

Abstract
Chemotherapy-induced peripheral neuropathy (CIPN) is a prevalent side effect of widely used platinum-based anticancer agents. There are few predictable risk factors with which to identify susceptible patients. Effective preventive measures or treatments are not available. Here, we have used a model of CIPN in Drosophila melanogaster to identify genetic changes that confer resistance to cisplatin-induced neuronal damage but not in the rapidly dividing cells of the ovary. The Drosophila strain attP40, used as a genetic background for the creation of RNAi lines, is resistant to cisplatin damage compared with the similar attP2 background strain. attP40 flies have reduced mRNA expression of ND-13A, a component of the mitochondria electron transport chain complex I. Reduction of ND-13A via neuron-specific RNAi leads to resistance to the dose-dependent climbing deficiencies and neuronal apoptosis observed in control flies. These flies are also resistant to acute oxidative stress, suggesting a mechanism for resistance to cisplatin. The mitochondria of attP40 flies function similarly to control attP2 mitochondria under normal conditions. Mitochondria are damaged by cisplatin, leading to reduced activity, but attP40 mitochondria are able to retain function and even increase basal respiration rates in response to this stress. This retained mitochondrial activity is likely mediated by Sirt1 and peroxisome proliferator-activated receptor gamma coactivator-1α, and is key to cisplatin resistance. Our findings represent the potential for both identification of susceptible patients and prevention of CIPN through the targeting of mitochondria.
SIGNIFICANCE STATEMENT Chemotherapy-induced peripheral neuropathy is a major, debilitating side effect of many platinum-based cancer drugs. There are few available screening tools to identify patients at risk, and there are no effective treatments. Here, we report a novel genetic change that confers resistance to cisplatin-induced neurotoxicity in a Drosophila model while preserving the toxic effect in rapidly dividing cells. This work has the potential to influence patient susceptibility testing and development of novel CIPN preventive treatments.
Keywords: CIPN, cisplatin, complex I, Drosophila, mitochondria
Introduction
Chemotherapy-induced peripheral neuropathy (CIPN) is a cumulative dose-related side effect of platinum-based cancer therapeutics. Cisplatin has been in clinical use since 1978 and is still widely used for germ line cancers. Carboplatin and oxaliplatin are very commonly used for treating breast, colon, and lung cancers. CIPN occurs in 30–40% of patients (Windebank and Grisold, 2008; Seretny et al., 2014; Shah et al., 2018). This lifelong side effect may result in dose reduction or cessation of treatment. It may also worsen for several months after treatment is suspended (Windebank and Grisold, 2008; Cavaletti and Marmiroli, 2010). Importantly, there are no preventive therapies or treatments available for CIPN (Albers et al., 2014; Piccolo and Kolesar, 2014; Staff et al., 2019). Many empirical treatment trials have failed to demonstrate benefit (Hu et al., 2019). In addition, there are no reliable predictive measures to identify those patients most likely to be susceptible to CIPN (Johnson et al., 2015; Staff et al., 2017). This is in part because the mechanism of cisplatin-induced neuronal cell death is poorly understood. Better understanding of the mechanism of neuronal death is essential for the development of new treatments or preventive measures for CIPN.
Cisplatin causes DNA damage by forming platinum-DNA adducts that subsequently trigger apoptosis in rapidly dividing cancer cells (Fischer et al., 2001; McDonald et al., 2005). In addition, cisplatin causes increased formation of reactive oxygen species (ROS), resulting in oxidative stress and general cellular damage. However, previous attempts to protect against CIPN in patients or animal models targeting these mechanisms with antioxidants like vitamin E or mitoprotective agents have not resulted in an effective preventive treatment in the clinic. Cisplatin also forms platinum adducts with mitochondrial DNA and causes damage to the mitochondria (Podratz et al., 2011a). This manifests as swollen mitochondria with disordered cristae, decreased respiratory function, and loss of membrane potential (Melli et al., 2008; Podratz et al., 2017). Mitochondrial health and function are central to a number of neurodegenerative diseases, including Parkinson's disease (Guo, 2012; Costa et al., 2013; Tufi et al., 2014) and Alzheimer's disease (Moreira et al., 2010; Onyango et al., 2016). Despite the focus on mitochondria in these diseases, as well as in aging, less is known about the importance of mitochondria to the cellular response to cisplatin. Drugs targeting the mitochondria have not been widely researched in the context of CIPN (Hu et al., 2019).
We have previously established a model of CIPN in Drosophila melanogaster and used the extensive genetic tools available to study cisplatin sensitivity and neuronal cell death (Podratz et al., 2011b). Drosophila treated with cisplatin exhibit dose-dependent climbing deficiencies as a result of neuronal apoptosis (Podratz et al., 2011b). The platinum concentration in the DNA of treated fly brains was physiologically relevant to rodent models and patients treated with cisplatin (Podratz et al., 2011b). Flies experience dose-dependent female fertility defects, an indication of cell death in rapidly dividing cells in the ovaries, but not in nondividing muscle cells. We have also shown that flies of different genetic backgrounds have different sensitivity to cisplatin in these different cell types, suggesting that the effects of cisplatin in cancer cells can be separated from those in neurons (Groen et al., 2018).
Here, we show that genetic reduction in mRNA expression of mitochondria complex I subunits is protective against cisplatin-induced neurotoxicity in Drosophila but not against cisplatin-induced fertility defects. Our results suggest that the reduction of complex I results in flies that are more resistant to acute oxidative stress and can retain mitochondrial activity when treated with cisplatin. These results identify a novel approach for the prediction and prevention of CIPN in patients.
Materials and Methods
Fly stocks.
All fly stocks were maintained on standard molasses-agar food (Archon Scientific) at 25°C in a 12 h light/dark cycle. Oregon-R flies were a gift from Amy Tang (Eastern Virginia Medical School in Norfolk, VA). The following stocks were obtained from the Bloomington Drosophila Stock Center: yv; attP2, yv; attP40, elavGal4, ND-13A RNAi, elavGal4 UAS Dicer, SDH-C RNAi, CytC1 RNAi, UQCR-Q RNAi, UQCR-C1 RNAi, COX-5A RNAi, COX-7A RNAi, COX-7C RNAi, cype RNAi, ND51 RNAi, ND-PDSW RNAi, ND-ACP RNAi, Spargel RNAi, and UAS Sirt1.
Cisplatin treatment.
Cisplatin stock solution (1 mg/ml in 1× PBS; Fresenius Kabi) was diluted in 10% sucrose (in 1× PBS) and fed to male and female flies in empty plastic vials. Forty flies per vial were starved for 6 h before initial cisplatin treatment. Flies were then fed 125 µl of sucrose/cisplatin solution per day for 3 d and maintained at 25°C. Where indicated, cisplatin was delivered in Nutrifly Instant solid instant food (Genesee Scientific). Cisplatin stock solution was diluted in 1× PBS to the indicated concentrations. Two milliliters of diluted cisplatin was then used to reconstitute 0.5 g of instant food. Flies were moved to cisplatin food and maintained at 25°C for 5 d before testing.
Negative geotaxis climbing assay.
The climbing assay was performed using our automated climbing apparatus (Podratz et al., 2013). After cisplatin treatment (as described above), male and female flies were scored for survival and moved to clean, empty vials (20–30 flies/vial). The climbing vials were loaded into racks holding 10 vials each, and the flies were allowed to acclimate to their environment for 1 h. Climbing racks were then placed in the climbing apparatus and again allowed to acclimate for ∼5 min. The standard climbing assay was then initiated: four successive taps, picture taken after 15 s, 45 s of recovery time, and then four additional cycles. Flies climbing over a set height of 2 cm were scored automatically by custom software (Podratz et al., 2013) and ImageJ (Abramoff et al., 2004). All experiments were performed a minimum of three times.
Quantitative RT-PCR.
Flies of the indicated genotypes were frozen and stored at −80°C. Frozen flies were vortexed to remove heads, and separated heads were sorted into clean nuclease-free 1.5 ml Eppendorf tubes. RNA was extracted using standard TRIzol (Thermo Fisher Scientific) and chloroform (Sigma-Aldrich) extraction and isopropanol precipitation. Quantitative PCR (qPCR) was performed using iTaq Universal SYBR Green One Step qRT-PCR kit (BIO-RAD). The following primers were used: RP49 (housekeeping control gene): forward, 5′-AGATCGTGAAGAAGCGCACCAAG-3′, reverse 5′-CACCAGGAACTTCTTGAATCCGG-3′; ND-13A: forward, 5′-CCAAGCGGTATGTGAACGAG-3′, reverse, 5′-GCAGAAGACAACTCGTTCGG-3′; Sirt1: forward, 5′-GATATATCCCGGCGAGTTTCAG-3′, reverse, 5′-GTGTCGATGTTCTGTGTGTAGT-3′; and PGC1: forward, 5′-CTCTTGGAGTCCGAGATCCGCAA-3′, reverse, 5′-GGGACCGCGAGCTGATGGTT-3′.
Fertility assay.
Female Drosophila treated with the indicated doses of cisplatin (3 d, in 10% sucrose) were mated to untreated OregonR males (three females and three males), allowed to lay eggs on standard food for 3 d, and maintained at 25°C. Progeny per female were counted 12 d later. Each assay was performed a minimum of three times for each genotype and treatment. For health span fertility measurements, females of the indicated genotypes and ages were mated to young (3- to 4-d-old) male OregonR flies.
Life-span analysis.
Flies of the indicated genotypes, 1–2 d old, were sorted into separate cohorts of males and females. Twenty-five flies were placed in each vial, and six vials of each genotype (150 flies total) were tested over two independent experiments. Flies were changed to fresh food and scored for survival every 3–4 d until all flies were dead.
Oxidative stress.
Hydrogen peroxide stock solution (30%; Thermo Fisher Scientific) was diluted to a final concentration of 2% in 10% sucrose (in 1× PBS) and fed to flies in empty plastic vials. Forty flies per vial were starved for 6 h before initial H2O2 treatment. Flies were then fed 125 µl of sucrose/H2O2 solution per day for 4 d and maintained at 25°. Survival was scored daily throughout H2O2 treatment. Each genotype was assayed a minimum of three independent times.
ATP quantification.
ATP was quantified using the Promega CellTiter-Glo 2.0 Assay Kit following the kit protocol. Lysates for the assay consisted of an individual fly head, dissected fresh, for each sample. Three separate biological replicates of the indicated genotypes were assayed in each of two independent experiments.
NAD+/NADH measurement.
The NAD+/NADH ratio was determined using the Promega NAD/NADH-Glo Assay Kit. Assay lysates consisted of individual fly heads of the indicated genotypes. Each genotype was analyzed with three individual biological replicates.
Mitochondrial respiration.
Flies of the indicated genotypes were treated with cisplatin as described above. Drosophila mitochondria isolation was adapted from a previously described protocol (Villa-Cuesta and Rand, 2015). Briefly, mitochondria were isolated from whole flies using a mitochondrial isolation buffer (70 mm sucrose, 210 mm mannitol, 5 mm HEPES, pH 7.2, 1 mm EGTA, and 0.5% fatty acid-free BSA, pH 7.2). One hundred whole flies were homogenized in a 1.5 ml Eppendorf tube with 1 ml of ice-cold isolation buffer using a plastic pestle. To remove cellular debris, the homogenate was centrifuged for 10 min at 300 × g at 4°C, and the supernatant was transferred to a clean 1.5 ml tube. The supernatant was then centrifuged at 3000 × g for 10 min at 4°C to pellet the mitochondria. The supernatant was discarded, and the pellet was resuspended in 1 ml of isolation buffer and centrifuged a second time at 3000 × g for 10 min at 4°C. The supernatant was again discarded, and the mitochondria pellet was resuspended in 100 µl of isolation buffer. Protein concentration was determined using the DC Protein Assay (BIO-RAD). Mitochondria were diluted to a concentration of 1 µg/µl (based on protein concentration) in mitochondria assay buffer (70 mm sucrose, 220 mm mannitol, 10 mm KH2PO4, 5 mm MgCl2, 2 mm HEPES, 1 mm EGTA, 0.2% BSA, 10 mm pyruvate, and 5 mm malate). The oxygen consumption rate (OCR) of the isolated mitochondria was then assessed using a Seahorse XF24 Extracellular Flux Analyzer (Agilent). Fifty microliters of mitochondria were plated in a 24-well Seahorse plate and centrifuged for 30 min at 3000 × g and 4°C to attach to the bottom of the well. The following four injection compounds were used to assess electron flow and oxygen consumption in isolated mitochondria: (1) ADP (substrate for ATP synthase); (2) oligomycin (ATP synthase inhibitor); (3) FCCP (carbonylcyanide-4-trifluoromethoxyphenylhydrazone; uncoupling agent); and (4) rotenone/antimycin A (complex I inhibitors). Seahorse injection compounds were diluted in assay buffer to the following final concentrations: 4 mm ADP, 4 μm oligomycin, 8 μm FCCP, and 2 μm rotenone/4 μm antimycin A. The Seahorse assay was performed on an Agilent XF24 Seahorse Extracellular Flux Analyzer at 29°C using the following injection protocol (looped twice) for each compound in sequence: compound injection (75 µl), 60 s mix, 2 min read, and 30 s mix.
Immunofluorescence.
Apoptosis was assessed using immunofluorescence analysis of fixed whole-mount Drosophila brains. Briefly, flies were treated with cisplatin for 3 d as described above. Brains were dissected in cold 1× PBS and fixed in 4% paraformaldehyde for 20 min at room temperature. Samples were permeabilized and blocked in 1× PBS with 0.1% Triton X-100 and 0.1% BSA. The following antibodies were used: rat anti-elav (1:5; Developmental Studies Hybridoma Bank) and rabbit anti-active caspase-3 (1:400; Cell Signaling Technology). The following secondary antibodies and concentrations were used: anti-rat Cy2 (1:400; Jackson ImmunoResearch) and anti-rabbit Cy3 (1:400; Jackson ImmunoResearch). Brains were mounted in Vectashield/DAPI and imaged on a confocal microscope (LSM 780, Zeiss).
Electron microscopy.
Drosophila were treated with cisplatin for 3 d as described above. Brains were dissected directly in SBFSEM fixative. Heavy metal staining, mounting in resin, and imaging on a VolumeScope Serial Block Face Scanning Electron Microscope (SBFSEM) or a 1400+ Transmission Electron Microscope (JEOL) was performed by the Mayo Clinic Microscopy and Cell Analysis Core. For image scoring, images were blind labeled and images were manually scored for mitochondria that were swollen, exhibited disordered cristae, or exhibited dense punctate staining. Five individual brains were fixed and sectioned for each treatment group, and at least two regions of interest (ROIs) per brain were scored for mitochondria abnormalities.
MitoSOX and TMRM imaging.
Cisplatin-treated Drosophila were dissected in HL6 buffer (Podratz et al., 2017). Live brains in HL6 buffer were stained with either MitoSOX (15 min) or TMRM (tetramethylrhodamine, methyl ester; 1 h). Live brains were mounted in HL6 buffer under a bridge coverslip and imaged live on a confocal microscope (LSM 780, Zeiss) as z-stacks using identical imaging settings. All figure panel images were brightened 40% to aid in visualization.
Image analysis.
Images of active caspase 3, TMRM, and MitoSOX were scored using CellProfiler automated analysis software. Objects (cell bodies or mitochondria) were automatically identified based on size and intensity with background threshold levels set globally for all images. Confocal z-stacks were scored as an image series, and a minimum of four separate brain z-stacks for each treatment group were scored for each dye or antibody. Data are reported as the mean signal intensity within positively identified objects.
Experimental design and statistical analyses.
All statistical analyses were performed using GraphPad Prism. Error bars on all graphs represent the SEM. Statistical significance for all climbing, fertility, and associated survival assays was calculated using a two-way multiple-comparisons ANOVA, comparing each strain to every other strain at each cisplatin dose and applying the Holm–Sidak method for multiple-comparisons corrections. Significance is reported as p values calculated in the ANOVAs. Life-span and oxidative stress survival assays were analyzed using Kaplan–Meier analysis with Mantel–Cox log-rank analysis curve comparisons. Statistical significance for image quantification data, qPCR, and mitochondria respiration data from the Seahorse Extracellular Flux Analyzer were determined using multiple unpaired t tests with Welch's correction to compare each genotype to its appropriate control genotype.
Results
attP40 flies are resistant to cisplatin
We established a Drosophila model of CIPN to use the genetic tools available to understand potential mechanisms of CIPN sensitivity in a semi-high-throughput screen. To control for genetic background we chose the TRiP RNAi collection (Ni et al., 2008, 2011), which was created using targeted insertion in only two background strains (yv; attP2 and yv; attP40). We have previously demonstrated the viability of the TRiP RNAi approach by driving elavGal4-dependent, neuron-specific knockdown of glutathione peroxidase (GPX), a gene known to be involved in cisplatin sensitivity in human patients. GPX RNAi caused increased sensitivity to cisplatin (Groen et al., 2018). During our initial analysis of this screen design, we observed that RNAi strains responded differently to cisplatin even in the absence of a Gal4 driver. RNAi strains created in the attP40 background were generally more resistant to cisplatin-induced climbing deficiencies than strains created in the attP2 background (data not shown). We followed up on this observation by assessing the sensitivity of attP2 and attP40 background flies with the elavGal4 driver to cisplatin. We treated flies with cisplatin doses ranging from 0 to 200 µg/ml in a 10% sucrose solution for 3 d and then assessed lethality and cisplatin-induced climbing deficiencies. Cisplatin-induced climbing deficiencies in the attP2 background strain were not statistically different from those of the wild-type control strain OregonR (Fig. 1A). In contrast, attP40 flies were more resistant to cisplatin-induced climbing deficiencies than attP2 flies in the presence of a Gal4 driver (Fig. 1A) or in the absence of a Gal4 driver, though the difference is less pronounced in the absence of Gal4 (Extended Data Fig. 1-1A). Cisplatin doses ranging from 50 to 150 µg/ml all elicited a significantly different climbing response between attP2 and attP40/elavGal4 flies. To control for the possible influence of a dietary change on the effect of cisplatin in these flies, we also delivered the cisplatin in a solid food medium. The difference in climbing response to cisplatin was similar whether the cisplatin was delivered in a 10% sucrose solution (Fig. 1) or mixed into solid instant food (Extended Data Fig. 1-1C).
Figure 1.
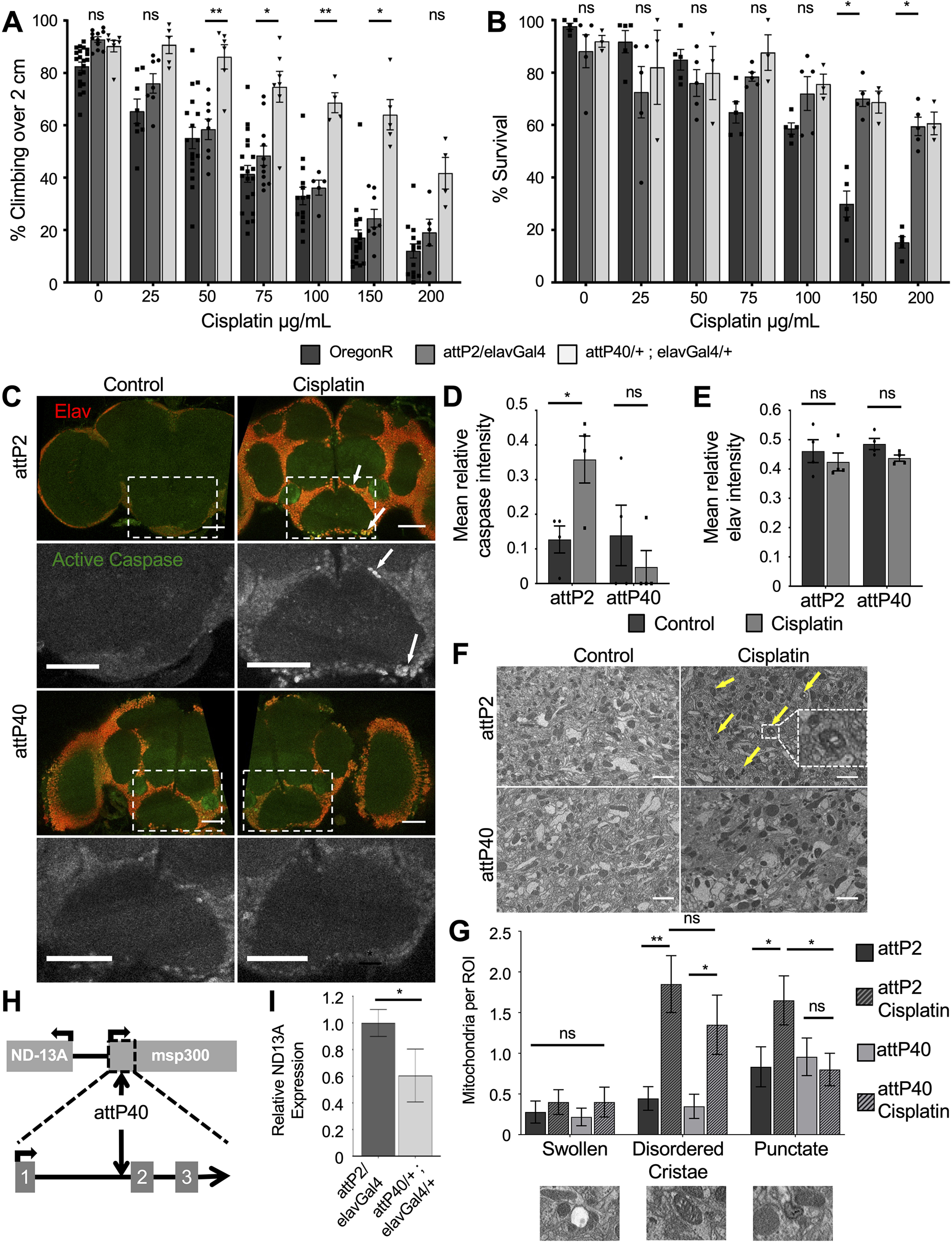
Resistance to cisplatin-induced climbing deficiencies in attP40 Drosophila. A, Bars indicate the percentage of flies of three genotypes treated with the indicated cisplatin doses able to climb above 2 cm in an automated climbing assay. Each bar represents a minimum of five individual assay vials. Each individual point indicates the mean result from a single vial of 20–30 flies. B, Graph displaying the mean survival percentage of cisplatin-treated flies of the same strains as in A. Individual points indicate the result from a single vial of 40 flies. C, Maximum projections of three confocal slices of whole-mount fixed Drosophila brains of the indicated genotypes with and without 100 µg/ml cisplatin treatment. Immunofluorescence marks neurons (anti-elav, red) and apoptotic cells (anti-active caspase 3, green). Scale bar, 50 μm. D, E, Graphs of mean active caspase intensity (D) and mean elav intensity (E) within objects (cell bodies) identified automatically by pixel size and background intensity thresholding. Each data point represents the mean object intensity for one Drosophila brain z-stack image. F, Single slice of Drosophila brains of the indicated genotypes with and without 100 µg/ml cisplatin treatment imaged by serial block face scanning electron microscopy. Scale bar, 1 μm. G, Quantification of mitochondria abnormalities identified via blind image scoring of electron microscopy images of Drosophila brains. H, Map of the Drosophila genome surrounding the attP40 insertion site on chromosome 2. The arrows indicate where attP40 inserts into the first three exons of the msp-300 gene and the position of the ND-13A gene. I, Graph of normalized ND-13A expression data in heads of attP2 and attP40 flies, as determined by qRT-PCR. Expression of ND-13A in attP40 is normalized to control attP2 flies. *p < 0.05, **p < 0.01, ns = not significant compared with noted control (two-way ANOVA with Holm–Sidak correction in A and B; unpaired Welch's t test in D, E, and G). Extended Data Figure 1-1 expands on A and B by demonstrating the same effect in attP2 and attP40 Drosophila lacking the Gal4 driver (Extended Data Fig. 1-1A,B) and with cisplatin delivered via solid food instead of sucrose (Extended Data Fig. 1-1C,D). Extended Data Figure 1-1E provides supporting data for the discussion of H and I.
Interestingly, both attP genotypes exhibited similar survival rates with cisplatin treatment (Fig. 1B, Extended Data Fig. 1-1B,D). In the presence of a Gal4 driver, both attP strains survived at a significantly higher rate than OregonR flies (Fig. 1B), while the attP strains without Gal4 survived at similar rates to OregonR (compare Extended Data Fig. 1-1B, 1B). These data demonstrate that cisplatin lethality and climbing deficiencies are not intrinsically linked in this Drosophila model, and that climbing deficiencies are not merely because of the use of flies that are near death. We have previously reported on fly strains that exhibit opposite changes in sensitivity to cisplatin in the neurons and in rapidly dividing cell types (Groen et al., 2018). These survival data further suggest that the effects of cisplatin in different cell types can be separated in our Drosophila model.
Imaging analysis of whole-mount Drosophila brains also demonstrated attP40 flies are resistant to cisplatin damage. We have previously shown that brains from Drosophila treated with cisplatin contain apoptotic neurons (Podratz et al., 2011b). We dissected whole brains from flies with and without 3 d of cisplatin treatment and stained with antibodies for neurons (elav) and apoptosis (active caspase-3). Cisplatin-treated attP2 flies contain numerous apoptotic neurons (Fig. 1C) similar to our previous observations in wild-type flies (Podratz et al., 2011b). In contrast, attP40 fly brains with cisplatin do not exhibit high levels of active caspase-3 staining (Fig. 1C). We quantified these images using CellProfiler software to automatically apply size and background signal thresholds to quantify individual objects (cell bodies) and the relative fluorescence intensity of each object. Image quantification revealed that cisplatin-treated attP2 brains exhibited significantly higher active caspase-3 fluorescence within positively identified cell bodies compared with untreated controls (Fig. 1D). Brains from cisplatin-treated attP40 flies did not show a difference in active caspase intensity (Fig. 1E). Quantification of elav immunofluorescence revealed that cisplatin treatment did not significantly alter mean fluorescence (Fig. 1E) or the number of positively identified cell bodies in the images analyzed (Table 1). These imaging data correlate with negative geotaxis assays to show that attP40 Drosophila brains do not accumulate neuronal cisplatin damage like their attP2 counterparts.
Table 1.
Object counts for active caspase and elav image analysis
| Genotype/treatment | Mean caspase count | Mean elav count |
|---|---|---|
| attP2/elavGal4 control | 0.29 ± 0.09 | 282.3 ± 43.8 |
| attP2/elavGal4 cisplatin | 21.63 ± 13.89 | 293 ± 139.2 |
| attP40/+ ; elavGal4/+ control | 1.19 ± 0.90 | 396 ± 36.4 |
| attP40/+ ; elavGal4/+ cisplatin | 0.09 ± 0.09 | 358 ± 46.0 |
The table shows the number of objects identified by CellProfiler image software for each genotype and treatment group. Numbers represent the mean number of identified objects per image slice. Positively identified objects meet criteria for object size and intensity over background threshold, automatically applied globally to all analyzed images.
We also analyzed Drosophila brains by electron microscopy. Damaged mitochondria in cisplatin-treated neurons can be visualized through electron microscopy as either swollen, vacuolized mitochondria with disordered cristae or areas of dense, punctate staining (Melli et al., 2008; Podratz et al., 2011a,b). Brains from attP2 flies treated with cisplatin contained mitochondria that contained dense punctate staining or disordered cristae (Fig. 1F, arrows, inset). Blinded scoring of these images revealed that control brains and attP40 brains with or without cisplatin contained fewer mitochondria abnormalities (Fig. 1G). We observed that cisplatin did not cause a significant increase in the number of swollen, vacuolized mitochondria per ROI in either genotype, though both trended higher (Fig. 1G). However, cisplatin caused a significant increase in the number of mitochondria with disordered cristae and a significant increase in the number of mitochondria exhibiting dense, punctate staining (Fig. 1G). In this final category, only attP2 brains with cisplatin exhibited the increase, not attP40 brains. These data suggest that cisplatin can damage mitochondria in both Drosophila strains and that attP2 mitochondria may have a small increase in damage compared with their attP40 counterparts. Interestingly, attP40 neurons do not progress to cell death (Fig. 1C,D) despite some evidence of damaged mitochondria (Fig. 1G).
attP40 flies have reduced ND-13A expression
We next wanted to follow-up on this significant observation that TRiP RNAi library background strains had different sensitivity to cisplatin. We hypothesized that a genetic difference between attP2 and attP40 flies accounted for the different response to cisplatin in the climbing assay. The TRiP RNAi background strains differ in the location of the attP insertion site: attP2 is located on chromosome 3 and attP40 is located on chromosome 2. As attP2 flies respond to cisplatin in a manner similar to wild-type flies, we hypothesized that a position insertion effect in attP40 flies lead to the observed protective effect. The attP insertion site in attP40 is located in the 25C6 locus of chromosome 2, within an intron of the msp-300 gene (Fig. 1H), a component of the LINC complex connecting the cytoskeleton to the nuclear lamina (Rothballer and Kutay, 2013). This insertion site suggests a possible disruption of gene expression. Knockdown of msp-300 via RNAi in the attP2 background did not reproduce the cisplatin resistance observed in attP40 flies (Extended Data Fig. 1-1E). Instead, msp-300 knockdown caused a significant increase in cisplatin sensitivity in our climbing assay, demonstrating that msp-300 and the LINC complex are in fact essential for neuronal survival during cisplatin treatment.
The attP40 site is also upstream of the NADH dehydrogenase 13 kDA subunit (ND-13A), a component of mitochondria complex I (Fig. 1H). Cisplatin is known to cause mitochondria damage and dysfunction (Podratz et al., 2011a, 2017). We hypothesized that the attP40 insertion site caused altered expression of ND-13A, leading to cisplatin resistance. We analyzed the expression of ND-13A using RNA isolated from fly heads. Quantitative RT-PCR revealed that attP40 flies express ND-13A mRNA transcript at ∼60% of the level observed in attP2 fly heads (Fig. 1I). This was an interesting finding because reduction of a complex I subunit would not necessarily be predicted to be protective against a stressor that damages mitochondria. However, genetic reduction of mitochondria complex I has been previously linked to increased stress resistance in multiple contexts (Copeland et al., 2009; Hur et al., 2014; Lionaki et al., 2015). We therefore hypothesized that reduction in ND-13A mRNA expression was protective against cisplatin-induced damage in Drosophila.
Knockdown of ND-13A specifically in neurons is protective against cisplatin
We next wanted to determine whether the protection against cisplatin observed in attP40 flies was indeed because of reduced ND-13A expression and whether this protection was specific to neurons. The TRiP collection includes ND-13A RNAi constructs in both the attP2 and attP40 backgrounds. We generated ND-13A knock-down flies using the pan-neuronal driver elavGal4. We were unable to generate any viable adult progeny when ND-13A RNAi was expressed from the attP40 background (data not shown). It is likely that the combination of a background already predisposed to low-level ND-13A mRNA expression and UAS-Gal4-driven RNAi resulted in such low-level expression of this subunit that the cells were not able to overcome the deficiency and thus were not able to develop into adulthood. ND-13A RNAi in the attP2 background produced viable offspring expressing ND-13A mRNA at ∼40% of control levels (Fig. 2C). Analysis of these flies in the negative geotaxis assay showed that ND-13A RNAi flies are resistant to cisplatin-induced climbing deficiencies, similar to attP40 flies (Fig. 2A). Comparison of dose response and calculation of cisplatin IC50 values in our climbing assay reveal that OregonR (IC50 = 58.6 µg/ml) and attP2 (IC50 = 65.3 µg/ml) strains have a similar response to cisplatin while attP40 (IC50 = 178.1 µg/ml) and ND-13A RNAi (IC50 = 195 µg/ml) flies require a significantly higher dose of cisplatin to cause a similar climbing defect (Table 2). ND-13A RNAi flies survive the 3 d of cisplatin treatment at approximately the same level as control attP2 flies (Fig. 2B), similar to results observed in attP40 flies.
Figure 2.

Neuron-specific knockdown of ND-13A is protective against cisplatin-induced climbing defects. A, D, Percentage of flies of the indicated genotypes treated with a range of cisplatin doses climbing over 2 cm in the automated assay. A represents the genes without UAS Dicer, and D represents genes with UAS Dicer. Each bar represents a minimum of five individual assay vials. Individual points indicate the mean result of a single vial of 20–30 flies. B, E, Mean percentage of survival of flies treated in the same way as in A and D. Individual points indicate the mean result of a single vial of 40 flies. C, Graph of relative ND-13A mRNA expression in fly heads of the indicated genotypes normalized to attP2. F, Mean number of progeny per female of indicated genotype with increasing doses of cisplatin. Individual data points represent the results of a single vial of three treated females mated to three untreated wild-type males. G, H, Percentage of flies of the indicated genotypes treated with a range of cisplatin doses climbing over 2 cm in the automated assay. G represents ND-13A knockdown in sensory neurons (ppkGal4), and H represents knockdown in motor neurons (D42Gal4). The statistical significance noted is ND13A RNAi,attP2/ppkGal4 compared with attP2/ppkGal4 or ND13A RNAi,attP2/D42Gal4 compared with attP2/D42Gal4. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns = not significant compared with control (two-way ANOVA with Holm–Sidak correction in A, B and D–H; unpaired Welch's t test in C).
Table 2.
Cisplatin IC50 values for automated climbing assay
| Genotype | IC50 cisplatin (µg/ml) |
|---|---|
| OregonR | 58.6 |
| attP2/elavGal4 | 65.3 |
| attP40/+ ; elavGal4/+ | 178.1 |
| ND13A RNAi, attP2/elavGal4 | 195.0 |
Table displays the dose of cisplatin required to block 50% of flies of the indicated genotype from climbing over a height of 2 cm in the automated climbing assay.
We further analyzed the protective effect of ND-13A knockdown using different Gal4 drivers. We induced an even greater reduction in ND-13A expression using elavGal4 and a UAS Dicer construct to enhance RNAi efficacy. This driver combination is neuron specific and reduced ND-13A transcript levels to ∼10% of control (Fig. 2C). Despite this very low level of ND-13A expression, these flies still exhibited high levels of resistance to cisplatin in our climbing assay (Fig. 2D), and the flies survived the cisplatin treatments at levels similar to attP2 controls (Fig. 2E). These data support our hypothesis that ND-13A knockdown is protective against cisplatin, and that this protection can be achieved through neuron-specific knockdown instead of a whole-organism genetic change (as in attP40 background strains).
While CIPN affects peripheral sensory neurons in human patients, we have demonstrated that neurons in the brain undergo apoptosis in our Drosophila model (Podratz et al., 2011b). It is not known whether specific types of neurons in Drosophila are primarily sensitive to cisplatin. The UAS-Gal4 system allows for targeted gene knockdown in specific subsets of neurons. We therefore asked whether protection against cisplatin-induced climbing deficiencies could be achieved through ND-13A knockdown in specific neuronal subtypes. We used the ppkGal4 driver to knock down ND-13A specifically in sensory neurons and the D42Gal4 driver for motor neurons. Crossing either of these drivers to the attP2 background resulted in increased resistance to cisplatin compared with attP2/elavGal4 flies (Fig. 2G,H). This suggests yet another genetic modification that can protect against CIPN. Importantly, knockdown of ND-13A with either driver demonstrated an additive protective effect. Flies expressing ND-13A RNAi in sensory (ppkGal4) or motor (D42Gal4) neurons climbed at a significantly higher rate than the attP2/ppkGal4 or attP2/D42Gal4 controls, respectively (Fig. 2G,H). Knockdown of ND-13A in sensory neurons (Fig. 2G) did produce flies that were more resistant to cisplatin than the ND-13A knockdown in motor neurons (Fig. 2H). The observation that resistance to cisplatin can be accomplished with either driver suggests that multiple types of Drosophila neurons are damaged by cisplatin, and that protecting a subset of neurons can still have beneficial effects for the organism as a whole.
Cisplatin also causes damage in rapidly dividing cell types, and maintaining this effect is essential to any potential treatment aimed at preventing cisplatin neurotoxicity. In Drosophila, damage to the rapidly dividing ovaries can be assessed using a fertility assay. We have previously shown that cisplatin causes dose-dependent fertility defects in treated flies, and that sensitivity to this effect varies depending on genotype (Groen et al., 2018). We therefore wanted to understand how changes in ND-13A expression affect the sensitivity of this rapidly dividing cell type. After a 3 d cisplatin treatment, female flies were mated to untreated OregonR males and allowed to lay eggs. Fertility was scored as the number of progeny per female. Flies with reduced ND-13A expression (attP40 and ND-13A RNAi) were more sensitive to cisplatin-induced fertility defects than control attP2 flies. Female attP2 flies produced the most viable progeny following increasing doses of cisplatin, while OregonR, attP40, and ND-13A RNAi flies all produced significantly fewer viable progeny at cisplatin doses as low as 25 µg/ml (Fig. 2F). These data show that reduction of ND-13A has a different effect in different cell types and that targeting of complex I may be specifically beneficial to neurons.
Knockdown of complex I subunits is protective against cisplatin
ND-13A is a component of mitochondria complex I, a large, multiprotein complex encoded by both nuclear and mitochondrial genes. We next determined whether protection against cisplatin required knockdown of this specific subunit, or whether knockdown of any component of complex I was protective. We knocked down expression of three other complex I subunits (all in the attP2 background) and subjected them to the cisplatin treatment and climbing assay. Knockdown of ND-51, ND-PDSW, or ND-ACP RNA expression produced the same protective effect observed in ND-13A RNAi flies (Fig. 3A,B), showing that protection against cisplatin derives from targeting complex I rather than one specific gene.
Figure 3.

RNAi knockdown of complex I subunits, but not complex IV, is protective against cisplatin. A, C, Percentage of flies of the indicated genotypes and treated with varying cisplatin doses that are able to climb over a height of 2 cm in the climbing assay. Each bar represents a minimum of five individual assay vials. Individual points indicate the mean result of a single vial of 20–30 flies. B, D, Mean percentage survival of the same fly strains under the same dose conditions as in A and C. Individual points indicate the result of a single vial of 40 flies. *p < 0.05, **p < 0.01, ns = not significant compared with control (two-way ANOVA with Holm–Sidak correction).
It was also important to address whether complex I is the key complex for cisplatin resistance or whether any component of the electron transport chain (ETC) could be targeted. We attempted to knock down subunits of complexes II, III, and IV in neurons using elavGal4. Crossing most of these RNAi lines to the elavGal4 driver line resulted in lethality at either the larval or pupal stages of development (Table 3). We were able to successfully generate a sufficient number of adult progeny expressing RNAi against the complex IV subunit COX-5A. Knockdown of COX-5A did not lead to significant protection against cisplatin in the climbing assay (Fig. 3C,D). This suggests that knockdown of complex I subunits is the key to providing protection against cisplatin.
Table 3.
Lethality of neuron-specific knockdown of complex II, III, and IV subunits
| Gene | Insertion Site | Complex | Result |
|---|---|---|---|
| Sdh-C | attP40 | 2 | Lethal |
| Cyt-C1 | attP2 | 3 | Lethal at pupal stage |
| UQCR-Q | attP2 | 3 | Lethal at pupal stage |
| UQCR-C1 | attP2 | 3 | Lethal |
| COX-5A | attP2 | 4 | Viable |
| COX-7A | attP40 | 4 | ∼95% lethal |
| COX-7C | attP40 | 4 | ∼85% lethal |
| Cype | attP2 | 4 | Lethal at pupal stage |
The listed RNAi lines were crossed to elavGal4 to drive neuron-specific knockdown of electron transport chain subunits. Survival of the resulting progeny is listed.
Flies with reduced ND-13A do not have improved health span
Reduced expression of complex I components has been linked to improved longevity and health span in other systems (Clancy, 2008; Miwa et al., 2014). One possible explanation for the resistance to cisplatin observed in attP40 and ND-13A RNAi flies is that they are generally healthier and more robust flies, as has been reported for other complex I RNAi flies (Copeland et al., 2009). We therefore determined whether our fly strains were more robust by analyzing life span and the following two parameters commonly used to assess Drosophila health span: climbing ability and female fertility in aging flies. Reduced ND-13A, either in the attP40 genetic background or driven by neuron-specific ND-13A RNAi in the attP2 background, did not improve Drosophila life span. Median life span in attP40 (45 d) and ND-13A RNAi (38 d) males was significantly reduced compared with attP2 (49 d) male flies (Fig. 4A). Female flies did not have any statistically significant difference in median life span (70 d) in any of the tested strains (Fig. 4B).
Figure 4.

ND-13A reduction is not beneficial to life span or health span. A, B, Kaplan–Meier survival curves depicting the life span of male (A) and female (B) flies of the indicated genotypes. C, D, Percentage of flies of the indicated genotypes and age that are able to climb over 2 cm in the climbing assay. C represents males, and D represents females. Each bar represents three individual assay vials. Individual points indicate the mean result of a single vial of 20–30 flies. E, Graph displaying the mean number of progeny per female of indicated genotype and age. Individual data points represent the results of a single vial of three treated females mated to three males. *p < 0.05, **p < 0.01, ***p < 0.001, ns = not significant compared with control (Kaplan–Meier Mantel–Cox survival analysis in A and B; two-way ANOVA with Holm–Sidak correction in C and D).
One commonly used measure of Drosophila health span is a decline in climbing ability during aging (Piper and Partridge, 2018). We assessed climbing ability every 15 d in aging flies. Climbing assays failed to show any significant differences in aging males (Fig. 4C). In female flies, attP40 and ND-13A RNAi flies had lower climbing ability than attP2 controls at 30 and 45 d and slightly higher climbing at 60 d. However, there was not a strong trend showing sustained improvement in climbing, suggesting that these differences were minor (Fig. 4D). The assessment of fertility during aging yielded similar results. The number of progeny per female was not significantly different between attP2 and attP40 flies at any age (Fig. 4E). ND-13A RNAi flies experienced a decline in fertility earlier than attP2 and attP40 flies, though these differences were not significant by two-way ANOVA (Fig. 4E). These data lead us to conclude that a reduction of ND-13A does not result in flies that are generally healthier and more robust.
Reduced ND-13A is protective against general oxidative stress
We next examined whether ND-13A reduction is beneficial in the context of other forms of more acute stress. Cisplatin is known to cause increased oxidative stress (Conklin, 2004; Areti et al., 2014) and mitochondrial damage (Podratz et al., 2017). Mitochondria are a primary source of reactive oxygen species in the cell, and complex I in particular generates high levels of ROS (Brookes, 2005). We hypothesized that knockdown of ND-13A protects against cisplatin in part by protecting cells against general oxidative stress. If this hypothesis is true, we reasoned that attP40 or ND-13A RNAi flies would then also be resistant to a general oxidative stress. To test this, we subjected flies to an oxidative stress survival assay. We treated attP2, attP40, and ND-13A RNAi flies with 2% hydrogen peroxide (H2O2) in 10% sucrose and monitored survival over a course of 4 d. We found that a reduction of ND-13A resulted in protection from acute oxidative stress. In male flies (Fig. 5A), the median survival of the H2O2 treatment was higher in attP40 (72 h) compared with attP2 controls (64 h), while ND-13A RNAi flies had the same median life span but a significantly improved survival curve (p = 0.0268). Similarly, both female ND-13A RNAi (70 h) and attP40 (88 h) flies had significantly higher median survival time than attP2 flies (64 h; Fig. 5B). These data suggest that reduced ND-13A protects against cisplatin-induced damage at least partly by protecting against oxidative damage.
Figure 5.

Reduced ND-13A expression is protective against general oxidative stress. A, B, Kaplan–Meier survival curve of flies of the indicated genotypes treated with 2% H2O2 in 10% sucrose and monitored for survival. A, Male flies. B, Female flies. C, E, Confocal imaging of live Drosophila brains incubated with TMRM (C) to show mitochondria with intact membrane potential or MitoSOX (E) to show reactive oxygen species generated by mitochondria. D, F, Graphs of mean TMRM intensity (D) and mean MitoSOX intensity (F) within objects (mitochondria) identified automatically by pixel size and background intensity thresholding. Each data point represents the mean object intensity for a single confocal plane image of a live Drosophila brain. Scale bar, 50 μm. *p < 0.05, **p < 0.01, ****p < 0.0001, ns = not significant compared with control (Kaplan–Meier Mantel–Cox survival analysis in A and B; unpaired Welch's t test in D, F).
In addition to general oxidative stress, cisplatin also causes damage in the mitochondria. This damage can be assessed using multiple measures of mitochondria health and function. Cisplatin-damaged mitochondria exhibit reduced membrane potential and increased production of reactive oxygen species. We hypothesized that attP40 flies would resist both of these damaging changes to the mitochondria. To assess mitochondrial damage specifically in the brain, we analyzed mitochondria via confocal microscopy using live-imaging dyes. Mitochondria were stained for membrane potential using TMRM, while mitochondria-generated ROSs were assessed using MitoSOX, as we have demonstrated previously (Podratz et al., 2017). Staining in live brains for TMRM mitochondria membrane potential revealed that flies treated with cisplatin exhibit decreased staining, and therefore have the loss of membrane potential that is indicative of cisplatin damage (Fig. 5C). We quantified these images using CellProfiler software to automatically identify individual objects (mitochondria) by particle size and background thresholding and measure relative fluorescence intensity. This analysis revealed that cisplatin treatment in attP2 and attP40 brains led to a significant decrease in TMRM intensity, though the magnitude of this decrease was greater in attP2 brains, suggesting more extensive damage in these flies (Fig. 5D).
When we stained live brains with MitoSOX to determine ROS production, we first observed that attP40 mitochondria exhibit less intense MitoSOX staining than attP2 mitochondria (Fig. 5E). When cisplatin was added, attP2 mitochondria increased mitochondrial ROS staining (Fig. 5E). attP40 mitochondria did not exhibit the same increase in mitochondria ROS marked by MitoSOX staining. Automatic quantification of these images showed that only attP2 brains treated with cisplatin exhibited increased MitoSOX signal (Fig. 5F). These live-imaging data reveal that attP40 mitochondria in the brain are resistant to increased mitochondrial ROS production but still exhibit some loss of membrane potential when exposed to cisplatin treatment. As we observed previously with our electron microscopy analysis, some measures of mitochondria damage are present in both attP2 and attP40 brains treated with cisplatin, while others are more extensive in attP2 brains. However, attP40 Drosophila exhibit a clear protective effect against cisplatin or general oxidative stress as evidenced by climbing assays, apoptosis analysis, and survival assays. This suggests that attP40 Drosophila neurons are not immune to cisplatin-induced mitochondrial damage but are better able to mitigate or compensate for this damage.
Mitochondria function in attP2 and attP40 flies
To understand how reduced ND-13A expression is protective against cisplatin/oxidative stress, we determined how mitochondria function was affected by this change. We assessed levels of electron transport chain products and substrate. ATP levels were determined from lysates of individual fly heads. Flies with reduced ND-13A did not have a statistically significant change in ATP (Fig. 6A), though levels trend slightly lower in these flies compared with controls. Flies with reduced ND-13A do, however, exhibit a change in NAD+/NADH levels. These flies have an increased NAD+/NADH ratio (Fig. 6B). NADH is a substrate in complex I, and altered NAD+/NADH ratios suggest that these flies may have altered mitochondrial respiration, particularly with regard to complex I activity.
Figure 6.

Mitochondria respiration in attP2 and attP40 Drosophila. A, Quantification of ATP from individual fly heads of the indicated genotypes. Bars represent the mean ATP fluorescence from five individual fly heads. Each data point represents the ATP fluorescence from a single fly head. B, Quantification of the ratio of NAD+/NADH in fly heads of the indicated genotypes. Bars represent the mean calculated ratio from three individual fly heads. Each data point represents the NAD+/NADH ratio from a single fly head. C, Extracellular flux analyzer oxygen consumption rate respiration curves. OCR of attP2 and attP40 mitochondria without (solid line) and with cisplatin treatment (dashed line). Lines represent the mean of four separate assays with five individual reaction wells each. Arrows indicate the time point at which the indicated compound was injected. D–K, Individual mitochondrial respiration parameters for attP2 and attP40 mitochondria with and without cisplatin. All values are calculated from the raw data depicted by the OCR line graph. D, Basal respiration (ADP state 3 OCR – nonmitochondrial OCR). E, Maximal respiration (FCCP uncoupled OCR – nonmitochondrial OCR). F, Reserve capacity (maximal respiration – basal respiration). G, ATP-linked respiration (basal respiration – proton leak). H, Proton leak (oligomycin state 4o OCR – nonmitochondrial OCR). I, Coupling efficiency (ATP-linked respiration/basal respiration). J, Respiratory control ratio (basal respiration/proton leak). K, State apparent [4-(basal respiration-oligomycin OCR)/(maximal respiration-oligomycin OCR)]. All bars represent the mean of four independent experiments each containing five separate assay wells. *p < 0.05, **p < 0.01, ns = not significant compared with control (unpaired Welch's t test).
We then assessed mitochondrial function by measuring OCR in the Agilent Seahorse XF Extracellular Flux Analyzer. We treated attP2 and attP40 flies with and without cisplatin and then isolated mitochondria from whole flies and plated the mitochondria on 24-well plates. We assayed the mitochondria for standard complex I-driven respiration using pyruvate and malate as substrates. ADP, oligomycin, FCCP, and rotenone/antimycin A were used as the injection compounds to assess oxygen consumption rates in an electron flow assay (Fig. 6C). The addition of ADP to isolated mitochondria measures basal respiration (state 3). Oligomycin blocks ATP synthase activity and reveals oxygen consumption that is linked to proton leak (state 4o). FCCP uncouples ATP synthesis from oxygen consumption by disrupting the proton gradient and reveals the maximal respiration capacity of the mitochondria. Rotenone and antimycin A inhibit complex I and block oxygen consumption in the system. The oxygen consumption rates revealed after the addition of each of these substrates can then be used to analyze mitochondrial function.
We observed that in untreated flies, attP2 and attP40 mitochondria do not exhibit strong differences in mitochondrial respiration (Fig. 6C). attP40 flies had slightly higher basal respiration, though this difference was not significant (Fig. 6D). There was no difference in maximal respiration in untreated flies (Fig. 6E). Reserve capacity, calculated by subtracting basal respiration from maximal respiration, trended down in attP40 flies, but the difference was not significant (Fig. 6F). Other measures of mitochondrial activity in untreated flies, including ATP-linked respiration (basal respiration–proton leak; Fig. 6G), proton leak (state 4o; Fig. 6H), coupling efficiency (ATP-linked respiration/basal respiration; Fig. 6I), respiratory control ratio (basal respiration/proton leak; Fig. 6J), and state apparent (a measure of how close to maximum capacity the mitochondria are operating; Fig. 6K) did not have any significant differences between attP2 and attP40 flies. The overall results demonstrate that the mitochondria from both types of fly operate similarly under control conditions and indicate that mitochondria in attP40 background have compensated for the genetic reduction of ND-13A mRNA expression to maintain normal mitochondrial function.
When mitochondria treated with cisplatin were analyzed, significantly greater differences between the strains were apparent. Overall oxygen consumption rates were reduced in attP2 mitochondria, as is expected of mitochondria that are damaged by cisplatin (Fig. 6C). Meanwhile, OCR was generally increased in cisplatin-treated attP40 mitochondria (Fig. 6C). Comparison of attP2 and attP40 mitochondria with cisplatin treatment revealed that attP2 mitochondria have decreased basal respiration (Fig. 6D), as well as reduced ATP-linked respiration (Fig. 6G) and proton leak (Fig. 6H) compared with the attP40 mitochondria. These data indicate that mitochondria in attP40 flies retain and exhibit a trend toward increasing their activity despite the presence of a compound known to be toxic to mitochondria, while attP2 mitochondria trend in the opposite direction. Mitochondria from attP40 flies are capable of maintaining the electron transport chain function in response to cisplatin stress, suggesting a mechanism for the protection of mitochondria from cisplatin in Drosophila.
Role of Sirt1 and peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) in protection against CIPN
Analysis of mitochondria respiration data suggests that the function of the electron transport chain in attP40 flies compared with attP2 flies is largely unchanged under normal conditions. This suggests that the function of the ETC is not directly the key to protection against cisplatin. However, the data showing the retention of mitochondrial activity, mild reduction of membrane potential, and mitochondrial ROS production levels suggest that cells have a mechanism to protect their mitochondria from damage. One mechanism by which cells maintain a healthy pool of mitochondria in response to stress is through biogenesis coupled with clearance of damaged organelles. This process is regulated at the transcriptional level through transcriptional coactivators such as peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α). PGC-1α is activated by Sirt1, which is in turn activated by increased NAD+ (for review, see Onyango et al., 2010). We hypothesized that this regulatory pathway for mitochondria biogenesis is critical for the resistance to cisplatin in attP40 flies.
To determine whether Sirt1 was essential to the protection in attP40 flies, we attempted to knock down Sirt1 in attP40 background using RNAi with the elavGal4 driver. Driving Sirt1 RNAi with elavGal4 in the attP40 background resulted in 100% lethality. There were no viable adult progeny to assess either the knock-down efficiency or sensitivity to cisplatin. Knockdown of Sirt1 in the attP2 background did produce viable progeny (data not shown). These data suggest that Sirt1 is essential for survival in attP40 flies. To further assess the role of Sirt1 in cisplatin sensitivity, we used overexpression (OE) of Sirt1 lines to express high levels of Sirt1 in the brain. OE of Sirt1 is strongly protective against cisplatin, as measured by the climbing assay (Fig. 7A,B). We determined mRNA expression levels of Sirt1 in attP2 compared with attP40 flies by qRT-PCR. Expression of Sirt1 is not elevated at the mRNA level in attP40, while the overexpression line has approximately a twofold increase in mRNA production compared with its background control, w1118 (Fig. 7C). Though Sirt1 mRNA expression is unchanged in untreated attP40 flies, it could still be regulated through post-translational means. Sirt1 is activated by increased NAD+/NADH ratios (Onyango et al., 2010; Anderson et al., 2017), and we observed that attP40 flies have an increase in the NAD+/NADH ratio. Therefore, our data are consistent with the model that increased Sirt1 activity plays a key mechanistic role in protection against cisplatin toxicity.
Figure 7.

Role of Sirt1 and PGC-1α in attP40 cisplatin resistance. A, D, Percentage of flies of the indicated genotypes treated with a range of cisplatin doses climbing over 2 cm in the automated assay. Each bar represents a minimum of five individual assay vials. Individual points indicate the mean result of a single vial of 20–30 flies. B, E, Mean percentage survival of flies treated in the same way as in A and D. Individual points indicate the mean result of a single vial of 40 flies. A, B, Overexpression of Sirt1 in a w1118 background. D, E, RNAi knockdown of PGC-1α (Drosophila spargel) in the attP40 background. C, F, Graph of normalized Sirt1 (C) or PGC-1α (F) expression data in heads from flies of the indicated genotypes, as determined by qRT-PCR. Expression is normalized to control attP2 flies. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns = not significant compared with control (two-way ANOVA with Holm–Sidak correction in A, B, D, and E; unpaired Welch's t test in C and F).
Activated Sirt1 regulates mitochondria biogenesis through the activation of the transcription factor PGC1α. We therefore tested whether PGC1α is important to attP40 cisplatin resistance. We were able to generate PGC1α knockdown flies in the attP40 background with the neuronal elavGal4 driver. This knockdown partially increased sensitivity to cisplatin in attP40 (Fig. 7D,E). We also analyzed mRNA expression of PGC-1α, and we observed that attP40 flies express elevated levels of PGC-1α compared with the control (Fig. 7F). The combination of elevated NAD+ level that can activate Sirt1 and elevated expression of PGC-1α provides key mechanistic insights into cisplatin resistance through increased positive regulation of mitochondria biogenesis and function at the transcriptional level.
Discussion
We have identified a genetic alteration that leads to CIPN resistance in Drosophila. Reduced expression of the electron transport chain complex I subunit ND-13A results in flies that are significantly resistant to cisplatin-induced climbing deficiencies. These flies are protected from the neurologic damage induced by cisplatin treatment but exhibit the same or increased sensitivity to cisplatin when assessing overall survival or damage to rapidly dividing cell types. The protection we observed does not appear to be dependent on one specific subunit of complex I, but, rather, complex I in general as multiple RNAi strains targeting complex I subunits have the same effect.
Flies with reduced ND-13A expression are no more robust than control flies when assessing life span and health span (climbing and fertility) but are significantly more resistant to acute oxidative stress. It will be critical in future studies to further elucidate the mechanism by which this genetic alteration protects neurons. We have reported here that the resistant strain exhibits altered NAD+/NADH ratios and modest changes in mitochondrial oxygen consumption rates when exposed to cisplatin. Complex I knockdown or inhibition has proven beneficial in multiple model systems of aging, disease, and acute oxidative stress (Copeland et al., 2009; Miwa et al., 2014; Zhang et al., 2015). The exact mechanism of this protection is still unclear, but there are many possibilities. The change to complex I may induce a rearrangement of the ETC super complexes to increase efficiency. Increased efficiency in the ETC can lead to increased coupling of oxygen consumption to ATP production and lowered proton leak, which reduces mitochondria-generated ROS. However, we have not observed increased coupling efficiency or reduced proton leak in our flies, suggesting a different mechanism.
Drosophila with reduced ND-13A contain mitochondria that respire in a similar manner as control flies under normal conditions. When exposed to cisplatin, however, basal respiration and ATP-linked respiration increase in response to treatment. The attP40 flies expressing lower levels of ND-13A increase their activity closer to maximum levels without an underlying increase in maximal respiration or reserve capacity, suggesting that attP40 flies maintain a healthy pool of mitochondria when exposed to cisplatin.
One possible mechanism for this protection is our observation that flies with reduced ND-13A have an altered NAD+/NADH ratio, which has been linked to mitochondria health and maintenance through multiple mechanisms (for review, see Ying, 2008). Increased NAD+ blocks ROS generation through NADPH and activation of antioxidant mechanisms like glutathione and catalase (Ying, 2008). Conversely, increased NADH leads directly to elevated ROS through complex I activity. Cisplatin-resistant attP40 flies have elevated NAD+/NADH ratio and increased resistance to acute oxidative stress, indicating that antioxidant capacity may be increased in these flies. The NAD+/NADH ratio is also significantly involved in the regulation of mitochondrial activity, as NAD+ acts as a coenzyme in three steps of the TCA cycle, NADH is a substrate for complex I, and the ratio of the two can influence membrane potential. NAD+ also affects cell signaling and gene expression through pathways such as the sirtuins, which are activated by NAD+ and are involved in regulating both mitochondria biogenesis and function through transcription factors such as PGC-1α (Onyango et al., 2010; Verdin et al., 2010; Anderson et al., 2017; Li et al., 2017).
We propose a model for the observed cisplatin resistance in attP40 Drosophila that is dependent on this mitochondrial regulatory pathway controlled by Sirt1 and PGC-1α (Fig. 8). The data presented here demonstrate that the expression of both Sirt1 and PGC-1α are critical in attP40 Drosophila. RNAi knockdown of Sirt1 in the attP40 background is lethal, while knockdown of PGC-1α reduces resistance to cisplatin. Meanwhile, overexpression of Sirt1 is strongly protective against cisplatin-induced climbing deficiencies. These data, along with the observed increase in NAD+/NADH ratio, a known activator of Sirt1, suggest a model in which increased activation of this pathway protects attP40 neurons from cisplatin mitochondrial damage.
Figure 8.
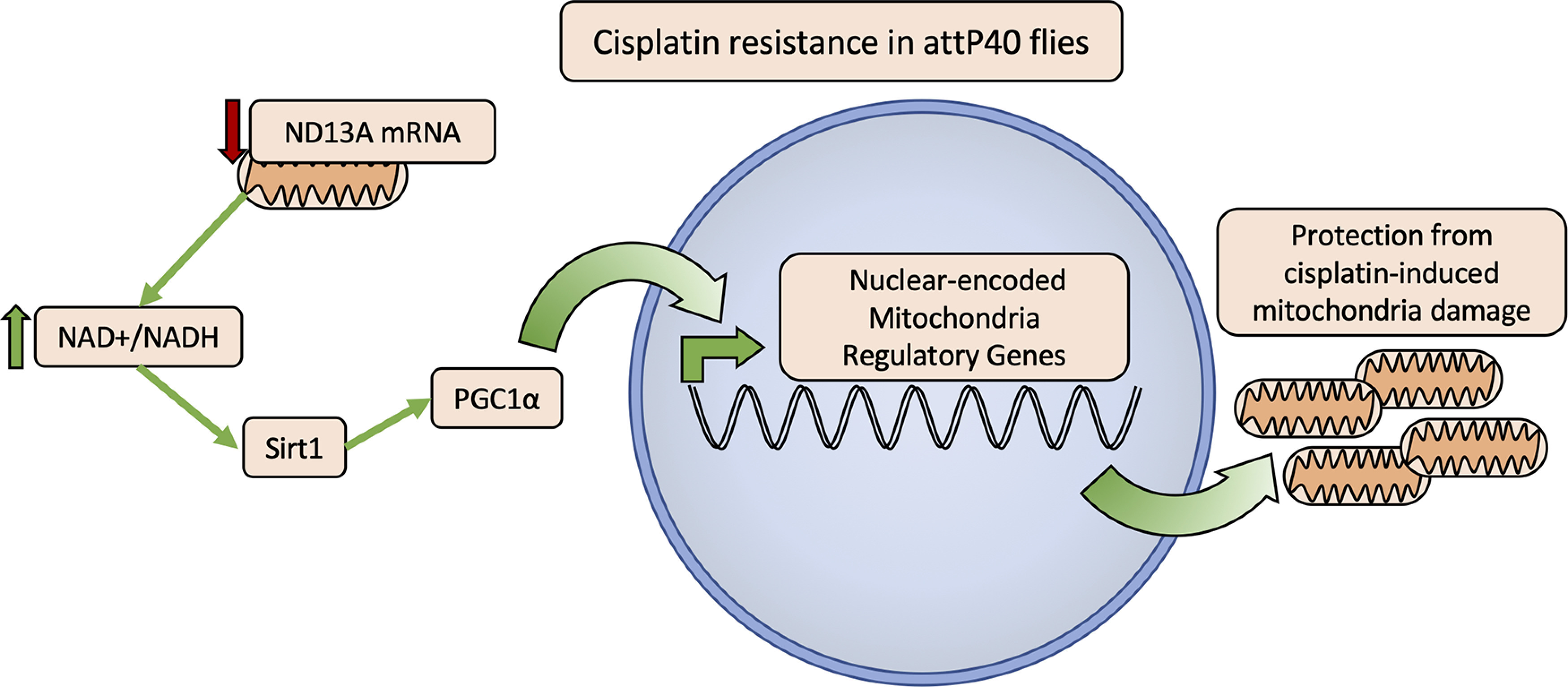
Model of the proposed mechanism of mitochondria protection from cisplatin damage in attP40 genetic background. Flies with reduced ND-13A expression (attP40 or ND-13A RNAi; Figs. 1, 2) have an elevated NAD+/NADH ratio (Fig. 6B). Data showing that overexpression of Sirt1 is protective against cisplatin (Fig. 7A), and PGC-1α knockdown sensitizes attP40 flies to cisplatin (Fig. 7D) suggest a model in which an elevated NAD+/NADH ratio leads to the activation of Sirt1 (Anderson et al., 2017), which activates PGC-1α activity to promote the expression of mitochondrial regulatory genes (Ventura-Clapier et al., 2008). These changes in mitochondrial regulatory genes prepare attP40 flies to respond to cisplatin mitochondria damage to maintain healthy functioning mitochondria.
Additional possible mechanisms for the observed mitochondrial protection remain possible, such as a mitohormetic stress response. Mitohormesis describes a positive adaptive response created by a low level of proton leak and ROS generation that ultimately results in mitochondria that are more resistant to stress (Islam et al., 2012; Ristow and Schmeisser, 2014). Normally, loss of membrane potential by leakage of protons back across the mitochondria membrane is detrimental because it wastes energy and can lead to increased ROS. However, mitochondria may leak protons in a controlled manner through uncoupler proteins (UCPs; Jastroch et al., 2010). Low-level proton leak through UCPs can produce a beneficial response to ROS (Brookes, 2005; Maiese, 2016; Cheng et al., 2017) by initiating a signaling cascade that triggers a mitohormetic stress response (Islam et al., 2012; Ristow and Schmeisser, 2014). Our data demonstrate that reduced levels of ND-13A leads to an adaptive response likely involving changes to mitochondrial function that creates flies that are better prepared to handle an acute stress like H2O2 or cisplatin treatment, though a possible mitohormetic response involving proton leak and ROS generation has yet to be fully explored in this model.
In addition to better understanding of the mechanism of cisplatin damage and resistance, our work here has the potential to affect CIPN research in the clinical setting. Previous efforts to identify susceptible patient populations likely to develop CIPN have identified genes involved with known mechanisms of cisplatin action and cellular clearance such as the glutathione pathway and DNA repair mechanisms (Johnson et al., 2015). Our findings here suggest another group of genes that may be associated with patient risk. Electron transport chain genes and other genes regulating mitochondria function are encoded by both nuclear and mitochondrial DNA. Instead of identifying patients who are at risk of CIPN, mutations such as single nucleotide polymorphisms in mitochondrial genes may show patients that are likely to be resistant to CIPN. Further research into the relationship between mitochondrial regulation and sensitivity or resistance to CIPN would be of great interest to clinical practice. A panel of mitochondrial genes has great potential as a useful screening tool for understanding CIPN susceptibility and resistance in patients.
The findings presented here also have potential to inform development for new preventive CIPN treatments. Cisplatin neurotoxicity is dose dependent (Windebank and Grisold, 2008; Seretny et al., 2014). Therefore, even a partial increase in resistance will reduce the number of patients developing symptomatic neuropathy. A number of drugs either directly target the mitochondria and the ETC or target signaling pathways that affect mitochondrial regulation and function, and may be useful as preventive measures against CIPN. Drugs such as metformin (Yamamoto and Egashira, 2020) and resveratrol (Vrailas-Mortimer et al., 2012) are two such examples that could prove beneficial in CIPN. Targeting mitochondria as a preventive therapy may be more promising than other CIPN treatments that have been unsuccessful in clinical trials. We have reported here that the neuronal defects in flies (climbing ability) are protected by reduced ND-13A, but that cisplatin toxicity in rapidly dividing cells (ovaries) is in fact increased. This separation of protective effects is essential because in order for a CIPN treatment to be effective, it must not protect cancer cells from cisplatin.
The data presented here also suggest the possibility of a nonpharmacologic approach to CIPN prevention. Genetic reduction of complex I may be triggering a mild stress response in the mitochondria of attP40 flies that then prepares the cells for the greater stress of cisplatin treatment. Replicating this mild, protective stress offers possibilities for protection against cisplatin that do not involve drug treatments. Exercise and intermittent fasting are nonpharmacologic stressors for the mitochondria, and both are linked to increased longevity and stress resistance in Drosophila (Magwere et al., 2004; Grandison et al., 2009; Zid et al., 2009; Laye et al., 2015) and a wide range of other experimental models (Anton and Leeuwenburgh, 2013; Madeo et al., 2019). Our findings in Drosophila suggest that research into the protective behavioral changes for CIPN may have value.
The work presented here demonstrates the potential of the CIPN Drosophila model to uncover novel genes affecting cisplatin sensitivity and potential treatment options for CIPN. As we described previously, this work also highlights the critical role of genetic background in any study of cisplatin sensitivity. Further work is necessary to identify the mechanism of protection in complex I knock-down flies and to assess its potential as a therapeutic option for CIPN prevention. In conclusion, we have shown that knockdown of complex I subunits has a strong potential to prevent cisplatin neurotoxicity and to inform new treatments for CIPN.
Resistance to cisplatin-induced climbing deficiencies in attP40 Drosophila. A, C, E, Bars indicate the percentage of flies of each genotype treated with the indicated cisplatin doses able to climb above 2 cm in an automated climbing assay. Each bar in A and C represents the mean of a minimum of five individual assay vials, and each bar in E represents the mean of three individual assay vials. Each individual point indicates the mean result from a single vial of 20–30 flies. A and E represent flies treated with cisplatin in 10% sucrose, and C represents flies treated with cisplatin in solid instant food. B, D, Graph displaying the mean percentage survival of cisplatin-treated flies of the same strains as in A and C, respectively. Individual points indicate the result from a single vial of 40 flies. *p < 0.05, **p < 0.01, ***p < 0.001 compared with control (two-way ANOVA with Holm–Sidak correction). Download Figure 1-1, TIF file (32.1MB, tif) .
Footnotes
We thank Dr. Eugenia Trushina, Dr. Sergey Trushin, and Dr. Padraig Flannery for technical assistance in the measurement of ATP, NAD+/NADH, and mitochondria respiration. We also thank Trace Christensen and Adam Jasperson from the Mayo Clinic Microscopy and Cell Analysis Core for assistance in imaging studies.
The authors declare no competing financial interests.
References
- Abramoff MD, Magalhaes PJ, Ram SJ (2004) Image processing with ImageJ. Biophotonics Int 11:36–42. [Google Scholar]
- Albers JW, Chaudhry V, Cavaletti G, Donehower RC (2014) Interventions for preventing neuropathy caused by cisplatin and related compounds. Cochrane Database Syst Rev (3):CD005228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KA, Madsen AS, Olsen CA, Hirschey MD (2017) Metabolic control by sirtuins and other enzymes that sense NAD(+), NADH, or their ratio. Biochim Biophys Acta Bioenerg 1858:991–998. 10.1016/j.bbabio.2017.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton S, Leeuwenburgh C (2013) Fasting or caloric restriction for healthy aging. Exp Gerontol 48:1003–1005. 10.1016/j.exger.2013.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Areti A, Yerra VG, Naidu V, Kumar A (2014) Oxidative stress and nerve damage: role in chemotherapy induced peripheral neuropathy. Redox Biol 2:289–295. 10.1016/j.redox.2014.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes PS (2005) Mitochondrial H(+) leak and ROS generation: an odd couple. Free Radic Biol Med 38:12–23. 10.1016/j.freeradbiomed.2004.10.016 [DOI] [PubMed] [Google Scholar]
- Cavaletti G, Marmiroli P (2010) Chemotherapy-induced peripheral neurotoxicity. Nat Rev Neurol 6:657–666. 10.1038/nrneurol.2010.160 [DOI] [PubMed] [Google Scholar]
- Cheng J, Nanayakkara G, Shao Y, Cueto R, Wang L, Yang WY, Tian Y, Wang H, Yang X (2017) Mitochondrial proton leak plays a critical role in pathogenesis of cardiovascular diseases. Adv Exp Med Biol 982:359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy DJ (2008) Variation in mitochondrial genotype has substantial lifespan effects which may be modulated by nuclear background. Aging Cell 7:795–804. 10.1111/j.1474-9726.2008.00428.x [DOI] [PubMed] [Google Scholar]
- Conklin KA (2004) Chemotherapy-associated oxidative stress: impact on chemotherapeutic effectiveness. Integr Cancer Ther 3:294–300. 10.1177/1534735404270335 [DOI] [PubMed] [Google Scholar]
- Copeland JM, Cho J, Lo T Jr, Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW (2009) Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol 19:1591–1598. 10.1016/j.cub.2009.08.016 [DOI] [PubMed] [Google Scholar]
- Costa AC, Loh SH, Martins LM (2013) Drosophila Trap1 protects against mitochondrial dysfunction in a PINK1/parkin model of Parkinson's disease. Cell Death Dis 4:e467. 10.1038/cddis.2012.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer SJ, Podratz JL, Windebank AJ (2001) Nerve growth factor rescue of cisplatin neurotoxicity is mediated through the high affinity receptor: studies in PC12 cells and p75 null mouse dorsal root ganglia. Neurosci Lett 308:1–4. 10.1016/s0304-3940(01)01956-5 [DOI] [PubMed] [Google Scholar]
- Grandison RC, Piper MD, Partridge L (2009) Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nature 462:1061–1064. 10.1038/nature08619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groen CM, Podratz JL, Treb K, Windebank AJ (2018) Drosophila strain specific response to cisplatin neurotoxicity. Fly (Austin) 12:174–182. 10.1080/19336934.2019.1565257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M (2012) Drosophila as a model to study mitochondrial dysfunction in Parkinson's disease. Cold Spring Harb Perspect Med 2:a009944. 10.1101/cshperspect.a009944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu LY, Mi WL, Wu GC, Wang YQ, Mao-Ying QL (2019) Prevention and treatment for chemotherapy-induced peripheral neuropathy: therapies based on CIPN mechanisms. Curr Neuropharmacol 17:184–196. 10.2174/1570159X15666170915143217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur JH, Stork DA, Walker DW (2014) Complex-I-ty in aging. J Bioenerg Biomembr 46:329–335. 10.1007/s10863-014-9553-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam R, Yang L, Sah M, Kannan K, Anamani D, Vijayan C, Kwok J, Cantino ME, Beal MF, Fridell YW (2012) A neuroprotective role of the human uncoupling protein 2 (hUCP2) in a Drosophila Parkinson's disease model. Neurobiol Dis 46:137–146. 10.1016/j.nbd.2011.12.055 [DOI] [PubMed] [Google Scholar]
- Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD (2010) Mitochondrial proton and electron leaks. Essays Biochem 47:53–67. 10.1042/bse0470053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C, Pankratz VS, Velazquez AI, Aakre JA, Loprinzi CL, Staff NP, Windebank AJ, Yang P (2015) Candidate pathway-based genetic association study of platinum and platinum-taxane related toxicity in a cohort of primary lung cancer patients. J Neurol Sci 349:124–128. 10.1016/j.jns.2014.12.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laye MJ, Tran V, Jones DP, Kapahi P, Promislow DEL (2015) The effects of age and dietary restriction on the tissue-specific metabolome of Drosophila. Aging Cell 14:797–808. 10.1111/acel.12358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Liu Y, Burns N, Zhao K-s, Song R (2017) SIRT1 is required for mitochondrial biogenesis reprogramming in hypoxic human pulmonary arteriolar smooth muscle cells. Int J Mol Med 39:1127–1136. 10.3892/ijmm.2017.2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionaki E, Markaki M, Palikaras K, Tavernarakis N (2015) Mitochondria, autophagy and age-associated neurodegenerative diseases: new insights into a complex interplay. Biochim Biophys Acta 1847:1412–1423. 10.1016/j.bbabio.2015.04.010 [DOI] [PubMed] [Google Scholar]
- Madeo F, Carmona-Gutierrez D, Hofer SJ, Kroemer G (2019) Caloric restriction mimetics against age-associated disease: targets, mechanisms, and therapeutic potential. Cell Metab 29:592–610. 10.1016/j.cmet.2019.01.018 [DOI] [PubMed] [Google Scholar]
- Magwere T, Chapman T, Partridge L (2004) Sex differences in the effect of dietary restriction on life span and mortality rates in female and male Drosophila melanogaster. J Gerontol 59:3–9. [DOI] [PubMed] [Google Scholar]
- Maiese K (2016) The bright side of reactive oxygen species: lifespan extension without cellular demise. J Transl Sci 2:185–187. 10.15761/JTS.1000138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald ES, Randon KR, Knight A, Windebank AJ (2005) Cisplatin preferentially binds to DNA in dorsal root ganglion neurons in vitro and in vivo: a potential mechanism for neurotoxicity. Neurobiol Dis 18:305–313. 10.1016/j.nbd.2004.09.013 [DOI] [PubMed] [Google Scholar]
- Melli G, Taiana M, Camozzi F, Triolo D, Podini P, Quattrini A, Taroni F, Lauria G (2008) Alpha-lipoic acid prevents mitochondrial damage and neurotoxicity in experimental chemotherapy neuropathy. Exp Neurol 214:276–284. 10.1016/j.expneurol.2008.08.013 [DOI] [PubMed] [Google Scholar]
- Miwa S, Jow H, Baty K, Johnson A, Czapiewski R, Saretzki G, Treumann A, von Zglinicki T (2014) Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice. Nat Commun 5:3837. 10.1038/ncomms4837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira PI, Carvalho C, Zhu X, Smith MA, Perry G (2010) Mitochondrial dysfunction is a trigger of Alzheimer's disease pathophysiology. Biochim Biophys Acta 1802:2–10. 10.1016/j.bbadis.2009.10.006 [DOI] [PubMed] [Google Scholar]
- Ni JQ, Markstein M, Binari R, Pfeiffer B, Liu LP, Villalta C, Booker M, Perkins L, Perrimon N (2008) Vector and parameters for targeted transgenic RNA interference in Drosophila melanogaster. Nat Methods 5:49–51. 10.1038/nmeth1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni JQ, Zhou R, Czech B, Liu LP, Holderbaum L, Yang-Zhou D, Shim HS, Tao R, Handler D, Karpowicz P, Binari R, Booker M, Brennecke J, Perkins LA, Hannon GJ, Perrimon N (2011) A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat Methods 8:405–407. 10.1038/nmeth.1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyango IG, Lu J, Rodova M, Lezi E, Crafter AB, Swerdlow RH (2010) Regulation of neuron mitochondrial biogenesis and relevance to brain health. Biochim Biophys Acta 1802:228–234. 10.1016/j.bbadis.2009.07.014 [DOI] [PubMed] [Google Scholar]
- Onyango IG, Dennis J, Khan SM (2016) Mitochondrial dysfunction in Alzheimer's disease and the rationale for bioenergetics based therapies. Aging Dis 7:201–214. 10.14336/AD.2015.1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccolo J, Kolesar JM (2014) Prevention and treatment of chemotherapy-induced peripheral neuropathy. Am J Health Syst Pharm 71:19–25. 10.2146/ajhp130126 [DOI] [PubMed] [Google Scholar]
- Piper MDW, Partridge L (2018) Drosophila as a model for ageing. Biochim Biophys Acta Mol Basis Dis 1864:2707–2717. 10.1016/j.bbadis.2017.09.016 [DOI] [PubMed] [Google Scholar]
- Podratz JL, Knight AM, Ta LE, Staff NP, Gass JM, Genelin K, Schlattau A, Lathroum L, Windebank AJ (2011a) Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol Dis 41:661–668. 10.1016/j.nbd.2010.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podratz JL, Staff NP, Froemel D, Wallner A, Wabnig F, Bieber AJ, Tang A, Windebank AJ (2011b) Drosophila melanogaster: a new model to study cisplatin-induced neurotoxicity. Neurobiol Dis 43:330–337. 10.1016/j.nbd.2011.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podratz JL, Staff NP, Boesche JB, Giorno NJ, Hainy ME, Herring SA, Klennert MT, Milaster C, Nowakowski SE, Krug RG 2nd, Peng Y, Windebank AJ (2013) An automated climbing apparatus to measure chemotherapy-induced neurotoxicity in Drosophila melanogaster. Fly (Austin) 7:187–192. 10.4161/fly.24789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podratz JL, Lee H, Knorr P, Koehler S, Forsythe S, Lambrecht K, Arias S, Schmidt K, Steinhoff G, Yudintsev G, Yang A, Trushina E, Windebank A (2017) Cisplatin induces mitochondrial deficits in Drosophila larval segmental nerve. Neurobiol Dis 97:60–69. 10.1016/j.nbd.2016.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M, Schmeisser K (2014) Mitohormesis: promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose Response 12:288–341. 10.2203/dose-response.13-035.Ristow [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothballer A, Kutay U (2013) The diverse functional LINCs of the nuclear envelope to the cytoskeleton and chromatin. Chromosoma 122:415–429. 10.1007/s00412-013-0417-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seretny M, Currie GL, Sena ES, Ramnarine S, Grant R, MacLeod MR, Colvin LA, Fallon M (2014) Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: a systematic review and meta-analysis. Pain 155:2461–2470. 10.1016/j.pain.2014.09.020 [DOI] [PubMed] [Google Scholar]
- Shah A, Hoffman EM, Mauermann ML, Loprinzi CL, Windebank AJ, Klein CJ, Staff NP (2018) Incidence and disease burden of chemotherapy-induced peripheral neuropathy in a population-based cohort. J Neurol Neurosurg Psychiatry 89:636–641. 10.1136/jnnp-2017-317215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staff NP, Grisold A, Grisold W, Windebank AJ (2017) Chemotherapy-induced peripheral neuropathy: a current review. Ann Neurol 81:772–781. 10.1002/ana.24951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staff NP, Cavaletti G, Islam B, Lustberg M, Psimaras D, Tamburin S (2019) Platinum-induced peripheral neurotoxicity: from pathogenesis to treatment. J Peripher Nerv Syst 24 [Suppl 2]:S26–S39. 10.1111/jns.12335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufi R, Gandhi S, de Castro IP, Lehmann S, Angelova PR, Dinsdale D, Deas E, Plun-Favreau H, Nicotera P, Abramov AY, Willis AE, Mallucci GR, Loh SH, Martins LM (2014) Enhancing nucleotide metabolism protects against mitochondrial dysfunction and neurodegeneration in a PINK1 model of Parkinson's disease. Nat Cell Biol 16:157–166. 10.1038/ncb2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura-Clapier R, Garnier A, Veksler V (2008) Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res 79:208–217. 10.1093/cvr/cvn098 [DOI] [PubMed] [Google Scholar]
- Verdin E, Hirschey MD, Finley LW, Haigis MC (2010) Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci 35:669–675. 10.1016/j.tibs.2010.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa-Cuesta E, Rand DM (2015) Preparation of mitochondrial enriched fractions for metabolic analysis in Drosophila. J Vis Exp (103):53149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrailas-Mortimer A, Gomez R, Dowse H, Sanyal S (2012) A survey of the protective effects of some commercially available antioxidant supplements in genetically and chemically induced models of oxidative stress in Drosophila melanogaster. Exp Gerontol 47:712–722. 10.1016/j.exger.2012.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windebank AJ, Grisold W (2008) Chemotherapy-induced neuropathy. J Peripher Nerv Syst 13:27–46. 10.1111/j.1529-8027.2008.00156.x [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Egashira N (2020) Drug repositioning for the prevention and treatment of chemotherapy-induced peripheral neuropathy: a mechanism- and screening-based strategy. Front Pharmacol 11:607780. 10.3389/fphar.2020.607780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W (2008) NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal 10:179–206. 10.1089/ars.2007.1672 [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhang S, Maezawa I, Trushin S, Minhas P, Pinto M, Jin LW, Prasain K, Nguyen TD, Yamazaki Y, Kanekiyo T, Bu G, Gateno B, Chang KO, Nath KA, Nemutlu E, Dzeja P, Pang YP, Hua DH, Trushina E (2015) Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer's disease. EBioMedicine 2:294–305. 10.1016/j.ebiom.2015.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zid BM, Rogers AN, Katewa SD, Vargas MA, Kolipinski MC, Lu TA, Benzer S, Kapahi P (2009) 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell 139:149–160. 10.1016/j.cell.2009.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Resistance to cisplatin-induced climbing deficiencies in attP40 Drosophila. A, C, E, Bars indicate the percentage of flies of each genotype treated with the indicated cisplatin doses able to climb above 2 cm in an automated climbing assay. Each bar in A and C represents the mean of a minimum of five individual assay vials, and each bar in E represents the mean of three individual assay vials. Each individual point indicates the mean result from a single vial of 20–30 flies. A and E represent flies treated with cisplatin in 10% sucrose, and C represents flies treated with cisplatin in solid instant food. B, D, Graph displaying the mean percentage survival of cisplatin-treated flies of the same strains as in A and C, respectively. Individual points indicate the result from a single vial of 40 flies. *p < 0.05, **p < 0.01, ***p < 0.001 compared with control (two-way ANOVA with Holm–Sidak correction). Download Figure 1-1, TIF file (32.1MB, tif) .