

Abstract
Purpose of review
Despite decades of scientific attention, chronic obstructive pulmonary disease (COPD) remains a major cause of both morbidity and mortality worldwide with strikingly few effective drug classes available. This may be in part because COPD is actually a syndrome composed of distinct diseases with varying pathophysiology (endotypes), and therapies have not been designed to target the causal pathological processes specific to an endotype.
Recent findings
Recent work has begun to clarify the nature of these endotypes and characterize them. One promising field focuses on the central role of the neutrophil and the tripeptide matrikine proline-glycine-proline (PGP) in a subset of COPD patients. Two drugs with mechanisms of action novel to the COPD therapeutic arena (azithromycin and roflumilast) have been shown to reduce acute exacerbations of COPD. Intriguingly, recent evidence has linked both of these agents to modulation of the PGP/neutrophil pathway in concert with this exacerbation reduction, suggesting that a neutrophilic endotype is present and amenable to pharmacological targeting.
Summary
Further work characterizing COPD endotypes, including this neutrophilic endotype, will be important as we strive to understand the mechanistic roots of this disease in the hope of creating more effective therapies.
Keywords: chronic obstructive pulmonary disease, endotype, personalized medicine, proline-glycine-proline, phenotype
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is a syndrome defined by permanently impaired expiratory airflow and characterized by cough, wheezing and exertional dyspnea. Evidence is now mounting that this syndrome we call COPD is composed of multiple distinct disorders, or ‘endotypes’ [1■■,2–10]. Anderson [11] describes an endotype as “a subtype of a condition, which is defined by a distinct functional or pathophysiological mechanism”. Further complicating matters, it is also evident that a given patient with COPD can have an endotype in isolation or in combination with another endotype and thus could be considered to comprise a unique ‘overlap’ endotype (e.g. the now well-recognized Asthma and COPD Overlap Syndrome or ‘ACOS’) [1■■,12]. This is in contrast to clinical phenotypes, which classify patients into groups that share meaningful characteristics predictive of clinical outcomes. The clinical phenotype of chronic bronchitis, for example, can occur both from chronic irreversible asthma and from an airway-predominant neutrophilic inflammatory process [12]. These clinical phenotypes may be enriched for a certain endotype, but are not necessarily descriptive of underlying pathophysiology and therefore are not necessarily predictive of response to potential (ideally curative) therapies.
Despite this disease heterogeneity, the traditional approach to COPD therapeutic development has been to treat patients using an approach derived from studies of the group as a whole [9]. This does have sound rationale; therapies that target disease aspects that are universal in the syndrome (such as airway obstruction) have a broad potential scope. However, as our knowledge deepens of the fundamental differences between endotypes, it is becoming increasingly apparent that such an approach is likely to limit us to disease management rather than disease modification, which is the ultimate goal. In light of this historical approach, we should not be surprised that no drug has been yet found to affect mortality in this common disease, and only a few drug classes exist with any proven efficacy, despite decades of scientific attention [1■■]. This disappointing truth supports the endotyping paradigm as a more promising way to achieve impactful progress in treating this syndrome. However, COPD endotypes have not yet been well classified and therapies specifically targeting the patients suffering from them are generally lacking; for this reason, some have gone so far as to call for COPD to be designated an orphan disease composed of several ‘small COPDs’ [11].
Endotyping can be used not only to predict response to therapy (and ideally to choose optimally tailored therapies) but also to identify risk of clinically relevant outcomes and inform prognosis [1■■]. The perfect endotype would:
Be readily identifiable (ideally using noninvasive or minimally invasive tests) [1■■,6].
Allow for detailed prognostication of clinically meaningful outcomes (symptoms, exacerbation, mortality and so on) [1■■,3].
Define pathophysiology that allows us to accurately predict response to specific therapies and/or design others [1■■,6].
Therefore, we must employ strategies designed to differentiate patients into meaningful groups using the tools at our disposal [1■■,3]. Such strategies include physiologic criteria, clinical characteristics, radiographic techniques, biomarker assays, genotype surveys including genotype/phenotype correlations, as well as metabolomics and proteomic analysis [1■■,2–6,12]. These strategies must also include longitudinal outcomes, as cross-sectional approaches cannot inform us as to whether a group of patients appears distinct because they suffer from a different disease subtype or because they are at different stages or states of disease activity within the same endotype [2,7,9,13■]. Although work has been done and much more is ongoing to categorize COPD patients using approaches such as cluster analysis and longitudinal observation, the number and nature of COPD endotypes is presently unknown. However, early returns on such techniques [3,8,10,14■■] are sufficiently encouraging that such subclassification will be possible and may lead to useful endotypes that meet the afore-mentioned criteria.
ENDOTYPING CHRONIC OBSTRUCTIVE PULMONARY DISEASE
A number of COPD subgroupings have been described and are summarized in Table 1. It is interesting to note that, though patient groups can be parsed in many ways, only one subgroup (α−1 antitrypsin deficiency or A1AT) has been indisputably characterized to meet all three of the criteria above for an ideal endotype. Indeed, some of these subphenotypes are likely composed of patients with varied pathophysiology; that is some categories may be useful but not necessarily descriptive of the underlying disease process or endotype from which a patient suffer [6,12]. As shown in Fig. 1, A1AT serves as a useful prototype for our understanding of this concept. Although A1AT disease is unified by its reasonably well-delineated underlying pathophysiology, its manifestations vary between patients. It is classically characterized by lower lobe predominant emphysema with bronchiectasis. However, we now know that its presentation can vary because of exposures, risk factors including age and patient idiosyncrasies. A1AT deficiency can and often does present with apical emphysema, with spirometric airflow obstruction in the absence of significant radiographic emphysema, and, especially in younger patients, with bronchodilator-reversible airflow obstruction similar to asthma [15]. These discoveries underscore the difficulty in differentiating the underlying pathophysiology from the clinical phenotype of a patient.
Table 1.
Chronic obstructive pulmonary disease phenotypes. Numerous methods of patient phenotyping have been pursued, with varying levels of success. Although some methods do seem to distinguish between endotypes, some of them merely enrich for certain endotypes and some of them may select for subpopulations within multiple endotypes, thus ultimately failing to define useful underlying pathophysiology or direct therapy
| Phenotype | Identifying characteristic | Predictive of response to specific therapy | Specific to COPD endotype (i.e. pathophysiology of airflow obstruction) | Predictive of clinical course? |
|---|---|---|---|---|
| A1AT deficiency | Genotype or low Al AT activity | A1AT replacement | Yes | Yes |
| Frequent exacerbator | Clinical | Azithromycin, PDE4 inhibition | No | ↑ Exacerbations by definition |
| Upper lobe predominant emphysema | Radiographic | Lung volume reduction | No | ↑ Survival (if LVRS) |
| Pure emphysema | Low sputum volume or purulence, ↓ BW:AW | No | No | ↑ Spirometric decline |
| Chronic bronchitis | High sputum volume or purulence, ↑ BW:AW | PDE4 inhibition for FEV<50% | No | ↑ Symptoms and mortality |
| Pulmonary vascular | ↑ PAPm, PA:A | Unknown | No | ↑ Exacerbations |
| High systemic inflammation | ↑ Laboratory markers of serum inflammation | Unknown | Possibly | ↑ Symptoms and mortality |
| Steroid responsive | May be predicted by: FEV1 variability, BDR, eosinophilia and other TH2 markers, ↑ Bronchial BM thickness | ICS, by definition | ? Irreversible asthma | Unknown |
| ACOS | Varied definitions; often early onset fixed airflow obstruction | Disputed | May enrich for asthma/ICS responsive + early-onset COPD | ↑ Symptoms and mortality |
A1 AT, alpha 1 antitrypsin; ACOS, asthma-COPD overlap syndrome; BDR, bronchodilator responsiveness; BM, basement membrane; BW:AW, bronchial wall: artery wall thickness; CT, computed tomography; FEV1, forced xpiratory volume in 1-s; ICS, inhaled corticosteroid; LVRS, lung volume reduction surgery; PAPm, mean pulmonary arterial pressure; TH2, type-2 helper T-cell.
FIGURE 1.
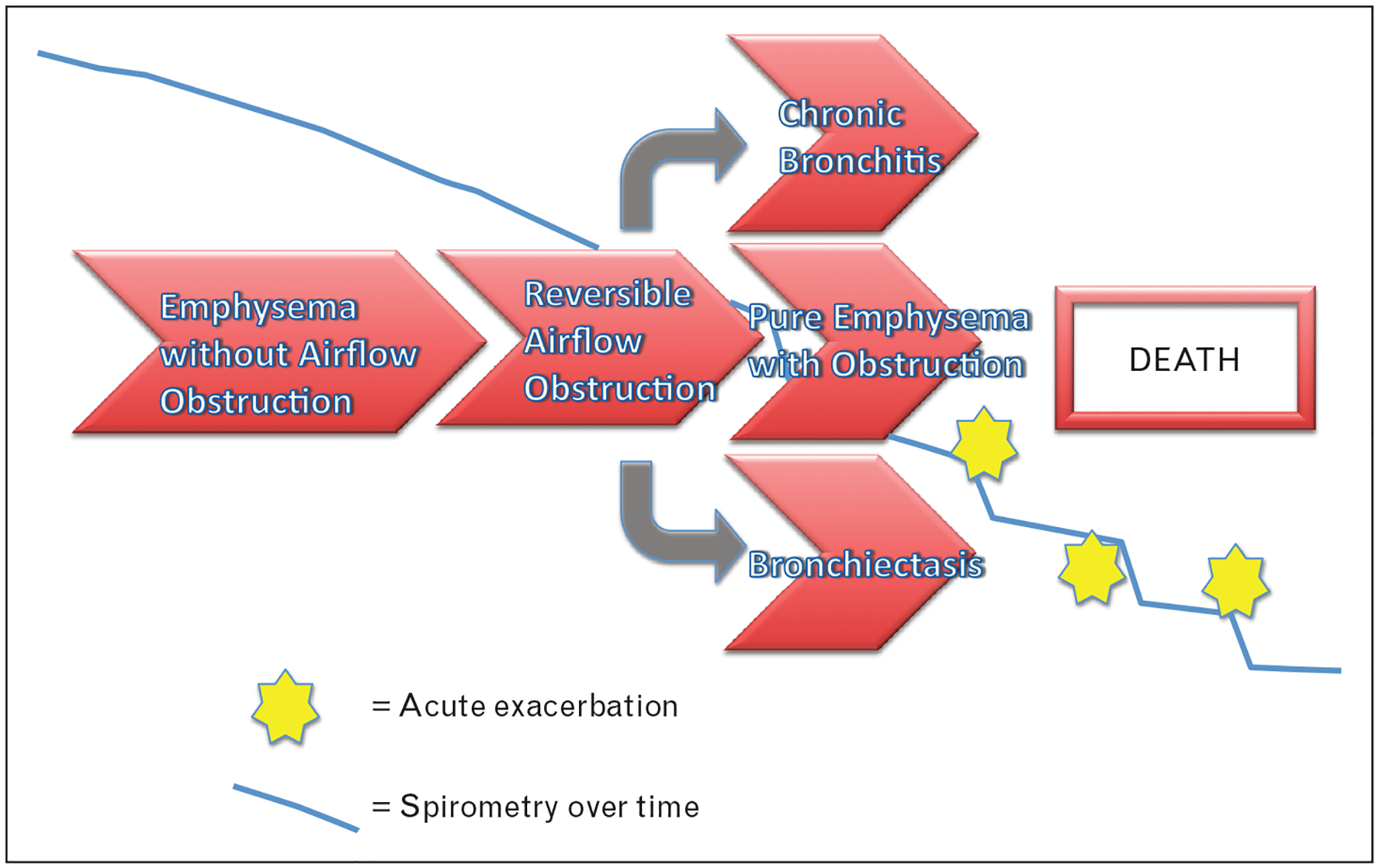
Alpha-1 antitrypsin-deficiency endotype. Alpha-1 antitrypsin deficiency is the first well-defined COPD endotype. Although the underlying pathophysiology of reduced or absent alpha-1 antitrypsin activity leading to unchecked proteolytic activity unifies the disease, its presentation can vary widely. It is now known that patients can present with reversible airflow obstruction (mimicking asthma), with upper-lobe predominant emphysema (which can be spirometrically silent), or with the phenotypes more classically associated with this endotype such as bronchiectasis and lower-lobe dominant pan-lobular emphysema. The interplay between numerous factors including patient age, smoking status, history of exacerbations and bacterial colonization is thought to influence the presentation and rate of progression despite the fact that these patients all share the same genetic defect. This illustrates the difference between clinical phenotype and endotype.
This interaction of endotype, risk factors and time course/stage of disease is complex as illustrated in Fig. 2. Although not fully characterized, a few putative COPD endotypes do appear as themes in the literature. One is the TH2-driven group, which appears to display many features in common with eosinophilic asthma and may represent an advanced stage of the latter disease with or without concurrent smoking-related emphysema. The TH2 endotype appears to have a more pronounced response to corticosteroid therapy and promising TH2-targeted therapies designed for asthma could potentially lead to breakthroughs in this cohort [7,12]. Another is the subgroup with high systemic inflammation (as measured by any number of biomarkers especially serum fibrinogen), which may lend itself to further study of immunotherapy directed at suppressing what is thought to be a state of pathologic immune activation or perhaps autoimmunity [7,14■■,16–21]. An additional important category of patient in terms of healthcare burden seems to be the highly symptomatic COPD patient with relatively intact spirometric lung function but a high number and severity of comorbid conditions [8,22]. This group suffers from high symptom burden and mortality, but whether it constitutes an endotype of COPD with unique pulmonary pathophysiology is not clear as their clinical course is not as well predicted by degree of airflow limitation as the other groups and could be driven by the comorbid conditions which happen to share risk factors with COPD [6]. Yet, another very important COPD subphenotype is chronic bronchitis. This group also suffers disproportionately from frequent exacerbations as well as a high symptom burden and high mortality [10,23]. Chronic bronchitis is not felt to be an endotype per se; it seems likely to represent a common presentation of multiple airway-centric endotypes including irreversible asthma [12], but this phenotype does appear to be enriched for responsiveness to a novel-targeted therapy, the phosphodiesterase 4 inhibitor roflumilast [24,25], which suggests the presence of an embedded endotype with unique pathophysiology. Emerging evidence suggests that the activity of roflumilast may be mediated by modulation of neutrophilic inflammation [26■] begging the question of whether this roflumilast-responsive group may be an endotype with prominent neutrophilic inflammation driving the disease. Indeed, neutrophils have long been known to be important drivers of COPD pathophysiology [27–32], and that they may be especially critical mediators in a subset of COPD patients has been speculated for some time [33]. As recent data suggest that activity against neutrophilic processes may account for the effectiveness of two of the most important recent pharmacological breakthroughs in COPD, roflumilast and azithromycin [26■,34,35], this does seem likely to meet the qualifications of an important endotype when the underlying pathophysiology is characterized further. The remainder of this discussion will focus on what is known of this potential endotype and how it might be targeted.
FIGURE 2.
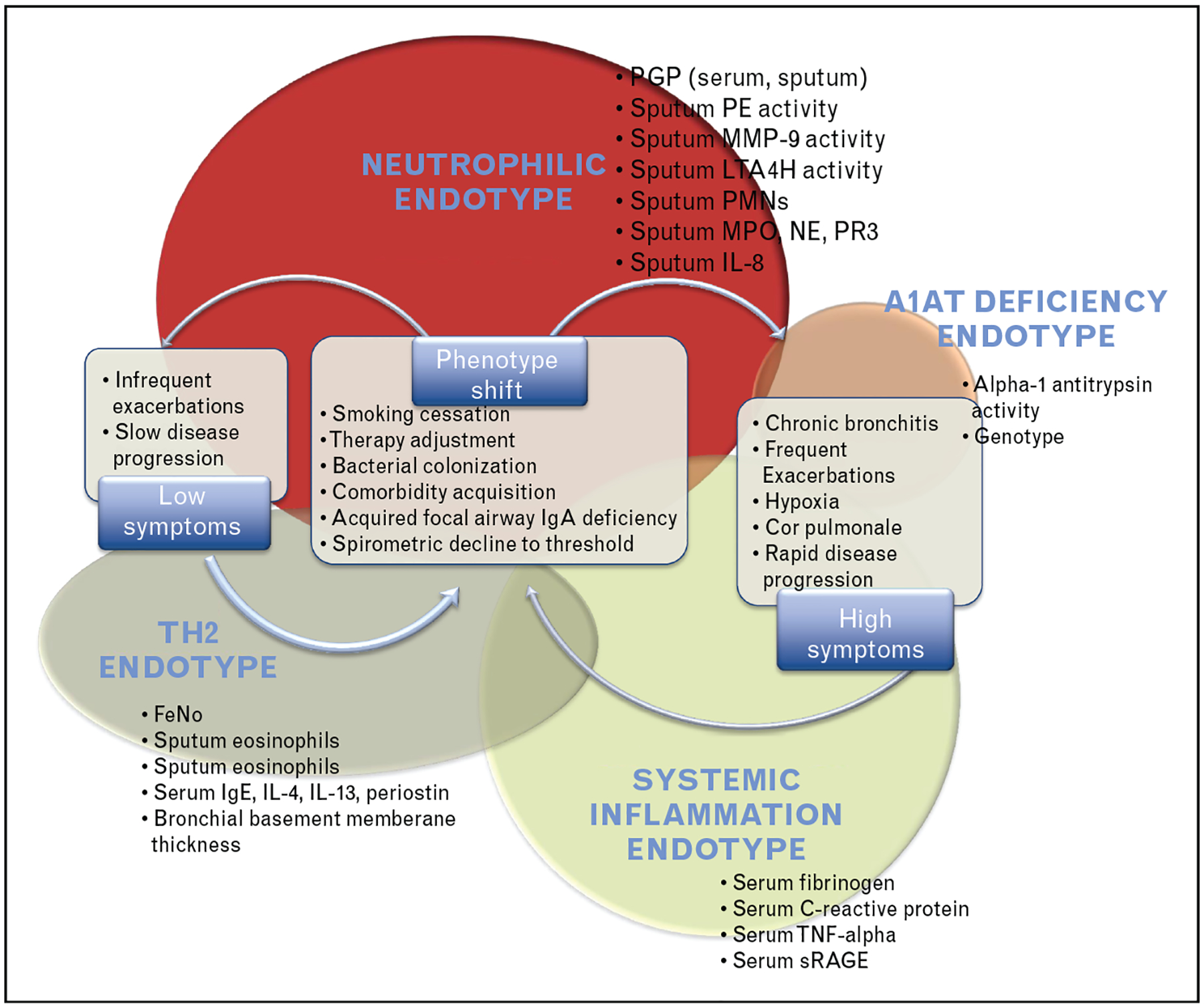
Many endotypes of COPD. In the new paradigm of COPD, the many ‘small COPDs’ are largely separate diseases with distinct mechanisms but they share phenotypic characteristics. Phenotype varies within an endotype in part because of patient factors such as smoking status, bacterial colonization and comorbidities. Here are depicted a few candidate endotypes and some proposed biomarkers for each. Note that this is not a comprehensive list of endotypes, but the total number and relative prevalences are unknown so these are chosen as examples. Note also that there are patients who suffer from multiple endotypes (‘overlap syndromes’); these subgroups have not yet been defined adequately to know how much overlap there is and between which endotypes. Improved biological assays hold promise in allowing us to identify not only which endotype a patient belongs to, but also which subphenotypes they currently possess. As we unravel the underlying pathophysiology of these endotypes, we become increasingly able to identify a patient’s endotype and thus address their disease, with the ultimate goal of shifting their phenotype to a lower level of symptoms and slower rate of progression.
THE NEUTROPHILIC ENDOTYPE
Emphysema is thought to ultimately be a result of imbalance between tissue destruction (especially proteolysis) and tissue repair leading to excessive extracellular matrix (ECM) digestion [36]. This aberrant proteolysis could be driven by any of multiple cell types or a combination thereof, and increased number and activity of several different cell types with capacity for such activity has been noted in COPD patients [36,37■]. In many COPD patients, this disease appears to be driven in large part by neutrophilic inflammation [27–32]. It has long been recognized that many COPD patients demonstrate increased sputum and peripheral blood neutrophilia and that a subset of patients with COPD has elevated markers of neutrophilic inflammation including the classic neutrophil chemokine interleukin 8 (IL-8) in blood or sputum [7]. There is evidence that neutrophils of these patients are, by many measures, in an abnormal state of excitation both locally (i.e. in the lungs) and systemically [33,38–40].
It is perhaps intuitive that such an inflammatory state could be driven by the noxious stimulus of smoking; however, this neutrophilic inflammation appears to persist after tobacco cessation in some COPD patients [41]. The impetus of this inflammation is unknown, but an intriguing hypothesis has been posited that this could be the result of parenchymal destruction (presumably initiated by smoke-induced local inflammation but self-perpetuating) leading to production of connective tissue breakdown products with activity at cytokine receptors (i.e. matrikines) [37■,42]. Such a pathway has been demonstrated for the tripeptide collagen breakdown product proline-glycine-proline (PGP) and its more potent acetylated product (AcPGP) [43–47]. These matrikines are potent stimuli of the CXC chemokine receptors CXCR1/2 (classically associated with IL-8) and carry the potential to perpetuate neutrophilic inflammation in areas of collagen destruction such as the emphysematous lung [48]. This powerful feed-forward process leads to recruitment of neutrophils to the area of ECM breakdown where the initial injury occurred (Fig. 3) [43]. The release of PGP from collagen requires initial cleavage of large protein or protein fragments by matrix metalloproteinases (such as MMP-8 and MMP-9), followed subsequently by peptide digestion by a unique serine protease called prolyl endopeptidase into the active tripeptide fragment [43,49]. PGP acts as a chemotactic stimulus to the neutrophil. These cells contain all the enzymes required for this cascade to generate further PGP, neutrophil elastase, and other proteases from collagen, thereby increasing locally destructive potential [43–46]. The PGP is ultimately degraded in the healthy state by an aminopeptidase site found on the enzyme leukotriene A4 hydrolase (LTA4H) [50]. In the smoker, LTA4H aminopeptidase activity is abrogated by an aldehyde component of cigarette smoke called acrolein (and possibly other components), leading to the accumulation of PGP in the smoker’s lungs, feeding the positive feedback loop of neutrophilic inflammation [37■,51]. Acrolein can also acetylate PGP forming AcPGP which increases its neutrophil chemotactic potency and further protects it from LTA4H degradation [44,51]. Intriguingly, acrolein is also formed endogenously in states of inflammation from oxidation of serine and threonine, which could account for the observation that LTA4H aminopeptidase activity remains depressed and AcPGP levels remain elevated in COPD patients who no longer smoke [37■,51].
FIGURE 3.
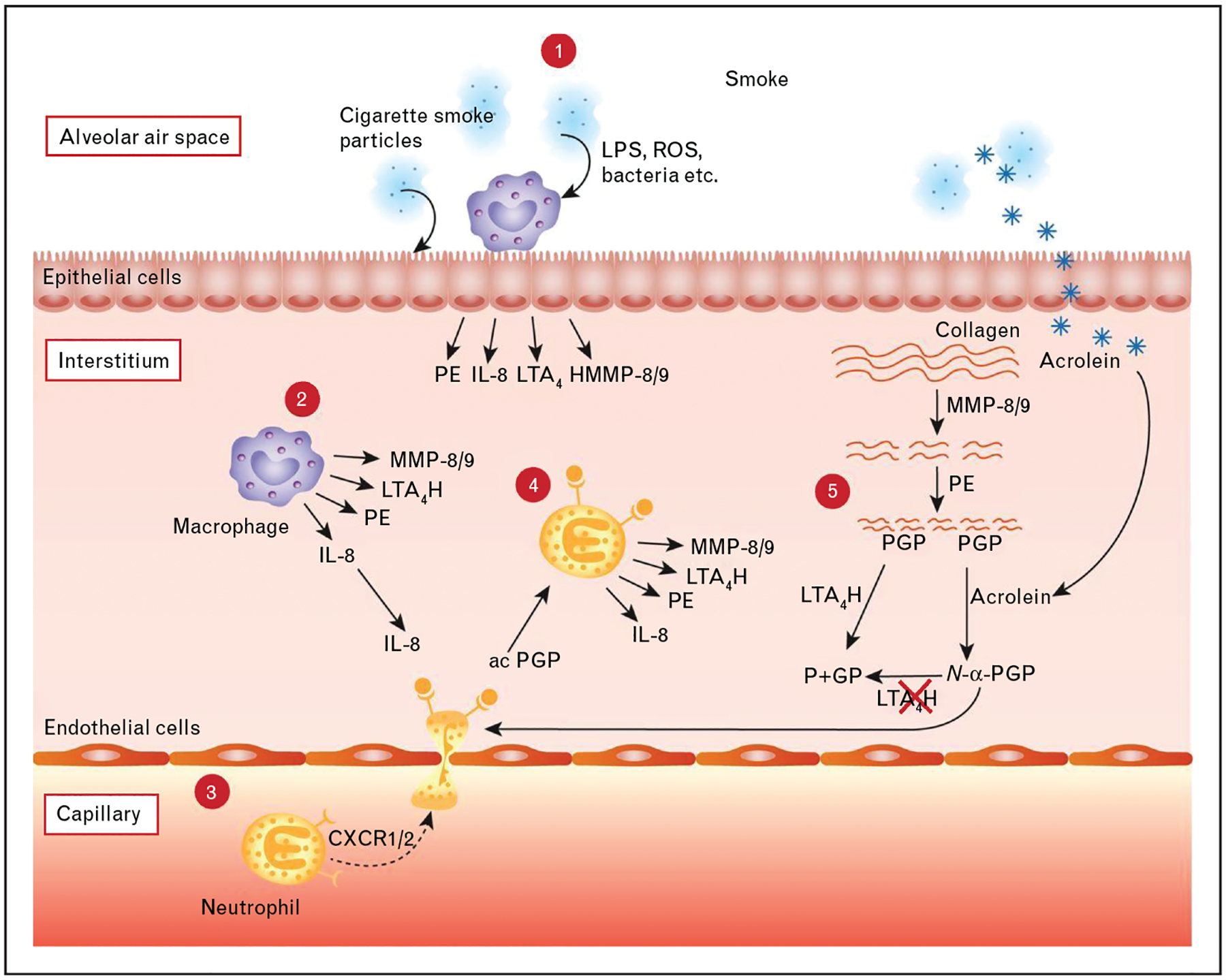
PGP pathway. 1: Cigarette smoke inhalation causes tissue resident cells such as macrophages and epithelial cells to release several mediators, including prolyl endopeptidase (PE), IL-8, leukotriene A4 hydrolase (LTA4H) and matrix metalloproteinase (MMP)8/9. 2 and 3: The neutrophilic chemokine IL-8 attracts neutrophils from the capillary via binding with CXCR1/2. 4 and 5: Neutrophils subsequently release MMPs and PE, which cleave collagen from the extracellular matrix to release the tripeptide and neutrophil chemoattractant PGP. LTA4H can cleave and inactivate PGP. However, components of cigarette smoke, such as acrolein, inhibit LTA4H and can acetylate the PGP to form the more potent acetylated form of PGP (N-α-PGP). Moreover, N-α-PGP cannot be cleaved by LTA4H. PGP-generating enzymes can now be released by neutrophils after recruitment and activation by PGP: a self-sustaining neutrophilic inflammation. ROS reactive oxygen species; P+GP, proline + glycine-proline. IL-8, interleukin-8; PGP, proline-glycine-proline. Figure and legend reproduced from [43].
Supporting a pathogenic role for this matrikine-driven neutrophilic pathway, chronic intratracheal instillation of PGP leads to an emphysematous phenotype with right ventricular hypertrophy (RVH) in a mouse model, recapitulating findings of chronic smoke exposure [52]. In similar chronic smoke exposure models, selective inhibition of PGP with the complementary peptide arginine-threonine-arginine reduced neutrophilic influx and prevented emphysema and RVH [53], providing robust evidence that this pathway is crucial in the development of COPD.
There have been two recent drug advances shown to reduce the rate of exacerbations in COPD, azithromycin and roflumilast. In a randomized, controlled trial, chronic azithromycin therapy was shown to reduce the rate of exacerbations in patients with COPD [34]. An interesting ancillary study by O’Reilly et al. [35] using biological samples from this trial showed that patients treated with azithromycin had lower sputum PGP and myeloperoxidase levels than patients treated with placebo. Most intriguingly, PGP levels were found to increase prior to an acute exacerbation and decrease again (though with a higher nadir than before exacerbating) following resolution of the acute event. This is a unique observation among known COPD biomarkers and underscores the importance of this pathway in the pathogenesis of COPD exacerbations. Azithromycin has been shown to have a number of effects on neutrophilic inflammation and also has some MMP-9 inhibitory activity, though O’Reilly’s group did not detect a statistically significant reduction in MMP-9 activity with azithromycin treatment. Roflumilast, the selective phosphodiesterase 4 inhibitor, has also been recently shown to reduce acute exacerbations of COPD [25]. This effect could be mediated through the PGP pathway as well. Wells et al. [26■] recently published a randomized-controlled trial of COPD patients with chronic bronchitis showing that roflumilast therapy reduced sputum AcPGP levels and prolyl endopeptidase activity by about 50% compared with placebo along with a reduction in other markers of neutrophilic inflammation. Although this trial was limited to 12 weeks, potential for benefit from modulation of this pathway is not lost on the authors, as they hypothesized that roflumilast might ‘alter the natural history of the disease’ through cessation of the feed-forward mechanism of neutrophilic inflammation driven by the PGP pathway.
Although exciting, the neutrophilic endotype needs prospective longitudinal validation and is currently in a primitive state of characterization. There is much left to study, but many aspects of the PGP pathway offer promise as means of defining and treating those COPD patients in whom neutrophilic inflammation may be driving exacerbations and/or disease progression as shown in Table 2. As with the other putative COPD endotypes, more research is needed. Breakthroughs are possible as new techniques allow for study of new and interesting aspects of this endotype. Of particular intrigue are metabolomic–phenotype and proteomic–phenotype interactions, pathogen colonization/microbiome effects and immunological mechanisms including immune microparticle/exosome pathway characterization, which all carry some promise because of recent advances in our ability to study them.
Table 2.
Candidate biomarkers in the neutrophilic chronic obstructive pulmonary disease endotype. Study of the neutrophilic endotype is currently in a primitive state of development, but many candidate biomarkers have been identified that offer promise as means of predicting various patient-centered outcomes and as potential therapeutic targets. Although some of these have been studied to some degree in COPD at large, study of these biomarkers among those patients who prove to have the neutrophilic endotype is lacking but offers a great deal of potential. Proline-glycine-proline and N-acetyl proline-glycine-proline are the best studied of these biomarkers and show great promise
| Candidate biomarker | Marker of neutrophilic inflammation | Genotypic relationship with COPD | Predicts exacerbations |
Predicts symptoms |
Predicts mortality |
Potential therapeutic target |
|---|---|---|---|---|---|---|
| Sputum PMNs | ✓ | n/a | ? | ✓ | ? | n/a |
| MPO | ✓ | ? | ? | ? | ? | ✓ |
| NE | ✓ | ? | ? | ? | ? | X |
| PGP/AcPGP | ✓ | n/a | ✓ | ✓ | ? | ✓ |
| MMP-9 | ✓ | Yes | ? | ? | ? | ✓ |
| PE | ✓ | ? | ? | ? | ? | ✓ |
| LTA4H | ✓ | Possibly | ? | ? | ? | ✓ |
AcPGP, N-acetyl proline-glycine-proline; LTA4H, leukotriene A4 hydrolase; MMP-9, matrix metalloproteinase 9; MPO, myeloperoxidase; NE, neutrophil elastase; PE, prolyl endopeptidase; PGP, proline-glycine-proline; PMNs, polymorphonuclear cell count.
CONCLUSION
COPD is a complex syndrome composed of multiple ‘small COPDs’ or endotypes with distinct patho-physiologies. Current understanding of these endotypes is primitive and is hampered by the complexity of interactions of the underlying endotype with other aspects of disease such as risk factors, disease activity and stage of progression. A few putative endotypes are emerging from recent literature, with only one endotype currently being clearly defined (A1AT deficiency) which can serve as a prototype for study of other endotypes. The long-suspected central role of the neutrophil in the pathogenesis of some cases of COPD may explain the response of a subset of patients to recently studied drugs such as azithromycin and roflumilast, which hints at the presence of an important neutrophilic endotype. This endotype may be driven by the potentially self-perpetuating cycle of PGP generation from pulmonary collagen destruction leading to neutrophil influx and subsequent elaboration of more PGP, which is increased during exacerbations. Further prospective study is merited in characterization of this and other endotypes. New technology may allow for breakthroughs as new aspects of neutrophil biology can be studied and be applied to this important disease with an ultimate goal of modifying or ceasing disease progression rather than merely controlling its manifestations.
KEY POINTS.
COPD is a syndrome within which exist different endotypes.
Interactions of endotype with phenotype are complex and poorly understood.
The neutrophilic endotype has emerged as an important, if poorly defined, endotype that deserves further study.
Financial support and sponsorship
The authors of this publication are supported in part by the NIH grants NIH/NHLBI K08 123940, R01HL114439, R01HL110950 and R01HL126596; Cystic Fibrosis Foundation grant RDP SORSCH15R; and the Family Smoking Prevention and Tobacco Control Act. The content is solely the responsibility of the authors and does not necessarily represent the view of the NIH, CFF or the Food and Drug Administration.
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
- 1.■■.Woodruff PG, Agusti A, Roche N, et al. Current concepts in targeting chronic obstructive pulmonary disease pharmacotherapy: making progress towards personalised management. Lancet 2015; 385:1789–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]; Woodruff et al. explore the complex interactions between clinical phenotypes and endotypes in COPD and call for action to achieve true personalized medicine in this syndrome. This is an important review although it did not include the emerging neutrophilic endotype discussed in this article, perhaps in part because of the newness of some of these data.
- 2.Speizer FE, Ware JH. Exploring different phenotypes of COPD. N Engl J Med 2015; 373:185–186. [DOI] [PubMed] [Google Scholar]
- 3.Burgel PR, Paillasseur JL, Roche N. Identification of clinical phenotypes using cluster analyses in COPD patients with multiple comorbidities. Biomed Res Int 2014; 2014:420134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agusti A, Sin DD. Biomarkers in COPD. Clin Chest Med 2014; 35:131–141. [DOI] [PubMed] [Google Scholar]
- 5.Agusti A. The path to personalised medicine in COPD. Thorax 2014; 69:857–864. [DOI] [PubMed] [Google Scholar]
- 6.Agusti A. Phenotypes and disease characterization in chronic obstructive pulmonary disease. Toward the extinction of phenotypes? Ann Am Thorac Soc 2013; 10 (Suppl):S125–S130. [DOI] [PubMed] [Google Scholar]
- 7.Roy K, Smith J, Kolsum U, et al. COPD phenotype description using principal components analysis. Respir Res 2009; 10:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia-Aymerich J, Gomez FP, Anto JM, et al. Phenotypic characterization and course of chronic obstructive pulmonary disease in the PAC-COPD Study: design and methods. Arch Bronconeumol 2009; 45:4–11. [DOI] [PubMed] [Google Scholar]
- 9.Rennard SI, Vestbo J. The many “small COPDs”: COPD should be an orphan disease. Chest 2008; 134:623–627. [DOI] [PubMed] [Google Scholar]
- 10.Pistolesi M, Camiciottoli G, Paoletti M, et al. Identification of a predominant COPD phenotype in clinical practice. Respir Med 2008; 102:367–376. [DOI] [PubMed] [Google Scholar]
- 11.Anderson GP. Endotyping asthma: new insights into key pathogenic mechanisms in a complex, heterogeneous disease. Lancet 2008; 372:1107–1119. [DOI] [PubMed] [Google Scholar]
- 12.Al-Kassimi FA, Alhamad EH. A challenge to the seven widely believed concepts of COPD. Int J Chron Obstruct Pulmon Dis 2013; 8:21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.■.Lange P, Celli B, Agusti A, et al. Lung-function trajectories leading to chronic obstructive pulmonary disease. N Engl J Med 2015; 373:111–122. [DOI] [PubMed] [Google Scholar]; This study tracks the spirometric trajectory of patients over time; substantial heterogeneity was found and it was noted that a significant proportion of patients to ultimately develop airflow obstruction did not suffer from a rapid decline of FEV1 but rather had low-normal spirometry originally and developed frank airflow obstruction as their spirometry declined at the usual rate. This again corroborates the notion that COPD is quite heterogeneous.
- 14.■■.Bhavani S, Tsai CL, Perusich S, et al. Clinical and immunological factors in emphysema progression: 5-year prospective Longitudinal Exacerbation Study of Chronic Obstructive Pulmonary Disease (LES-COPD). Am J Respir Crit Care Med 2015; 192:1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bhavani et al. take a crucial step forward in the untangling of COPD endotypes in this prospectively collected study linking increased systemic inflammation and autoreactive T cells with emphysema progression in smokers but not in nonsmokers. These observations corroborate the notion of COPD as inflammatory, autoimmune disorder, which is an important paradigm shift with far-reaching implications for future study.
- 15.Stockley RA. Alpha1-antitrypsin review. Clin Chest Med 2014; 35:39–50. [DOI] [PubMed] [Google Scholar]
- 16.Mannino DM, Tal-Singer R, Lomas DA, et al. Plasma fibrinogen as a biomarker for mortality and hospitalized exacerbations in people with COPD. Chronic Obstr Pulm Dis (Miami) 2015; 2:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duvoix A, Dickens J, Haq I, et al. Blood fibrinogen as a biomarker of chronic obstructive pulmonary disease. Thorax 2013; 68:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valvi D, Mannino DM, Mullerova H, Tal-Singer R. Fibrinogen, chronic obstructive pulmonary disease (COPD) and outcomes in two United States cohorts. Int J Chron Obstruct Pulmon Dis 2012; 7:173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mannino DM, Valvi D, Mullerova H, Tal-Singer R. Fibrinogen, COPD and mortality in a nationally representative U.S. cohort. COPD 2012; 9:359–366. [DOI] [PubMed] [Google Scholar]
- 20.Agusti A, Edwards LD, Rennard SI, et al. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS One 2012; 7:e37483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Polatli M, Cakir A, Cildag O, et al. Microalbuminuria, von Willebrand factor and fibrinogen levels as markers of the severity in COPD exacerbation. J Thromb Thrombolysis 2008; 26:97–102. [DOI] [PubMed] [Google Scholar]
- 22.Burgel PR, Paillasseur JL, Peene B, et al. Two distinct chronic obstructive pulmonary disease (COPD) phenotypes are associated with high risk of mortality. PLoS One 2012; 7:e51048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim V, Han MK, Vance GB, et al. The chronic bronchitic phenotype of COPD: an analysis of the COPDGene Study. Chest 2011; 140:626–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wedzicha JA, Rabe KF, Martinez FJ, et al. Efficacy of roflumilast in the COPD frequent exacerbator phenotype. Chest 2013; 143:1302–1311. [DOI] [PubMed] [Google Scholar]
- 25.Garnock-Jones KP. Roflumilast: a review in COPD. Drugs 2015; 75:1645–1656. [DOI] [PubMed] [Google Scholar]
- 26.■.Wells JM, Jackson PL, Viera L, et al. A randomized, placebo-controlled trial of roflumilast. Effect on proline-glycine-proline and neutrophilic inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2015; 192:934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wells et al. prospectively demonstrate a reduction in PGP levels with roflumilast administration in concert with a reduction in exacerbations.
- 27.Singh D, Edwards L, Tal-Singer R, Rennard S. Sputum neutrophils as a biomarker in COPD: findings from the ECLIPSE study. Respir Res 2010; 11:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vlahos R, Bozinovski S, Hamilton JA, Anderson GP. Therapeutic potential of treating chronic obstructive pulmonary disease (COPD) by neutralising granulocyte macrophage-colony stimulating factor (GM-CSF). Pharmacol Ther 2006; 112:106–115. [DOI] [PubMed] [Google Scholar]
- 29.van Beurden WJ, Wielders PL, Scheepers PJ, et al. Superoxide production by peripheral polymorphonuclear leukocytes in patients with COPD. Respir Med 2003; 97:401–406. [DOI] [PubMed] [Google Scholar]
- 30.Qiu Y, Zhu J, Bandi V, et al. Biopsy neutrophilia, neutrophil chemokine and receptor gene expression in severe exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2003; 168:968–975. [DOI] [PubMed] [Google Scholar]
- 31.Beeh KM, Kornmann O, Buhl R, et al. Neutrophil chemotactic activity of sputum from patients with COPD: role of interleukin 8 and leukotriene B4. Chest 2003; 123:1240–1247. [DOI] [PubMed] [Google Scholar]
- 32.Aaron SD, Angel JB, Lunau M, et al. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 163:349–355. [DOI] [PubMed] [Google Scholar]
- 33.Sin D, van Eeden SF. Neutrophil-mediated lung damage: a new COPD phenotype? Respiration 2012; 83:103–105. [DOI] [PubMed] [Google Scholar]
- 34.Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Reilly PJ, Jackson PL, Wells JM, et al. Sputum PGP is reduced by azithromycin treatment in patients with COPD and correlates with exacerbations. BMJ Open 2013; 3:e004140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Djekic UV, Gaggar A, Weathington NM. Attacking the multitiered proteolytic pathology of COPD: new insights from basic and translational studies. Pharmacol Ther 2009; 121:132–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.■.Wells JM, Gaggar A, Blalock JE. MMP generated matrikines. Matrix Biol 2015; 44–46:122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]; The increasing appreciation that breakdown products of extracellular matrix components cause potentially self-perpetuating inflammatory changes relevant to COPD disease led to this review by Wells et al. which summarizes the current state of knowledge in this field.
- 38.Hoonhorst SJ, Timens W, Koenderman L, et al. Increased activation of blood neutrophils after cigarette smoking in young individuals susceptible to COPD. Respir Res 2014; 15:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, He J, Xia J, et al. Delayed apoptosis by neutrophils from COPD patients is associated with altered Bak, Bcl-xl, and Mcl-1 mRNA expression. Diagn Pathol 2012; 7:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makris D, Vrekoussis T, Izoldi M, et al. Increased apoptosis of neutrophils in induced sputum of COPD patients. Respir Med 2009; 103:1130–1135. [DOI] [PubMed] [Google Scholar]
- 41.Polosukhin VV, Cates JM, Lawson WE, et al. Bronchial secretory immuno-globulin a deficiency correlates with airway inflammation and progression of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2011; 184:317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Overbeek SA, Braber S, Koelink PJ, et al. Cigarette smoke-induced collagen destruction; key to chronic neutrophilic airway inflammation? PLoS One 2013; 8:e55612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abdul Roda M, Fernstrand AM, Redegeld FA, et al. The matrikine PGP as a potential biomarker in COPD. Am J Physiol Lung Cell Mol Physiol 2015; 308:L1095–L1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hardison MT, Brown MD, Snelgrove RJ, et al. Cigarette smoke enhances chemotaxis via acetylation of proline-glycine-proline. Front Biosci (Elite Ed) 2012; 4:2402–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ten Hoeve AL, Roda MA, Redegeld F, et al. Proline-glycine-proline as a potential biomarker in chronic obstructive pulmonary disease and cystic fibrosis. Tanaffos 2012; 11:12–15. [PMC free article] [PubMed] [Google Scholar]
- 46.Gaggar A, Rowe SM, Matthew H, Blalock JE. Proline-glycine-proline (PGP) and high mobility group box protein-1 (HMGB1): potential mediators of cystic fibrosis airway inflammation. Open Respir Med J 2010; 4:32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weathington NM, van Houwelingen AH, Noerager BD, et al. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med 2006; 12:317–323. [DOI] [PubMed] [Google Scholar]
- 48.O’Reilly P, Jackson PL, Noerager B, et al. N-alpha-PGP and PGP, potential biomarkers and therapeutic targets for COPD. Respir Res 2009; 10:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abdul Roda M, Sadik M, Gaggar A, et al. Targeting prolyl endopeptidase with valproic acid as a potential modulator of neutrophilic inflammation. PLoS One 2014; 9:e97594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Snelgrove RJ, Jackson PL, Hardison MT, et al. A critical role for LTA4H in limiting chronic pulmonary neutrophilic inflammation. Science 2010; 330:90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wells JM, O’Reilly PJ, Szul T, et al. An aberrant leukotriene A4 hydrolase-proline-glycine-proline pathway in the pathogenesis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2014; 190:51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Houwelingen AH, Weathington NM, Verweij V, et al. Induction of lung emphysema is prevented by L-arginine-threonine-arginine. FASEB J 2008; 22:3403–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Braber S, Koelink PJ, Henricks PA, et al. Cigarette smoke-induced lung emphysema in mice is associated with prolyl endopeptidase, an enzyme involved in collagen breakdown. Am J Physiol Lung Cell Mol Physiol 2011; 300:L255–L265. [DOI] [PMC free article] [PubMed] [Google Scholar]