

Abstract
Direct reprogramming of fibroblasts into cardiomyocytes (CMs) represents a promising strategy to regenerate CMs lost after ischemic heart injury. Overexpression of GATA4, HAND2, MEF2C, TBX5, miR-1, and miR-133 (GHMT2m) along with transforming growth factor beta (TGF-β) inhibition efficiently promote reprogramming. However, the mechanisms by which TGF-β blockade promotes cardiac reprogramming remain unknown. Here, we identify interactions between the histone H3 lysine 27 trimethylation (H3K27me3) demethylase JMJD3, the SWI/SNF remodeling complex subunit BRG1, and cardiac transcription factors. Furthermore, canonical TGF-β signaling regulates the interaction between GATA4 and JMJD3. TGF-β activation impairs the ability of GATA4 to bind target genes and prevents demethylation of H3K27 at cardiac gene promoters during cardiac reprogramming. Finally, a mutation in GATA4 (V267M) that is associated with congenital heart disease exhibits reduced binding to JMJD3 and impairs cardiomyogenesis. Thus, we have identified an epigenetic mechanism wherein canonical TGF-β pathway activation impairs cardiac gene programming, in part by interfering with GATA4-JMJD3 interactions.
1. Introduction
Ischemic heart disease (IHD) accounts for nearly 9 million annual deaths worldwide and the mortality rate associated IHD has begun to plateau in developed countries despite advances in clinical intervention [1]. Ischemic injury causes loss of cardiomyocytes (CMs), which leads to fibrotic remodeling and impaired heart function [2-4]. CMs in the adult heart have limited (<1% annual) turnover [5], and current clinical therapies, while effective at slowing progression of disease, do not promote regeneration of lost CMs [6]. The landmark studies demonstrating conversion of fibroblasts to induced pluripotent stem cells via transcription factor overexpression [7,8] inspired the development of transcription factor cocktails that reprogram fibroblasts into terminally differentiated cell types including neurons [9], hepatocytes [10], and CMs [11]. Overexpression of cardiac transcription factors GATA4, MEF2C, and TBX5 converted fibroblasts into induced CMs (iCMs), albeit with low efficiency [11]. Since this initial report, many studies have focused on optimizing the efficiency of reprogramming by overexpressing additional cardiac transcription factors [12-16], epigenetic modifiers [17], microRNAs [18,19], or AKT [20]. Additionally, knockdown of repressive epigenetic complex components [21,22] or splicing factors [23] have also demonstrated improved reprogramming efficiency. Cardiac reprogramming has also been achieved exclusively by microRNA overexpression [24] or treatment with small molecules [25]. More recently, optimized protocols have led to reprogramming efficiencies in excess of 50% [15,20,22,26] and have identified signaling pathways that impair the reprogramming process including inflammatory [24,26], Notch [27], Wnt [13,28], and pro-fibrotic signaling pathways [13,15,28]. However, the mechanisms by which these diverse pathways impair cardiac reprogramming remain poorly understood.
Widespread re-patterning of epigenetic modifications including histone tail methylation and acetylation are associated with cellular reprogramming [29]. Previous studies have shown that histone H3 lysine 27 trimethylation (H3K27me3) is highly enriched at cardiac gene loci in non-myocytes but is removed during cardiomyogenesis [21,30,31]. Moreover, previous studies demonstrated that cardiac transcription factors physically associate with H3K27me3-specific demethylases; Isl1 interacts with Jumonji Domain-Containing Protein 3 (JMJD3) [32] whereas GATA4, NKX2.5, and TBX5 interact with Ubiquitously-Transcribed X Chromosome Tetratricopeptide Repeat Protein (UTX) [33]. Knockdown of either Kdm6a (UTX) or Kdm6b (JMJD3) profoundly disrupts cardiomyogenesis. Kdm6a knockout prevented cardiac differentiation of embryonic stem cells (ESCs) and largely disrupted cardiac looping in female embryonic mice [33]. Similarly, depletion of Kdm6b impaired cardiac differentiation of ESCs, similar to Isl1 knockdown [32]. Furthermore, double knockdown of Kdm6a and Kdm6b modestly reduced expression of Gata4, Mef2c, and Tbx5 in fibroblasts reprogrammed by microRNA cocktail [21]. In addition to cardiac transcription factors, other transcription factors including transforming growth factor beta (TGF-β) signaling effectors SMAD2/3 are known to interact with H3K27me3 demethylases [34]. Moreover, activation of pro-fibrotic genes via TGF-β signaling impedes cardiac reprogramming [13,15]. Therefore, while previous studies demonstrate the necessity of H3K27me3 demethylases in heart development, mechanisms that regulate transcription factor-mediated recruitment of these demethylases to chromatin during cardiomyogenesis remain elusive.
In addition to interacting with cardiac transcription factors, UTX was also found to bridge the interaction between cardiac transcription factors and the SWItch/Sucrose Non-Fermentable (SWI/SNF) ATP-dependent chromatin remodeling complex subunit Brahma-related gene 1 (BRG1, encoded by the Smarca4 gene) [33]. Myocardial depletion of BRG1 led to embryonic lethality and was accompanied by a thinning of the myocardium, failure to form the interventricular septum, and reduced cardiomyocyte proliferation [35]. Moreover, silencing of Smarcd3, which encodes a heart-enriched subunit of the SWI/SNF complex, BAF60C, also led to embryonic lethality and impaired heart development and cardiomyocyte differentiation [36].
Our previous report demonstrated that inhibition of pro-fibrotic signaling pathways including the TGF-β pathway alongside overexpression of GATA4, HAND2, MEF2C, TBX5, miR-1, and miR-133 (GHMT2m) reprogrammed mouse embryonic fibroblasts (MEFs) with approximately 60% efficiency [15]. Here, we identify novel interactions between JMJD3 and cardiac transcription factors GATA4, HAND2, and TBX5. Depletion of JMJD3 or inhibition of its H3K27me3 demethylase activity dramatically reduces GHMT2m-mediated reprogramming efficiency. In addition, we show that the canonical TGF-β signaling effector SMAD2 disrupts the interactions between GATA4 and chromatin modifiers JMJD3 and BRG1. Furthermore, SMAD2 overexpression or TGF-β stimulation results in inefficient demethylation of H3K27me3 at cardiac genes corresponding with decreased transcription and reduced reprogramming efficiency. A point mutation in GATA4 (c.799G > A, [37,38]) associated with congenital heart disease (CHD) disrupts the interaction between GATA4 and JMJD3. Human induced pluripotent stem cells (hiPSCs) carrying this mutation exhibit increased H3K27me3 levels at cardiac loci and impaired cardiogenesis. Thus, our data establish a novel mechanism through which canonical TGF-β signaling controls cardiomyogenesis.
2. Results
2.1. Canonical TGF-β signaling effector SMAD2 impairs cardiac reprogramming
Our previous reports indicate that pro-fibrotic signaling pathways including TGF-β are potent endogenous barriers to cardiac reprogramming [15,39]. Canonical TGF-β signaling results in phosphorylation of downstream effectors SMAD2 and SMAD3, which then translocate to the nucleus via SMAD4 to activate TGF-β responsive genes. We therefore overexpressed GFP, SMAD2, or SMAD7 (an endogenous inhibitor of SMAD2 and SMAD3 phosphorylation) in GHMT2m-reprogrammed cells. Overexpression of SMAD2 robustly increased levels of phosphorylated SMAD2, as did TGF-β1 stimulation (Fig. 1A and B). In contrast, SMAD7 overexpression reduced levels of phosphorylated SMAD2, as did treatment with the type I TGF-β receptor inhibitor A-83-01 (Fig. 1A and B). Overexpression of SMAD7 or treatment with A-83-01 significantly increased the number of beating iCMs at days 10 and 13 post-transduction whereas overexpression of SMAD2 dramatically reduced the number of beating iCMs at these time points, comparable to TGF-β1 treatment (Fig. 1C and Movies S1-5). Interestingly, A-83-01 treatment generated the greatest number of beating cells, which may be due to the higher efficiency in blocking phosphorylation of SMAD2 than SMAD7 overexpression (Fig. 1A). Strikingly, A-83-01 treatment of reprogrammed cells overexpressing SMAD2 produced similar numbers of beating iCMs to GFP control, which was accompanied by nearly undetectable levels of phosphorylated SMAD2 levels in both conditions (Fig. 1D and S1A), suggesting that A-83-01 enhances cardiac reprogramming through inhibiting canonical TGF-β signaling. SMAD7 overexpression resulted in two- to four-fold upregulation of cardiac genes Myh6, Actc1, and Pln, comparable to A-83-01 treatment (Fig. 1E). In contrast, SMAD2 overexpression and TGF-β1 treatment resulted in reduced expression of these genes compared to GFP control (Fig. 1E). To solidify the inhibitory role of SMAD2 in cardiac reprogramming, we utilized short hairpin RNA (shRNA)-mediated gene silencing to knock down SMAD2 expression in GHMT2m-reprogrammed fibroblasts. Knockdown of Smad2 resulted in approximately four times more beating cells and increased cardiac gene expression compared to control shRNA delivery at Days 5 and 13 post-transduction (Fig. S1B-D). Finally, while no condition significantly altered the number of α-actinin positive cells (Fig. 1F and S1E), SMAD7 overexpression promoted better sarcomere organization by Day 9 compared to GFP control whereas SMAD2 overexpression resulted in significantly more sarcomere disarray (Fig. 1G-H), which correlated to the efficiency of generating beating cells by Day 13 (Fig. 1C and S1E).
Fig. 1. Activation of Canonical TGF-β signaling impairs cardiac reprogramming.

A and B) Immunoblot (A) and quantification (B) of phosphorylated SMAD2 (p-SMAD2) normalized to GAPDH from whole cell extracts of Day 12 GHMT2m-reprogrammed cells co-infected with GFP, SMAD2, or SMAD7, or treated with 5 ng/mL TGF-β1 or 0.5 μM A-83-01. N = 3 per group. C) Beating cell counts per field (0.89 mm2) from Day 10 and Day 13 GHMT2m-reprogrammed cells co-infected with GFP, SMAD2, or SMAD7, or treated with 5 ng/mL TGF-β1 or 0.5 μM A-83-01. N = 3 per group. D) Beating cell counts per field (0.89mm2) in Day 10 GHMT2m-reprogrammed cells co-transduced with GFP or SMAD2. Reprogrammed cells were maintained in 0.5 μM A-83-01 starting at Day 3. N = 3 per group. E) Messenger RNA expression of cardiac genes Myh6, Actc1, and Pln harvested from Day 13 GHMT2m-reprogrammed MEFs co-infected with GFP, SMAD2, or SMAD7, or treated with 5 ng/mL TGF-β1 or 0.5 μM A-83-01. All data were normalized to the GFP group. N = 3 per group. F) Quantification of the percentage of cells expressing α-actinin protein from Day 9 GHMT2m-reprogrammed MEFs co-infected with GFP, SMAD2, or SMAD7. 10 fields of view per dish were collected across 3 individual experiments per group to analyze sarcomere organization. G) Representative fluorescent images of indicated levels of sarcomere organization in Day 9 GHMT2m-reprogrammed MEFs. Red: α-actinin; Green: Troponin I; Blue: Hoechst. Scale bar = 25 μM. H) Quantification of sarcomere organization in Day 9 GHMT2m-reprogrammed MEFs co-infected with GFP, SMAD2, or SMAD7. 10 fields of view per dish were collected across 3 individual experiments per group to analyze sarcomere organization.
All data shown as mean ± SEM. * p < 0.05 by one-way ANOVA with Tukey’s multiple comparison test vs the GFP group unless otherwise specified. # p < 0.05 by Student’s t-test vs the GFP group.
Taken together, these results indicate that activation of canonical TGF-β signaling not only impedes cardiac differentiation but also plays a significant role in myocyte myofibrillogenesis and generation of functional iCMs.
2.2. Cardiac transcription factors physically interact with JMJD3
Removal of the transcriptionally repressive epigenetic mark H3K27me3 has been observed during cardiac reprogramming [21,30]. We therefore tested the dynamics of H3K27me3 removal in our system by performing chromatin immunoprecipitation (ChIP) followed by quantitative PCR (ChIP-qPCR). In agreement with previous reports, we observed significant reduction of H3K27me3 at the Gata4 promoter by Day 3 in GHMT reprogrammed cells compared to undifferentiated MEFs (Fig. 2A). H3K27me3 removal proceeded through Day 7, but not to the extent observed in neonatal mouse CMs (NMCMs) (Fig. 2A). Previous reports indicate that the H3K27me3 demethylase JMJD3 physically interacts with SMAD2/3 [34] whereas the H3K27me3 demethylase UTX interacts with GATA4, NKX2.5, and TBX5 [33]. To investigate these interactions, we overexpressed HA-tagged UTX or JMJD3 alongside MYC-tagged GHMT in HEK293T cells and performed co-immunoprecipitations (Co-IPs). In agreement with previous studies, our data confirm JMJD3 physically interacts with SMAD2/3 and UTX weakly interacts with GATA4 and TBX5 (Fig. 2B and S2A). In addition, we observed strong interactions between JMJD3 and GATA4, TBX5 and HAND2 (Fig. 2B-D).
Fig. 2. GHMT Reprogramming Factors Interact with JMJD3 to promote demethylation of H3K27me3.
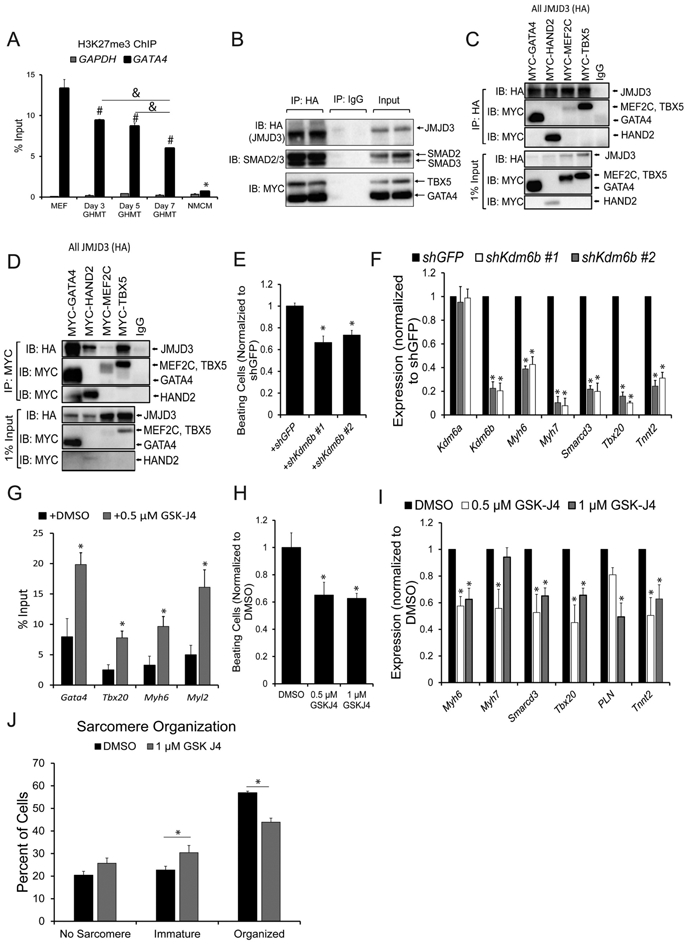
A) H3K27me3 levels in Day 3, 5, and 7 GHMT-reprogrammed cells at the Gata4 and Gapdh promoters. Undifferentiated MEFs and neonatal mouse cardiomyocytes (NMCMs) were used as positive and negative controls respectively for Gata4 H3K27me3 levels. Gapdh served as a negative control for H3K27me3 in all samples. All data are presented as percentage of input. *, #, and & p < 0.05 by one-way ANOVA with Tukey’s multiple comparison test vs all groups (*), MEFs (#), or specified comparisons (&). B) Co-IPs using nuclear lysates harvested from HEK293T cells transfected with MYC-GHMT and HA-JMJD3. Following Co-IP with anti-HA, proteins were resolved by SDS-PAGE and immunoblotted for MYC, HA, and SMAD2/3. C and D) Co-IPs using nuclear lysates harvested from HEK293T cells transfected with HA-JMJD3 and individual MYC-G, -H, –M, or -T transcription factors. Following Co-IP with anti-HA (C) or anti-MYC (D), proteins were resolved by SDS-PAGE and immunoblotted for MYC and HA. E) Beating cell counts per field (0.89 mm2) from Day 12 GHMT2m-reprogrammed cells co-infected with shGFP or two different shRNA sequences targeting Kdm6b. Reprogrammed cells were maintained in 0.5 μM A-83-01 starting at Day 3. All data were normalized to the shGFP group N = 3 per group. F) Messenger RNA expression of Kdm6a, Kdm6b, and cardiac genes Myh6, Myh7, Smarcd3, Tbx20, and Tnnt2 harvested from Day 13 GHMT2m-reprogrammed MEFs co-infected with shGFP or two different shRNAs targeting Kdm6b. Reprogrammed cells were maintained in 0.5 μM A-83-01 starting at Day 3. All data were normalized to the shGFP group. N = 3 per group. G) H3K27me3 levels at cardiac gene promoters Gata4, Tbx20, Myh6, and Myl2 from Day 7 GHMT2m-reprogrammed cells. Reprogrammed cells were treated with DMSO or 0.5 μM GSK-J4 between Days 3 and 5 and maintained in 0.5 μM A-83-01 starting at Day 3. Data are presented as percentage input. N = 4 per group. H) Beating cell counts per field (0.89 mm2) in Day 12 GHMT2m-reprogrammed cells treated with DMSO, 0.5 μM, or 1 μM GSK-J4. GSK-J4 was added to cells between Days 3 and 5. Reprogrammed cells were maintained in 0.5 μM A-83-01 starting at Day 3. All data were normalized to the DMSO group. N = 3 per group. I) Messenger RNA expression of cardiac genes Myh6, Myh7, Smarcd3, Tbx20, Pln, and Tnnt2 harvested from Day 13 GHMT2m-reprogrammed MEFs treated with DMSO, 0.5 μM, or 1 μM GSK-J4. GSK-J4 was added to cells between Days 3 and 5. Reprogrammed cells were maintained in 0.5 μM A-83-01 starting at Day 3. All data were normalized to the DMSO group. N = 3 per group. J) Quantification of sarcomere organization in Day 12 GHMT2m-reprogrammed MEFs treated with DMSO or 1 μM GSK-J4. GSK-J4 was administered between Days 3 and 5. Reprogrammed cells were maintained in 0.5 μM A-83-01 starting at Day 3. 10 fields of view per dish were collected across 3 individual experiments per group to analyze sarcomere organization. * p < 0.05 by Student’s t-test. All data shown as mean ± SEM. * p < 0.05 by one-way ANOVA with Tukey’s multiple comparison test vs the control (shGFP or DMSO) groups.
Together, our data indicate that cardiac transcription factors GATA4, HAND2, and TBX5 as well as TGF-β signaling effectors SMAD2 and SMAD3 physically interact with the H3K27me3 demethylase JMJD3.
2.3. JMJD3 is required for cardiac reprogramming
We next sought to determine the role of JMJD3 in cardiac reprogramming. Expression of shRNAs targeting Kdm6b (JMJD3) decreased expression of Kdm6b by approximately 80% and decreased the number of beating cells by approximately 40% in GHMT2m-reprogrammed cells, compared to GFP shRNA control (Fig. 2E and F). Moreover, Kdm6b knockdown in reprogrammed cells resulted in significantly reduced mRNA expression of the cardiac genes Myh6, Myh7, Smarcd3, Tbx20, and Tnnt2 (Fig. 2F). In contrast, cells expressing Kdm6a (UTX) shRNAs yielded fewer beating cells during early stages of reprogramming (Day 9) but were indistinguishable from cells expressing shGFP by Day 13 (Fig. S2B-D). To determine whether H3K27me3 demethylase activity is essential in cardiac reprogramming, we utilized the H3K27me3 demethylase inhibitor GSK-J4 [40]. Treatment with 0.5 μM GSK-J4 resulted in two- to three-fold increased H3K27me3 levels at cardiac gene promoters compared to DMSO treatment (Fig. 2G). Importantly, treatment with GSK-J4 did not interfere with the ability of GATA4 to bind target gene promoters Tbx20, Myh6, or Nppa (Fig. S3B) or the ability of GATA4 to bind JMJD3 (Fig. S3C). Treatment of GHMT2m-reprogrammed cells with 0.5 or 1 μM GSK-J4 yielded approximately 40% fewer beating cells compared to DMSO treatment (Fig. 2H), similar to Kdm6b knockdown. Additionally, treatment of GHMT2m-reprogrammed cells with 0.5 or 1 μM GSK-J4 significantly reduced expression of Myh6, Smarcd3, Tbx20, and Tnnt2, but only 1 μM GSK-J4 significantly reduced expression of Pln (Fig. 2I). Finally, GHMT2m-reprogrammed cells treated with 1 μM GSK-J4 resulted in increased sarcomere disarray compared to DMSO control (Fig. 2J). These results demonstrate that the H3K27me3 demethylase activity of JMJD3 is essential for cardiac reprogramming.
2.4. Activated TGF-β signaling prevents demethylation of H3K27me3 and reduces the binding of GATA4 to target genes
Our data indicate GATA4, HAND2, TBX5, SMAD2, and SMAD3 physically interact with JMJD3 (Fig. 2B-D) and that the H3K27me3 demethylase activity of JMJD3 is required for cardiac reprogramming. Therefore, we inquired whether TGF-β pathway activation alters the H3K27me3 landscape during reprogramming. We profiled the global H3K27me3 landscape by performing H3K27me3 ChIP followed by massively parallel Next Generation sequencing (ChIP-seq) in Day 7 GHMT2m-reprogrammed cells and undifferentiated MEFs (Fig. 3A). We manipulated TGF-β signaling in reprogrammed cells by either overexpressing SMAD2 or treating with 0.5 μM A-83-01 as these conditions led to the greatest changes in phosphorylation of SMAD2 and numbers of beating cells (Fig. 1A-C). All reprogramming conditions exhibited reduced H3K27me3 levels at cardiogenic promoters including Gata4 and Tbx20 compared to undifferentiated MEFs (Fig. 3B-C). Our previous RNA-seq analysis identified 509 genes upregulated >two-fold in reprogrammed cells treated with A-83-01 vs those treated with DMSO which corresponded with cardiac muscle development and mitochondrial function pathways by Gene Ontology (GO) analysis [15]. We selected one of the top enriched cardiac-specific GO terms (Regulation of Heart Contraction, GO:0008016) and quantified the levels of H3K27me3 at all promoters within this pathway (Fig. S4A and S4B). All reprogrammed cells exhibited reduced H3K27me3 signal compared to undifferentiated MEFs. Additionally, cardiac gene promoters in reprogrammed cells overexpressing SMAD2 contained higher H3K27me3 levels than reprogrammed cells treated with DMSO or A-83-01 (Fig. S4A and S4B). We then further analyzed the RNA-seq data (Zhao et al., 2015, deposited in the NCBI Gene Expression Omnibus, GSE71405) and selected genes upregulated >two-fold in GHMT2m reprogrammed cells treated with A-83-01 compared to undifferentiated MEFs. GO analysis revealed a number of pathways associated with muscle differentiation, ion channel physiology, metabolism, and cardiac muscle (Fig. S4C). We evaluated the top enriched pathway by GO analysis, Muscle System Process (GO: GO:0003012). All three reprogramming conditions displayed significantly reduced H3K27me3 levels compared to undifferentiated MEFs (Fig. 3D). Strikingly, reprogrammed cells overexpressing SMAD2 had significantly increased H3K27me3 levels at muscle-related genes compared to DMSO or A-83-01 treated reprogrammed cells (Fig. 3D). We observed similar trends when plotting individual candidate cardiac genes (Fig. 3E and S4G).
Fig. 3. TGF-β signaling impairs demethylation of H3K27me3 and recruitment of GATA4 to target genes.

A) Global ChIP-seq density heatmap of H3K27me3 in Day 7 GHMT2m-reprogrammed cells with indicated TGF-β pathway manipulation and undifferentiated MEFs at ±2 kb from annotated transcription start sites (TSS). B and C) Representative IGV tracks depicting H3K27me3 intensity at Gata4 (B) and Tbx20 (C) promoters from Day 7 GHMT2m-reprogrammed cells with indicated TGF-β pathway manipulation and undifferentiated MEFs. D and E) Logarithmic transformation of H3K27me3 levels in relation to undifferentiated MEFs at ±2 kb from the annotated TSS of genes within the Muscle System Process Gene Ontology pathway (GO:0003012) that were also upregulated >2 fold in GHMT2m + A-83-01 reprogrammed cells compared to undifferentiated MEFs as identified by RNA-seq. (D) and at candidate cardiac genes selected from GO:0003012 (E). F) H3K27me3 levels at cardiac gene promoters Gata4, Tbx20, Actn2, Des, and Myh6 from Day 7 GHMT2m-reprogrammed cells co-infected with GFP, SMAD2, or SMAD7 or treated with DMSO, 5 ng/mL TGF-β1, or 0.5 μM A-83-01 analyzed by ChIP-qPCR. Data are presented as percentage of input. N = 6 for GHMT2m + GFP + DMSO, N = 6 for GHMT2m + GFP + TGF-β1, N = 5 for GHMT2m + GFP + A-83-01, N = 4 for GHMT2m + SMAD2, and N = 5 for GHMT2m + SMAD7. #, $, & p < 0.05 by Student’s t-test vs DMSO, SMAD7, or at specified comparison, respectively. G) GATA4 levels at cardiac gene promoters Nppa, Tbx20, and Myh6 from Day 5 GHMT2m-reprogrammed cells treated with DMSO, 5 ng/mL TGF-β1, or 0.5 μM A-83-01 analyzed by ChIP-qPCR. Data are presented as enrichment to IgG control. N = 5 for GHMT2m + DMSO, N = 4 for GHMT2m + TGF-β1, N = 5 for GHMT2m + A-83-01.
All data shown as mean ± SEM. * p < 0.05 by one-way ANOVA with Tukey’s multiple comparison test at specified comparisons.
We then performed ChIP-qPCR to validate our ChIP-seq model at candidate cardiac genes. We observed H3K27me3 demethylation in all reprogrammed cells at Gata4, Tbx20, and Des promoters (Fig. S4D) compared to uninfected MEFs. Strikingly, reprogrammed cells treated with A-83-01 displayed lower levels of H3K27me3 compared to DMSO treatment at all promoters investigated aside from Des, indicating highly efficient removal (Fig. 3F). Intriguingly, while H3K27me3 levels in reprogrammed cells overexpressing SMAD7 did not differ significantly from DMSO treated cells at any of the five promoters tested, A-83-01 treated cells displayed significantly reduced H3K27me3 compared to SMAD7 overexpressing cells only at the Gata4 and Tbx20 promoters. In contrast, reprogrammed cells treated with TGF-β1 or overexpressing SMAD2 had consistently elevated levels of H3K27me3 compared to other reprogrammed conditions (Fig. 3F). Notably, demethylation of H3K27me3 did not occur in reprogrammed cells treated with TGF-β1 or overexpressing SMAD2 at the Actn2 and Myh6 promoters (Fig. S4D) compared to uninfected MEFs. H3K27me3 levels negatively correlated to gene expression in reprogrammed cells with various TGF-β pathway activation states (Fig. 1E and S4E). Interestingly, vehicle treated reprogrammed cells had indistinguishable levels of H3K27me3 compared to A-83-01 treated reprogrammed cells by Day 9, suggesting that H3K27me3 demethylation at early stages is crucial for cardiac reprogramming (Fig. S4F). In contrast, H3K27me3 levels in reprogrammed cells treated with TGF-β1 remained significantly elevated at Day 9 (Fig. S4F). GATA4 and SMAD2/3/4 are known to form a complex [41]; therefore, we hypothesized that activation of TGF-β signaling may recruit GATA4 away from cardiac genes. Indeed, we observed reduced binding of GATA4 to the Nppa promoter in Day 5 reprogrammed cells treated with TGF-β1 (Fig. 3G). In contrast, reprogrammed cells treated with A-83-01 displayed significantly increased binding of GATA4 to Tbx20 and Myh6 promoters compared to TGF-β1 treated or vehicle treated reprogrammed cells (Fig. 3G). Basal TGF-β signaling appeared to have intermediate GATA4 binding at these promoters; GATA4 binding to Nppa was indistinguishable from A-83-01 treated reprogrammed cells whereas GATA4 binding to Tbx20 and Myh6 was significantly lower than A-83-01 treated reprogrammed cells (Fig. 3G).
Taken together, our data indicate that canonical activation of TGF-β signaling significantly impairs demethylation of H3K27me3 and binding of GATA4 to cardiac gene loci and correlates with decreased cardiac gene expression and reprogramming efficiency (Fig 1).
2.5. Canonical TGF-β signaling disrupts the interactions between GATA4 and JMJD3/BRG1, thereby reducing gene expression
It has been shown that JMJD3 also acts as a scaffolding protein by bridging the interactions between BRG1 and T-box transcription factors including T-bet [42]. Therefore, we hypothesized that JMJD3 and cardiac transcription factors could interact with other nuclear factors to form a complex and efficiently activate gene expression. Thus, we transduced MEFs with GHMT, harvested nuclear extracts of reprogrammed cells at Day 5, performed Co-IP followed by SDS-PAGE, stained gels with SPYRO Ruby protein gel stain, and excised individual bands to performed unbiased mass spectrometry. Our mass spectrometry analysis revealed that several components of the SWI/SNF chromatin remodeling complex including BRG1, BAF180, BAF170, and BAF155 co-immunoprecipitated with GHMT reprogramming factors (Fig. S5A and S5B). We confirmed by Co-IP assays that both JMJD3 and GHMT reprogramming factors interact with BRG1 (Fig. 4A and B).
Fig. 4. GATA4, JMJD3, and BRG1 form a large epigenetic complex which is disrupted by canonical TGF-β signaling.
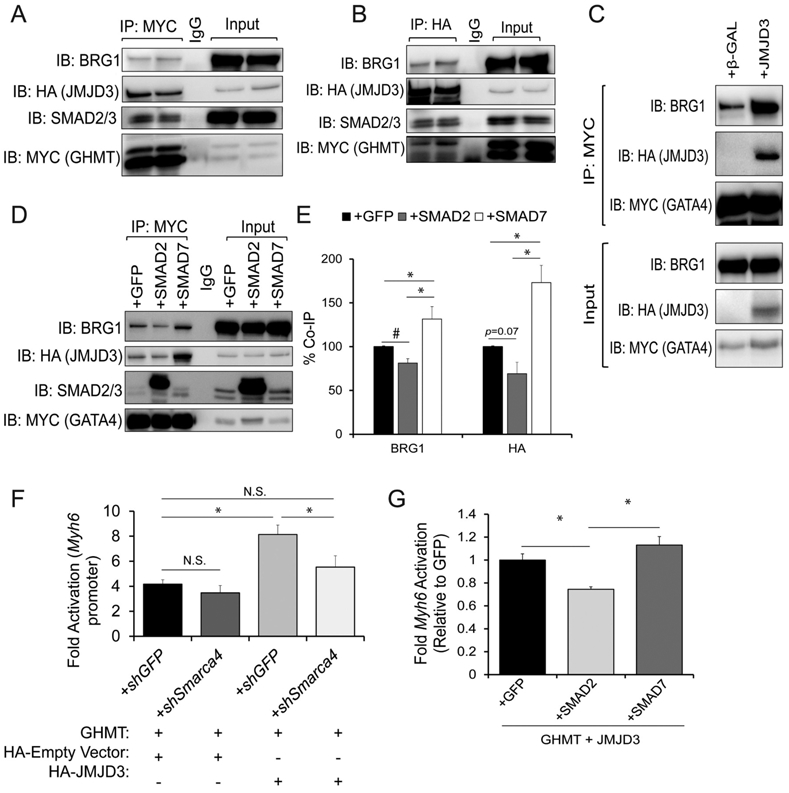
A and B) Co-IPs using nuclear lysates harvested from HEK293T cells transfected with MYC-GHMT and HA-JMJD3. Following Co-IP with anti-MYC (A) or anti-HA (B), proteins were resolved by SDS-PAGE and immunoblotted for BRG1, HA, MYC and SMAD2/3. C) Co-IPs using nuclear lysates harvested from HEK293T cells co-transfected with MYC-GATA4 and either β-GAL (control) or HA-JMJD3. Following Co-IP with anti-MYC, proteins were resolved by SDS-PAGE and immunoblotted for BRG1, HA, and MYC. D) Co-IPs using nuclear lysates harvested from HEK293Ts co-transfected with HA-JMJD3, MYC-GATA4, and GFP, SMAD2, or SMAD7. Following Co-IP with anti-MYC, lysates were resolved by SDS-PAGE and immunoblotted for BRG1, HA, SMAD2/3, and MYC. E) Quantification of interactions between GATA4 and JMJD3 or BRG1 shown in panel D. N = 3 per group. F) Luciferase reporter assay performed in HEK293Ts co-transfected with Myh6 promoter vector, Renilla vector, GHMT, pCMV-HA (empty vector control) or HA-JMJD3, and shGFP or shSmarca4 (BRG1). Reporter activation was determined as a ratio of firefly luciferase to Renilla luciferase and normalized to HEK293Ts transfected with Myh6 promoter vector, Renilla vector, and pCMV-HA alone. N = 3 per group. G) Luciferase reporter assay performed in HEK293Ts co-transfected with Myh6 promoter vector, Renilla vector, GHMT, HA-JMJD3, and GFP, SMAD2, or SMAD7. Reporter activation was determined as a ratio of firefly luciferase to Renilla luciferase and normalized to the GFP group. N = 3 per group.
All data shown as mean ± SEM. * p < 0.05 by one-way ANOVA with Tukey’s multiple comparison test at specified comparisons. # p < 0.05 by Student’s t-test.
To reduce the complexity of our experiments, we opted to use only a single bait protein rather than a pool of MYC-tagged bait proteins (G/H/M/T). We therefore focused on identifying which reprogramming factor was indispensable for cardiac reprogramming. To this end, we serially removed one factor from our GHMT cocktail. Whereas the majority of cells stained positive for cardiac Troponin T (cTnT) and α-actinin in GHMT-transduced MEFs, removal of any single factor significantly reduced the number of positive cells for either marker (Fig. S6A-D). While removal of MEF2C greatly reduced the number of cells expressing α-actinin and modestly reduced the number of cells expressing cTnT, removal of GATA4 nearly abolished expression of cTnT and α-actinin in reprogrammed MEFs (Fig. S6A-D). Since GATA4 is indispensable in cardiac reprogramming, we specifically focused on the interactions between GATA4 and chromatin modifiers. We observed that by itself, GATA4 interacts with BRG1 (Fig. 4C) but that JMJD3 greatly enhances the binding of GATA4 to BRG1 (Fig. 4C).
GATA4 interacts with SMAD2/3/4 to synergistically activate gene expression in the gut epithelium [41]. Our data indicate TGF-β signaling prevents GATA4 from binding promoters of cardiac genes (Fig. 3G). Therefore, we hypothesized that nuclear SMAD2/3 perturbs the formation of the epigenetic complex containing GATA4, JMJD3, and BRG1. While TGF-β treatment did not induce a response in HEK293T cells, A-83-01 treatment potently suppressed basal phosphorylation of SMAD2 (Fig. S7A). Furthermore, overexpression of SMAD2 significantly increased SMAD2 phosphorylation whereas SMAD7 overexpression suppressed basal phosphorylation (Fig. S7A). Therefore, we overexpressed MYC-GATA4 and HA-JMJD3 in HEK293Ts and co-expressed GFP, SMAD2, or SMAD7. Overexpression of SMAD2 significantly decreased the association between GATA4 and BRG1 (Fig. 4D and E). While not significant, a similar trend was observed in the association between GATA4 and JMJD3 (p = 0.07). In contrast, overexpression of SMAD7 enhanced the interaction between GATA4 and both BRG1 and JMJD3, indicating that canonical TGF-β signaling perturbs the formation of this epigenetic complex (Fig. 4D and E). To exclude the possibility that strong overexpression of SMAD2 or SMAD7 artificially influences the interactions between GATA4 and epigenetic modifiers, we also co-expressed MYC-GATA4 and HA-JMJD3 in HEK293Ts and treated them with DMSO or A-83-01. As with SMAD7 overexpression, cells treated with A-83-01 exhibited enhanced interactions between GATA4 and BRG1/JMJD3 compared to vehicle treated cells (Fig. S7B and C). BRG1 has been previously shown to interact with RNA polymerase II [43]. Therefore, we hypothesized that JMJD3 promotes transcription independent of its H3K27me3 demethylase activity via its interaction with BRG1. Thus, we overexpressed GHMT and utilized a Myh6-luciferase reporter as a readout of transcriptional activity. Strikingly, addition of JMJD3 doubled GHMT-driven reporter activity compared to empty vector (HA) control (Fig. 4F). Moreover, knockdown of Smarca4 (BRG1) completely abrogated the effect of JMJD3 on reporter activity (Fig. 4F). Finally, we assessed Myh6-luciferase reporter activity in response to canonical TGF-β signaling. Overexpression of SMAD2 in conjunction with GHMT and JMJD3 significantly reduced reporter activity compared to overexpression of GFP or SMAD7 (Fig. 4G).
Altogether, our data show that GATA4 forms an epigenetic complex containing JMJD3 and BRG1 to promote transcription of cardiac genes. Additionally, the formation of this complex is disrupted by activation of canonical TGF-β signaling.
2.6. GATA4 interacts with JMJD3 via a linker sequence between zinc finger domains
Since canonical TGF-β signaling disrupts interactions between GATA4 and JMJD3/BRG1 (Fig 4D and E), we posited that SMAD2/3 and JMJD3 bind similar or adjacent regions of GATA4. To investigate this, we generated GATA4 truncations to map which domain(s) of GATA4 interact with BRG1, JMJD3, and SMAD2/3, respectively. Strikingly, SMAD2/3 and JMJD3 binding to GATA4 was completely abolished by removal of either zinc finger domain (Fig. 5A and B, lanes 3 and 5). In addition, these truncation constructs also displayed reduced binding to BRG1 (Fig. 5A and B, lanes 3 and 5). We noted that an 18 amino acid sequence (amino acids 252 to 269) residing between the two zinc finger domains of GATA4 did not overlap between truncation constructs #3 and #5 which removed the N terminal (NZF) or C terminal zinc fingers (CZF), respectively. We then generated a deletion of this 18-amino acid sequence (GATA4Δ252–269) but left both zinc finger domains intact. GATA4Δ252–269 did not exhibit impaired binding to SMAD2/3; however, binding to JMJD3 was completely abolished (Fig. 5A and B, lane 6). Therefore, we conclude that GATA4 binds to JMJD3 via a short sequence between its two zinc finger domains, which is flanked by SMAD2/3 binding elements. We next generated truncations to map which domain(s) of JMJD3 bind GATA4. JMJD3 truncations lacking the Jumonji C (JmjC) catalytic domain exhibited reduced binding to GATA4 (Fig. 5C and D). In contrast, the JmjC domain alone robustly co-immunoprecipitated with GATA4, suggesting the primary GATA4 binding domain resides in the JmjC domain of JMJD3 (Fig. 5C and D, lane 5).
Fig. 5. GATA4 interacts with JMJD3 via a short linker between Zinc Finger domains including residue V266.

A and B) Co-IPs (A) using nuclear lysates harvested from HEK293Ts transfected with full length HA-JMJD3 and MYC-GATA4 truncation constructs (Schematic, B). Following Co-IP with anti-MYC, proteins were resolved by SDS-PAGE and immunoblotted for BRG1, HA, SMAD2/3, and MYC. NZF, N Terminal zinc finger. CZF, C Terminal zinc finger. +++, ++, +, +/−, and – represent strong, moderate, mild, minimal, and no binding respectively. C and D) Co-IP (C) using nuclear lysates harvested from HEK293Ts transfected with full length MYC-GATA4 and HA-JMJD3 truncation constructs (Schematic, D). Following Co-IP with anti-MYC, proteins were resolved by SDS-PAGE and immunoblotted for HA and MYC. JmjC, Jumonji C domain. +++ and + represent strong and mild binding respectively. E) Co-IPs using nuclear lysates harvested from HEK293Ts transfected with HA-JMJD3 and MYC-GATA4 wildtype (WT) or MYC-GATA4 V266M mutant. Following Co-IP with anti-MYC, proteins were resolved by SDS-PAGE and immunoblotted for HA and MYC. F) Quantification of interactions between JMJD3 and GATA4 shown in panel E. N = 3 per group. * p < 0.05 by Student’s t-test. G) Electrophoretic mobility shift assay (EMSA) performed using nuclear extracts from HEK293Ts transfected with GFP, WT GATA4, GATA4Δ252–269, or GATA4(V266M). Arrows point to free probes, probes bound to GATA4, or probes bound to GATA4/MYC antibody (supershift) complex. H) Luciferase reporter assay performed in HeLa cells co-transfected with Nppa or Myh6 promoter vectors, Renilla vector, and WT GATA4, GATA4Δ252–269, or GATA4 V266M. Reporter activation was determined as a ratio of firefly luciferase to Renilla luciferase and normalized to the reporter alone group. N = 3 per group. Data shown as mean ± SEM. * p < 0.05 by one-way ANOVA with Tukey’s multiple comparison test vs WT GATA4.
2.7. Interaction between GATA4 and JMJD3 is required for GATA4 to promote cardiac reprogramming and human heart development
Mutations in GATA4 are associated both with familial and sporadic cardiomyopathies [44] and are also observed in patients with CHD [45,46]. Mechanistically, previous studies demonstrated that the GATA4(G296S) mutation caused cardiomyopathy or CHD by impairing the ability of GATA4 to bind DNA, transcriptional cofactors, and transcriptional co-repressors, which coincided with transcriptional dysregulation and epigenetic repatterning at endothelial and cardiogenic genes [47,48]. To determine whether the interaction between GATA4 and JMJD3 is crucial for GATA4 to promote cardiogenesis, we screened the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk) and identified several GATA4 mutations residing within the JMJD3 binding domain (amino acids 252–269). One such mutation (GATA4 C.799G > A, which encodes a missense from valine to methionine at residue 267) was associated with cardiac defects including ventricular-septal defect (VSD) and patent ductus arteriosus (PDA) [37,38]. We used site-directed mutagenesis to generate a homologous mutation in mouse GATA4, GATA4(V266M). GATA4(V266M) exhibited significantly impaired binding to JMJD3 (Fig. 5E and F). However, this mutation did not affect the ability of GATA4 to bind DNA or activate transcription of target genes including Myh6 and Nppa compared to wildtype in in vitro luciferase assays (Fig. 5G and H). In contrast, GATA4Δ252–269 was unable to bind DNA, resulting in significantly impaired transcriptional activation of target genes (Fig. 5G and H).
Since GATA4(V266M) exhibits reduced JMJD3 binding rather than altered DNA binding and/or transcriptional activity, it is an ideal mutant to determine the roles of the JMJD3-GATA4 interaction in cardiogenesis. We therefore reprogrammed MEFs with wildtype (WT), Δ252–269, or GATA4(V266M) in addition to HMT2m reprogramming factors. Since GATA4Δ252–269 is unable to bind DNA, we speculated MEFs transduced with this construct would fail to reprogram. Indeed, MEFs transduced with GATA4Δ252–269 displayed significantly reduced expression of cardiac genes at Day 5 and generated almost no beating cells by Days 10 or 13 (Fig. 6A and B). In contrast, MEFs transduced with GATA4(V266M) did generate beating CMs, but to a lesser extent than MEFs transduced with WT GATA4 (Fig. 6B). Moreover, expression of cardiac genes ACTC1, Tbx20, and Smarcd3 was impaired in MEFs transduced with GATA4(V266M) compared to WT GATA4 at Day 5 (Fig. 6A).
Fig. 6. GATA4 mutation associated with CHD impairs cardiac differentiation.

A) Messenger RNA expression of cardiac genes Myh6, Actc1, Tbx20, and Smarcd3 harvested from Day 5 GHMT2m-reprogrammed using WT, Δ252–269, or V266M GATA4. Reprogrammed cells were maintained in 0.5 μM A-83-01 starting at Day 3. All data were normalized to the WT GATA4 group. N = 3 per group. B) Beating cell counts per field (0.89mm2) from Day 10 and Day 13 GHMT2m-reprogrammed cells using WT, Δ252–269, or V266M GATA4. Reprogrammed cells were maintained in 0.5 μM A-83-01 starting at Day 3. All data were normalized to the WT GATA4 group. N = 3 per group. C) Messenger RNA expression of cardiac genes GATA4, TBX20, HAND1, ISL1, and MEF2C harvested from Day 5 WT (CUSO-2) or three isogenic GATA4(V267M) hiPS-CM lines. N = 4 for CUSO-2, V267M-1, and V267M-7 lines. N = 3 for the V267M-9 line. D) H3K27me3 levels at cardiac gene promoters Gata4 and Tbx20 from Day 7 WT or three isogenic GATA4(V267M) hiPS-CM lines. N = 4 per group. All data shown as mean ± SEM. * p < 0.05 by one-way ANOVA with Tukey’s multiple comparison test vs WT GATA4 groups. # p < 0.05 by Student’s t-test vs WT GATA4.
CHD arises from impaired heart development; therefore, we sought to study the GATA4(V267M) mutation in a human model that better reflects physiological development. We utilized CRISPR-Cas9 genome editing to generate a homozygous GATA4(V267M) mutation in hiPSCs (Fig. S8A and B). We then differentiated GATA4(V267M) and WT GATA4 (CUSO-2) hiPSC lines into CMs using previously published methods [49,50]. GATA4 protein levels in GATA4(V267M) lines were significantly reduced at Day 7 of differentiation (Fig. S8C and D). Moreover, mRNA expression of cardiac markers GATA4, TBX20, HAND1, and ISL1 was significantly reduced at Day 5 of differentiation in two GATA4(V267M) lines, with similar trends in the third line (Fig. 6C). Strikingly, H3K27me3 levels at GATA4 and TBX20 promoters were markedly increased in GATA4 V267M lines compared to WT control (Fig. 6D). Our findings reveal a novel mechanism by which mutant GATA4 causes impaired cardiac differentiation, which may translate to CHD in humans, by interfering with interactions between the transcription factor and epigenetic modifiers including JMJD3.
3. Discussion
Cardiac reprogramming represents a promising novel therapeutic approach to regenerate CMs lost after ischemic injury. While significant progress has been made in optimizing reprogramming efficiency [15,20,22,26] since the initial report [11], mechanisms governing cardiac reprogramming remain largely unknown. Here we report a novel mechanism by which canonical TGF-β pathway activation impairs cardiac reprogramming via disrupting interactions between GATA4 and epigenetic modifiers JMJD3 and BRG1. This results in elevated levels of H3K27me3 and lower levels of GATA4 bound to cardiac promoters. Furthermore, a mutation in GATA4 (V267M), which is associated with human CHD, disrupted binding to JMJD3 and impaired demethylation of H3K27me3 at cardiac loci. This was accompanied by reduced cardiac gene expression and impaired cardiomyogenesis in hiPSCs carrying the mutation (GATA4 C.799G > A) or reprogrammed fibroblasts overexpressing mutant GATA4(V266M) (Fig. 7). Our study therefore sheds new light on how recruitment of epigenetic complexes regulates cardiomyogenesis.
Fig. 7.

Recruitment mechanisms of epigenetic modifiers to cardiogenic genes by GATA4. Schematic summary of the regulation of interactions between GATA4 and epigenetic modifiers. Canonical TGF-β pathway activation or the congenital heart disease-associated mutation in GATA4 (V267M) impede the recruitment of epigenetic modifiers JMJD3 and BRG1 to cardiogenic genes by GATA4, resulting in impaired H3K27me3 demethylation and gene expression in cardiomyogenesis. TGF-β blockade prevents nuclear translocation of the SMAD2/3/4 complex, promoting the recruitment of epigenetic modifiers to cardiogenic genes by GATA4 resulting in efficient removal of H3K27me3 and gene expression.
TGF-β signaling results in increased deposition of H3K27me3 at genes silenced by epithelial-to-mesenchymal transition including Cdh1 [51]. Moreover, deposition of H3K27me3 was accompanied by elevated binding of Polycomb repressive complex 2 (PRC2) members Enhancer of zeste homolog 2 (EZH2) and Jumonji And AT-Rich Interaction Domain Containing 2 (JARID2) [51]. We observed similar trends towards elevated levels of H3K27me3 at the Myh6 promoter in GHMT2m-reprogrammed cells overexpressing SMAD2 compared to control MEFs. This suggests that canonical TGF-β signaling may also actively repress promoters, potentially via interactions with PRC2 components. Previous studies have shown that inhibition of EZH2 improves reprogramming efficiency [21,52]. Parenthetically, the EZH2 inhibitor Tazemetostat has shown promising results in clinical trials with limited toxicity [53] and was granted accelerated approval status by the FDA for the treatment of epithelioid sarcoma in early 2020. Therefore, inhibition of EZH2 may be a promising alternative to TGF-β inhibition, which despite showing favorable safety profiles in patients in more recent trials, remains a safety concern given the diverse roles TGF-β signaling plays in normal physiology [54-56].
Previous studies have demonstrated the importance of H3K27me3 demethylases in heart development and cardiomyogenesis [21,30,32,33]. However, to our knowledge, we are the first to report a defined mechanism by which canonical TGF-β signaling dramatically alters the epigenetic landscape of GHMT2m-reprogrammed cells. JMJD3 has been shown to interact with Isl1 and is critical for cardiac differentiation of ESCs [32]. GHMT-mediated reprogramming does not induce expression of Isl1 (data not shown), so it is not likely that the JMJD3 recruitment depends on ISL1 in cardiac reprogramming. Instead, our data show that JMJD3 interacts with additional transcription factors GATA4, HAND2, and TBX5, which are downstream of ISL1 in cardiomyogenesis. These data therefore suggest that JMJD3 is critical at multiple stages of cardiac development, from early mesodermal stages through immature CMs. We show that cardiac transcription factors strongly interact with JMJD3, but minimally interact with UTX (Fig. 2B-D, S2A). Consistently, knockdown of Kdm6a encoding UTX impaired generation of beating iCMs at early time points but beating iCM numbers were not significantly changed compared to shGFP control at later time points (Fig. S2B). In contrast, knockdown of Kdm6b encoding JMJD3 significantly reduced beating iCM counts at all time points investigated. This may suggest JMJD3 can compensate for UTX loss in cardiac reprogramming, but UTX cannot compensate for JMJD3 loss. However, we also note that the knockdown efficiency of Kdm6a was worse than the knockdown efficiency of Kdm6b in our study (Fig. 2F vs S2C). While it is unlikely that UTX influences cardiac reprogramming through the same mechanism as JMJD3 described in our study given the differences in interaction affinity to cardiac transcription factors (compare Fig. 2B with Fig. S2A), establishing the precise roles of Kdm6a in cardiac reprogramming requires further investigation. Treatment of mouse ESCs with a small molecule that induces degradation of the type II TGF-β receptor was previously reported to promote cardiac differentiation at early time points [57]. Since JMJD3 plays crucial roles in cardiac differentiation of ESCs [32], it is possible that the mechanism by which canonical TGF-β pathway activation disrupts recruitment of JMJD3 to cardiogenic genes also functions in physiological development in addition to fibroblast reprogramming.
Our data indicate that canonical TGF-β signaling is the primary mechanism through which TGF-β signaling impairs cardiomyogenesis. Overexpression of SMAD2 resulted in similar levels of H3K27me3 at cardiac promoters, similar expression of cardiac gene transcripts, and similar numbers of beating iCMs as treatment with TGF-β1. Moreover, overexpression of SMAD7 trended in the same direction as treatment with A-83-01 in all assays tested. However, treatment with A-83-01 still resulted in significantly increased beating iCM counts and trends in reduced H3K27me3 at cardiac promoters compared to SMAD7 overexpression. One possibility is that SMAD7 overexpression did not reduce levels of phosphorylated SMAD2 to the extent that A-83-01 treatment did (Fig. 1A). Alternatively, it is possible that blockade of non-canonical TGF-β signaling also positively regulates cardiac reprogramming. Therefore, downstream effectors of non-canonical TGF-β signaling should be evaluated in future studies. Additionally, SMAD7 enables activation of STAT3 [58], which has previously been linked to impairing cardiac reprogramming efficiency [24]. Paradoxically, JAK-STAT activation also upregulates SMAD7 expression [59], which may trigger a positive feedback loop to promote inflammatory signaling while also suppressing TGF-β signaling. Recently, Hashimoto et al. demonstrated that suppression of EGFR signaling or the downstream effectors JAK1/2 enhanced GHMT+AKT-reprogramming by 1.5− to 2-fold [60]. In contrast, we show here that suppression of TGF-β signaling by A-83-01 increases GHMT2m-mediated reprogramming by five- to six-fold. Additional downstream effectors of EGFR signaling, ERK1/2, have been observed to mediate SMAD2 phosphorylation [61]. Therefore, the relatively modest effects of EGFR inhibition could be due to opposing roles on the reprogramming process; suppression of inflammatory signaling promotes reprogramming, but activation of TGF-β signaling represses reprogramming. Future studies that focus on understanding the crosstalk between various signaling pathways will likely lead to improved reprogramming strategies.
While our ChIP-qPCR data indicate significantly reduced levels of H3K27me3 at four out of five validated promoters in A-83-01 treated reprogrammed cells compared to vehicle (DMSO), our ChIP-seq analysis did not reveal significant differences in H3K27me3 status between these two groups globally. A caveat of ChIP assays is that they only offer a snapshot of the epigenetic landscape, which is well known to dynamically change during cell differentiation. We initially chose to evaluate H3K27me3 levels in Day 7 reprogrammed cells since we observed the highest degree of demethylation by this time point in our time course assay (Fig. 2A) and therefore rationalized that we would see the largest differences between reprogrammed cells with activated TGF-β signaling and those with inhibited TGF-β signaling at this time point. We also noted that vehicle treated cells exhibited demethylation of H3K27 similar to A-83-01 treated cells by Day 9, suggesting that basal TGF-β signaling does not prevent but does slow the kinetics of H3K27 demethylation. Therefore, it is possible that larger differences in H3K27me3 levels between A-83-01 treated cells and vehicle treated cells would be observed at earlier time points than Day 7. Alternatively, it is possible that the reprogramming processes is sensitive to the expression of a handful of cardiac transcription factors. Consistent with this hypothesis, we observed that H3K27me3 levels at the Tbx20 and GATA4 promoter were significantly reduced in A-83-01 treated reprogrammed cells compared to vehicle treated cells (Fig. 3E and F). Finally, removal of a single repressive epigenetic mark like H3K27me3 does not necessarily guarantee expression of a gene. We did not assess deposition of active chromatin marks including H3K4me1, H3K4me3, or H3K27ac in this study. We chose to focus on H3K27me3 since its removal during reprogramming is rapid (Fig. 2A) whereas deposition of active chromatin marks in reprogrammed cells continues throughout the reprogramming process [30]. Moreover, H3K27me3 cannot exist on the same histone tail as H3K27ac. Conditions that prevent or delay demethylation of H3K27 necessarily prevent or delay acetylation of H3K27, thus it is possible that A-83-01 treatment facilitates rapid demethylation of H3K27me3 and a concomitant decondensed chromatin state to allow for sooner deposition of active chromatin marks to further enhance reprogramming efficiency. Future studies should therefore evaluate the dynamics and cross-talk of the epigenetic landscape beyond H3K27me3 in the context of cardiac reprogramming.
A previous report indicated that GATA4(V267M) displayed reduced binding to the mediator complex and BRG1, which significantly reduced its transcriptional activation of genes in hepatocellular carcinoma cells [62]. Our data indicate that the mouse analog, GATA4(V266M), does not disrupt DNA binding or transcriptional activity but does exhibit reduced binding to JMJD3. This reduced interaction with JMJD3 correlated with impaired fibroblast reprogramming to CMs and CM differentiation of hiPSCs. It is possible that in hepatocytes, GATA4 (V267M) cannot bind a critical cofactor and fails to recruit the mediator complex whereas in cardiac progenitor cells, this mutation can still bind cofactors that recruit the mediator complex. However, in cardiac progenitor cells, GATA4(V267M) does not efficiently recruit JMJD3 to cardiogenic genes, resulting in impaired H3K27me3 demethylation and gene expression, leading to CHD-associated phenotypes. Unraveling the myriad of protein-protein interactions involved in transcriptional activation in future studies will therefore further elucidate the specific contexts in which the same mutation can lead to different pathogenic mechanisms in different tissues.
To summarize, we have established a mechanism whereby the canonical TGF-β signaling effector SMAD2 disrupts interactions between GATA4 and epigenetic modifiers JMJD3 and BRG1, resulting in elevated levels of H3K27me3 at cardiac promoters, reduced cardiac gene transcription, and impaired binding of GATA4 to target genes. Interactions between GATA4 and these epigenetic modifiers are not only required for optimal cardiac reprogramming, but also normal human heart development.
4. Materials and methods
4.1. Isolation of mouse embryonic fibroblasts
Embryonic fibroblasts were isolated as described previously [39]. Briefly, pregnant C57BL/6 were purchased at E13 from Charles River Laboratories. Upon arrival, animals were sacrificed and embryos at E14.5 were harvested. Upper body, head, and internal organs were discarded. The body below the liver was minced into fine pieces and digested in 0.25% Trypsin/EDTA (Gibco) for 40 min at 37 °C with 5% CO2. Cells were resuspended in 25 mL DMEM/High Glucose (HyClone) containing 10% fetal bovine serum (FBS; Gemini), 1.1% Penicillin-Streptomycin (Gibco), and 1.1% GlutaMAX supplement (Gibco) and plated in a 15-cm dish. Medium was changed after 24 h. After 72 h, MEFs were collected and frozen for future use.
4.2. Plasmids and cloning
pBabe-X GATA4, Hand2, Mef2c, Tbx5, miR-1, and miR-133 were generated previously [14,15]. pCMV-HA-JMJD3 was a gift from Kristian Helin (Addgene plasmid #24167 [63]). pBabe-X-HA-UTX was subcloned from pCMV-HA-UTX, a gift from Kristian Helin (Addgene plasmid #24168 [63]). pBabe-X-HA-SMAD2 was subcloned from pCMV5B-HA-SMAD2, a gift from Jeff Wrana (Addgene plasmid #11734 [64]). pBabe-puro-SMAD7-HA was a gift from Sam Thiagalingam (Addgene plasmid #37044 [65]). Kdm6a, Kdm6b, and Smarca4 shRNA sequences (Table S1) were designed using the online webtool http://cancan.cshl.edu/RNAi_central/RNAi.cgi?type=shRNA and oligonucleotides were purchased from IDT, amplified by PCR, and ligated into a modified pShagMagic2 miR-based vector, MSCV-PM (Addgene plasmid #27021 [66]). pRS-Smad2 shRNA constructs were purchased from Origene (TR509694). Site directed mutagenesis to generate GATA4(V266M) was performed with the Q5 Site-Directed Mutagenesis Kit (NEB) as per the manufacturer’s instructions. Mutagenesis primers were designed using the NEBaseChanger web tool (http://nebasechanger.neb.com/). GATA4 and JMJD3 truncation plasmids were generated by serially removing domains from N or C termini by PCR. Primers used for site directed mutagenesis and domain truncation and shRNA sequences are listed in Table S2.
4.3. Generation of retrovirus and transduction of MEFs
Retroviral infection of MEFs was performed as previously described [39]. Approximately 5 × 106 Platinum E cells (PE cells; Cell Biolabs) were plated in 10-cm dishes. 20 h later, cells were transfected with 36 μL FuGENE 6 (Promega) or polyethylenimine (PEI, Sigma) and 12 μg of retroviral plasmid DNA. On the day of transfection, MEFs were thawed and plated into 60-mm dishes pre-coated with SureCoat (Cellutron). MEFs were allowed to settle overnight. Viral supernatant was collected from transfected PE cells at 24 and 48 h and filtered through a 0.45-μm cellulose filter. In experiments performed with PEI, culture medium was changed 16 h post-transfection and viral supernatant was instead collected from PE cells at 48 and 72 h post-transfection to avoid PEI toxicity to MEFs. Polybrene (Sigma) was added to viral supernatant at a concentration of 6 μg/mL and added to dishes containing MEFs. Following the first collection of viral supernatant, fresh medium (DMEM/High Glucose (HyClone) containing 10% FBS (Gemini), 1.1% Penicillin-Streptomycin (Gibco), and 1.1% GlutaMAX supplement (Gibco) was added to PE cells. Infected MEFs were maintained in induction medium composed of DMEM/High Glucose:Medium 199 (4,1, HyClone,Gibco), 10% FBS (Gemini), 5% horse serum (Gemini), antibiotics (Gibco), 1 × non-essential amino acids (Gibco), 1 × essential amino acids (Gibco), 1 × B-27 (Gibco), 1 × insulin-selenium-transferin (Gibco), 1× MEM vitamin solution (Gibco) and 1× sodium pyruvate (Gibco) starting at 72 h post-infection. DMSO (Thermo Scientific), 5 ng/mL TGF-β1 (Novoprotein CA59), 0.5 μM A-83-01 (Tocris 2939), or 0.5 μM or 1 μM GSK-J4 (Tocris 4594) were administered starting at 72 h post-infection. Induction medium was changed every 2 days.
4.4. RNA extraction and quantitative, real time PCR
Total RNA was extracted from D13 reprogrammed MEFs using TRI-zol reagent (Invitrogen) and chloroform. The aqueous layer containing RNA was transferred to a new tube and RNA was precipitated using isopropanol. RNA was washed with 70% ethanol and dried. cDNA was synthesized using the iScript Reverse Transcription Supermix (Biorad). Diluted cDNA was amplified using SYBR PowerUP master mix (Applied Biosystems) and expression was determined by StepOne Real-Time PCR System (Applied Biosystems). qPCR primer sequences are listed in Table S3.
4.5. Immunocytochemistry
MEFs were seeded and reprogrammed by GHMT or GHMT2m in 12 well plates. On days 9 and 12, cells were rinsed 1× in ice-cold PBS and fixed in 2% paraformaldehyde for 10 min at room temperature. Cells were washed 3× in PBS and permeabilized with 0.2% Triton X-100 for 15 min at room temperature. Cells were then blocked in 10% horse serum (Gemini). Primary antibodies (Cardiac Troponin T; Thermo Scientific ms-295-p - 1:400; α-actinin; Sigma A7811L - 1:400; Cardiac Troponin I; Phosphosolutions 2010-TNI – 1:400) were diluted in 10% horse serum (Gemini) and added to fixed cells for 1 h at room temperature. Cells were washed 3× in PBS and then incubated with diluted secondary antibodies (anti-mouse Alexa 555; Life Technologies A-21422 - 1:800; anti-mouse Alexa 488; Life Technologies A11034–1:800) and Hoechst (Life Technologies 62,249–1:10,000) for 1 h at room temperature in the dark. Cells were then washed 3× in PBS and imaged an EVOS FL Color Imaging System (Life Technologies).
4.6. Co-immunoprecipitation
HEK293T cells were split into 10 cm dishes and 12 μg DNA was transfected into cells using PEI at a 3:1 ratio of PEI:DNA. 48 h post-transfection, nuclear lysates were prepared using the NE-PER Nuclear and Cytosolic Extraction Kit (Thermo 78,835). Protein concentration was determined by BCA assay and 250 μg protein lysates were incubated with anti-HA (5 μg, UBPBio Y1071), anti-Myc (2.5 μg, BD Pharmingen 551,102), or anti-IgG (5 μg, Santa Cruz sc-2025) and 20 μL Protein G DynaBeads slurry (Invitrogen 10004D) overnight at 4 °C in an end-over-end mixer. The following morning, beads were washed 4 times in wash buffer (150mM NaCl, 50mM Tris-Cl pH 7.4, 1 mM EDTA, 1% Triton) and boiled in 1 × loading dye (75 mM Tris-HCl, pH 6.8, 2.4% SDS, 12% glycerol, 0.12 M DTT, 0.012% bromophenol blue). Lysates were run on a 10% polyacrylamide gel and transferred to a PVDF membrane (110 mA, 16 h, 4 °C).
4.7. Western blot
Cells were washed with ice-cold DPBS (Gibco) twice. Whole cell extracts were harvested with ice-cold lysis buffer (150mM NaCl, 50mM Tris-Cl pH 7.4, 1 mM EDTA, 1% Triton, and freshly added protease inhibitors (Complete mini tablet (Roche) and 1 mM phenylmethylsulphonyl fluoride (PMSF)). Protein concentration was determined by BCA assay and 30 μg protein lysates were loaded and resolved on a 10% polyacrylamide gel and transferred to a PVDF membrane (110 mA, 16 h, 4 °C). The following primary antibodies were used: anti-p-Smad2 (Cell Signaling 3108S, 1:1000), anti-total Smad2/3 (Cell Signaling 8685S, 1:1000), anti-HA (Rockland 600–401-384, 1:5000), anti-Myc (Santa Cruz sc-789, 1:500), anti-Brg1 (Santa Cruz sc-17,796, 1:500), and anti-GAPDH (Life Technologies AM4300, 1:5000). Secondary antibodies used include the following: Goat Anti-Mouse IgG (H + L; Southern Biotech, 1031–05, 1:2000) and Goat Anti-Rabbit IgG (Life Technologies, 65–6120, 1:2000). Band intensities were quantified by densitometry using ImageJ software.
4.8. Chromatin immunoprecipitation
Chromatin from D5 control MEFs or D7 reprogrammed MEFs was isolated using the Magna ChIP A/G Chromatin Immunoprecipitation kit (Millipore 17–10,085) as per the manufacturer’s instructions. Briefly, 10 cm dishes of cells were crosslinked with 1% paraformaldehyde for 9 min at room temperature. Cells were then quenched with glycine (125 mM final concentration) for 5 min at room temperature. Chromatin was sheared to approximately 300 bp fragments using a Diagenode Bioruptor 300 ultrasonicator (D5 control MEFs: 15 cycles, 30 s on/90 s off, high intensity; D7 reprogrammed MEFs: 25 cycles, 30 s on/30 s off, high intensity). Chromatin was diluted 10 times and incubated overnight at 4 °C with 20 μL Protein A/G MagnaChIP beads (Millipore) and 8 μg anti-H3K27me3 (Millipore, 07–449), 10 μg anti-GATA4 (Santa Cruz sc-1237×) or 10 μg control IgG (Millipore, PP64B) antibodies in an end-over-end mixer. The next day, immunoprecipitated chromatin was washed, eluted, and reverse crosslinked with Proteinase K for 2 h at 62 °C. For qPCR, ChIP DNA was amplified with SYBR PowerUP master mix (Applied Biosystems). Results are expressed as percentage of input. ChIP-qPCR primer sequences are listed in Table S4. For ChIP-seq, libraries were prepared from 1 ng ChIP DNA using the Ovation Ultralow Library System V2 1–16 (NuGEN). Libraries were sequenced using the NovaSEQ 6000 platform (Illumina). To remove Illumina adapters and quality-trim the read ends, reads were filtered using BBDuk (http://jgi.doe.gov/data-and-tools/bb-tools). Bowtie2 (v. 2.3.2) was used to align the 150-bp paired-end sequencing reads to the mm10 reference human genome. Samtools (v.1.5) was used to remove unmapped reads and to randomly extract the same number of reads for all samples. Peaks were called using MACS2 (v2.1.1.20160309) [67] with default parameters. Ngs.plot.r [68] was used for generating the heatmaps and average profiles related to the TSS. Peak locations were further annotated using the ChIPseeker R package [69]. ChIP-seq data have been deposited in the NCBI Gene Expression Omnibus under accession code GSE145290.
4.9. Luciferase reporter assay
HEK293T cells were plated into 6-well plates and transfected with PEI and two micrograms of DNA. pGL3 backbones containing a 2 kb promoter upstream of the Myh6 gene or a 640 bp promoter upstream of the Nppa gene were used to assess promoter activity. 48 h post-transfection, cells were rinsed twice in 1× PBS (Gibco) and lysed with 1× passive lysis buffer (Promega). Cells were snap-frozen in liquid nitrogen to facilitate lysis. 20 μL lysates were loaded into a 96 well plate and luminescence following administration of 50 μL Luciferase Assay Reagent II (Promega) and 50 μL Stop & Glo Reagent (Promega) was measured on a plate reader (BioTek). Firefly luciferase activity was normalized to Renilla luciferase activity and all data are normalized to pGL3-reporter construct alone.
4.10. Mass spectrometry identification of GHMT-interaction partners
Nuclear lysates from D5 GHMT2m reprogrammed MEFs were prepared using the NE-PER Nuclear and Cytosolic Extraction Kit (Thermo 78,835). Protein concentration was determined by BCA assay and 250 μg protein lysates were incubated with anti-Myc (2.5 μg, BD Pharmingen 551,102) or anti-IgG (2.5 μg, Santa Cruz sc-2025) and 20 μL Protein G DynaBeads slurry (Invitrogen 10004D) overnight at 4 °C in an end-over-end mixer. The following morning, beads were washed 4 times in wash buffer (150mM NaCl, 50mM Tris-Cl pH 7.4, 1 mM EDTA, 1% Triton) and boiled in 1 × loading dye (75 mM Tris-HCl, pH 6.8, 2.4% SDS, 12% glycerol, 0.12 M DTT, 0.012% bromophenol blue). Lysates were run on a 10% polyacrylamide gel and stained with SPYRO Ruby Protein Gel Stain (Thermo). Gels were imaged under UV light with a FluorChem 8900 imaging system (Alpha Innotech). Individual bands were excised from the gel with a sterile razor blade. Excised bands were reduced, alkylated, and digested with trypsin. Cleaved peptides were analyzed by nanoflow LC-ESI-MS/MS (Thermo Scientific LTQ Orbitrap Velos Pro). Peptide sequences were mapped using ProteinProspector and Mascot software packages.
4.11. Electrophoretic mobility shift assay (EMSA)
Nuclear lysates were prepared from HEK293T cells transfected with GFP, wildtype GATA4, GATA4Δ252–269, or GATA4(V266M) using the NE-PER Nuclear and Cytosolic Extraction Kit (Thermo 78,835). Protein concentration was determined by BCA assay and 10 μg protein was incubated with 1.25 μM annealed oligonucleotides (5’-TCGAGGTAATTAACTGATAATGGTGC-3′ [48,70]) and 5× binding buffer (20 mM HEPES, pH 7.5, 60 mM KCl, 0.5 mM DTT, 1 mM MgCl2, 4.8% glycerol, and 1.5 mg/mL BSA) for 30 min at room temperature. For supershift assays, 0.375 μg anti-Myc (BD Pharmingen 551,102) was also added to the reaction mixture. Reactions were separated on a 5% non-denaturing polyacrylamide gel, stained with 1:10,000 SYBR Gold nucleic acid gel stain (Invitrogen S11494) for 40 min, and imaged under 302 nm UV light with a FluorChem 8900 imaging system (Alpha Innotech).
4.12. CRISPR-Cas9 genome editing
The GATA4(V267M) Alt-R CRISPR-Cas9 crRNA targeting sequence was designed using the online tool at http://crispr.mit.edu and purchased from IDT. crRNA was mixed with Alt-R CRISPR-Cas9 tracrRNA (IDT) and annealed at 95 °C for 5 min before cooling to room temperature. Annealed gRNA complex was added to Alt-R S.p. HiFi Cas9 Nuclease, V3 (IDT) to form the RNP complex and incubated at room temperature for 20 min. RNP complex was added to ssDNA GATA4 (V267M) repair template (IDT) and delivered to wildtype iPSCs by electroporation with the Amaxa human stem cell nucleofector starter kit (Lonza). Following electroporation, single cell clones were selected and expanded for genotyping by PCR. Genomic GATA4(V267M) edits were confirmed by Sanger sequencing. GATA4(V267M) crRNA, ssDNA repair template, PCR genotyping, and sequencing oligonucleotide sequences are listed in Table S2.
4.13. iPSC culture and iPS-CM differentiation
Cardiac differentiation of hiPSCs was performed as previously described [49,50]. Briefly, hiPSCs were cultured until ~85% confluence in mTeSR1 Plus medium (STEMCELL Technologies). Cells were dissociated with Accumax (Innovative Cell Technologies) and seeded into 24 well plates pre-coated with Matrigel (Corning) at a density of 300,000 cells per well in mTeSR1 medium containing 10 μM Y27632 (APExBIO) (Day −4). mTeSR1 medium was replenished daily for 3 additional days. At Day 0, medium was changed to RPMI 1640 (Life Technologies) containing B-27 supplement minus insulin (Life Technologies) and cells were treated with 8 μM ChIR99021 (Cayman). After 24 h (Day 1), medium was changed to RPMI 1640 containing B-27 supplement minus insulin. After 72 h (Day 3), 0.5 mL fresh RPMI 1640 containing B-27 supplement minus insulin was added to 0.5 mL conditioned (old) medium and added to cells along with 5 μM IWP2 (Tocris). Medium was aspirated at Day 5 and replenished with fresh RPMI 1640 containing B-27 supplement minus insulin. WT and three GATA4(V267M) mutant lines were cultured and harvested at Day 5 for RNA extraction, Day 7 for protein extraction, and Day 7 for H3K27me3 ChIP.
4.14. Statistical analysis
Unless otherwise stated, statistical analysis was performed using ANOVA followed by Tukey post hoc test for multiple comparisons. If only two groups were compared, Student’s t-test (2 tailed, unequal variance) was performed instead. Data were regarded as significant at p < 0.05. All graphs are displayed as mean ± SEM.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors thank the University of Colorado School of Medicine Biological Mass Spectrometry Core Facility and the Genomics and Microarray Core Facility. A.S.R. was supported by predoctoral fellowships from the University of Colorado Consortium for Fibrosis Research & Translation, Colorado Clinical & Translational Sciences Institute (TL1 TR001081), and the American Heart Association (18PRE34030030). R. A.B. received a postdoctoral fellowship award from the Canadian Institutes of Health Research (FRN-216927). T.A.M. received funding from National Institute of Health by grants HL116848, HL147558, DK119594, HL127240, HL150225, and a grant from the American Heart Association (16SFRN31400013). K.S. was supported by funds from the Boettcher Foundation, American Heart Association (13SDG17400031), University of Colorado Department of Medicine Outstanding Early Career Scholar Program, Gates Frontiers Fund, and National Institute of Health HL133230. We thank Dr. Aaron Johnson and Dr. David Port for insightful discussion and Dr. Jennifer Major for critical reading and editing of the manuscript. Fig. 7 and the graphical abstract were created using Biorender.com.
Abbreviations:
- BRG1
Brahma Related Gene 1
- CHD
Congenital Heart Disease
- ChIP
Chromatin immunoprecipitation
- CM
Cardiomyocyte
- Co-IP
Coimmunoprecipitation
- cTnT
Cardiac Troponin T
- Ezh2
Enhancer of zeste homolog 2
- FDA
Food and Drug Administration
- GATA4
GATA Binding Protein 4
- GHMT
GATA4/HAND2/MEF2C/TBX5
- GHMT2m
GATA4/HAND2/MEF2C/TBX5/miR-1/miR-133
- H3K27me3
Histone H3 Lysine 27 trimethylation
- HAND2
Heart and Neural Crest Derivatives Expressed 2
- hiPSCs
human induced pluripotent stem cells
- iCM
induced Cardiomyocyte
- IHD
Ischemic Heart Disease
- ISL1
ISL LIM Homeobox 1
- JMJD3/Kdm6b
Jumonji-domain Containing Protein D3/Lysine demethylase 6B
- MEF2C
MADS box transcription enhancer factor 2 polypeptide C
- miR
microRNA
- qPCR
quantitative polymerase chain reacion
- shRNA
short hairpin ribonucleic acid
- SMAD
Sma/Mothers Against Decapentaplegic homolog
- SWI/SNF
SWItch/Sucrose Non-Fermentable
- TBX5
T-Box Transcription Factor 5
- TGF-β
Transforming Growth Factor β
- UTX/Kdm6a
Ubiquitously-transcribed X chromosome tetratricopeptide repeat protein/Lysine demethylase 6A
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.yjmcc.2020.12.005.
Declaration of Competing Interests
The authors declare no competing interests.
References
- [1].Roth GA, Johnson C, Abajobir A, Abd-Allah F, Abera SF, Abyu G, et al. , Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015, J. Am. Coll. Cardiol 70 (1) (2017) 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Laflamme MA, Murry CE, Regenerating the heart, Nat. Biotechnol 23 (7) (2005) 845–856. [DOI] [PubMed] [Google Scholar]
- [3].Mercola M, Ruiz-Lozano P, Schneider MD, Cardiac muscle regeneration: Lessons from development, Genes Dev. 25 (4) (2011) 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Talman V, Ruskoaho H, Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration, Cell Tissue Res. 365 (3) (2016) 563–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, et al. , Evidence for cardiomyocyte renewal in humans, Science 324 (5923) (2009) 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hashimoto H, Olson EN, Bassel-Duby R, Therapeutic approaches for cardiac regeneration and repair, Nat. Rev. Cardiol 15 (10) (2018) 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. , Induction of pluripotent stem cells from adult human fibroblasts by defined factors, Cell 131 (5) (2007) 861–872. [DOI] [PubMed] [Google Scholar]
- [8].Takahashi K, Yamanaka S, Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors, Cell 126 (4) (2006) 663–676. [DOI] [PubMed] [Google Scholar]
- [9].Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M, Direct conversion of fibroblasts to functional neurons by defined factors, Nature 463 (7284) (2010) 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Huang P, He Z, Ji S, Sun H, Xiang D, Liu C, et al. , Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors, Nature 475 (7356) (2011) 386–389. [DOI] [PubMed] [Google Scholar]
- [11].Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, et al. , Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors, Cell 142 (3) (2010) 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Addis RC, Ifkovits JL, Pinto F, Kellam LD, Esteso P, Rentschler S, et al. , Optimization of direct fibroblast reprogramming to cardiomyocytes using calcium activity as a functional measure of success, J. Mol. Cell. Cardiol 60 (2013) 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ifkovits JL, Addis RC, Epstein JA, Gearhart JD, Inhibition of TGFbeta signaling increases direct conversion of fibroblasts to induced cardiomyocytes, PLoS One 9 (2) (2014), e89678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN, et al. , Heart repair by reprogramming non-myocytes with cardiac transcription factors, Nature 485 (7400) (2012) 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhao Y, Londono P, Cao Y, Sharpe EJ, Proenza C, O’Rourke R, et al. , High-efficiency reprogramming of fibroblasts into cardiomyocytes requires suppression of pro-fibrotic signalling, Nat. Commun 6 (2015) 8243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hirai H, Katoku-Kikyo N, Keirstead SA, Kikyo N, Accelerated direct reprogramming of fibroblasts into cardiomyocyte-like cells with the MyoD transactivation domain, Cardiovasc. Res 100 (1) (2013) 105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Christoforou N, Chellappan M, Adler AF, Kirkton RD, Wu T, Addis RC, et al. , Transcription factors MYOCD, SRF, Mesp1 and SMARCD3 enhance the cardio-inducing effect of GATA4, TBX5, and MEF2C during direct cellular reprogramming, PLoS One 8 (5) (2013), e63577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Muraoka N, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Isomi M, et al. , MiR-133 promotes cardiac reprogramming by directly repressing Snai1 and silencing fibroblast signatures, EMBO J. 33 (14, 2014) 1565–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nam YJ, Song K, Luo X, Daniel E, Lambeth K, West K, et al. , Reprogramming of human fibroblasts toward a cardiac fate, Proc. Natl. Acad. Sci. U. S. A 110 (14) (2013) 5588–5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhou H, Dickson ME, Kim MS, Bassel-Duby R, Olson EN, Akt1/protein kinase B enhances transcriptional reprogramming of fibroblasts to functional cardiomyocytes, Proc. Natl. Acad. Sci. U. S. A 112 (38) (2015) 11864–11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dal-Pra S, Hodgkinson CP, Mirotsou M, Kirste I, Dzau VJ, Demethylation of H3K27 is essential for the induction of direct cardiac reprogramming by miR combo, Circ. Res 120 (9) (2017) 1403–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhou Y, Wang L, Vaseghi HR, Liu Z, Lu R, Alimohamadi S, et al. , Bmi1 is a key epigenetic barrier to direct cardiac reprogramming, Cell Stem Cell 18 (3) (2016) 382–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu Z, Wang L, Welch JD, Ma H, Zhou Y, Vaseghi HR, et al. , Single-cell transcriptomics reconstructs fate conversion from fibroblast to cardiomyocyte, Nature 551 (7678) (2017) 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jayawardena TM, Egemnazarov B, Finch EA, Zhang L, Payne JA, Pandya K, et al. , MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes, Circ. Res 110 (11, 2012) 1465–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cao N, Huang Y, Zheng J, Spencer CI, Zhang Y, Fu JD, et al. , Conversion of human fibroblasts into functional cardiomyocytes by small molecules, Science 352 (6290) (2016) 1216–1220. [DOI] [PubMed] [Google Scholar]
- [26].Zhou H, Morales MG, Hashimoto H, Dickson ME, Song K, Ye W, et al. , ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression, Genes Dev. 31 (17, 2017) 1770–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Abad M, Hashimoto H, Zhou H, Morales MG, Chen B, Bassel-Duby R, et al. , Notch inhibition enhances cardiac reprogramming by increasing MEF2C transcriptional activity, Stem Cell Rep. 8 (3) (2017) 548–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mohamed TM, Stone NR, Berry EC, Radzinsky E, Huang Y, Pratt K, et al. , Chemical enhancement of in vitro and in vivo direct cardiac reprogramming, Circulation 135 (10, 2017) 978–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Theunissen TW, Jaenisch R, Molecular control of induced pluripotency, Cell Stem Cell 14 (6) (2014) 720–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liu Z, Chen O, Zheng M, Wang L, Zhou Y, Yin C, et al. , Re-patterning of H3K27me3, H3K4me3 and DNA methylation during fibroblast conversion into induced cardiomyocytes, Stem Cell Res. 16 (2) (2016) 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Paige SL, Thomas S, Stoick-Cooper CL, Wang H, Maves L, Sandstrom R, et al. , A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development, Cell 151 (1) (2012) 221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang Y, Li Y, Guo C, Lu Q, Wang W, Jia Z, et al. , ISL1 and JMJD3 synergistically control cardiac differentiation of embryonic stem cells, Nucleic Acids Res. 44 (14, 2016) 6741–6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lee S, Lee JW, Lee SK, UTX, a histone H3-lysine 27 demethylase, acts as a critical switch to activate the cardiac developmental program, Dev. Cell 22 (1) (2012) 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Estaras C, Akizu N, Garcia A, Beltran S, de la Cruz X, Martinez-Balbas MA, Genome-wide analysis reveals that Smad3 and JMJD3 HDM co-activate the neural developmental program, Development 139 (15, 2012) 2681–2691. [DOI] [PubMed] [Google Scholar]
- [35].Hang CT, Yang J, Han P, Cheng HL, Shang C, Ashley E, et al. , Chromatin regulation by Brg1 underlies heart muscle development and disease, Nature 466 (7302) (2010) 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lickert H, Takeuchi JK, Von Both I, Walls JR, McAuliffe F, Adamson SL, et al. , Baf60c is essential for function of BAF chromatin remodelling complexes in heart development, Nature 432 (7013) (2004) 107–112. [DOI] [PubMed] [Google Scholar]
- [37].Tang ZH, Xia L, Chang W, Li H, Shen F, Liu JY, et al. , Two novel missense mutations of GATA4 gene in Chinese patients with sporadic congenital heart defects, Zhonghua Yi Xue Yi Chuan Xue Za Zhi 23 (2) (2006) 134–137. [PubMed] [Google Scholar]
- [38].Wang E, Sun S, Qiao B, Duan W, Huang G, An Y, et al. , Identification of functional mutations in GATA4 in patients with congenital heart disease, PLoS One 8 (4) (2013), e62138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Riching AS, Zhao Y, Cao Y, Londono P, Xu H, Song K, Suppression of pro-fibrotic signaling potentiates factor-mediated reprogramming of mouse embryonic fibroblasts into induced cardiomyocytes, J. Vis. Exp (136) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, et al. , A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response, Nature 488 (7411) (2012) 404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Belaguli NS, Zhang M, Rigi M, Aftab M, Berger DH, Cooperation between GATA4 and TGF-beta signaling regulates intestinal epithelial gene expression, Am. J. Physiol. Gastrointest. Liver Physiol 292 (6) (2007) G1520–G1533. [DOI] [PubMed] [Google Scholar]
- [42].Miller SA, Mohn SE, Weinmann AS, Jmjd3 and UTX play a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression, Mol. Cell 40 (4) (2010) 594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhao LH, Ba XQ, Wang XG, Zhu XJ, Wang L, Zeng XL, BAF complex is closely related to and interacts with NF1/CTF and RNA polymerase II in gene transcriptional activation, Acta Biochim. Biophys. Sin. Shanghai 37 (7) (2005) 440–446. [DOI] [PubMed] [Google Scholar]
- [44].Li J, Liu WD, Yang ZL, Yuan F, Xu L, Li RG, et al. , Prevalence and spectrum of GATA4 mutations associated with sporadic dilated cardiomyopathy, Gene 548 (2) (2014) 174–181. [DOI] [PubMed] [Google Scholar]
- [45].Srivastava D, Making or breaking the heart: from lineage determination to morphogenesis, Cell 126 (6) (2006) 1037–1048. [DOI] [PubMed] [Google Scholar]
- [46].Tomita-Mitchell A, Maslen CL, Morris CD, Garg V, Goldmuntz E, GATA4 sequence variants in patients with congenital heart disease, J. Med. Genet 44 (12) (2007) 779–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ang YS, Rivas RN, Ribeiro AJS, Srivas R, Rivera J, Stone NR, et al. , Disease model of GATA4 mutation reveals transcription factor cooperativity in human cardiogenesis, Cell 167 (7) (2016) 1734–1749, e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, et al. , GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5, Nature 424 (6947) (2003) 443–447. [DOI] [PubMed] [Google Scholar]
- [49].Chi C, Leonard A, Knight WE, Beussman KM, Zhao Y, Cao Y, et al. , LAMP-2B regulates human cardiomyocyte function by mediating autophagosome-lysosome fusion, Proc. Natl. Acad. Sci. U. S. A 116 (2) (2019) 556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, et al. , Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions, Nat. Protoc 8 (1) (2013) 162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Tange S, Oktyabri D, Terashima M, Ishimura A, Suzuki T, JARID2 is involved in transforming growth factor-beta-induced epithelial-mesenchymal transition of lung and colon cancer cell lines, PLoS One 9 (12) (2014), e115684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hirai H, Kikyo N, Inhibitors of suppressive histone modification promote direct reprogramming of fibroblasts to cardiomyocyte-like cells, Cardiovasc. Res 102 (1) (2014) 188–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Tremblay-LeMay R, Rastgoo N, Pourabdollah M, Chang H, EZH2 as a therapeutic target for multiple myeloma and other haematological malignancies, Biomark Res. 6 (2018) 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Akhurst RJ, Targeting TGF-beta signaling for therapeutic gain, Cold Spring Harb. Perspect. Biol 9 (10) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Connolly EC, Freimuth J, Akhurst RJ, Complexities of TGF-beta targeted cancer therapy, Int. J. Biol. Sci 8 (7) (2012) 964–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ganesh K, Massague J, TGF-beta inhibition and immunotherapy: Checkmate, Immunity 48 (4) (2018) 626–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Willems E, Cabral-Teixeira J, Schade D, Cai W, Reeves P, Bushway PJ, et al. , Small molecule-mediated TGF-beta type II receptor degradation promotes cardiomyogenesis in embryonic stem cells, Cell Stem Cell 11 (2) (2012) 242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yu Y, Gu S, Li W, Sun C, Chen F, Xiao M, et al. , Smad7 enables STAT3 activation and promotes pluripotency independent of TGF-beta signaling, Proc. Natl. Acad. Sci. U. S. A 114 (38, 2017) 10113–10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ulloa L, Doody J, Massague J, Inhibition of transforming growth factor-beta/ SMAD signalling by the interferon-gamma/STAT pathway, Nature 397 (6721) (1999) 710–713. [DOI] [PubMed] [Google Scholar]
- [60].Hashimoto H, Wang Z, Garry GA, Malladi VS, Botten GA, Ye W, et al. , Cardiac reprogramming factors synergistically activate genome-wide cardiogenic stage-specific enhancers, Cell Stem Cell 25 (1) (2019) 69–86, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Funaba M, Zimmerman CM, Mathews LS, Modulation of Smad2-mediated signaling by extracellular signal-regulated kinase, J. Biol. Chem 277 (44) (2002) 41361–41368. [DOI] [PubMed] [Google Scholar]
- [62].Enane FO, Shuen WH, Gu X, Quteba E, Przychodzen B, Makishima H, et al. , GATA4 loss of function in liver cancer impedes precursor to hepatocyte transition, J. Clin. Invest 127 (9) (2017) 3527–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, et al. , UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development, Nature 449 (7163) (2007) 731–734. [DOI] [PubMed] [Google Scholar]
- [64].Eppert K, Scherer SW, Ozcelik H, Pirone R, Hoodless P, Kim H, et al. , MADR2 maps to 18q21 and encodes a TGFbeta-regulated MAD-related protein that is functionally mutated in colorectal carcinoma, Cell 86 (4) (1996) 543–552. [DOI] [PubMed] [Google Scholar]
- [65].Papageorgis P, Lambert AW, Ozturk S, Gao F, Pan H, Manne U, et al. , Smad signaling is required to maintain epigenetic silencing during breast cancer progression, Cancer Res. 70 (3) (2010) 968–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Nguyen DX, Chiang AC, Zhang XH, Kim JY, Kris MG, Ladanyi M, et al. , WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis, Cell 138 (1) (2009) 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. , Model-based analysis of ChIP-Seq (MACS), Genome Biol. 9 (9) (2008) R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Shen L, Shao N, Liu X, Nesder E, ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases, BMC Genomics 15 (2014) 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yu G, Wang LG, He QY, ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization, Bioinformatics 31 (14) (2015) 2382–2383. [DOI] [PubMed] [Google Scholar]
- [70].McFadden DG, Charite J, Richardson JA, Srivastava D, Firulli AB, Olson EN, A GATA-dependent right ventricular enhancer controls dHAND transcription in the developing heart, Development 127 (24) (2000) 5331–5341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.