3d RIXS planes of all trivalent lanthanide cations (except Pm and Gd, reported in Figure S7) embedded in an isostructural complex, a valuable guide for further fundamental and applied research studies devoted to the 4f elements.
Acknowledgments
The authors acknowledge the ESRF for the beamtime allocation at the ID26 beamline (proposal number HC-1993).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.1c01525.
Additional electronic structure calculations, Lorentzian broadening parameters, and details of the atomic parameter computations (PDF)
Author Contributions
R.C. and K.O.K. planned the project to study the trivalent lanthanide cations by the Ln L3 HERFD and RIXS methods. O.W. performed the synthesis of the Ln compounds. K.O.K. and R.C. carried out HERFD and RIXS measurements at the Ln L3 edge at the ID26 beamline of the ESRF. L.A., P.V.Z., and M.R. did theoretical calculations of the HERFD and RIXS data. P.V.Z., L.A. and K.O.K. cowrote the paper. All authors discussed the results and contributed to the final manuscript.
L.A. and K.O.K. acknowledge the support from the European Research Council (ERC) (grant agreement no. 759696). P.V.Z. acknowledges support by the Russian Ministry of Science and Education under grant no. 075-15-2019-1891.
The authors declare no competing financial interest.
Supplementary Material
References
- Bonnelle C.; Spector N.. Rare-Earths and Actinides in High Energy Spectroscopy, Progress in Theoretical Chemistry and Physics; Springer Netherlands: Dordrecht, Netherlands, 2015; p 380. [Google Scholar]
- Balaram V. Rare earth elements: A review of applications, occurrence, exploration, analysis, recycling, and environmental impact. Geosci. Front. 2019, 10, 1285–1303. 10.1016/j.gsf.2018.12.005. [DOI] [Google Scholar]
- Dushyantha N.; Batapola N.; Ilankoon I. M. S. K.; Rohitha S.; Premasiri R.; Abeysinghe B.; Ratnayake N.; Dissanayake K. The story of rare earth elements (REEs): occurrences, global distribution, genesis, geology, mineralogy and global production. Ore Geol. Rev. 2020, 122, 103521. 10.1016/j.oregeorev.2020.103521. [DOI] [Google Scholar]
- De Groot F.; Kotani A.. Core Level Spectroscopy of Solids; CRC press, 2008; p 512. [Google Scholar]
- Van Bokhoven J. A.; Lamberti C.. X-ray Absorption and X-ray Emission Spectroscopy: Theory and Applications; John Wiley & Sons, 2016; p 890. [Google Scholar]
- Jhans H.; Croft M. Systematic trends in the LIII-edge absorption spectra of the rare-earth based systems. J. Magn. Magn. Mater. 1985, 47–48, 203–205. 10.1016/0304-8853(85)90394-4. [DOI] [Google Scholar]
- Jeon Y.; Lu F.; Jhans H.; Shaheen S. A.; Croft M.; Ansari P. H. X-ray absorption spectroscopy studies of high-Tc superconductors. J. Appl. Phys. 1988, 63, 4190–4192. 10.1063/1.340221. [DOI] [Google Scholar]
- Kvashnina K. O.; Butorin S. M.; Glatzel P. Direct study of the f-electron configuration in lanthanide systems. J. Anal. At. Spectrom. 2011, 26, 1265–1272. 10.1039/c0ja00142b. [DOI] [Google Scholar]
- Asakura H.; Hosokawa S.; Teramura K.; Tanaka T. Local Structure Study of Lanthanide Elements by X-Ray Absorption Near Edge Structure Spectroscopy. Chem. Rec. 2019, 19, 1420–1431. 10.1002/tcr.201800164. [DOI] [PubMed] [Google Scholar]
- Bartolomé F.; Tonnerre J. M.; Sève L.; Raoux D.; Chaboy J.; García L. M.; Krisch M.; Kao C. C. Identification of Quadrupolar Excitation Channels at the L3 Edge of Rare-Earth Compounds. Phys. Rev. Lett. 1997, 79, 3775–3778. 10.1103/physrevlett.79.3775. [DOI] [Google Scholar]
- Bartolomé F.; Krisch M. H.; Raoux D.; Tonnerre J.-M. Quadrupolar excitation channels at the L3 edge of rare-earth ions probed by resonant inelastic x-ray scattering. Phys. Rev. B: Condens. Matter Mater. Phys. 1999, 60, 13497–13506. 10.1103/physrevb.60.13497. [DOI] [Google Scholar]
- Dallera C.; Krisch M. H.; Rogalev A.; Gauthier C.; Goulon J.; Sette F.; Sole A. Resonant inelastic x-ray scattering at the L2 and L3 edge of terbium in TbCo2 and TbF3. Phys. Rev. B: Condens. Matter Mater. Phys. 2000, 62, 7093–7097. 10.1103/physrevb.62.7093. [DOI] [Google Scholar]
- Journel L.; Mariot J. M.; Rueff J. P.; Hague C. F.; Krill G.; Nakazawa M.; Kotani A.; Rogalev A.; Wilhelm F.; Kappler J.-P.; Schmerber G. Resonant inelastic x-ray scattering at the lanthanum L3 edge. Phys. Rev. B: Condens. Matter Mater. Phys. 2002, 66, 045106. 10.1103/physrevb.66.045106. [DOI] [Google Scholar]
- Nakai S.; Ohkawa K.; Takada Y.; Odaka M.; Kashiwakura T.; Yamazaki T. Quadrupole transition spectra of rare-earth fluorides observed by resonant X-ray emission spectroscopy. J. Electron Spectrosc. Relat. Phenom. 2004, 137, 363–368. 10.1016/j.elspec.2004.02.123. [DOI] [Google Scholar]
- Kotani A.; Shin S. Resonant inelastic x-ray scattering spectra for electrons in solids. Rev. Mod. Phys. 2001, 73, 203–246. 10.1103/revmodphys.73.203. [DOI] [Google Scholar]
- Wende H. Recent advances in x-ray absorption spectroscopy. Rep. Prog. Phys. 2004, 67, 2105–2181. 10.1088/0034-4885/67/12/r01. [DOI] [Google Scholar]
- Kotani A. Resonant inelastic X-ray scattering in d and f electron systems. Eur. Phys. J. B 2005, 47, 3–27. 10.1140/epjb/e2005-00303-4. [DOI] [Google Scholar]
- Hirsch O.; Kvashnina K.; Willa C.; Koziej D. Hard X-Ray Photon-in Photon-out Spectroscopy as a Probe of the Temperature-Induced Delocalization of Electrons in Nanoscale Semiconductors. Chem. Mater. 2017, 29, 1461–1466. 10.1021/acs.chemmater.6b05218. [DOI] [Google Scholar]
- Park K.-H.; Oh S.-J. Electron-spectroscopy study of rare-earth trihalides. Phys. Rev. B: Condens. Matter Mater. Phys. 1993, 48, 14833–14842. 10.1103/physrevb.48.14833. [DOI] [PubMed] [Google Scholar]
- Parlebas J. C.; Asakura K.; Fujiwara A.; Harada I.; Kotani A. X-ray magnetic circular dichroism at rare-earth L2,3 absorption edges in various compounds and alloys. Phys. Rep. 2006, 431, 1–38. 10.1016/j.physrep.2006.05.002. [DOI] [Google Scholar]
- Rogalev A.; Wilhelm F. Magnetic circular dichroism in the hard X-ray range. Phys. Met. Metallogr. 2015, 116, 1285–1336. 10.1134/s0031918x15130013. [DOI] [Google Scholar]
- Hämäläinen K.; Siddons D. P.; Hastings J. B.; Berman L. E. Elimination of the inner-shell lifetime broadening in x-ray-absorption spectroscopy. Phys. Rev. Lett. 1991, 67, 2850–2853. 10.1103/physrevlett.67.2850. [DOI] [PubMed] [Google Scholar]
- Glatzel P.; Bergmann U. High resolution 1s core hole X-ray spectroscopy in 3d transition metal complexes—electronic and structural information. Coord. Chem. Rev. 2005, 249, 65–95. 10.1016/j.ccr.2004.04.011. [DOI] [Google Scholar]
- Kvashnina K. O.; Kvashnin Y. O.; Butorin S. M. Role of resonant inelastic X-ray scattering in high-resolution core-level spectroscopy of actinide materials. J. Electron Spectrosc. Relat. Phenom. 2014, 194, 27–36. 10.1016/j.elspec.2014.01.016. [DOI] [Google Scholar]
- Gerkin R. E.; Reppart W. J. The structures of the lanthanide ethyl sulfate enneahydrates, M(C2H5SO4)3·9H2O [M = La-Lu (except Pm)], at 171 K. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1984, 40, 781–786. 10.1107/s0108270184005710. [DOI] [Google Scholar]
- Bernhardt P. V.; Flanagan B. M.; Riley M. J. Isomorphous lanthanide complexes of a tripodal N4O3 ligand. Aust. J. Chem. 2000, 53, 229–231. 10.1071/ch99175. [DOI] [Google Scholar]
- Bernhardt P. V.; Flanagan B. M.; Riley M. J. Rapid communication: completion of the isomorphous Ln(trensal) series. Aust. J. Chem. 2001, 54, 229–232. 10.1071/ch01076. [DOI] [Google Scholar]
- Matsumoto K.; Suzuki K.; Tsukuda T.; Tsubomura T. A chiral 2, 6-bis (oxazolinyl) pyridine ligand with amide groups to form isomorphous complexes through all the lanthanoid series. Inorg. Chem. 2010, 49, 4717–4719. 10.1021/ic901743q. [DOI] [PubMed] [Google Scholar]
- Seitz M.; Oliver A. G.; Raymond K. N. The lanthanide contraction revisited. J. Am. Chem. Soc. 2007, 129, 11153–11160. 10.1021/ja072750f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilà D.; Barrios L. A.; Velasco V.; Arnedo L.; Aliaga-Alcalde N.; Menelaou M.; Teat S. J.; Roubeau O.; Luis F.; Aromí G. Lanthanide contraction within a series of asymmetric dinuclear [Ln2] complexes. Chem.—Eur. J. 2013, 19, 5881–5891. 10.1002/chem.201204451. [DOI] [PubMed] [Google Scholar]
- Aguilà D.; Velasco V.; Barrios L. A.; González-Fabra J.; Bo C.; Teat S. J.; Roubeau O.; Aromí G. Selective Lanthanide Distribution within a Comprehensive Series of Heterometallic [LnPr] Complexes. Inorg. Chem. 2018, 57, 8429–8439. 10.1021/acs.inorgchem.8b01112. [DOI] [PubMed] [Google Scholar]
- Santos I.; Marques N. Recent advances in the chemistry of f-element poly(pyrazol)borate complexes. New J. Chem. 1995, 19, 551–571. [Google Scholar]
- Marques N.; Sella A.; Takats J. Chemistry of the lanthanides using pyrazolylborate ligands. Chem. Rev. 2002, 102, 2137–2160. 10.1021/cr010327y. [DOI] [PubMed] [Google Scholar]
- Apostolidis C.; Rebizant J.; Kanellakopulos B.; von Ammon R.; Dornberger E.; Müller J.; Powietzka B.; Nuber B. Homoscorpionates (hydridotris(1-pyrazolyl)borato complexes) of the trivalent 4f ions. The crystal and molecular structure of [(HB(N2C3H3)3]3 LnIII,(Ln = Pr, Nd). Polyhedron 1997, 16, 1057–1068. 10.1016/s0277-5387(96)00391-9. [DOI] [Google Scholar]
- Apostolidis C.; Rebizant J.; Walter O.; Kanellakopulos B.; Reddmann H.; Amberger H.-D. Zur Elektronenstruktur hochsymmetrischer Verbindungen der f-Elemente. 35 [1] Kristall-und Molekülstrukturen von Tris (hydrotris (1-pyrazolyl) borato)-lanthanid (III) (LnTp3; Ln= La, Eu) sowie Elektronenstruktur von EuTp3. Z. Anorg. Allg. Chem. 2002, 628, 2013–2025. 10.1002/1521-3749(200209)628:9/10<2013::aid-zaac2013>3.0.co;2-k. [DOI] [Google Scholar]
- Amberger H.-D.; Reddmann H.; Apostolidis C.; Kanellakopulos B. Zur Elektronenstruktur hochsymmetrischer Verbindungen der f-Elemente. 36 [1] Parametrische Analyse der optischen Spektren eines orientierten Tris(hydrotris(1-pyrazolyl)borato)praseodym(III)-Einkristalls. Z. Anorg. Allg. Chem. 2003, 629, 147–160. 10.1002/zaac.200390008. [DOI] [Google Scholar]
- Reddmann H.; Apostolidis C.; Walter O.; Rebizant J.; Amberger H.-D. Zur Elektronenstruktur hochsymmetrischer Verbindungen der f-Elemente. 38 [1] Kristall-, Molekül- und Elektronenstruktur von Tris (hydrotris (1-pyrazolyl) borato)-samarium (III). Z. Anorg. Allg. Chem. 2005, 631, 1487–1496. 10.1002/zaac.200500004. [DOI] [Google Scholar]
- Stainer M. V. R.; Takats J. X-ray crystal and molecular structure of Tris[hydridotris(pyrazol-1-yl)borato]ytterbium(III), Yb(HBPz3)3. Inorg. Chem. 1982, 21, 4050–4053. 10.1021/ic00141a034. [DOI] [Google Scholar]
- Faltynek R. A. Lanthanide coordination chemistry: spectroscopic properties of terbium and europium poly(pyrazol-1-yl)- and poly(imidazole-1-yl)borate complexes. J. Coord. Chem. 1989, 20, 73–80. 10.1080/00958978909408850. [DOI] [Google Scholar]
- Seminara A.; Musumeci A. Absorption and emission spectra of neodymium(III) and europium(III) complexes. Inorg. Chim. Acta 1984, 95, 291–307. 10.1016/s0020-1693(00)84898-7. [DOI] [Google Scholar]
- Apostolidis C.; Kovács A.; Walter O.; Colineau E.; Griveau J. C.; Morgenstern A.; Rebizant J.; Caciuffo R.; Panak P. J.; Rabung T.; Schimmelpfennig B.; Perfetti M. Tris-{hydridotris(1-pyrazolyl)borato}actinide Complexes: Synthesis, Spectroscopy, Crystal Structure, Bonding Properties and Magnetic Behaviour. Chem.—Eur. J. 2020, 26, 11293–11306. 10.1002/chem.202001095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolidis C.; Kovács A.; Morgenstern A.; Rebizant J.; Walter O. Tris-{hydridotris(1-pyrazolyl)borato}lanthanide Complexes: Synthesis, Spectroscopy, Crystal Structure and Bonding Properties. Inorganics 2021, 9, 44. 10.3390/inorganics9060044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier C.; Solé V. A.; Signorato R.; Goulon J.; Moguiline E. The ESRF beamline ID26: X-ray absorption on ultra dilute sample. J. Synchrotron Radiat. 1999, 6, 164–166. 10.1107/s0909049598016835. [DOI] [PubMed] [Google Scholar]
- Glatzel P.; Harris A.; Marion P.; Sikora M.; Weng T.-C.; Guilloud C.; Lafuerza S.; Rovezzi M.; Detlefs B.; Ducotté L. The five-analyzer point-to-point scanning crystal spectrometer at ESRF ID26. J. Synchrotron Radiat. 2021, 28, 362–371. 10.1107/s1600577520015416. [DOI] [PubMed] [Google Scholar]
- Bunau O.; Joly Y. Self-consistent aspects of x-ray absorption calculations. J. Phys.: Condens. Matter 2009, 21, 345501. 10.1088/0953-8984/21/34/345501. [DOI] [PubMed] [Google Scholar]
- Wood J. H.; Boring A. M. Improved Pauli Hamiltonian for local-potential problems. Phys. Rev. B: Condens. Matter Mater. Phys. 1978, 18, 2701–2711. 10.1103/physrevb.18.2701. [DOI] [Google Scholar]
- Haverkort M. W.; Zwierzycki M.; Andersen O. K. Multiplet ligand-field theory using Wannier orbitals. Phys. Rev. B: Condens. Matter Mater. Phys. 2012, 85, 165113. 10.1103/physrevb.85.165113. [DOI] [Google Scholar]; http://www.quanty.org.
- Haverkort M. W. Quanty for core-level spectroscopy – excitons, resonances and band excitations in time and frequency domain. J. Phys.: Conf. Ser. 2016, 712, 012001. 10.1088/1742-6596/712/1/012001. [DOI] [Google Scholar]
- Zimmermann P.; Green R. J.; Haverkort M. W.; de Groot F. M. F. Quanty4RIXS: a program for crystal field multiplet calculations of RIXS and RIXS-MCD spectra usingQuanty. J. Synchrotron Radiat. 2018, 25, 899–905. 10.1107/s1600577518004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann P.; Hunault M. O. J. Y.; de Groot F. M. F. 1s2p RIXS Calculations for 3d Transition Metal Ions in Octahedral Symmetry. J. Spectrosc. 2018, 2018, 3618463. 10.1155/2018/3618463. [DOI] [Google Scholar]
- Retegan M.Crispy v0.7.3; Zenodo, 2019.
- Cowan R. D.The Theory of Atomic Structure and Spectra; Los Alamos Series in Basic and Applied Sciences; University of California Press: Berkeley, Calif, 1981; p 752. [Google Scholar]
- Krause M. O.; Oliver J. H. Natural Widths of Atomic K and L Levels, Kα X-ray Lines and Several KLL Auger Lines. J. Phys. Chem. Ref. Data 1979, 8, 329–338. 10.1063/1.555595. [DOI] [Google Scholar]
- Keski-Rahkonen O.; Krause M. O. Total and partial atomic-level widths. At. Data Nucl. Data Tables 1974, 14, 139–146. 10.1016/s0092-640x(74)80020-3. [DOI] [Google Scholar]
- Kutzler F. W.; Hodgson K. O.; Misemer D. K.; Doniach S. Theory of white lines in the x-ray absorption spectra of lanthanide complexes. Chem. Phys. Lett. 1982, 92, 626–630. 10.1016/0009-2614(82)83662-2. [DOI] [Google Scholar]
- Nakazawa M.; Fukui K.; Ogasawara H.; Kotani A.; Hague C. F. X-ray absorption and resonant x-ray emission spectra by electric quadrupole excitation in light rare-earth systems. Phys. Rev. B: Condens. Matter Mater. Phys. 2002, 66, 113104. 10.1103/physrevb.66.113104. [DOI] [Google Scholar]
- Nakazawa M.; Fukui K.; Kotani A. Theory of X-ray absorption and resonant X-ray emission spectra by electric quadrupole excitation in light rare-earth systems. J. Solid State Chem. 2003, 171, 295–298. 10.1016/s0022-4596(02)00180-9. [DOI] [Google Scholar]
- Butorin S. M. 3d-4f Resonant Inelastic X-ray Scattering of Actinide Dioxides: Crystal-Field Multiplet Description. Inorg. Chem. 2020, 59, 16251–16264. 10.1021/acs.inorgchem.0c02032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.