

Abstract
Congenital dyserythropoietic anemias (CDAs) are characterized by ineffective erythropoiesis and distinctive erythroblast abnormalities; the diagnosis is often missed or delayed due to significant phenotypic heterogeneity. We established the CDA Registry of North America (CDAR) to study the natural history of CDA and create a biorepository to investigate the pathobiology of this heterogeneous disease. Seven of 47 patients enrolled so far on CDAR have CDA-I due to biallelic CDAN1 mutations. They all presented with perinatal anemia and required transfusions during infancy. Anemia spontaneously improved during infancy in three patients; two became transfusion-independent rapidly after starting interferon-α2; and two remain transfusion-dependent at last follow-up at ages 5 and 30 y.o. One of the transfusion-dependent patients underwent splenectomy at 11 y.o due to misdiagnosis and returned to medical attention at 27 y.o with severe hemolytic anemia and pulmonary hypertension. All patients developed iron overload even without transfusions; four were treated with chelation. Genetic testing allowed for more rapid and accurate diagnosis; the median age of confirmed diagnosis in our cohort was 3 y.o compared to 17.3 y.o historically. In conclusion, CDAR provides an organized research network for multidisciplinary clinical and research collaboration to conduct natural history and biologic studies in CDA.
Keywords: congenital dyserythropoietic anemia, CDAN1, anemia, erythropoiesis, rare disease registry
Introduction
Congenital dyserythropoietic anemias (CDAs) are rare inborn errors of erythropoiesis. The principal mechanism shared by all types of CDA is ineffective erythropoiesis, leading to varying degrees of anemia and progressive iron overload.1 CDA is defined by the presence of distinct morphologic abnormalities of the erythroblasts in the bone marrow.2 Patients present typically with hemolytic anemia and inappropriate reticulocyte response, jaundice, poikilocytosis, and frequently, splenomegaly.3 The clinical presentation overlaps with more common hematologic diseases, such as red blood cell (RBC) membrane disorders and thalassemias, which often complicates and delays accurate diagnosis. Classification of CDA has mainly been morphologic, based on the classification proposed by Heimpel and Weindt in 1968, in which three major subtypes of CDA were identified based on morphologic similarities.2 Recently, more subgroups and variants of CDA have been identified and added to the classification.4,5 CDA type II (CDA-II) is the most common type followed by CDA type I (CDA-I).6 The phenotypic and genetic variability of CDA is remarkable, including intra-family variability, suggesting a role for other modifiers in determining disease phenotype.7,8
Pathogenetic mutations causing the common CDA types have been identified. CDA-I is a recessive disease caused by biallelic mutations in either CDAN1 or CDIN1 (CDAN1-interacting nuclease 1, previously known as C15orf41); the molecular cause is still unidentified in 10–20% of cases.9,10 CDA-II is an autosomal recessive disease caused by mutations in SEC23B gene; however, in several patients, only one SEC23B variant is identified, suggesting the presence of as yet unidentified, non-coding mutations that result in reduced SEC23B expression.3 Dominant mutations in KIF23 underlie familial CDA-III; the KLF1 mutation p.E325K cause dominant CDA-IV, while mutations in GATA1 cause X-linked thrombocytopenia with dyserythropoietic anemia.5,6,11–13 These genetic discoveries have improved our understanding of CDA; however, more work is needed to elucidate the molecular pathogenetic mechanisms and the basis for the significant heterogeneity seen in the different types of CDA.
CDA-I is characterized clinically by macrocytic anemia. Bone marrow studies characteristically demonstrate binucleated erythroblasts with chromatin bridges by light microscopy, and spongy heterochromatin in intermediate and late erythroblasts ultrastructurally.14 Non-hematological features, especially skeletal features affecting distal extremities, have been reported in 4–25% of CDA-I patients.15,16 Because of its remarkable clinical variability and its overlap with other hematologic conditions, misdiagnosis or delayed diagnosis of CDA-I was common, with a median age of 17.3 years at the time of diagnosis, historically.7
The rarity of CDA is a significant impediment to continued investigation of this group of diseases. Findings from the European registries of patients with CDA-I and II have expanded our knowledge about the natural history of these subtypes of CDA.5,7,17,18 Nonetheless, there are still significant gaps in our understanding of CDA pathophysiology, clinical heterogeneity, natural history, optimal treatment, and genotype-phenotype correlations.14 We established the CDA Registry (CDAR) in North America in 2016 (NCT02964494) to provide a platform for the systematic study of CDA in patients in the United States, Canada, and Mexico. The objectives of CDAR are twofold: 1) to longitudinally collect demographic, clinical, and treatment data on patients with CDA, and 2) to create a biorepository of de-identified samples that can be used as a tool to investigate the biology and molecular pathology of CDA. This study describes CDAR’s structure and current findings and focuses on the clinical data and disease course of participants with CDA-I due to CDAN1 mutations, enrolled to date in CDAR.
Methods
Study design and patient enrollment
We established CDAR in August 2016 at Cincinnati Children’s Hospital Medical Center (ClinicalTrials.gov Identifier: NCT02964494) as a collaborative effort between the investigators and the referring physicians. Participants were identified by their treating physician and referred to CDAR for enrollment. Patients or family members can also contact CDAR directly for enrollment. Patients of any age who have a phenotypic or genotypic diagnosis of CDA are eligible for enrollment.1 Family members of patients with CDA are also eligible to enroll to facilitate genetic studies. Exclusion criteria include the presence of cancer or myelodysplastic syndrome diagnosis, explaining a new onset of dyserythropoiesis, or other causes of acquired dyserythropoiesis, e.g., vitamin B12 deficiency or medications. The current status of CDAR and the data of participants with CDA-I, including two previously reported cases,19,20 are presented in this study.
Procedures, data, and sample collection
The study was approved by the Institutional Review Board (IRB) at Cincinnati Children’s Hospital Medical Center. Adult participants and parents/legal guardians of minors provided written informed consent and permission, respectively, and minors 11 years of age or older provided written assent. Patients or legal guardians and family members were consented to participate in person or by teleconference by the investigators and a clinical research professional as a phone-consent witness. After informed consent is obtained, participants and their physicians are invited to complete standardized clinical forms or share pertinent medical records to capture demographic information, medical history, family pedigree and history, diagnostic test results, treatments, and complications. Annual follow up information is solicited from patients and physicians. Pathology reports and bone marrow slides, when available, are centrally reviewed by an expert hematopathologist for confirmation and classification of the diagnosis.
Participants may elect to provide biological samples to the CDAR biorepository, such as blood, bone marrow, or tissue samples collected only when clinically indicated procedures are performed. Written informed consent is sought to collect DNA and genetic studies, and the generation of induced pluripotent stem cells (iPSC) and immortalized B-cells for biological studies.
Genetic studies
For participants without a genetic diagnosis at time of referral, high throughput next-generation sequencing and deletion/duplication panels for the known causative CDA genes (CDAN1, CDIN1 [or C15orf41], SEC23B, KIF23, KLF1, GATA1) are used to identify pathogenic or potentially pathogenic variants. In addition, genes known to be associated with other hereditary hemolytic anemias that may cause reactive dyserythropoiesis were also investigated. All substitution and small indel mutations were subsequently confirmed by Sanger sequencing. If a causative mutation is not identified, whole-exome or genome sequencing (WES or WGS) is performed to identify candidate genes for further validation and investigation of CDA pathogenesis.
Results
General Characteristics of CDAR Participants
Between August 2016 and July 2020, 84 individuals (47 affected and 37 asymptomatic family members, from 42 different families) were enrolled on CDAR (Figure 1A). Nine individuals who had been previously diagnosed with CDA based on bone marrow morphology were excluded because pathogenic mutations for other diseases were identified per CDAR protocol’s genetic evaluation: unstable hemoglobin [n=4], pyruvate kinase deficiency [n=2], hereditary spherocytosis due to biallelic SPTA1 mutations [n=1], Diamond-Blackfan anemia due to RPL35A de novo variant [n=1], and primary myelofibrosis due to compound heterozygosity of MPIG6B mutations [n=1]. Among the 38 patients remaining, the pathologic diagnosis was confirmed genetically for 18 participants: 7 (18%) with CDA-I, 8 (21%) with CDA-II, 1 (2.5%) with CDA-III, and 2 (5%) with CDA-IV. Twenty (53%) have untypable CDA, i.e., pathologic diagnosis of CDA but no known CDA-causing pathogenic variant (Figure 1B). In three patients with untypable syndromic CDA associated with severe neurodevelopmental disorder, we identified mutations in VPS4A, a gene encoding an ATPase that regulates the ESCRT III machinery in a variety of cellular processes, including abscission during cytokinesis and endosomal vesicle trafficking.21 We also identified a heterozygous missense variant in PRDX2 gene that encodes the antioxidant enzyme peroxiredoxin II in a family with atypical CDA.5,22
Figure 1. The general characteristics of CDAR participants.

(A) Cumulative enrollment on CDAR by year. The proportion of affected individuals, family members, and misdiagnosed participants is shown in each year. (B) The distribution of CDA subtypes based on genetic results in the 38 affected individuals with CDA in CDAR.
Characteristics and Presentation of CDA-I Participants
Seven participants had a diagnosis of CDA-I due to biallelic CDAN1 mutations. The median age of genetic diagnosis was 3 y.o (range 0.1 to 27 years).
All participants had characteristic blood and bone marrow findings. A representative example of these findings in patients with CDA-I is demonstrated in figure 2. Blood smears showed anisopoikilocytosis, RBC fragmentation, and basophilic stippling (Figure 2A). Bone marrow examination by light microscopy showed erythroid hyperplasia (Figure 2B), binucleated erythroid progenitors in ≤10% of erythroblasts, chromatin bridges between nuclei, and nuclear lobation (Figure 2C). Ultrastructural evaluation of erythroblasts confirmed the presence of nuclear heterochromatin with a characteristic “spongy” appearance of the nuclear heterochromatin and widening of pore membrane dilations (Figure 2D).
Figure 2. Peripheral blood and bone marrow abnormalities in CDA-I.
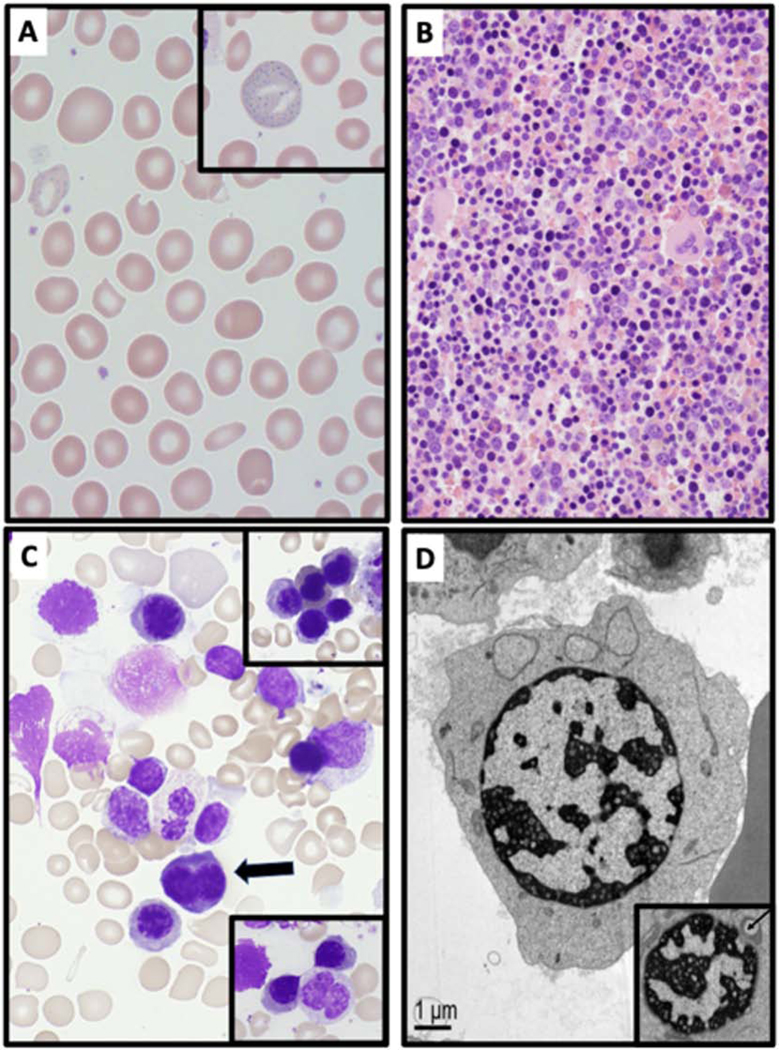
(A) Peripheral blood smear of CDA-I patient showing marked anisopoikilocytosis, RBC fragmentation and basophilic stippling (inset). (B) Bone marrow biopsy (H&E, 400x) showing erythroid hyperplasia in a patient with CDA-I. (C) Bone marrow aspirate (100x) demonstrating binucleated erythroblast (arrow). The lower inset shows erythroid cells with a chromatin bridge, and the upper inset showing nuclear lobation. (D) Electron microscopy image of erythroblasts in CDA-I showing the characteristic “spongy” or “swiss cheese” appearance of nuclear heterochromatin and widened pore membrane dilations (arrow in inset).
All CDA-I patients presented early in life with varying degrees of non-immune hemolytic anemia. One was diagnosed prenatally with fetal anemia and started intrauterine transfusions at 24 weeks of gestation; 3 presented with severe neonatal anemia and signs of hydrops, transient pulmonary hypertension, transaminitis, severe hyperbilirubinemia, and thrombocytopenia; and 3 presented with neonatal jaundice and only moderate anemia (Table 1). Two had family history of stillbirth or fetal demise in older siblings due to hydrops fetalis.
Table 1.
Characteristics of participants with CDA-I due to CDAN1 mutations
| Pt # | CDAN1 mutations (compound heterozygous) | Age, Sex, & Ethnic Background | Laboratory findings | Age at first transfusion | Age at last transfusion | Other treatments | Non-hematologic findings | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Hb | ARC | MCV | Ferritin/LIC | |||||||
| 1 | c.3025dupG (p. E1009Gfs*10) and c.3389C>T (p.P1130L) | 8 y.o., M, Hispanic-American | 9.4 | 74 | 88 | F 410 | Neonate | 4 m.o. | None | Macrocephaly and dolichocephaly, one café au lait spot, midline hyperpigmented macule |
| 2 | c.2015C>T(p.P672L) and c.2173C>T(p.R725W) | 5 y.o., F, Japanese and European-American | 10.3 | 91 | 89 | F 809 | Neonate | 4 m.o. | None | curved toenails, one café au lait spot |
| 3 | c.1003C>T (p.R335W) and c.2174G>A (p.R725Q) | 2 y.o., F, European-American | 8.2–10.7 | 11–45 | 88–95 | F 1189→361 LIC 9.3→3.26 | Neonate | 13 m.o. | Deferasirox (1–2 y.o) | Transient pulmonary hypertension (as neonate), hepatomegaly |
| 4 | c.156C>G (p.F52L) and c.3024_3025insTT (p.E1009Lfs*24) | 7 y.o., M, European-American | 7.8–10.1 | 50–88 | 89–91 | F 397 | Neonate | 11 m.o. | IFNα2b (1–3 y.o) | Hepatomegaly, transient pulmonary hypertension (as neonate), thalamic infarction |
| 5 | c.2072dupT (p.T692Hfs*13) and c.2093A>T (p.E698V) | 5 y.o., F, European-American | 7–8 | 96 | 87 | F 1162 LIC 8.2 | Intrauterine | Ongoing at 5 y.o | Deferasirox, (EPO-no response) | web between 4th and 5th toe |
| 6 | c.156C>G (p.F52L) and c.2015C>T (p.P672L) | 20 y.o., F, European-American and Ashkenazi Jewish | 10.1–12.4 | 34.9–120.4 | 90.9–93.4 | F 2200→680 LIC 6 cardiac T2* 27–39 (normal > 20 ms) | Neonate | 19 y.o. | Deferoxamine, Deferasirox, IFNα2b | Transient pulmonary hypertension (as neonate), digital clubbing, joint laxity, osteoarthritis |
| 7 | c.2868+1G>A and c.3389C>T (p.P1130L) | 30 y.o., M, African-American | 7.4 | 365 | 93 | F 2012 LIC 33.5 cardiac T2* 17.8 (normal > 20 ms) | Neonate | 11 y.o. before splenectomy, now again on chronic transfusions since 27 y.o. | Deferasirox, Hydroxyurea for extramedullary erythropoiesis | Short stature, scoliosis, pulmonary hypertension post splenectomy, paraspinal extramedullary erythropoiesis causing kyphosis and obstructive lung disease |
Abbreviations and units: Hb: hemoglobin concentration (g/dL); ARC: absolute reticulocyte count (K/μL); MCV: mean corpuscular volume (fL); F: serum ferritin (ng/mL); LIC: liver iron content (mg/g dry liver weight); IFNα2: interferon-alpha; EPO: erythropoietin.
Transfusion and Treatment History
All participants required blood transfusions in the neonatal period. Three had spontaneous improvement of anemia and did not require transfusions beyond infancy. Two patients became transfusion-independent after initiation of interferon-alpha (IFN-α2): patient #4 became transfusion-independent after starting IFN-α2 at 1 year of age and remained transfusion-independent after discontinuation at age 3 y.o., while patient #6 was chronically transfused up to 19 years of age when she was started on IFN-α2 resulting in transfusion-independence for the last year. Two patients (#5 and #7) remain transfusion-dependent at last follow up at ages 5 and 30 y.o, respectively. Patient #7 was initially misdiagnosed with hereditary spherocytosis and underwent splenectomy at 11 y.o. He was lost to follow up and returned to medical care in adulthood, presenting with hemolytic anemia and pulmonary hypertension and was diagnosed with CDA-I by genetic sequencing.20
Iron overload
Serum ferritin was elevated in all participants at the last follow-up (range from 361 to 2012 ng/mL), and 4 received iron chelation. Chelation was discontinued in one patient after one year due to improved liver iron concentration and serum ferritin (Table 1). All four participants who had hepatic iron assessment by MRI (range 2–30 years of age) showed hepatic hemosiderosis (liver iron concentration range: 6 to 33.5 mg/g dry liver weight), and the oldest patient, who underwent splenectomy and had an extended lapse in medical care until 27 years of age, had evidence of myocardial hemosiderosis (T2* 17.8 ms; normal >20 ms). Notably, serum ferritin elevation was progressive over time even in the absence of transfusions. For example, patient #2 received only four transfusions in the neonatal period; however, her serum ferritin increased from 408 ng/dL at 1 y.o to 809 ng/dL over 4 years despite not receiving any blood transfusion beyond infancy, consistent with spontaneous iron-loading independent of blood transfusions.
Non-hematological features
All participants had one or more non-hematological manifestations including curved toenails, syndactyly, café-au-lait spots, skin pigmentation, macrocephaly, dolichocephaly, spinal fusion, scoliosis, and short stature. Three participants developed transient neonatal pulmonary hypertension in the setting of severe anemia, requiring mechanical ventilation and inhaled nitric oxide; one of them also suffered a thalamic stroke during this period. One participant developed pulmonary hypertension post-splenectomy in adulthood (Table 1).
Discussion
The registry for patients with CDA in North America, CDAR, provides a collaborative platform to study the natural history, genetics, and biology of CDA. Registries facilitate the study of rare diseases and can improve clinical outcomes when carefully planned and implemented. In this first report from CDAR, CDA-I cases due to biallelic CDAN1 mutations represented approximately 40% of genetically confirmed cases of CDA. CDA-I patients presented with early-onset anemia, even prenatally, with varying hemolysis severity ranging from hydrops to mild neonatal jaundice and anemia. Non-hematological manifestations, mainly distal skeletal, nail, and skin abnormalities, were more common in our study than previously reported. Importantly, molecular testing expedited the definitive diagnosis of CDA in this cohort of patients.
The characteristics of successful registries and the main challenges facing hematology registries were recently discussed in a comprehensive review.23 The registries for CDA-I and CDA-II in Europe and Israel have been instrumental in delineating these diseases’ clinical characteristics, identifying causative genes (CDAN1 for most patients with CDA-I7 and SEC23B for CDA-II18,24), and establishing phenotype-genotype correlations.25,26 Learning from those successful precedent CDA registries and similar registries for rare hematologic disorders,27,28 we established CDAR with two specific objectives: 1) to collect longitudinal data that allow us to study the natural history of CDA, and 2) to establish a biorepository that will allow for novel candidate gene discovery and mechanistic biologic studies. We have intentionally designed CDAR with broad inclusion criteria to capture a diverse CDA population and not miss eligible candidates before thorough evaluation. Concurrently, we instituted a central review process to ensure the accuracy of the diagnosis and the quality of data. Maintaining a cooperative effort between CDAR investigators, the referring and collaborating physicians, and the patients will be essential for the success of long-term natural history studies in CDAR. The elective CDAR biorepository is one of the main strengths of our registry. It provides a tool for physicians and scientists to collaborate on biologic and genetic studies in CDA. Results of such studies can improve CDA treatment and expand our understanding of erythropoiesis in general. Using the CDAR biorepository, novel candidate genes have been identified in participants who do not conform to a known form of CDA and are being validated by multidisciplinary teams, such as the VPS4A variants, which were recently shown to cause syndromic CDA associated with neurodevelopmental disorder.21
Due to the phenotypic heterogeneity of CDAs and their clinical overlap with other more common hematologic diseases, delayed diagnosis or misdiagnosis is not infrequent.7 In CDAR, 9 out of 47 enrolled patients (19%) carried a misdiagnosis of CDA, sometimes for decades. Misdiagnosis in such cases likely reflects the prominent dyspoiesis that may be a feature of the stress erythropoiesis associated with severe hemolytic anemias due to globin, erythrocyte membrane or enzyme disorders that can be confused for CDA. Besides, one patient with CDA-I (patient #7) and another with CDA-II enrolled in CDAR were initially misdiagnosed with hereditary spherocytosis, before genetic testing revealed biallelic CDAN1 and SEC23B variants, respectively. The importance of early use of genetic testing in diagnosing CDA was recently emphasized.5,29–31 In our study, genetic testing significantly expedited the accurate diagnosis compared to historical data (median age of 3 y.o vs. 17.3 y.o).7 A timely, accurate, genetically confirmed CDA diagnosis has several advantages. First, It can obviate the need for invasive studies such as bone marrow aspiration and biopsy. In a recent study, up to 55% of patients were estimated to have avoided unnecessary testing if gene testing was employed early enough.31 Second, an accurate diagnosis informs counseling about the disease and its complications and impacts treatment decisions such as the use of IFNα2, avoidance of splenectomy in patients with CDA-I, and monitoring and management of iron overload. Finally, as shown in this study, accurate diagnosis based on phenotypic evaluation alone may not be possible, especially in transfusion-dependent patients. Our results underscore the importance of early genetic testing and support recently proposed diagnostic algorithms for hereditary hemolytic and dyserythropoietic anemias.5,32,33
Consistent with previous studies,8 we observed significant variability in the clinical course of patients with CDA-I enrolled in CDAR. All patients presented with prenatal or neonatal macrocytic anemia of varying severity ranging from hydrops fetalis to neonatal jaundice and anemia. All of them required blood transfusions during infancy. Neonatal or perinatal anemia is reported in up to 60% of CDA-I patients with most requiring blood transfusion in the neonatal period.16,34 Four out of seven patients had improvement in anemia during infancy or childhood, while the remaining three manifested a more severe disease course requiring ongoing transfusions or treatment with IFN-α2, consistent with the known phenotypic heterogeneity of CDA-I.6,7,35
IFNα2 has been shown to be an effective treatment for CDA-I. It was first noted to normalize hemoglobin in an adult with transfusion-dependent CDA-I and chronic hepatitis C.36 Since this serendipitous discovery, several reports have confirmed the efficacy of IFNα2 in ameliorating anemia and reducing iron overload in patients with CDA-I.7,37–39 The mechanism of this salutary effect of IFNα2 in CDA-I is unclear; however, IFN appears to attenuate the nuclear structural abnormalities and ineffective erythropoiesis characteristic of CDA-I.40 EBV-transformed B-cells derived from CDA-I patients produced less IFNα in vitro,41 and erythroblasts cultured in IFNα demonstrated significant reduction in the “swiss-cheese” nuclear appearance evident by EM.42 One patient in our study received IFNα2 for ~ 2 years with amelioration of anemia within three months of starting treatment and another patient who was transfusion-dependent for 19 years achieved transfusion-independence after starting IFNα2. Importantly, IFNα2 should be used cautiously in infancy due to the risk of spastic diplegia.43 Some studies suggest that this devastating complication is secondary to benzyl alcohol, a preservative in some preparations of IFNα2, and that alcohol-free preparations are safer.44,45 IFN may also have inherent toxicities including peripheral neuropathy. Considering these concerns, initiation of IFN therapy should be delayed, when possible, beyond early childhood, especially since many patients may spontaneously improve after the first few years of life. Recently, long-acting pegylated-IFNα2 was used successfully in patients with CDA-I. In a recent study, five out of seven patients became transfusion-independent after receiving pegylated-IFNα2. However, one patient had to stop treatment due to side-effects (“moon face” appearance, abdominal distention, weight gain and generalized weakness), and a sixth patient had partial response with decreased transfusion requirement.46
Iron overload is a recognized, progressive complication of CDA and is the most common cause of early mortality in CDA-I.7 Iron overload was thought to appear in the second or third decade of life; however, evidence of iron overload, including hepatic and cardiac siderosis, may be observed in younger patients.47,48 In our cohort comprised of relatively young patients, high serum ferritin or evidence of hepatic hemosiderosis, was detected in all patients as early as 14 months of age. Three patients are receiving ongoing chelation while a fourth was treated for 2 years and was able to discontinue treatment for now. These results confirm that iron overload, due to ineffective erythropoiesis and enhanced iron absorption,49 develops early in patients with CDA-I. Therefore, all children with CDA-I should be vigilantly monitored for evidence of iron overload, including transfusion-independent patients, to guide appropriate initiation of chelation therapy.
Unlike CDA-II, where splenectomy may be effective in reducing transfusion requirement although not iron overload,17 splenectomy is contraindicated in CDA-I due to poor hematologic efficacy and the potential for significant morbidity. In one study, splenectomy was ineffective in patients with CDA-I.7 In another study, five of six patients with CDA-I reportedly had improvement in hemoglobin concentration following splenectomy (but with no specifics about response or duration), while the sixth remained transfusion-dependent.8 Importantly, however, significant morbidity and early mortality were noted in this study and attributed to complications of splenectomy (pulmonary hypertension and sepsis).8 Similarly, patient #7 in our study who underwent splenectomy at a young age developed pulmonary hypertension in his 3rd decade of life,20 a known complication of splenectomy,50 and continues to require transfusions and chelation for iron overload. This high rate of complications and low efficacy highlight the importance of diagnostic accuracy, especially if splenectomy is contemplated.
The outcome of stem cell transplant as a curative therapy for transfusion-dependent patients with CDA is guarded. In a recent report of the outcome of stem cell transplant in 39 patients with CDA, including 10 patients with CDA-I, the overall survival and event-free survival at 36 months were 71% and 45%, respectively, and 12% experienced graft failure. Iron overload was a poor prognostic factor on outcome of stem cell transplant in this study.51
Non-hematologic features are reported in 4–25% of CDA-I patients.15,16 In our study, non-hematologic findings were common including skin and skeletal features, especially in distal extremities, and short stature. Additionally, findings likely due to complications of severe anemia rather than CDAN1 defects included transient pulmonary hypertension which was noted in 3 of our cohort, as previously reported,16,52–54 and perinatal ischemic stroke (thalamic infarction), a complication not previously reported with CDA-I.
The cause of skeletal features in CDA-I is unknown. CDAN1 mutations in CDA-I result in defects in Codanin-1 protein whose function is not yet well-understood. Codanin-1 is known to interact with the histone chaperone Asf1 (anti-silencing function 1) which provides histones during the S-phase for replication-coupled nucleosome assembly. Codanin-1 depletion disrupts chromatin assembly and accelerates DNA replication disrupting chromatin assembly and DNA synthesis,55 which may interfere with the normal silencing and packing of large parts of the erythroblast nucleus during the late terminal erythropoiesis. While Codanin-1 is ubiquitously expressed in most tissues, its expression is cell-cycle-dependent, which could explain the predilection of the rapidly dividing late erythroblasts to Codanin-1 abnormalities in CDA-I.31 Alterations in cell division may also affect the rapidly dividing cells of the developing limb buds causing the characteristic limb abnormalities of CDA-I.31,56 The high rates of neonatal pulmonary hypertension and non-hematologic features in CDA-I, especially limb and skin abnormalities, should provide a clue to the diagnosis.
In summary, CDA-I due to CDAN1 mutations presents with early-onset macrocytic anemia and high rates of distal skeletal and skin dysmorphic features, with or without pulmonary hypertension of the newborn. Anemia improves in about half of the patients over time, but the rest remain in need of treatment, which may include chronic transfusions, iron chelation, or IFNα. Historically, CDA was frequently misdiagnosed, or the accurate diagnosis was significantly delayed. Genetic testing improved the accuracy and speed of diagnosis in our study and should be considered a first-line test as it may obviate the need for invasive diagnostic procedures and could impact treatment decisions, prognosis, and family counseling. CDAR allows for a systematic study of this rare disease and provides a collaborative platform for physicians, scientists, and patients to study the natural history of CDA and explore molecular and genetic pathways of CDA and erythropoiesis.
Highlights.
The CDA registry (CDAR) is an organized network for the collaborative study of CDAs
Genetic testing allows for a rapid and accurate diagnosis of CDA type I (CDA-I)
Anemia severity in CDA-I is variable and iron overload develops even without transfusions
Non-erythroid features, especially skeletal features affecting distal extremities, point to CDA-I diagnosis
Interferon-α2 improves anemia and iron overload in at least some patients with severe CDA-I
Acknowledgement:
We thank our patients and their families for their generous participation in CDAR. We thank Megan Reynolds, Tyler James, and Amy Shova for their valuable role in patient enrollment and data collection.
Funding: This work was supported by the National Institutes of Health, National Heart, Lung, and Blood Institute grant R01 HL152099 (TAK), by the National Institutes of Health, National Center for Advancing Translational Sciences (award 1UL1TR001425) through a CCTST-funded T1 award to TAK, and the Cincinnati Children’s Hospital CPG (Center for Pediatric Genomics) and CuSTOM (Center for Stem Cell & Organoid Medicine) funding awards (TAK). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
CDAR COLLABORATORS (CDAR Consortium)
Cincinnati Children’s Hospital Medical Center, Cincinnati, OH
Ammar Husami, PhD
Theodosia Kalfa, MD, PhD
Robert Lorsbach, MD, PhD
Carolyn Lutzko, PhD
Adam Nelson, MD
Omar Niss, MD
Charles Quinn, MD, MS
Katie G. Seu, PhD
Wenying Zhang, PhD
Children’s Healthcare of Atlanta, Atlanta, GA Satheesh Chonat, MD
CHOC Children’s Hospital, Orange, CA David Buchbinder, MD
Cook Children’s Hospital, Fort Worth, TX
Clarissa Johnson, MD
Timothy McCavit, MD
Dell Children’s Medical Center, Austin, TX Linda G. Shaffer, MD
Duke University, Division of Pediatric Hematology/Oncology, Durham, North Carolina Jennifer A. Rothman, MD
Indiana Hemophilia and Thrombosis Center, Indianapolis, IN Sweta Gupta, MD, MS
Instituto Nacional de Pediatria, Mexico City, Mexico Mara Nuñez Toscano, MD
Madigan Army Medical Center, Tacoma, WA Melissa Forouhar, MD
Methodist Children’s Hospital of South Texas, San Antonio, TX Vinod K. Gidvani-Diaz, MD
Rocky Mountain Pediatric Hematology Oncology, Denver, CO James B. Ball, MD
Ronald Reagan UCLA Medical Center, Los Angeles, CA Gavin D. Roach, MD
Sanford Children’s Hospital, ND KayeLyn Wagner, MD
Sam Milanovich, MD
Sentara Martha Jefferson Medical Group, Charlottesville, VA James Boyer, MD
Texas Oncology, Austin, TX Jane Chawla, MD
Vanderbilt University, Department of Pediatrics, Nashville, TN Christine Moore Smith, MD
University of Calgary, Calgary, Alberta, Canada
Adrienne Lee, MD
University of Florida Health Pediatric Subspecialty Program in Pensacola, Pensacola, FL
Jessica Simpson, RN
Jeffrey H. Schwartz, MD
University of Kentucky Children’s Hospital, Pediatric Hematology-Oncology, Lexington, KY Vlad C. Radulescu, MD
University of North Carolina Hospitals, Chapel Hill, NC Yasmina L. Abajas, MD
University of Pittsburgh, Department of Pediatrics, Pittsburgh, PA
A. Kim Ritchey, MD
University of Utah, Department of Pediatrics, Salt Lake City, UT
Jessica Meznarich, MD
Hunter R. Underhill, MD, PhD
Wayne State University, Detroit, MI Yaddanapudi Ravindranath, MD
Winship Cancer Institute, Emory University School of Medicine, Department of Hematology and Medical Oncology, Atlanta, GA Morgan McLemore, MD
Yale University, Pediatric Stem Cell Transplant and Cellular Therapy Program Niketa C Shah MD
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Heimpel H. Congenital dyserythropoietic anemias: epidemiology, clinical significance, and progress in understanding their pathogenesis. Ann Hematol. 2004;83(10):613–621. [DOI] [PubMed] [Google Scholar]
- 2.Heimpel H, Wendt F. Congenital dyserythropoietic anemia with karyorrhexis and multinuclearity of erythroblasts. Helv Med Acta. 1968;34(2):103–115. [PubMed] [Google Scholar]
- 3.Iolascon A, Esposito MR, Russo R. Clinical aspects and pathogenesis of congenital dyserythropoietic anemias: from morphology to molecular approach. Haematologica. 2012;97(12):1786–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wickramasinghe SN, Wood WG. Advances in the understanding of the congenital dyserythropoietic anaemias. Br J Haematol. 2005;131(4):431–446. [DOI] [PubMed] [Google Scholar]
- 5.Iolascon A, Andolfo I, Russo R. Congenital dyserythropoietic anemias. Blood. 2020;136(11):1274–1283. [DOI] [PubMed] [Google Scholar]
- 6.Iolascon A, Heimpel H, Wahlin A, Tamary H. Congenital dyserythropoietic anemias: molecular insights and diagnostic approach. Blood. 2013;122(13):2162–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heimpel H, Schwarz K, Ebnother M, et al. Congenital dyserythropoietic anemia type I (CDA I): molecular genetics, clinical appearance, and prognosis based on long-term observation. Blood. 2006;107(1):334–340. [DOI] [PubMed] [Google Scholar]
- 8.Shalev H, Al-Athamen K, Levi I, Levitas A, Tamary H. Morbidity and mortality of adult patients with congenital dyserythropoietic anemia type I. European journal of haematology. 2017;98(1):13–18. [DOI] [PubMed] [Google Scholar]
- 9.Dgany O, Avidan N, Delaunay J, et al. Congenital dyserythropoietic anemia type I is caused by mutations in codanin-1. Am J Hum Genet. 2002;71(6):1467–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Babbs C, Roberts NA, Sanchez-Pulido L, et al. Homozygous mutations in a predicted endonuclease are a novel cause of congenital dyserythropoietic anemia type I. Haematologica. 2013;98(9):1383–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liljeholm M, Irvine AF, Vikberg AL, et al. Congenital dyserythropoietic anemia type III (CDA III) is caused by a mutation in kinesin family member, KIF23. Blood. 2013;121(23):4791–4799. [DOI] [PubMed] [Google Scholar]
- 12.Risinger M, Emberesh M, Kalfa TA. Rare Hereditary Hemolytic Anemias: Diagnostic Approach and Considerations in Management. Hematol Oncol Clin North Am. 2019;33(3):373–392. [DOI] [PubMed] [Google Scholar]
- 13.Wakabayashi A, Ulirsch JC, Ludwig LS, et al. Insight into GATA1 transcriptional activity through interrogation of cis elements disrupted in human erythroid disorders. Proc Natl Acad Sci U S A. 2016;113(16):4434–4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iolascon A, Delaunay J. Close to unraveling the secrets of congenital dyserythropoietic anemia types I and II. Haematologica. 2009;94(5):599–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wickramasinghe SN. Dyserythropoiesis and congenital dyserythropoietic anaemias. British journal of haematology. 1997;98(4):785–797. [DOI] [PubMed] [Google Scholar]
- 16.Shalev H, Kapelushnik J, Moser A, Dgany O, Krasnov T, Tamary H. A comprehensive study of the neonatal manifestations of congenital dyserythropoietic anemia type I. J Pediatr Hematol Oncol. 2004;26(11):746–748. [DOI] [PubMed] [Google Scholar]
- 17.Heimpel H, Anselstetter V, Chrobak L, et al. Congenital dyserythropoietic anemia type II: epidemiology, clinical appearance, and prognosis based on long-term observation. Blood. 2003;102(13):4576–4581. [DOI] [PubMed] [Google Scholar]
- 18.Iolascon A, Delaunay J, Wickramasinghe SN, Perrotta S, Gigante M, Camaschella C. Natural history of congenital dyserythropoietic anemia type II. Blood. 2001;98(4):1258–1260. [DOI] [PubMed] [Google Scholar]
- 19.Meznarich JA, Draper L, Christensen RD, et al. Fetal presentation of congenital dyserythropoietic anemia type 1 with novel compound heterozygous CDAN1 mutations. Blood Cells Mol Dis. 2018;71:63–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chonat S, McLemore ML, Bunting ST, Nortman S, Zhang K, Kalfa TA. Congenital dyserythropoietic anaemia type I diagnosed in a young adult with a history of splenectomy in childhood for presumed haemolytic anaemia. Br J Haematol. 2018;182(1):10. [DOI] [PubMed] [Google Scholar]
- 21.Seu KG, Trump LR, Emberesh S, et al. VPS4A mutations in humans cause syndromic congenital dyserythropoietic anemia due to cytokinesis and trafficking defects. Am J Hum Genet. 2020;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emberesh M, Giger Seu K, Emberesh S, et al. Peroxiredoxin II (PRDX2) Is a Novel Candidate Gene for Congenital Dyserythropoietic Anemia. Blood. 2018;132(Supplement 1):3605–3605. [Google Scholar]
- 23.Dale DC, Bolyard AA, Steele LA, Zeidler C, Welte K, Severe Chronic Neutropenia International R. Registries for study of nonmalignant hematological diseases: the example of the Severe Chronic Neutropenia International Registry. Curr Opin Hematol. 2020;27(1):18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwarz K, Iolascon A, Verissimo F, et al. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nature genetics. 2009;41(8):936–940. [DOI] [PubMed] [Google Scholar]
- 25.Tamary H, Dgany O, Proust A, et al. Clinical and molecular variability in congenital dyserythropoietic anaemia type I. Br J Haematol. 2005;130(4):628–634. [DOI] [PubMed] [Google Scholar]
- 26.Iolascon A, Russo R, Esposito MR, et al. Molecular analysis of 42 patients with congenital dyserythropoietic anemia type II: new mutations in the SEC23B gene and a search for a genotype-phenotype relationship. Haematologica. 2010;95(5):708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dale DC, Bolyard AA, Schwinzer BG, et al. The Severe Chronic Neutropenia International Registry: 10-Year Follow-up Report. Supportive cancer therapy. 2006;3(4):220–231. [DOI] [PubMed] [Google Scholar]
- 28.Vlachos A, Klein GW, Lipton JM. The Diamond Blackfan Anemia Registry: tool for investigating the epidemiology and biology of Diamond-Blackfan anemia. Journal of pediatric hematology/oncology. 2001;23(6):377–382. [DOI] [PubMed] [Google Scholar]
- 29.Moreno-Carralero MI, Horta-Herrera S, Morado-Arias M, et al. Clinical and genetic features of congenital dyserythropoietic anemia (CDA). Eur J Haematol. 2018;101(3):368–378. [DOI] [PubMed] [Google Scholar]
- 30.Russo R, Andolfo I, Manna F, et al. Multi-gene panel testing improves diagnosis and management of patients with hereditary anemias. Am J Hematol. 2018;93(5):672–682. [DOI] [PubMed] [Google Scholar]
- 31.Roy NBA, Babbs C. The pathogenesis, diagnosis and management of congenital dyserythropoietic anaemia type I. British journal of haematology. 2019;185(3):436–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rothman JA, Stevens JL, Gray FL, Kalfa TA. How I approach hereditary hemolytic anemia and splenectomy. Pediatr Blood Cancer. 2020:e28337. [DOI] [PubMed] [Google Scholar]
- 33.Risinger M, Kalfa TA. Red cell membrane disorders: structure meets function. Blood. 2020;136(11):1250–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin SM, Chen M, Ma ESK, Lam YH, Wong KY, Tang MHY. Intrauterine therapy in a fetus with congenital dyserythropoietic anaemia type I. Journal of obstetrics and gynaecology : the journal of the Institute of Obstetrics and Gynaecology. 2014;34(4):352–353. [DOI] [PubMed] [Google Scholar]
- 35.al-Fawaz IM, al-Mashhadani SA. Congenital dyserythropoietic anaemia type I. Report of two siblings from Saudi Arabia. Acta haematologica. 1995;93(1):50–53. [DOI] [PubMed] [Google Scholar]
- 36.Lavabre-Bertrand T, Blanc P, Navarro R, et al. alpha-Interferon therapy for congenital dyserythropoiesis type I. British journal of haematology. 1995;89(4):929–932. [DOI] [PubMed] [Google Scholar]
- 37.Lavabre-Bertrand T, Ramos J, Delfour C, et al. Long-term alpha interferon treatment is effective on anaemia and significantly reduces iron overload in congenital dyserythropoiesis type I. European journal of haematology. 2004;73(5):380–383. [DOI] [PubMed] [Google Scholar]
- 38.Bader-Meunier B, Leverger G, Tchernia G, et al. Clinical and laboratory manifestations of congenital dyserythropoietic anemia type I in a cohort of French children. Journal of pediatric hematology/oncology. 2005;27(8):416–419. [DOI] [PubMed] [Google Scholar]
- 39.Rathe M, Møller MB, Greisen PW, Fisker N. Successful management of transfusion-dependent congenital dyserythropoietic anemia type 1b with interferon alfa-2a. Pediatric blood & cancer. 2018;65(3): 10.1002/pbc.26866. [DOI] [PubMed] [Google Scholar]
- 40.Lavabre-Bertrand T, Blanc P, Navarro R, et al. alpha-Interferon therapy for congenital dyserythropoiesis type I. Br J Haematol. 1995;89(4):929–932. [DOI] [PubMed] [Google Scholar]
- 41.Wickramasinghe SN, Hasan R, Smythe J. Reduced interferon-alpha production by Epstein-Barr virus transformed B-lymphoblastoid cell lines and lectin-stimulated lymphocytes in congenital dyserythropoietic anaemia type I. British journal of haematology. 1997;98(2):295–298. [DOI] [PubMed] [Google Scholar]
- 42.Menike D, Wickramasinghe SN. Effects of four species of interferon-alpha on cultured erythroid progenitors from congenital dyserythropoietic anaemia type I. British journal of haematology. 1998;103(3):825–830. [DOI] [PubMed] [Google Scholar]
- 43.Barlow CF, Priebe CJ, Mulliken JB, et al. Spastic diplegia as a complication of interferon Alfa-2a treatment of hemangiomas of infancy. The Journal of pediatrics. 1998;132(3 Pt 1):527–530. [DOI] [PubMed] [Google Scholar]
- 44.Ezekowitz RA, Mulliken JB, Folkman J. Interferon alfa-2a therapy for life-threatening hemangiomas of infancy. The New England journal of medicine. 1992;326(22):1456–1463. [DOI] [PubMed] [Google Scholar]
- 45.Deb G, Jenkner A, Donfrancesco A. Spastic diplegia and interferon. J Pediatr. 1999;134(3):382. [DOI] [PubMed] [Google Scholar]
- 46.Abu-Quider A, Asleh M, Shalev H, et al. Treatment of transfusion-dependent congenital dyserythropoietic anemia Type I patients with pegylated interferon alpha-2a. Eur J Haematol. 2020;105(2):216–222. [DOI] [PubMed] [Google Scholar]
- 47.Shalev H, Kapleushnik Y, Haeskelzon L, et al. Clinical and laboratory manifestations of congenital dyserythropoietic anemia type I in young adults. European journal of haematology. 2002;68(3):170–174. [DOI] [PubMed] [Google Scholar]
- 48.Berdoukas V, Nord A, Carson S, et al. Tissue iron evaluation in chronically transfused children shows significant levels of iron loading at a very young age. American Journal of Hematology. 2013;88(11):E283–E285. [DOI] [PubMed] [Google Scholar]
- 49.Tamary H, Shalev H, Perez-Avraham G, et al. Elevated growth differentiation factor 15 expression in patients with congenital dyserythropoietic anemia type I. Blood. 2008;112(13):5241–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoeper MM, Niedermeyer J, Hoffmeyer F, Flemming P, Fabel H. Pulmonary Hypertension after Splenectomy? Annals of Internal Medicine. 1999;130(6):506–509. [DOI] [PubMed] [Google Scholar]
- 51.Miano M, Eikema DJ, Aljurf M, et al. Stem cell transplantation for congenital dyserythropoietic anemia: an analysis from the European Society for Blood and Marrow Transplantation. Haematologica. 2019;104(8):e335–e339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.El-Sheikh AA, Hashem H, Holman C, Vyas YM. Congenital dyserythropoietic anemia type I presenting as persistent pulmonary hypertension with pigeon chest deformity. Pediatric blood & cancer. 2014;61(8):1460–1462. [DOI] [PubMed] [Google Scholar]
- 53.Shalev H, Moser A, Kapelushnik J, et al. Congenital dyserythropoietic anemia type I presenting as persistent pulmonary hypertension of the newborn. The Journal of pediatrics. 2000;136(4):553–555. [DOI] [PubMed] [Google Scholar]
- 54.Landau D, Kapelushnik J, Harush MB, Marks K, Shalev H. Persistent pulmonary hypertension of the newborn associated with severe congenital anemia of various etiologies. J Pediatr Hematol Oncol. 2015;37(1):60–62. [DOI] [PubMed] [Google Scholar]
- 55.Ask K, Jasencakova Z, Menard P, Feng Y, Almouzni G, Groth A. Codanin-1, mutated in the anaemic disease CDAI, regulates Asf1 function in S-phase histone supply. The EMBO journal. 2012;31(8):2013–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tickle C. How the embryo makes a limb: determination, polarity and identity. Journal of anatomy. 2015;227(4):418–430. [DOI] [PMC free article] [PubMed] [Google Scholar]