

Abstract
Histopathologic examination of lungs afflicted by chronic lung allograft dysfunction (CLAD) consistently show both mononuclear cell (MNC) inflammation and mesenchymal cell (MC) fibroproliferation. We hypothesize that interleukin 6 (IL-6) trans-signaling may be a critical mediator of MNC-MC crosstalk and necessary for the pathogenesis of CLAD. Bronchoalveolar lavage (BAL) fluid obtained after the diagnosis of CLAD has approximately 2-fold higher IL-6 and soluble IL-6 receptor (sIL-6R) levels compared to matched pre-CLAD samples. Human BAL-derived MCs do not respond to treatment with IL-6 alone but have rapid and prolonged JAK2-mediated STAT3 Tyr705 phosphorylation when exposed to the combination of IL-6 and sIL-6R. STAT3 phosphorylation within MCs upregulates numerous genes causing increased invasion and fibrotic differentiation. MNC, a key source of both IL-6 and sIL-6R, produce minimal amounts of these proteins at baseline but significantly upregulate production when cocultured with MCs. Finally, the use of an IL-6 deficient recipient in a murine orthotopic transplant model of CLAD reduces allograft fibrosis by over 50%. Taken together these results support a mechanism where infiltrating MNCs are stimulated by resident MCs to release large quantities of IL-6 and sIL-6R which then feedback onto the MCs to increase invasion and fibrotic differentiation.
INTRODUCTION
A major complication after lung transplantation is progressive and irreversible decline in graft function, termed chronic lung allograft dysfunction (CLAD). The prevalence and outcome of CLAD has not changed significantly over the past decade with CLAD developing in 50% of transplant recipients by five years and accounting for over 40% of deaths after the first year of transplantation (1). Histopathologic examination of lungs with CLAD exhibit mononuclear cell (MNC) infiltrates and intraluminal/peribronchial fibrosis with prominent fibroblast proliferation and collagen scarring (2, 3). Consistent with this clinical observation, prior mechanistic studies demonstrate that CLAD is associated with an increase in allo-reactive CD4+ T-cells (4, 5) and mononuclear phagocytic cells (i.e. monocytes and macrophages) (6, 7). Our prior research has shown that donor-derived mesenchymal stromal cells (MCs) within CLAD allografts have upregulation of autocrine lysophosphatidic acid(LPA)–autotaxin(ATX) autocrine signaling (8), elevated cap-dependent translation (9) and resistance to molecular target of rapamycin (mTOR) inhibitors (10). This abnormal signaling results in increased proliferation, mobilization, and fibrotic gene expression (8, 11, 12). While significant progress has been made in understanding the immunologic injury and fibrotic transformation associated with CLAD, crosstalk between infiltrating immune cells and resident mesenchymal cells within the transplanted lung is still obscure.
Interleukin 6 (IL-6) is a pleomorphic cytokine which is produced by multiple cell types to influence a variety of cellular processes including proliferation, cell survival, invasion, fibrotic gene expression, and immune cell differentiation (reviewed in (13)). IL-6 signaling occurs via two similar but mechanistically distinct pathways. The classical pathway or cis-signaling occurs when IL-6 binds the heterodimer of IL-6 receptor (IL-6R) and glycoprotein 130 (gp130) to activate downstream janus kinases (JAKs). IL-6R is exclusively expressed on a small subset of cells (leukocytes, megakaryocytes, hepatocytes) allowing this pathway to regulate acute-phase inflammation, hematopoiesis, and immune cell proliferation/differentiation (14). In contrast, trans-signaling occurs when a soluble form of IL-6R (sIL-6R), produced either by alternative mRNA splicing or proteolytic cleavage by A disintegrin and metalloproteinase 17 (ADAM17), binds to IL-6 in the extracellular space (13, 15–17). The complex then binds gp130 alone to activate JAK and downstream signaling. Since gp130 is ubiquitously expressed, IL-6 trans-signaling can stimulate numerous cells types resulting in diverse physiologic effects. Both classical and trans-signaling pathways result in JAK-mediated phosphorylation of signal transducer and activator of transcription (STAT)-family proteins which dimerize and translocate into the nucleus to influence gene expression (18). STAT3 is the predominant target of IL-6 signaling however, phosphorylation of STAT5 and other targets has been reported (19). STAT3 is known to regulate genes controlling cell proliferation (cyclin B1, cyclin D1), survival (B-cell lymphoma-extra-large, survivin), invasion (matrix metalloproteases 6 and 9), angiogenesis (vascular endothelial growth factor), fibrosis (transforming growth factor beta, collagen I, ATX), and immunosuppression (interleukin 10) (20–22).
Several studies have alluded to a role for IL-6 signaling in the pathogenesis of CLAD. IL-6 is upregulated in bronchoalveolar lavage (BAL) fluid (23, 24), BAL-derived macrophages (25) and serum (26) of patients undergoing acute cellular rejection, a known risk factor for CLAD (1). Lu and colleagues demonstrated that the presence of a high-expression polymorphism in the promoter of the IL-6 gene is associated with an increased risk of CLAD (27) which has been confirmed in at least one other patient cohort (28). Additionally, one study of 43 patients demonstrated elevated IL-6 levels in the BAL fluid of patients with bronchiolitis obliterans (29). In this manuscript, we confirm upregulation of IL-6 trans-signaling within the lungs of CLAD patients and describe a synergistic relationship between infiltrating immune cells and resident MCs which generates IL-6 and sIL-6R leading to allograft fibrosis.
MATERIALS AND METHODS
Patient population and sample collection
Lung transplant recipients at the University of Michigan underwent spirometry every three months and were diagnosed with CLAD following three months of sustained spirometric decline (FEV1 fall of 20% from post-transplant baseline) in accordance with the 2019 International Society for Heart and Lung Transplantation guidelines (1). BAL fluid from surveillance and “for cause” bronchoscopies was collected as previously described (30). The BAL cell pellet was used for cell culture while the supernatant was aliquoted and stored at −80°C. Blood samples were obtained by venipuncture in tubes containing ethylenediaminetetraacetic acid (EDTA; Sigma-Aldrich, St. Louis, MO). This study was approved by the University of Michigan Institutional Research Board and all participants signed informed consent.
Human primary cell isolation and culture
Human lung resident MCs were cultured by resuspending the BAL cell pellet in high-glucose Dulbecco’s modified Eagle medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 100 U/ml penicillin/streptomycin, and 0.5% amphotericin B (Invitrogen) and plated onto standard cell culture dishes (11, 12). Colonies were harvested after 14 days and passaged at least twice prior to use. For all experiments, MCs were grown to 80% confluence and then serum deprived 24 hours. Culture media was removed after 24 hours and used as conditioned media where indicated. Peripheral blood MNCs were isolated via centrifugations through Ficoll-Plaque PLUS (GE Healthcare, Chicago, IL) per established protocol based on (31). Cells were plated in unsupplemented DMEM and all non-adherent cells were washed away after six hours. For coculture experiments, MNCs were seeded into the top chamber of a 0.4μm transwell (Fisher scientific, Waltham, MA), washed twice after six hours to remove all non-adherent cells and then incubated with MCs for 24 hours. Cells were treated with 50ng/mL human recombinant IL-6 (R&D Systems, Minneapolis, MO), 200ng/mL human recombinant sIL-6R (R&D Systems), 1μg/mL neutralizing human IL-6 antibody (R&D Systems), ruxolitinib (Selleckchem, Houston, TX), NVP-BSK805 (Selleckchem), WHP154 (Selleckchem), SH-4–54 (Selleckchem) and 1μM GW280264X (Aobious, Gloucester, MA) as indicated in the text.
Murine orthotopic lung transplant model, histopathologic evaluation, and hydroxyproline assay
Transplantation, histology and hydroxyproline quantification were all conducted as previously described (32). Allogeneic transplants (i.e. allografts) were created by transplanting the left lung of a B6D2F1/J donor mouse (first generation offspring between a DBA/2J and C57BL/6J) into a C57BL/6J or C57BL/6J IL6−/− recipient; syngeneic transplants (i.e. isografts) were created by transplanting the left lung of B6D2F1/J donor mice into a B6D2F1/J recipient. No immunosuppressive or antibiotic medications were utilized pre or post-transplant. For histology, formalin-fixed paraffin-embedded tissue sections were stained with hematoxylin & eosin and Masson’s trichrome (IHC World, Woodstock, MD) per manufacturer’s protocol. Images were collected using an Olympus BX41 microscope and processed using Fiji/ImageJ. For hydroxyproline quantification (described in (8, 12)), the left lung was homogenized in phosphate buffered saline and hydrolyzed in 12N hydrochloric acid for 24 hours at 120°C. Samples were then combined with citrate/acetate buffer, chloramine T solution, and Ehrlich’s reagent before having their absorbance measured at 550nm. All animal studies were approved by the University of Michigan’s Institutional Animal Care and Use Committee.
Statistical Analysis
All data are presented as means ± standard deviation unless otherwise noted. Except where noted in the text, variables were compared using a Students’ t-test (two groups) or one-way analysis of variance with a Tukey post-hoc test (three or more groups). Survival was analyzed using the Kaplan-Meier approach. All statistical analyses were done using Prism 8 (GraphPad, LaJolla, CA) with a p-value less than 0.05 considered significant.
Additional Materials and Methods can be found online in the Supporting Information section.
RESULTS
BAL fluid from CLAD patients exhibit higher levels of IL-6 and sIL-6R.
To determine whether IL-6 trans-signaling was important for the pathogenesis of CLAD, we first compared IL-6 and sIL-6R levels in BAL samples obtained before and after CLAD onset in a cohort of 22 lung transplant recipients (demographics outlined in Table I). The majority of patients (n=16, 72.7%) had higher level of IL-6 in post-CLAD BAL samples compared to matched pre-CLAD lavage fluid with the entire cohort having an average 1.8-fold greater post-CLAD IL-6 levels (p=0.0251, Figure 1A, B). Similarly, 63.6% of patients (n=14) had higher levels of sIL-6R in their post-CLAD BAL samples which was approximately 2-fold higher than pre-CLAD (p=0.0074, Figure 1C, D). Significant correlation was noted between BAL IL-6 and sIL-6R levels (Spearman correlation=0.6085; Figure 1E). To investigate whether IL-6 concentrations could predict development of CLAD, BAL fluid from 40 one-year surveillance bronchoscopies negative for both infection and acute rejection were analyzed. The surveillance BAL cohort was divided into three tertiles based on IL-6 levels and patients in the lowest tertile were noted to have significantly longer CLAD free survival (Figure 1F).
Table I:
Patient demographics and transplant statistics.
| Entire cohort (n=22) | ||
|---|---|---|
| Variable | Pre-CLAD BAL | Post-CLAD BAL |
| Age in years, n (%) | ||
| <30 | 6 (27.2%) | |
| 30–40 | 2 (9.1%) | |
| 40–50 | 3 (13.6%) | |
| 50–60 | 8 (36.4%) | |
| >60 | 3 (13.6%) | |
| Female sex, n (%) | 9 (40.9%) | |
| Pre-transplant diagnosis, n (%) | ||
| COPD/emphysema | 4 (18.1%) | |
| ILD | 8 (36.4%) | |
| Cystic fibrosis | 6 (27.2%) | |
| Other | 4 (18.1%) | |
| Bilateral lung transplant, n (%) | 15 (68.1%) | |
| Days BAL after lung transplant, mean±SD | 418±260 | 1548±1148 |
Abbreviations: COPD-chronic obstructive pulmonary disease, ILD-interstitial lung disease, BAL-bronchoalveolar lavage
Figure 1. BAL fluid obtained from patients with CLAD demonstrate elevated levels of IL-6 and sIL-6R when compared to CLAD free controls.
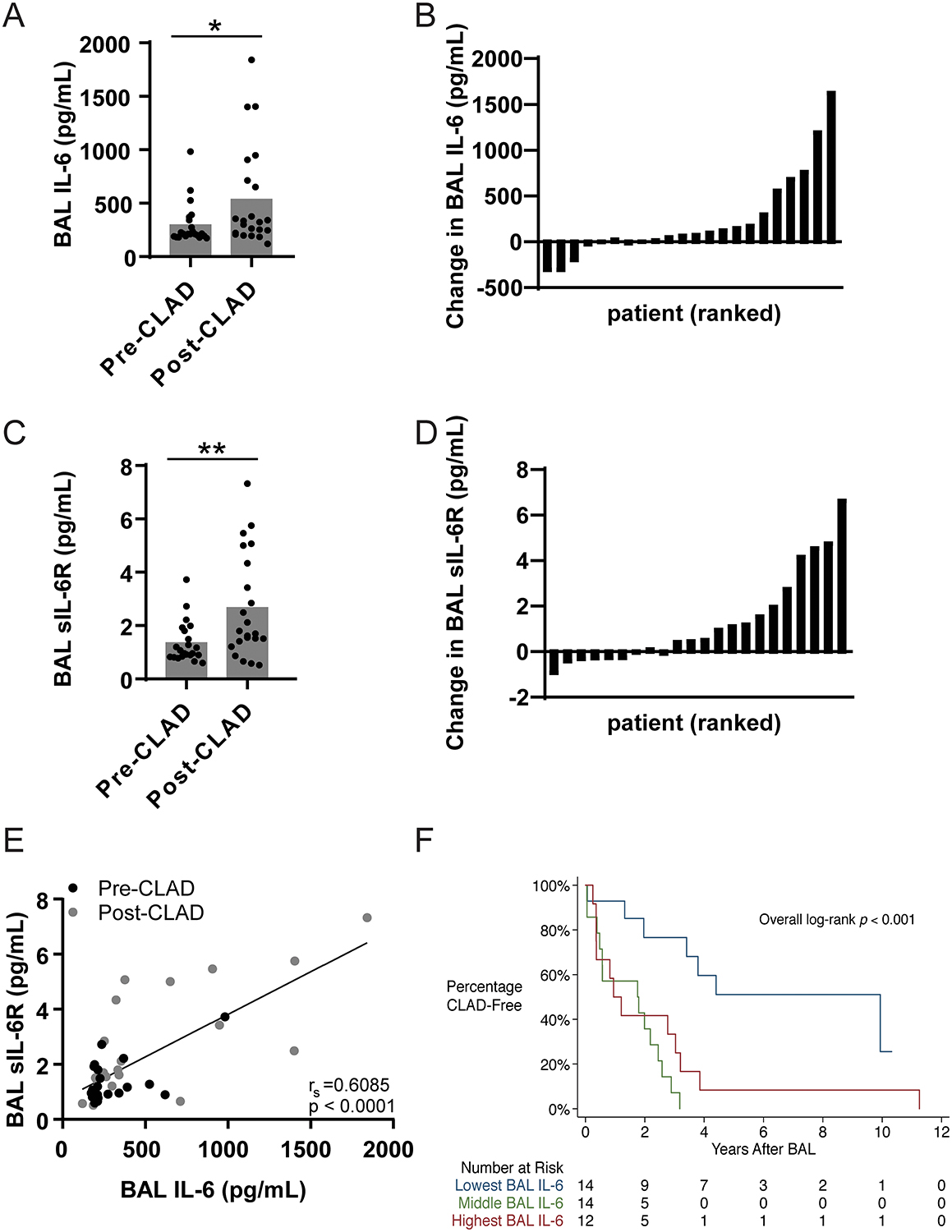
(A) BAL concentrations of IL-6 were measured by ELISA for 22 matched pre-CLAD and post-CLAD samples and were compared using a matched pair Wilcoxon test. Post-CLAD samples had significantly higher IL-6 levels (535.2±476.7pg/mL) compared to CLAD free controls (296.0±193.2pg/mL, Wilcoxon test p=0.0251). (B) Absolute change in BAL IL-6 concentration per patient ranked in ascending order. (C) Post-CLAD BAL samples also had higher levels of sIL-6R (2.670±1.96pg/mL), measured by ELISA, compared to CLAD free controls (1.385±0.77pg/mL; Wilcoxon test p=0.0074). (D) Absolute change in BAL sIL-6R concentration per patient ranked in ascending order. (E) There is a significant linear correlation between IL-6 and sIL-6R levels in both pre-CLAD (Spearman’s correlation=0.3056, p=0.0076), post-CLAD (Spearman’s correlation=0.5011, p=0.0002), and combined groups (shown). (F) The parent cohort of 40 one-year surveillance bronchoscopies was subdivided into tertiles based on BAL IL-6 levels (<180 pg/mL, 180–212 pg/mL, >212 pg/mL) and compared using standard Kaplan-Meyer analysis. Patients in the lowest tertile had significantly longer time to CLAD onset compared to patients in the middle or upper tertiles (media survival 4.402 vs. 1.777 vs. 1.077 years respectively, p=0.001).
JAK2-dependent IL-6 trans-signaling in human lung BAL-derived MCs.
Previous studies have demonstrated that bone marrow-derived MCs produce low levels of IL-6 (33). However, it is unknown whether lung-resident MCs express either IL-6 or sIL-6R. To investigate this further, conditioned media from trypsin-digested cadaveric human lung fibroblasts and BAL-derived MCs from lung transplant recipients were analyzed for IL-6 and sIL-6R concentrations using ELISA. MCs isolated from human lung allografts expressed approximately 10-fold more IL-6 as compared to lung fibroblasts (Figure 2A). However, none of the conditioned media contained any appreciable amount of sIL-6R (Figure 2B).
Figure 2. IL-6 trans-signaling stimulates prolonged STAT3 phosphorylation via a JAK2-dependent mechanism in human BAL-derived MCs.
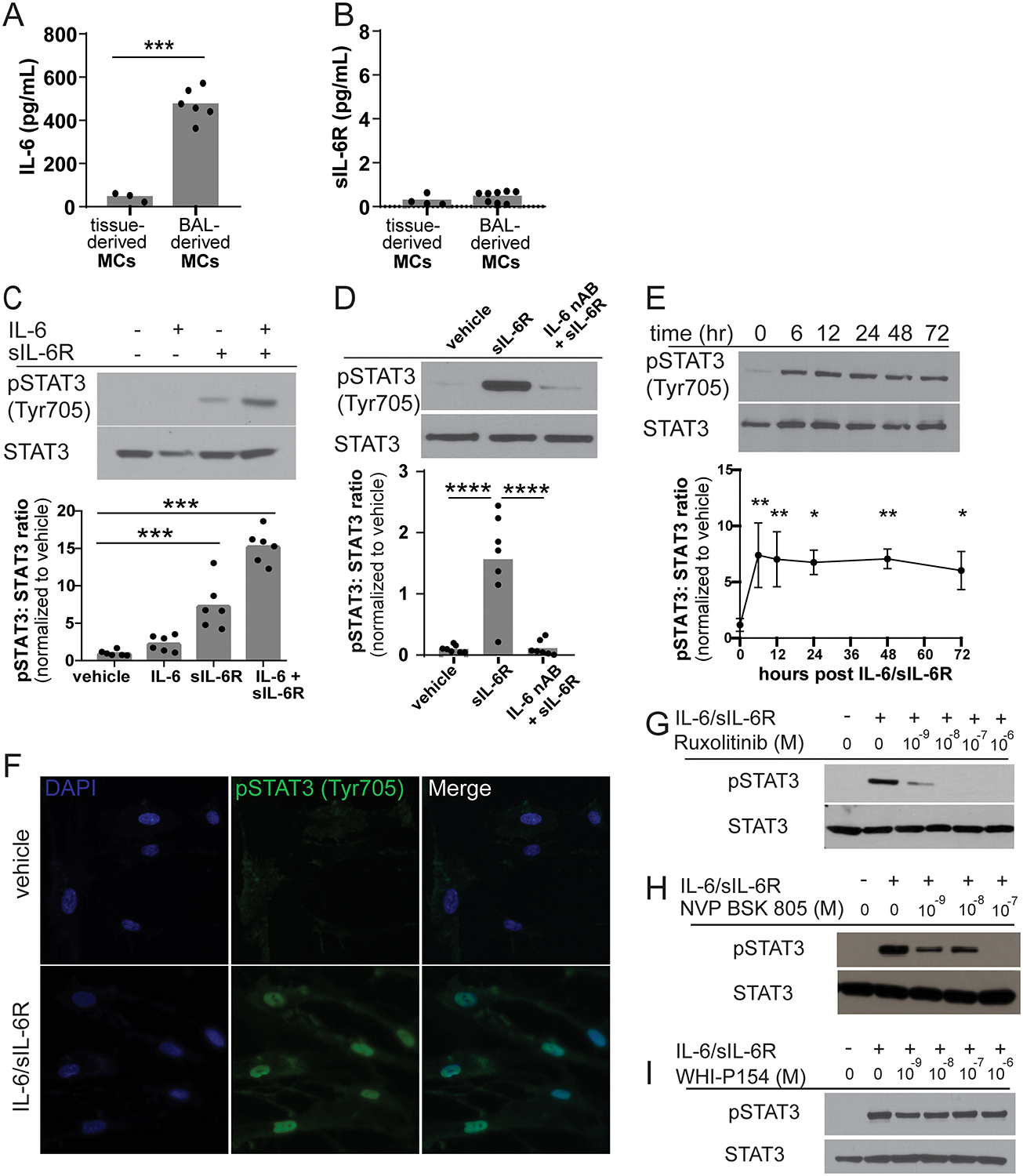
(A) Confluent cultures of pulmonary tissue-derived fibroblasts and BAL-derived MC were grown in serum free media for 24 hours. The media was removed and IL-6 and sIL-6R concentrations were measured by ELISA. Tissue-derived fibroblasts secreted approximately one-tenth of the IL-6 (44.75±20.49pg/mL, n=3) produced by BAL-derived MCs cells (474.5±74.11pg/mL, n=6, p<0.0001). (B) Neither tissue-derived fibroblasts or BAL-derived MCs secrete significant amounts of sIL-6R (0.2749±0.24 pg/mL, n=4 and 0.4600±0.25 pg/mL, n=8 respectively). (C) MCs were treated with vehicle, 50 ng/mL IL-6, 200 ng/mL sIL-6R or the combination of IL-6 and sIL-6R for 30 minutes. Representative western blot and quantification of STAT3 Tyr705 phosphorylation. sIL-6R alone stimulates a 7.5-fold increase in STAT3 Tyr705 phosphorylation while the combination of IL-6 and sIL-6R stimulates approximately 15-fold increase in STAT3 Tyr705 phosphorylation compared to vehicle control (n=6, ***p=0.001). (D) Reduction in endogenously produced IL-6 by addition of an IL-6 neutralizing antibody completely blocks sIL-6R-stimulated STAT3 phosphorylation (0.824±0.54 vs 11.707±7.53 vs 0.400±0.93, ***p < 0.001, ****p<0.0001, n=7). (E) IL-6/sIL-6R treatment induces significant prolonged STAT3 Tyr705 phosphorylation (n=3, *p<0.05 **p<0.01 compared to time 0). (F) MCs grown on coverslips were treated with either vehicle or the combination of IL-6 and sIL-6R for one hour and then labeled by immunofluorescence phospho-STAT3 Tyr705 and DAPI (n=3, representative images shown). IL-6/sIL-6R stimulated phospho-STAT3 was predominantly observed within the nucleus of the cell. MCs were co-treated with IL-6/sIL-6R and various concentrations of the JAK inhibitors (G) ruxolitinib, (H) NVP-BSK805 and (I) WHI-P154. Ruxolitinib (JAK 1/2 pan inhibitor; JAK1 IC50 3.3 nM, JAK2 IC50 2.8 nM) and NVP-BSK805 (JAK 2 specific inhibitor; JAK1 IC50 31.6nM, JAK2 IC50 0.5nM) were able to block IL-6/sIL-6R-mediated STAT3 phosphorylation in a dose dependent manner. WHI-P154 (JAK 3 inhibitor; JAK3 IC50 1.8μM) had no effect on IL-6/sIL-6R stimulated STAT3 phosphorylation (n=3, representative images shown).
We next sought to investigate how lung resident MCs would respond to IL-6 stimulation. MCs from CLAD free patients were treated with vehicle, 50ng/mL IL-6, 200ng/mL sIL-6R or the combination of IL-6 and sIL-6R for 30 minutes. Treatment with both IL-6 and sIL-6R (i.e. full activation of IL-6 trans-signaling) resulted in robust STAT3 phosphorylation on tyrosine 705 (Figure 2C). Treatment with sIL-6R alone was able to stimulate STAT3 Tyr705 phosphorylation, although to a lesser degree. We hypothesized that sIL-6R was interacting with endogenously secreted IL-6 to induce STAT3 phosphorylation. Consistent with this hypothesis, addition of an IL-6 neutralizing antibody was able to prevent sIL-6R-induced STAT3 phosphorylation (Figure 2D). IL-6 expression appeared to be independent of IL-6 trans-signaling as cells treated with vehicle, IL-6 or sIL-6R expressed similar levels of IL-6 mRNA (0.877±0.25 vs 0.906±0.53 vs 1.205±0.64, p > 0.05). Previous studies have demonstrated that the duration of STAT activation is biologically meaningful and trans-signaling is prolonged due to slow sIL-6R internalization (34, 35). Consistent with those observations, IL-6 trans-signaling resulted in sustained STAT3 Tyr705 phosphorylation for up to 72 hours (Figure 2E). While phosphorylation is necessary for STAT3 activation, it is not sufficient as STAT3 needs to dimerize and enter the nucleus to alter gene expression. To evaluate IL-6/sIL-6R-mediated nuclear import, MCs grown on coverslips were stimulated with IL-6 alone or the combination of IL-6/sIL-6R and subjected to immunofluorescent staining for phospho-STAT3 Tyr705. As shown in Figure 2F, treatment with IL-6/sIL-6R stimulates STAT3 Tyr705 phosphorylation, with STAT3 almost exclusively located in the nucleus.
While several JAKs have been shown to mediate IL-6 signaling in different cells types, IL-6 dependent STAT3 phosphorylation is most commonly reported to be mediated via JAK2 (18, 21, 22). To determine which JAK promotes trans-signaling-mediated STAT3 activation in lung MCs, cells were pretreated with various JAK inhibitors. Pretreatment with ruxolitinib eliminated IL-6/sIL-6R-induced STAT3 phosphorylation implicating either JAK1 or JAK2 (Figure 2G). Treatment with the JAK2 specific inhibitor NVP-BSK805 eliminated over 50% of STAT3 phosphorylation at a concentration of 1nM and completely eliminated all STAT3 phosphorylation at a concentration of 100nM suggested that the majority of STAT3 phosphorylation is dependent on JAK2 but there is also a small contribution by JAK1 (Figure 2H). WHI-P154, a JAK3 inhibitor, had no effect (Figure 2I). In summary, IL-6 trans-signaling triggers robust and prolonged STAT3 Tyr705 phosphorylation and nuclear translocation in a predominant JAK2-dependent manner.
IL-6 trans-signaling promotes MC invasion and fibrotic gene expression.
STAT3 transcriptionally activates genes involved in numerous cellular processes including proliferation, survival/resistance to apoptosis, migration/invasion, and fibrosis (20–22). To better characterize the role of STAT3 within MCs, we selected representative genes from known pathways regulated by STAT3 and assessed how their expression changed after stimulation with IL-6/sIL-6R. ATX, collagen 1A and matrix metallopeptidase 9 all showed significant upregulation following treatment with either sIL-6R or the combination IL-6/sIL-6R suggesting activation of migration/invasion and fibrosis pathways (Figure 3A). Despite similar effects on gene expression, only the combination of IL-6 and sIL-6R increased invasion of MCs across a Matrigel-coated transwell compared to vehicle control (Figure 3B–C). In contrast both sIL-6R alone and IL-6/sIL-6R were able to increase collagen protein expression by approximately 1.6-fold and 2.5-fold respectively (Figure 3D–E). While CCND1 (cyclin D1) mRNA was downregulated by IL-6 trans-signaling there was no significant change in cellular proliferation (data not shown).
Figure 3. IL-6 trans-signaling stimulates cellular invasion and fibrotic differentiation.
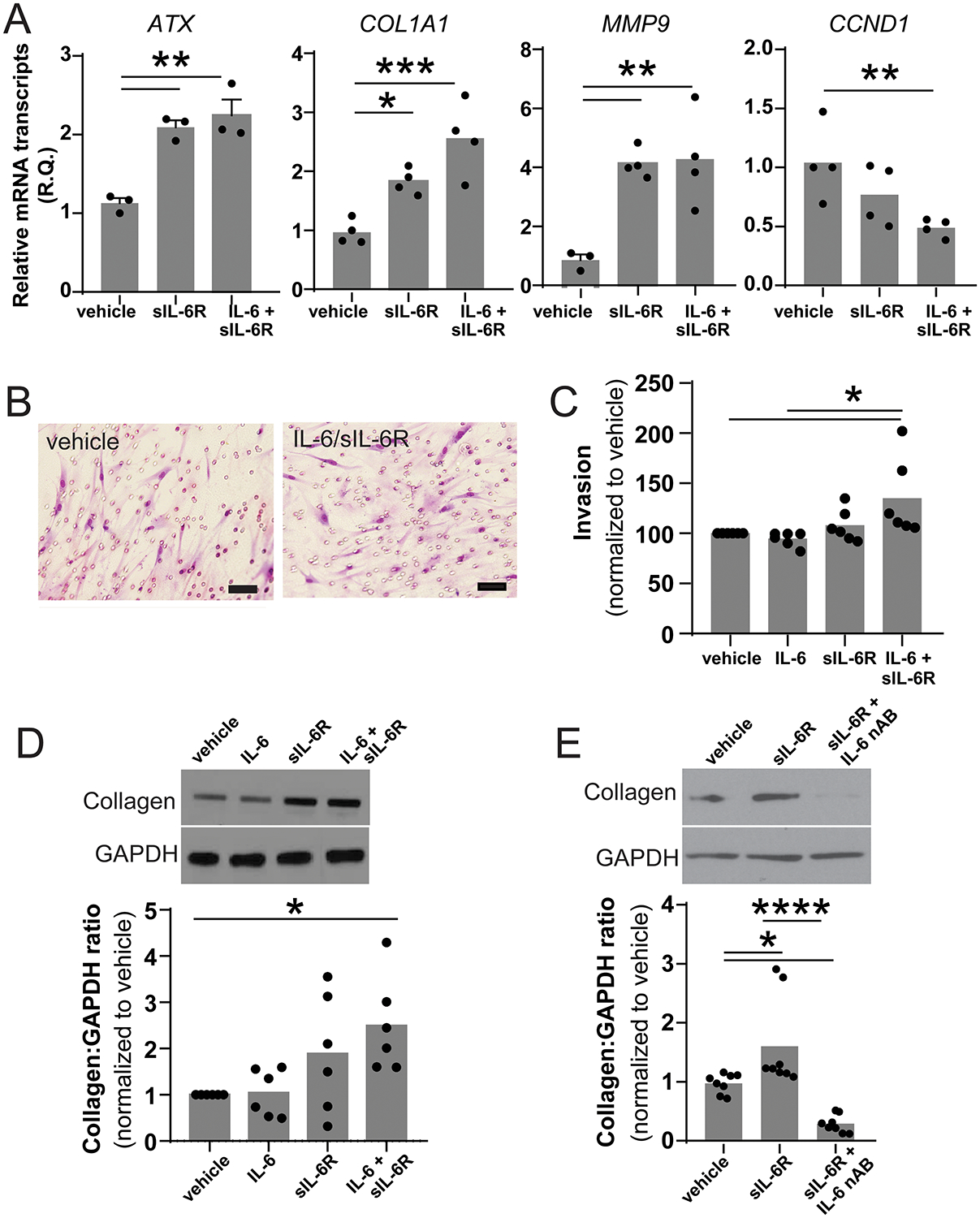
(A) mRNA was harvested from MCs treated with either vehicle or the combination of IL-6 and sIL-6R for 24 hours. Expression of known STAT3-regulated genes was assessed using qPCR (n=4, * p<0.05 ** p<0.01 *** p<0.001). (B) The invasiveness of MC treated with vehicle, IL-6, sIL-6R or IL-6/sIL-6R was measured using a standard matrigel transwell invasion assay and qualitatively assessed by cytology and quantitatively evaluated by MTT assay. Representative image of MC which migrated through the matrigel to the underside of the transwell membrane. (scale bar represents 50 μm). (C) The combination of IL-6/sIL-6R caused a 37% increase in invasion compared to vehicle control (100.0±25.9 versus 136.9±23.14, n=6, p<0.05) (D) Co-treatment with IL-6 and sIL-6R stimulated significant increase in collagen protein expression (1.0 vs 2.492±1.03, n=6, *p<0.05). (E) sIL-6R-induced collagen expression was completed blocked by the addition of an IL-6 neutralizing antibody (1.00±0.18 vs 1.642±0.85 vs 0.295±0.17, n=4, *p<0.05 ****p<0.0001).
MCs stimulate sIL-6R shedding and IL-6 production in peripheral blood-derived MNCs.
Taken together, our studies demonstrate that both IL-6 and sIL-6R are upregulated in the lungs of patients with CLAD and IL-6 trans-signaling strongly stimulates MC invasion and fibrotic differentiation. Since MCs do not express appreciable levels of sIL-6R (Figure 2B), the necessary sIL-6R must be produced by some other cell type in the local microenvironment. Prior studies in mice have shown that over 70% of all circulating sIL-6R is derived from myeloid cells (36) and CLAD lungs show significant infiltration by myeloid MNC cells (15). This led us to investigate the interaction of lung resident MCs with peripheral blood-derived MNCs. Freshly isolated MNCs grown alone express a low level of sIL-6R (Figure 4A). A coculture system of both MNCs and CLAD free MC produced over 40-fold more sIL-6R (Figure 4A). Since MCs do not express the IL-6 receptor, we attributed this increase in sIL-6R exclusively to the MNC population. To explicitly test this assumption, we treated MNC with MC-conditioned media which we previously confirmed lacked sIL-6R (Figure 2B). As seen in Figure 4A, MC-conditioned media was able to stimulate a 7-fold increase in sIL-6R concentration which was higher than baseline but significantly lower than in the co-culture system. Taken together the coculture and conditioned media experiments prove that MC constitutively release a stable signaling molecule which stimulates MNC to increase sIL-6R generation. When cocultured with MCs or exposed to MC-conditioned media, MNCs had no significant change in their IL-6R mRNA expression (data not shown) but did have significant upregulation in ADAM17 mRNA level (Figure 4B). This suggests that sIL-6R may be generated at least partially by cell surface shedding. To investigate this hypothesis further we treated MNC with MC-conditioned media in the presence of GW280264X, an ADAM17 inhibitor. Inhibition of ADAM17 decreased sIL-6R production by approximately 51.4% confirming that ADAM17 is important for sIL-6R generation but is likely not the only mechanism (Figure 4C). Interestingly, MNCs cocultured with MCs also had a greater than 50-fold increase in IL-6 mRNA expression (Figure 4D). Given our prior observation that the combination of IL-6/sIL-6R stimulated a higher degree of STAT3 phosphorylation compared to sIL-6R alone (Figure 2C), increased secretion of IL-6 by MNC should maximize IL-6 trans-signaling in the MC population. Consistent with this hypothesis, MCs in coculture with MNC demonstrated strong persistent STAT3 phosphorylation (Figure 4E, lanes 1 versus 4). This phosphorylation was inhibited by both an IL-6 neutralizing antibody and ruxolitinib confirming it was downstream of IL-6-mediated JAK1/2 signaling (Figure 4E).
Figure 4. Coculture of peripheral blood MNCs and MCs activates both cell types.
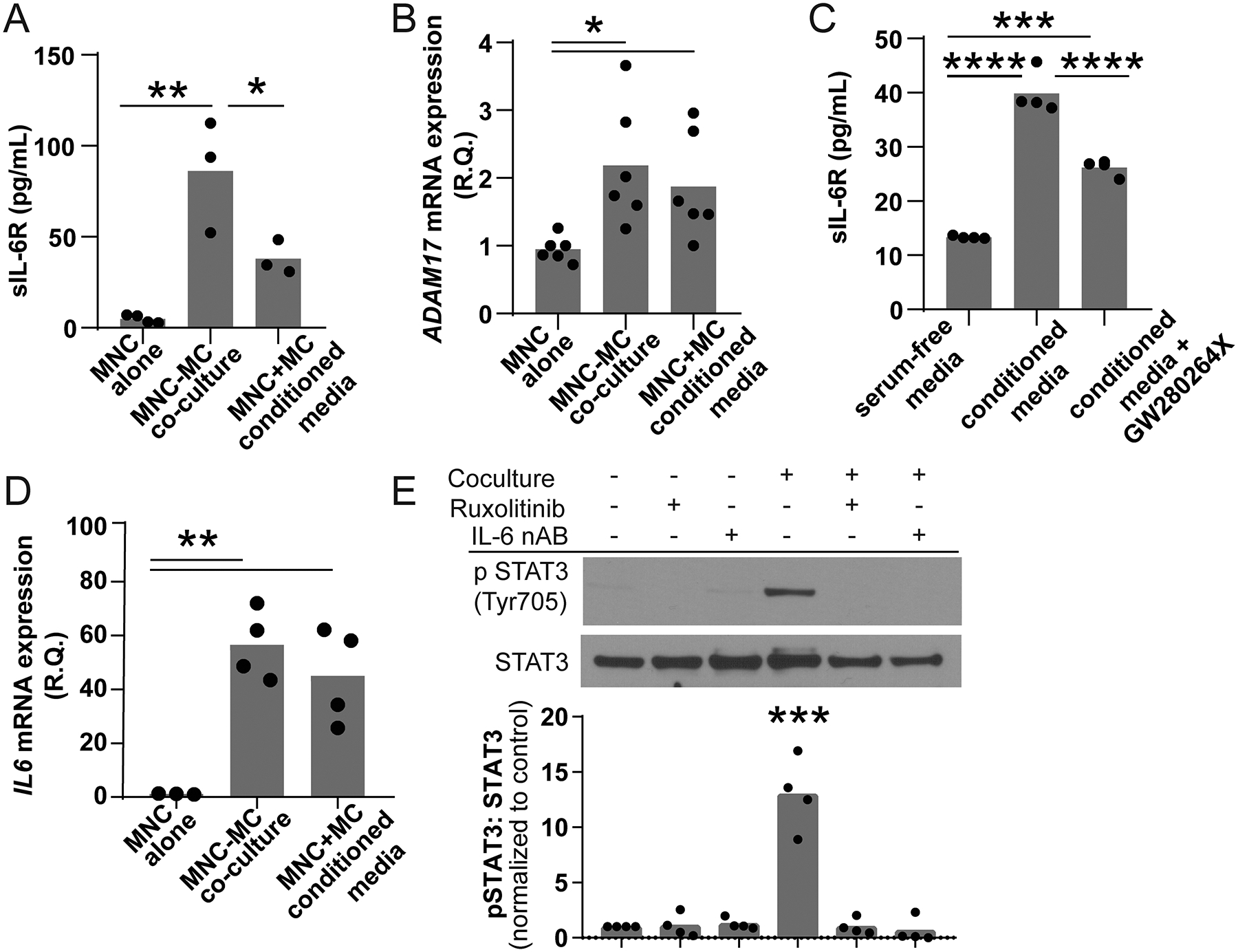
Peripheral blood MNCs (top chamber) were grown in the presence or absence of MCs (bottom chamber) in a transwell containing serum free media for 24 hours. (A) MNCs cocultured with MC produced significantly higher levels of sIL-6R (86.20±30.82, n=3) compared to PBMCs grown alone (4.88±2.20, n=4, **p<0.01). MNCs treated with conditioned media also had an increase in sIL-6R generation that was lower in the coculture model (37.96±9.30, n=3, *p<0.05). (B) Both MNC cocultured with MCs and treated with MC-conditioned media had significant upregulation of ADAM17 mRNA (0.95±0.19 vs 1.875±0.77 vs 2.183±0.9, n=4, **p<0.001). (C) The addition of the ADAM17 inhibitor GW280264X reduced the amount of sIL-6R stimulated by MC-conditioned media by approximately 51% suggesting that membrane shedding was a significant source of media sIL-6R (13.32+0.25 vs 39.85+3.19 vs 26.22+1.47, n=4, ***p<0.001 ****p<0.0001). (D) The presence of MCs also induced a 50-fold increase in MNC IL-6 mRNA expression (1.158±0.14 versus 56.50±12.92, n=3–4, p=0.0008). (E) The lysates of MCs grown in the presence or absence of MNCs, 10 nM ruxolitinib and/or 0.1 μg/mL IL-6 neutralizing antibody were probed for phospho-STAT3 Tyr705. Coculture stimulated robust STAT3 phosphorylation consistent with IL-6 trans-signaling (*** p<0.001, n=4). This phosphorylation was blocked by inhibition of JAK and neutralization of IL-6.
Orthotopic lung transplantation into an IL-6 deficient recipient ameliorates allograft fibrosis.
After demonstrating that human MNCs release IL-6/sIL-6R and stimulate MC invasion and fibrosis, we sought to evaluate the effect of IL-6 signaling in an established mouse model of allograft fibrosis. First, we wanted to confirm that murine cells responded to IL-6 trans-signaling similar to their human counterparts. Primary mouse pulmonary MCs increase STAT3 phosphorylation in response to stimulation with IL-6 and sIL-6R (Figure 5A). Furthermore, coculture of murine MNCs with MCs produces a robust upregulation of IL-6 mRNA within the MNCs (Figure 5B) as well as collagen 1 and ATX within the MCs (Figure 5C).
Figure 5. Blockade IL-6 signaling reduces fibrosis in a murine model of CLAD.
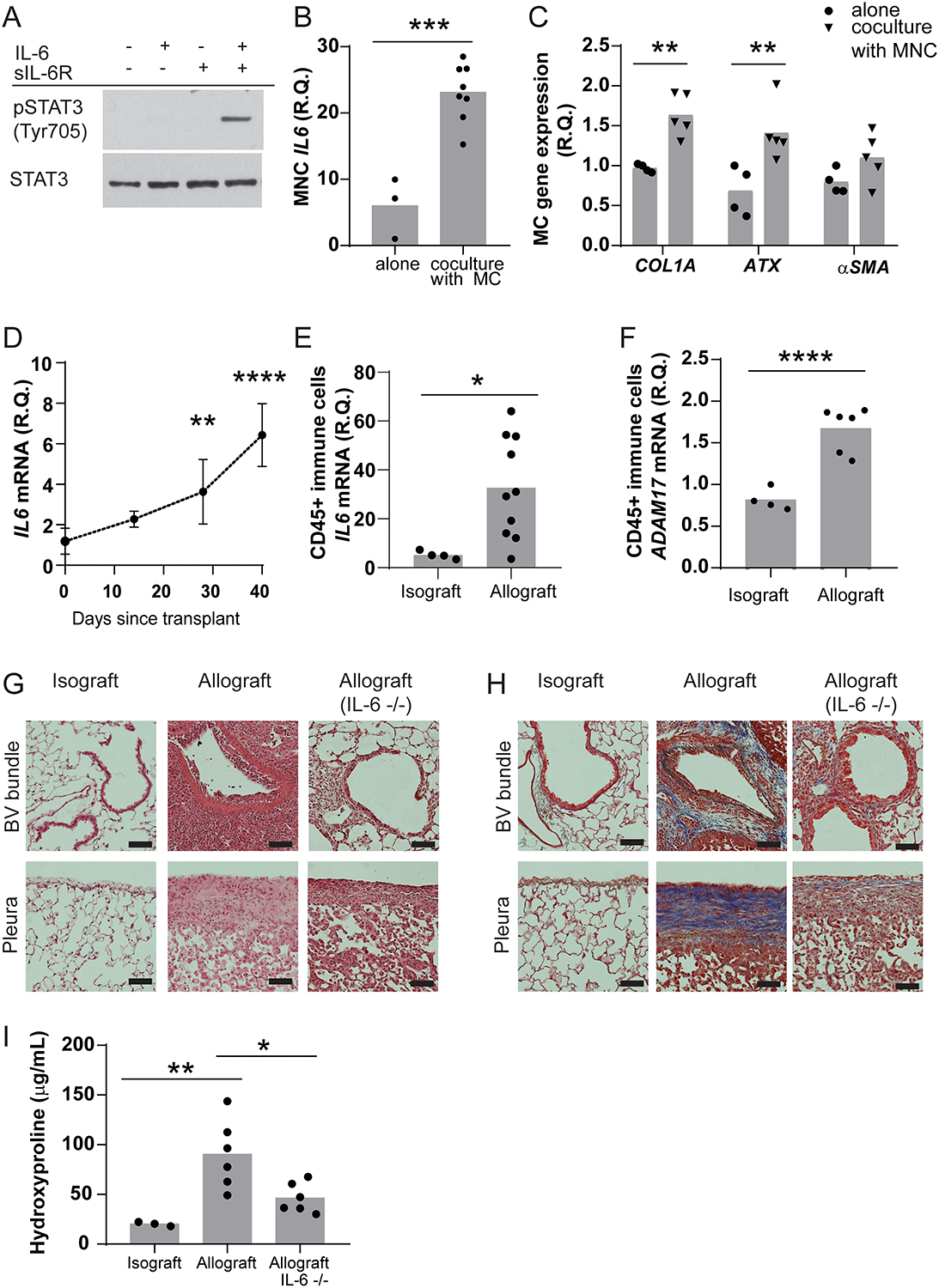
(A) Confluent primary MCs isolated from the lung of B6D2F1/J mice were grown in serum free media for 24 hours and then treated with vehicle, 50 ng/mL IL-6, 200 ng/mL sIL-6R or the combination of IL-6 and sIL-6R for 30 minutes. Only the combination of both IL-6 and sIL-6R induced STAT3 Tyr705 phosphorylation (n=4, representative image shown). (B) Adherent MNCs were isolated from the bone marrow of C57BL/6J and grown in coculture with primary pulmonary MCs for 48 hours and then mRNA was harvested from both compartments. MNC grown in coculture with MCs expressed significantly higher IL-6 mRNA compared to MNC grown alone (6.023±4.58 vs 23.13±4.34, n=3–8, ***p=0.007). (C) The presence of MNC causes MC to express 1.7-fold greater levels of collagen 1 mRNA (0.97±0.05 vs 1.63±0.27, n=4, **p<0.01) and 2.1-fold greater levels of ATX mRNA (0.68±0.31 vs 1.41±0.35, n=4, **p<0.01). Coculture had no effect on α-SMA mRNA expression. (D) The left lung of a B6D2F1/J donor mouse was transplanted into B6D2F1/J recipient (isograft) or a C57BL/6J recipient (allograft). Following transplantation, there was progressive increase in total lung homogenate IL-6 mRNA (n=3–6). At 28 and 40 days post-transplant, IL-6 mRNA expression was 3-fold and approximately 5-fold higher than baseline respectively (1.185±0.65 vs 3.629±1.59 vs 6.427±1.55, *** p=0.008 **** p<0.0001). (E) RNA isolation from CD45+ positive cells derived from either isografts or day 28 allografts showed a significant difference in IL-6 expression (5.211±1.618 vs 32.78±20.80, n=4–10, *p=0.0239). (F) There was also a significant increase in ADAM17 mRNA expression in CD45 positive cells isolated from allografts compared to matched isografts (0.8216±0.13 vs 1.673±0.27, ****p=0.0001). (G) Representative hematoxylin and eosin stained paraffin sections of isograft, allograft and allograft using an IL-6−/− recipient 28 days after transplantation. Allografts have extensive inflammation around the bronchovascular bundle (endothelitis and lymphocytic bronchitis) associated alveolar mononuclear cell infiltration. A mononuclear cell pleuritis was also present. Transplantation into an IL-6−/− recipient resulted in reduced parenchymal and pleural inflammation. (n=3, representative images shown, scale bar represents 500 μm) (H) Isograft, allograft and IL-6−/− allograft paraffin sections stained with trichrome. Compared to isografts, allografts show dense fibrosis around the bronchovascular bundle and along the pleura. This fibrosis was significantly reduced in allografts using an IL-6−/− recipient. (n=3, representative images shown, scale bar represents 500 μm) (I) Allografts had 4.5-fold greater hydroxyproline compared to isografts (20.1±2.1 vs. 90.3±34.7, n=6, **p<0.01). IL-6−/− recipient mice had 51% reduction in hydroxyproline when compared with wildtype allografts (46.3±15.0 vs 90.3±34.7, n=6, *p<0.01).
We have recently characterized an orthotopic lung transplant model of CLAD where transplantation of lung from an B6D2F1/J donor into a C57BL/6J recipient causes progressive peribronchial and pleural fibrosis with high reproducibility (37). In addition to fibrosis, our mouse model recapitulates specific histopathologic aspects noted in a particularly fulminant phenotype of CLAD termed restrictive allograft syndrome (RAS) including lymphocytic infiltration around the bronchovascular bundle, endothelialitis, MNC influx, and fibrinous exudates associated with alveolar damage. Similar to the human results presented in Figure 1, transplanted murine lungs show progressive increases in IL-6 expression as the mouse develops allograft fibrosis (Figure 5D). This increase in IL-6 mRNA expression was also seen in sorted CD45+ immune cells (Figure 5E). Interestingly CD45+ cells also show an increase in ADAM17 expression making it probable that there is a concurrent increase in sIL-6R (Figure 5F). Histology from 28-day old allografts show marked lymphocytic bronchiolitis and proliferation of peribronchial MCs which invade into the adjacent alveolar space (Figure 5G). Similar inflammation and MC proliferation can be seen along the visceral pleura (Figure 5G). This is in stark contrast to the bronchovascular bundle and pleura of isograft lungs which are histologically normal. Overlapping the inflammation in the allograft lungs are dense collagen deposits (Masson’s Trichrome stain, Figure 5H). Transplantation of a donor lung into an IL-6−/− C57BL/6J recipient produces significantly less MC proliferation and fibrotic scarring (Figure 5G, H). Quantification of mature collagen using hydroxyproline assay demonstrated a 50% decrease in collagen content in IL-6−/− recipients (Figure 5I). These data strongly support a role of IL-6 signaling in the progression of lung allograft fibrogenesis.
DISCUSSION
Fibrosis, a key pathologic feature of CLAD, develops as a remodeling response by the donor mesenchymal cells to recipient-derived immune cell inflammation following an alloimmune insult. Despite the interconnected nature of this pathogenic process, the exact crosstalk between different cell populations within the allograft remains poorly understood. In this manuscript, we present a longitudinal series of studies which demonstrates that trans IL-6 signaling is a critical mediator of mesenchymal cell–immune cell crosstalk and CLAD progression. First, we demonstrate that in human lung transplant recipients, BAL levels of IL-6 and sIL-6R consistently increase following the establishment of CLAD. Furthermore, high BAL levels of IL-6 predicts future development of CLAD. Next, we establish that MCs promote IL-6 secretion and sIL-6R shedding by MNCs. This in turn induces vigorous IL-6 trans-signaling in MCs stimulating JAK2-dependent STAT3 phosphorylation and nuclear translocation. Nuclear STAT3 promotes fibrotic activation of the MC by upregulating key fibrotic factors such as ATX and collagen. Finally, in a murine orthotopic lung transplant model, elimination of recipient immune cell IL-6 signaling results in a significant decrease in allograft fibrosis. Overall, these studies support a synergistic model where graft-resident MC and recipient-derived MNCs stimulate each other resulting in IL-6 trans-signaling, MC invasion, and allograft fibrosis.
sIL-6R has been implicated in the development of several autoimmune diseases (e.g. rheumatoid arthritis (38), systemic sclerosis (39)), immune-mediated lung diseases (e.g. asthma (40), COPD (41)), and fibrotic lung disease (42). Our study is the first to link sIL-6R and IL-6 trans-signaling with CLAD. Longitudinal investigation of BAL fluid from lung transplant recipients demonstrated a significant increase in both IL-6 and sIL-6R after CLAD onset. Similar changes were noted in the murine model where IL-6 and ADAM17, a surrogate for sIL-6R, increase over time. As expected, infiltrating immune cells were the predominant source of both IL-6 and sIL-6R. The exact signaling pathway which stimulates immune cells to upregulate IL-6 secretion and sIL-6R shedding is unclear. However, the process that we observed in the lungs was remarkably similar to prior studies which cocultured peripheral blood macrophages and bone marrow-derived mesenchymal stem cells (43, 44). These mesenchymal stem cell-educated macrophages (MEMs) increased the expression of IL-6 and promoted wound healing (43) which is comparable to our activated MNCs which induce invasion and scar formation. Further study of the signaling cascades upstream of IL-6 and sIL-6R generation will not only advance our understanding of CLAD pathogenesis but will also produce prime therapeutic targets.
A key finding of this study is that IL-6 trans-signaling directly activates allograft MCs and upregulates genes associated with invasion and fibrosis. For our studies, we used human primary MCs derived from BAL-fluid which we have previously shown are of donor origin and are the primary contributors to allograft fibrogenesis (12, 30). At baseline, these cells express moderate quantities of IL-6 comparable to mesenchymal stromal cells isolated from other organs (33, 45). Since they lack IL-6R expression there is no STAT3 phosphorylation or autocrine signaling at baseline. However, the production of IL-6 means they are “primed” and exposure to even small amounts of sIL-6R alone can produce rapid and prolonged signaling. This is in contrast to pulmonary fibroblasts obtained via trypsin digestion which do not express IL-6 and require both IL-6 and sIL-6R for STAT3 activation (46–49). This differential response to IL-6 trans-signaling may explain some of the spatial heterogeneity associated with allograft fibrosis. MCs responded to IL-6 trans-signaling with prolonged STAT3 phosphorylation resulting in increased invasion and collagen deposition. Notable and unique among genes upregulated by STAT3 is ATX. ATX secretion by MCs drives their migration and fibrotic differentiation in an autocrine manner via generation of pro-fibrotic lipid mediator LPA. We have previously demonstrated a key role for ATX/LPA signaling in development of CLAD. The potential cooperativity and interplay between these two pathways in driving MC activation and fibrotic differentiation leaves an exciting avenue for future studies aimed at deciphering and targeting interconnected mechanisms of MC activation and allograft fibrogenesis.
In this paper, we propose a model of MC-MNC interactions where the MNCs migrate into the MC microenvironment and are stimulated to release both IL-6 and sIL-6R which feeds back onto the MC to increase the expression of invasive and fibrotic genes. Superficially this model explains why transplantation of a donor lung into an IL-6 deficient recipient results in a reduction of collagen deposition in the allograft lung. However, IL-6 is a pleomorphic cytokine which is known to regulate multiple aspect of immune cell function including B cell maturation, suppression of regulator T-cells, and inhibition of natural killer cells (13, 50). Therefore, IL-6 likely contributes to allograft fibrogenesis via direct stimulation of MCs and indirect alteration of the immune cell populations. Further studies are needed to delineate the effect of IL-6 in regulating other key cellular and biological mediators of graft injury and remodeling.
The critical role of IL-6 signaling in the pathogenesis of both acute pulmonary illness (e.g. COVID-19 infection) and chronic pulmonary disease (e.g. idiopathic pulmonary fibrosis) is increasingly recognized. Our manuscript adds to this growing body of literature by establishing the role of trans IL-6 signaling in progression of allograft fibrosis following lung transplant. While we are the first to establish a role of IL-6 signaling in chronic lung disease following lung transplantation it should be noted that hematologists have been studying the role of IL-6 in chronic lung disease following bone marrow transplant since 2013(51, 52). Currently, there are several FDA-approved medications designed to target parts of the IL-6 signaling pathway including trans-signaling blocker (olamkicept), IL-6 neutralizing antibody (sirukumab, olokzumab, siltiximab), IL-6R neutralizing antibody (tocilizumab, sarilumab) and STAT inhibitors (tofacitinib, ruxolitinib)(13). We believe that our data support further investigations into the role of anti-IL-6 therapies as treatment for CLAD.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health Grants R01 HL118017 (VNL), R01 HL094622 (VNL), T32 HL007749-26 (DSW, MPC), Cystic Fibrosis Foundation Grant LAMA16XX0 (VNL) and the Brian and Mary Campbell and Elizabeth Campbell-Carr research gift fund (VNL).
Abbreviations:
- ADAM17
A disintegrin and metalloproteinase 17
- ATX
autotaxin
- BAL
bronchoalveolar lavage
- CLAD
chronic lung allograft dysfunction
- DMEM
Dulbecco’s modified Eagle medium
- GP130
glycoprotein 130
- IL-6
interleukin 6
- IL-6R
interleukin 6 receptor
- JAK
janus kinases
- LPA
lysophosphatidic acid
- MC
mesenchymal cells
- MNC
mononuclear cells
- mTOR
molecular target of rapamycin
- sIL-6R
soluble interleukin 6 receptor
- STAT
signal transducer and activator of transcription
Footnotes
DISCLOSURE
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- 1.Verleden GM, Glanville AR, Lease ED, Fisher AJ, Calabrese F, Corris PA et al. Chronic lung allograft dysfunction: Definition, diagnostic criteria, and approaches to treatment-A consensus report from the Pulmonary Council of the ISHLT. J Heart Lung Transplant 2019;38(5):493–503. [DOI] [PubMed] [Google Scholar]
- 2.von der Thusen JH, Vandermeulen E, Vos R, Weynand B, Verbeken EK, Verleden SE. The histomorphological spectrum of restrictive chronic lung allograft dysfunction and implications for prognosis. Mod Pathol 2018;31(5):780–790. [DOI] [PubMed] [Google Scholar]
- 3.Martinu T, Howell DN, Davis RD, Steele MP, Palmer SM. Pathologic correlates of bronchiolitis obliterans syndrome in pulmonary retransplant recipients. Chest 2006;129(4):1016–1023. [DOI] [PubMed] [Google Scholar]
- 4.Heng D, Sharples LD, McNeil K, Stewart S, Wreghitt T, Wallwork J. Bronchiolitis obliterans syndrome: incidence, natural history, prognosis, and risk factors. J Heart Lung Transplant 1998;17(12):1255–1263. [PubMed] [Google Scholar]
- 5.Sharples LD, McNeil K, Stewart S, Wallwork J. Risk factors for bronchiolitis obliterans: a systematic review of recent publications. J Heart Lung Transplant 2002;21(2):271–281. [DOI] [PubMed] [Google Scholar]
- 6.Elssner A, Jaumann F, Dobmann S, Behr J, Schwaiblmair M, Reichenspurner H et al. Elevated levels of interleukin-8 and transforming growth factor-beta in bronchoalveolar lavage fluid from patients with bronchiolitis obliterans syndrome: proinflammatory role of bronchial epithelial cells. Munich Lung Transplant Group. Transplantation 2000;70(2):362–367. [DOI] [PubMed] [Google Scholar]
- 7.Magnan A, Mege JL, Escallier JC, Brisse J, Capo C, Reynaud M et al. Balance between alveolar macrophage IL-6 and TGF-beta in lung-transplant recipients. Marseille and Montreal Lung Transplantation Group. Am J Respir Crit Care Med 1996;153(4 Pt 1):1431–1436. [DOI] [PubMed] [Google Scholar]
- 8.Cao P, Aoki Y, Badri L, Walker NM, Manning CM, Lagstein A et al. Autocrine lysophosphatidic acid signaling activates beta-catenin and promotes lung allograft fibrosis. J Clin Invest 2017;127(4):1517–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walker NM, Mazzoni SM, Vittal R, Fingar DC, Lama VN. c-Jun N-terminal kinase (JNK)-mediated induction of mSin1 expression and mTORC2 activation in mesenchymal cells during fibrosis. J Biol Chem 2018;293(44):17229–17239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walker NM, Belloli EA, Stuckey L, Chan KM, Lin J, Lynch W et al. Mechanistic Target of Rapamycin Complex 1 (mTORC1) and mTORC2 as Key Signaling Intermediates in Mesenchymal Cell Activation. J Biol Chem 2016;291(12):6262–6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Badri L, Murray S, Liu LX, Walker NM, Flint A, Wadhwa A et al. Mesenchymal stromal cells in bronchoalveolar lavage as predictors of bronchiolitis obliterans syndrome. Am J Respir Crit Care Med 2011;183(8):1062–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walker N, Badri L, Wettlaufer S, Flint A, Sajjan U, Krebsbach PH et al. Resident tissue-specific mesenchymal progenitor cells contribute to fibrogenesis in human lung allografts. Am J Pathol 2011;178(6):2461–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rose-John S, Winthrop K, Calabrese L. The role of IL-6 in host defence against infections: immunobiology and clinical implications. Nat Rev Rheumatol 2017;13(7):399–409. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol 2014;6(10):a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horiuchi S, Koyanagi Y, Zhou Y, Miyamoto H, Tanaka Y, Waki M et al. Soluble interleukin-6 receptors released from T cell or granulocyte/macrophage cell lines and human peripheral blood mononuclear cells are generated through an alternative splicing mechanism. Eur J Immunol 1994;24(8):1945–1948. [DOI] [PubMed] [Google Scholar]
- 16.Lust JA, Donovan KA, Kline MP, Greipp PR, Kyle RA, Maihle NJ. Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine 1992;4(2):96–100. [DOI] [PubMed] [Google Scholar]
- 17.Mullberg J, Oberthur W, Lottspeich F, Mehl E, Dittrich E, Graeve L et al. The soluble human IL-6 receptor. Mutational characterization of the proteolytic cleavage site. J Immunol 1994;152(10):4958–4968. [PubMed] [Google Scholar]
- 18.Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem 2007;282(28):20059–20063. [DOI] [PubMed] [Google Scholar]
- 19.Tormo AJ, Letellier MC, Sharma M, Elson G, Crabe S, Gauchat JF. IL-6 activates STAT5 in T cells. Cytokine 2012;60(2):575–582. [DOI] [PubMed] [Google Scholar]
- 20.Huynh J, Chand A, Gough D, Ernst M. Therapeutically exploiting STAT3 activity in cancer - using tissue repair as a road map. Nat Rev Cancer 2019;19(2):82–96. [DOI] [PubMed] [Google Scholar]
- 21.Chakraborty D, Sumova B, Mallano T, Chen CW, Distler A, Bergmann C et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat Commun 2017;8(1):1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jarnicki A, Putoczki T, Ernst M. Stat3: linking inflammation to epithelial cancer - more than a “gut” feeling? Cell Div 2010;5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whitehead BF, Stoehr C, Wu CJ, Patterson G, Burchard EG, Theodore J et al. Cytokine gene expression in human lung transplant recipients. Transplantation 1993;56(4):956–961. [DOI] [PubMed] [Google Scholar]
- 24.Iacono A, Dauber J, Keenan R, Spichty K, Cai J, Grgurich W et al. Interleukin 6 and interferon-gamma gene expression in lung transplant recipients with refractory acute cellular rejection: implications for monitoring and inhibition by treatment with aerosolized cyclosporine. Transplantation 1997;64(2):263–269. [DOI] [PubMed] [Google Scholar]
- 25.Rizzo M, SivaSai KS, Smith MA, Trulock EP, Lynch JP, Patterson GA et al. Increased expression of inflammatory cytokines and adhesion molecules by alveolar macrophages of human lung allograft recipients with acute rejection: decline with resolution of rejection. J Heart Lung Transplant 2000;19(9):858–865. [DOI] [PubMed] [Google Scholar]
- 26.Yoshida Y, Iwaki Y, Pham S, Dauber JH, Yousem SA, Zeevi A et al. Benefits of posttransplantation monitoring of interleukin 6 in lung transplantation. Ann Thorac Surg 1993;55(1):89–93. [DOI] [PubMed] [Google Scholar]
- 27.Lu KC, Jaramillo A, Lecha RL, Schuessler RB, Aloush A, Trulock EP et al. Interleukin-6 and interferon-gamma gene polymorphisms in the development of bronchiolitis obliterans syndrome after lung transplantation. Transplantation 2002;74(9):1297–1302. [DOI] [PubMed] [Google Scholar]
- 28.Snyder LD, Hartwig MG, Ganous T, Davis RD, Herczyk WF, Reinsmoen NL et al. Cytokine gene polymorphisms are not associated with bronchiolitis obliterans syndrome or survival after lung transplant. J Heart Lung Transplant 2006;25(11):1330–1335. [DOI] [PubMed] [Google Scholar]
- 29.Scholma J, Slebos DJ, Boezen HM, van den Berg JW, van der Bij W, de Boer WJ et al. Eosinophilic granulocytes and interleukin-6 level in bronchoalveolar lavage fluid are associated with the development of obliterative bronchiolitis after lung transplantation. Am J Respir Crit Care Med 2000;162(6):2221–2225. [DOI] [PubMed] [Google Scholar]
- 30.Lama VN, Smith L, Badri L, Flint A, Andrei AC, Murray S et al. Evidence for tissue-resident mesenchymal stem cells in human adult lung from studies of transplanted allografts. J Clin Invest 2007;117(4):989–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boyum A Isolation of mononuclear cells and granulocytes from human blood. Isolation of monuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand J Clin Lab Invest Suppl 1968;97:77–89. [PubMed] [Google Scholar]
- 32.Mimura T, Walker N, Aoki Y, Manning CM, Murdock BJ, Myers JL et al. Local origin of mesenchymal cells in a murine orthotopic lung transplantation model of bronchiolitis obliterans. Am J Pathol 2015;185(6):1564–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mi F, Gong L. Secretion of interleukin-6 by bone marrow mesenchymal stem cells promotes metastasis in hepatocellular carcinoma. Biosci Rep 2017;37(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peters M, Blinn G, Solem F, Fischer M, Meyer zum Buschenfelde KH, Rose-John S. In vivo and in vitro activities of the gp130-stimulating designer cytokine Hyper-IL-6. J Immunol 1998;161(7):3575–3581. [PubMed] [Google Scholar]
- 35.Groner B Determinants of the extent and duration of STAT3 signaling. JAKSTAT 2012;1(3):211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McFarland-Mancini MM, Funk HM, Paluch AM, Zhou M, Giridhar PV, Mercer CA et al. Differences in wound healing in mice with deficiency of IL-6 versus IL-6 receptor. J Immunol 2010;184(12):7219–7228. [DOI] [PubMed] [Google Scholar]
- 37.Misumi K, Aoki Y, Wheeler DS, Mimura T, Kreisel D, Walker N et al. The Role of B-Cells in an Experimental Murine of Restrictive Allograft Syndrome. AJRCCM 2019;199:A2885. [Google Scholar]
- 38.Polgar A, Brozik M, Toth S, Holub M, Hegyi K, Kadar A et al. Soluble interleukin-6 receptor in plasma and in lymphocyte culture supernatants of healthy individuals and patients with systemic lupus erythematosus and rheumatoid arthritis. Med Sci Monit 2000;6(1):13–18. [PubMed] [Google Scholar]
- 39.Hasegawa M, Sato S, Ihn H, Takehara K. Enhanced production of interleukin-6 (IL-6), oncostatin M and soluble IL-6 receptor by cultured peripheral blood mononuclear cells from patients with systemic sclerosis. Rheumatology (Oxford) 1999;38(7):612–617. [DOI] [PubMed] [Google Scholar]
- 40.Yokoyama A, Kohno N, Sakai K, Kondo K, Hirasawa Y, Hiwada K. Circulating levels of soluble interleukin-6 receptor in patients with bronchial asthma. Am J Respir Crit Care Med 1997;156(5):1688–1691. [DOI] [PubMed] [Google Scholar]
- 41.Ravi AK, Khurana S, Lemon J, Plumb J, Booth G, Healy L et al. Increased levels of soluble interleukin-6 receptor and CCL3 in COPD sputum. Respir Res 2014;15:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yokoyama A, Kohno N, Hirasawa Y, Kondo K, Abe M, Inoue Y et al. Evaluation of soluble IL-6 receptor concentration in serum and epithelial lining fluid from patients with interstitial lung diseases. Clin Exp Immunol 1995;100(2):325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eggenhofer E, Hoogduijn MJ. Mesenchymal stem cell-educated macrophages. Transplant Res 2012;1(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim J, Hematti P. Mesenchymal stem cell-educated macrophages: a novel type of alternatively activated macrophages. Exp Hematol 2009;37(12):1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen L, Tredget EE, Wu PY, Wu Y. Paracrine factors of mesenchymal stem cells recruit macrophages and endothelial lineage cells and enhance wound healing. PLoS One 2008;3(4):e1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moodley YP, Misso NL, Scaffidi AK, Fogel-Petrovic M, McAnulty RJ, Laurent GJ et al. Inverse effects of interleukin-6 on apoptosis of fibroblasts from pulmonary fibrosis and normal lungs. Am J Respir Cell Mol Biol 2003;29(4):490–498. [DOI] [PubMed] [Google Scholar]
- 47.Willis RA, Nussler AK, Fries KM, Geller DA, Phipps RP. Induction of nitric oxide synthase in subsets of murine pulmonary fibroblasts: effect on fibroblast interleukin-6 production. Clin Immunol Immunopathol 1994;71(2):231–239. [DOI] [PubMed] [Google Scholar]
- 48.Eickelberg O, Pansky A, Mussmann R, Bihl M, Tamm M, Hildebrand P et al. Transforming growth factor-beta1 induces interleukin-6 expression via activating protein-1 consisting of JunD homodimers in primary human lung fibroblasts. J Biol Chem 1999;274(18):12933–12938. [DOI] [PubMed] [Google Scholar]
- 49.Fries KM, Felch ME, Phipps RP. Interleukin-6 is an autocrine growth factor for murine lung fibroblast subsets. Am J Respir Cell Mol Biol 1994;11(5):552–560. [DOI] [PubMed] [Google Scholar]
- 50.Cifaldi L, Prencipe G, Caiello I, Bracaglia C, Locatelli F, De Benedetti F et al. Inhibition of natural killer cell cytotoxicity by interleukin-6: implications for the pathogenesis of macrophage activation syndrome. Arthritis Rheumatol 2015;67(11):3037–3046. [DOI] [PubMed] [Google Scholar]
- 51.MacDonald KP, Blazar BR, Hill GR. Cytokine mediators of chronic graft-versus-host disease. J Clin Invest 2017;127(7):2452–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tvedt THA, Skaarud KJ, Tjonnfjord GE, Gedde-Dahl T, Iversen PO, Bruserud O. The Systemic Metabolic Profile Early after Allogeneic Stem Cell Transplantation: Effects of Adequate Energy Support Administered through Enteral Feeding Tube. Biol Blood Marrow Transplant 2020;26(2):380–391. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.