

Abstract
The neurobiology of sleep and narcolepsy is reviewed. Non-rapid eye movement (NREM) sleep is generated by neurons in the preoptic region of the hypothalamus and adjacent basal forebrain. Lesions in these regions cause insomnia. Stimulation of these regions rapidly produces sleep onset. The key brain structure for generating REM sleep is the pons and adjacent portions of the midbrain. Damage to the pons and/or caudal midbrain can cause abnormalities in REM sleep. The persistent sleepiness of narcolepsy is a result of a loss of hypocretin function.
Keywords: REM sleep, brainstem, pons, midbrain, glutamate, acetylcholine, norepinephrine, serotonin, hypocretin, orexin
Non-rapid eye movement (NREM) sleep is generated by neurons in the preoptic region of the hypothalamus and adjacent basal forebrain. Lesions in these regions cause a profound insomnia, whereas stimulation rapidly produces sleep onset. Two populations of gamma-aminobutyric acid (GABA)-ergic neurons are responsible for these effects. A population in the ventrolateral preoptic region is active during spontaneous sleep (Fig. 1). A population of neurons in the median preoptic region is active during sleep and is also active during waking in sleep-deprived animals, suggesting that this cell population mediates sleep debt. The key brain structure for generating REM sleep is the pons and adjacent portions of the midbrain. These areas and the hypothalamus contain cells that are maximally active in REM sleep, called REM-on cells, and cells that are minimally active in REM sleep, called REM-off cells. Subgroups of REM-on cells use the transmitter GABA, acetylcholine, glutamate, or glycine. Subgroups of REM-off cells use the transmitter norepinephrine, epinephrine, serotonin, histamine, and GABA. Destruction of large regions within the midbrain and pons can prevent the occurrence of REM sleep. Damage to portions of the brainstem can cause abnormalities in certain aspects of REM sleep. Hypocretin neurons have an important role in the regulation of sleep–wake phenomena.
Figure 1.
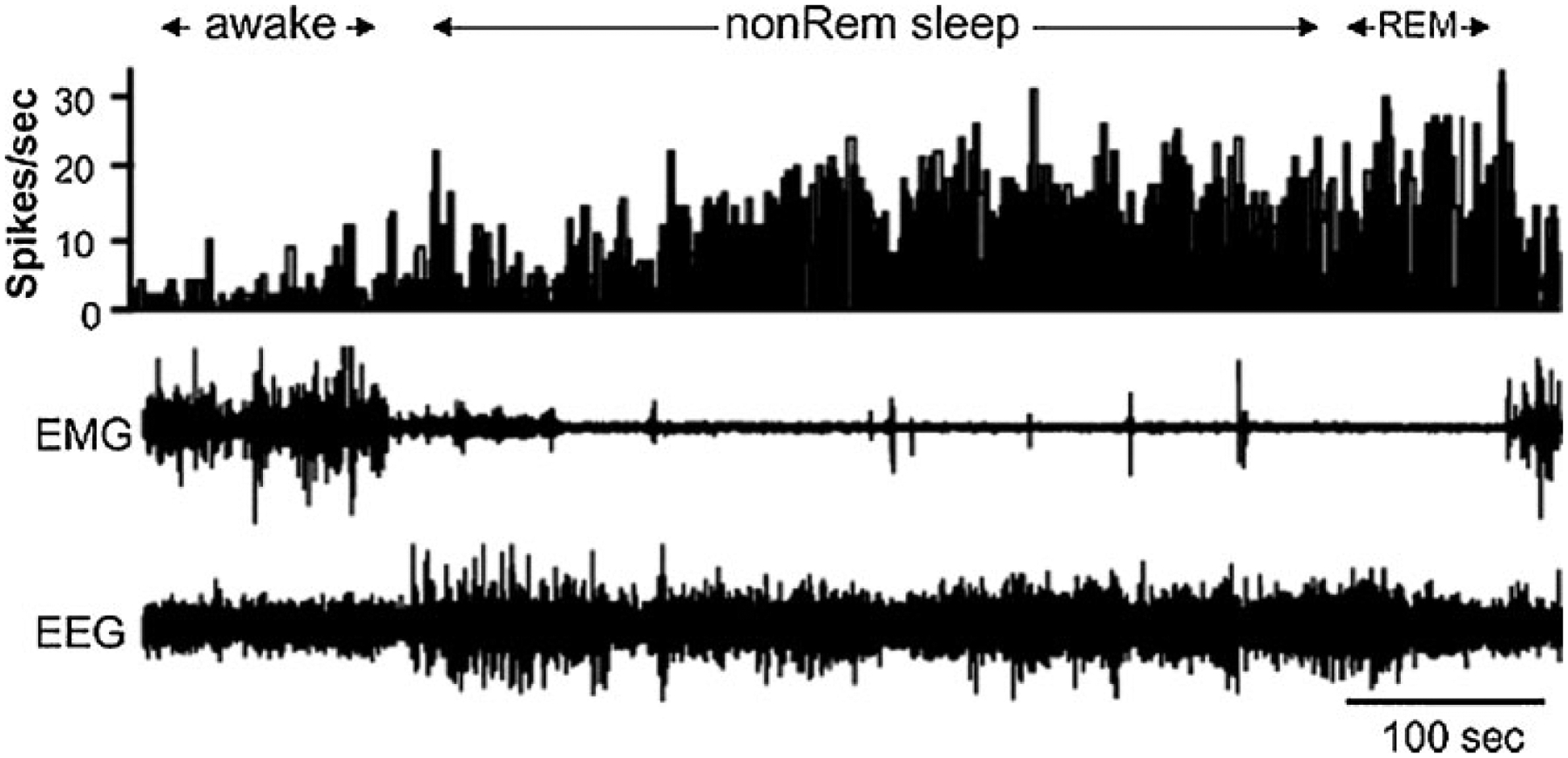
Sleep active neuron recorded in the ventrolateral preoptic region. Top channel is histogram indicating the number of action potentials each 1-second period. Note the increased activity in both non-rapid eye movement (REM) and REM sleep. EEG, electroencephalogram; EMG, electromyogram. (From Szymusiak et al,15 with permission.)
NON-REM SLEEP
NREM sleep is controlled by hypothalamic mechanisms that modulate thalamic and cortical activity as well as controlling brainstem arousal systems. Hypothalamic mechanisms were first implicated in sleep control by von-Economo’s studies of patients with encephalitis lethargica.1,2 He concluded from studies of human autopsy material that damage to the posterior hypothalamus resulted in excessive sleepiness, whereas damage to the anterior hypothalamus resulted in insomnia. Subsequently, similar observations were made from experiments in the rat3 and cat.4
Further evidence for the role of the anterior hypothalamus and adjacent basal forebrain were derived from stimulation studies. It was found that electrical stimulation of the anterior hypothalamus could rapidly induce relatively normal looking sleep.5 Studies of the expression of Fos, a protein produced by many neurons during periods of maximal activity,6 revealed that most neuronal expression of Fos greatly decreases shortly after sleep onset.7 However, Fos expression in median preoptic and ventrolateral preoptic regions increases, indicating that these cells increase activity with sleep onset. These “sleep active neurons” are GABAergic.8,9 Neuronal activity recording shows that cells in this region become maximally active during sleep10–13 and are temperature sensitive, increasing discharge with brain heating. The latter observation suggests a functional mechanism for NREM sleep whereby increases in brain temperature increase sleep propensity and depth, possibly related to brain thermoregulation.14,15 Sleep active preoptic neurons may also respond to sleep-inducing substances that accumulate during waking, including adenosine, prostaglandin D2, interleukin 1β, and growth hormone releasing hormone.16 Median preoptic sleep active neurons have been hypothesized to control the transitions from wake to NREM sleep, and have been hypothesized to mediate sleepiness because they become active in waking in the sleep-deprived animal and increase activity prior to sleep onset. Cells in the ventrolateral preoptic neurons have been hypothesized to be particularly important in maintaining sleep continuity and in the homeostatic control of REM sleep. They are inactive during waking, even in sleep-deprived animals and maintain elevated levels of activity throughout NREM sleep. One subgroup of median and ventrolateral preoptic neurons maintains their NREM sleep activity in REM sleep whereas the remaining sleep active neurons are maximally active in NREM and have greatly reduced activity in REM sleep.8,16
Sleep active neurons in the median and lateral preoptic and adjacent basal forebrain regions have projections to brainstem regions that can induce other aspects of NREM sleep. Inhibition of brainstem monoaminergic cell groups and of forebrain and brainstem cholinergic arousal-related cell groups by the activity of sleep active neurons disfacilitates thalamic neurons.
The electroencephalogram (EEG; brain waves) recorded from the cerebral cortex result from the synchronized occurrence of excitatory and inhibitory postsynaptic potentials (EPSPs and IPSPs) in cortical neurons. The generation of “sleep spindles” and “slow waves” has been shown to result from the activity of neurons that are able to discharge rhythmically because of their membrane properties. Steriade, McCormick, and their colleagues17–22 have demonstrated that the spindle waves that characterize the sleep EEG are generated by interactions between the nucleus reticularis, which forms a shell surrounding the thalamus, and the thalamic nuclei. The nucleus reticularis contains GABAergic cells. These cells fire in a 7 to 14 Hz rhythm due to the membrane time course of their low threshold calcium spikes, so named because calcium rather than sodium is admitted through voltage sensitive channels that open only when the cell is relatively hyperpolarized. After the calcium spikes, membrane currents return the cell to the hyperpolarized state, restarting the process. Through GABA release by their projections into the thalamus, reticularis neurons synchronize rhythmically recurring hyperpolarizations in large populations of thalamocortical neurons. Reticularis-induced IPSPs result in rebound depolarizations in the thalamocortical cells because the hyperpolarization “turns on” a low threshold calcium current in these cells also. These depolarizations of thalamocortical cells produce action potentials and cortical EPSPs and IPSPs, which cause the waves recorded as sleep spindles. Delta waves are produced by a similar process occurring at higher levels of membrane hyperpolarization, which produce a slower membrane oscillation.
The histamine-containing neurons of the posterior hypothalamus have been shown to be important in maintaining the waking state. These cells are tonically active in waking, greatly reduce discharge in NREM sleep and become nearly silent in REM sleep. This discharge profile is shared by noradrenergic neurons of the locus coeruleus, serotonergic neurons of the raphe nuclei, and hypocretin-containing neurons of the hypothalamus,23–26 all of which have been shown to increase waking when activated.16 It is likely that the inactivity of these neurons in sleep, a discharge pattern reciprocal to that of the sleep active GABAergic neurons which project to them, have an important role in NREM sleep regulation. The sleep active neurons in the preoptic area have direct projections to these areas and it has been shown that GABA, possibly originating in the sleep active neurons as well as in local GABA neurons, is released during sleep at several of these sites.16,27–29 A potent role in the maintenance of waking has also been shown for the cholinergic wake active neurons of the basal forebrain. However, a recent study has shown that destruction of hypocretin, histamine, and basal forebrain cholinergic waking active neurons is without major effect of sleep state organization.30 Together these findings indicate that other elements of the classic ascending activating system, including especially glutamatergic cells of the midbrain and pons, are sufficient to maintain waking at normal levels after recovery from such lesions.31–35
REM SLEEP
Most early work on REM sleep control was done in cats. Figure 2, top, shows the principal electrical signs of REM sleep. These include a reduction in cortical EEG amplitude, particularly in the power of its lower frequency components. A theta rhythm is generated in the hippocampus during REM sleep.24 REM sleep is also characterized by a suppression of muscle tone (called atonia), visible in the electromyogram (EMG). Erections tend to occur in males.36 Thermoregulation (e.g., sweating and shivering) largely ceases in most animals and body temperatures drift toward environmental temperatures, as in reptiles.37 Pupils constrict, reflecting a parasympathetic dominance in the control of the iris.38 These changes that are present throughout the REM sleep period have been termed its “tonic” features.
Figure 2.
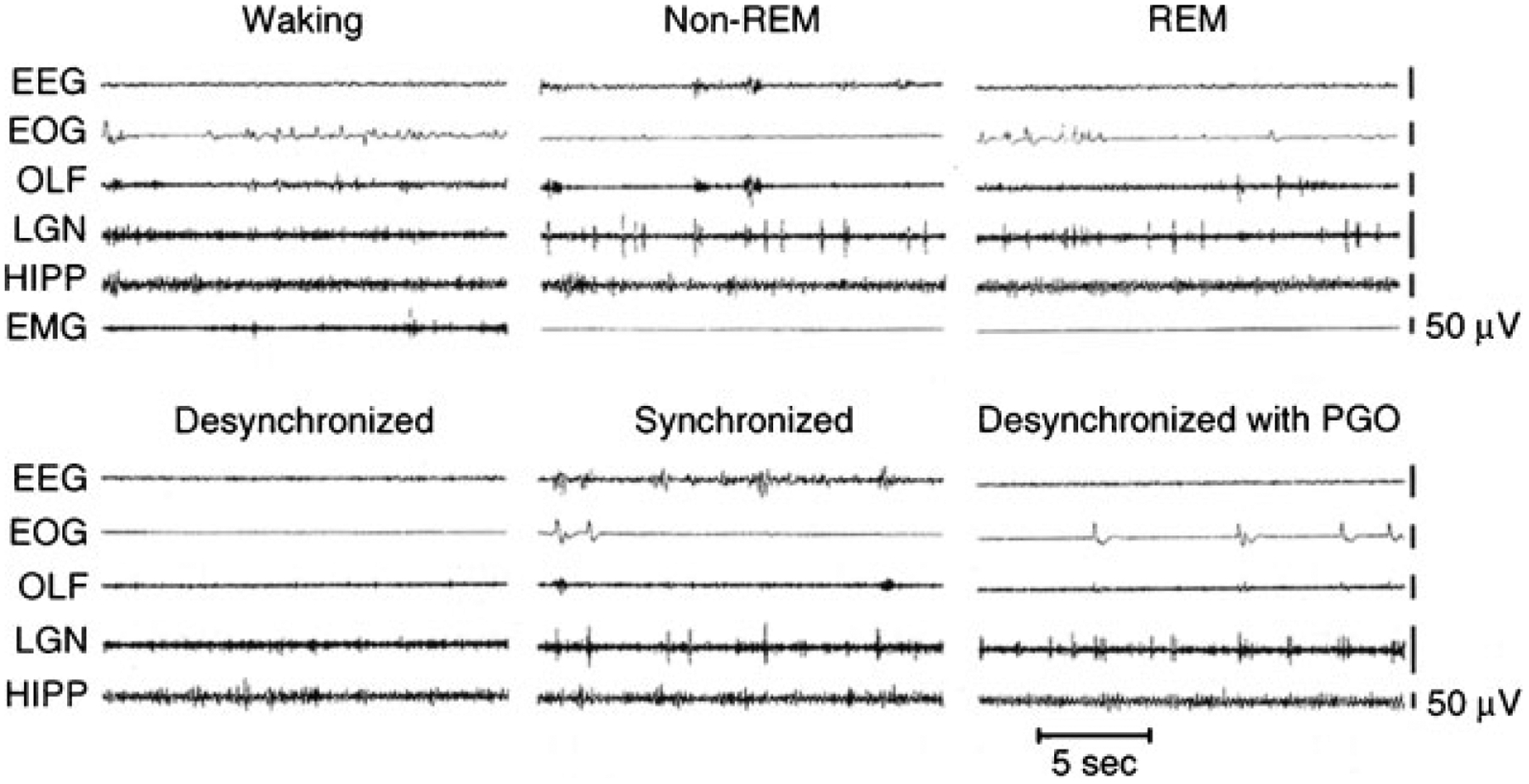
Top: polygraph tracings of states seen in the intact cat. Bottom: states seen in the forebrain 4 days after transection at the pontomedullary junction. REM, rapid eye movement; EEG, sensorimotor electroencephalogram; EOG, electrooculogram; OLF, olfactory bulb; LGN, lateral geniculate nucleus; HIPP, hippocampus; EMG, dorsal neck electromyogram; PGO, pontogeniculo-occipital spikes.
Also visible are electrical potentials that can be most easily recorded in the lateral geniculate nucleus of the cat.39 These potentials originate in the pons, appear after a few milliseconds in the lateral geniculate nucleus, and can be observed with further delay in the occipital cortex, leading to the name ponto-geniculo-occipital (PGO) spikes. They occur as large amplitude, isolated potentials 30 or more seconds before the onset of REM sleep as defined by EEG and EMG criteria. After REM sleep begins, they arrive in bursts of 3 to 10 waves, usually correlated with rapid eye movements. Ponto-geniculo-occipital linked potentials can also be recorded in the motor nuclei of the extraocular muscles, where they trigger the rapid eye movements of REM sleep. They are also present in thalamic nuclei other than the geniculate and in neocortical regions other than the occipital cortex. In humans, rapid eye movements are loosely correlated with contractions of the middle ear muscles of the sort that accompany speech generation and that are part of the protective response to loud noise.40 Other muscles also contract during periods of rapid eye movement, briefly breaking through the muscle atonia of REM sleep. There are periods of marked irregularity in respiratory and heart rates during REM sleep, in contrast with NREM sleep, during which respiration and heart rate are highly regular. No single pacemaker for all of this irregular activity has been identified. Rather, the signals producing twitches of the peripheral or middle ear muscles may lead or follow PGO spikes and rapid eye movements. Bursts of brainstem neuronal activity may likewise lead or follow the activity of any particular recorded muscle.41–43 These changes that occur episodically in REM sleep have been called its “phasic” features.
Transection Studies
The most radical types of lesion studies are those that slice through the brainstem, severing the connections between regions rostral and caudal to the cut. Sherrington discovered that animals in which the forebrain is removed after transecting the neuraxis in the coronal plane at the rostral border of the superior colliculus, showed tonic excitation of the “antigravity muscles” or extensors (Fig. 3, level A). This decerebrate rigidity was visible as soon as anesthesia was discontinued. Bard and Macht reported in 1958 that animals with decerebrate rigidity would show periodic limb relaxation.44 We now know that Bard and Macht were observing the periodic muscle atonia of REM sleep.
Figure 3.
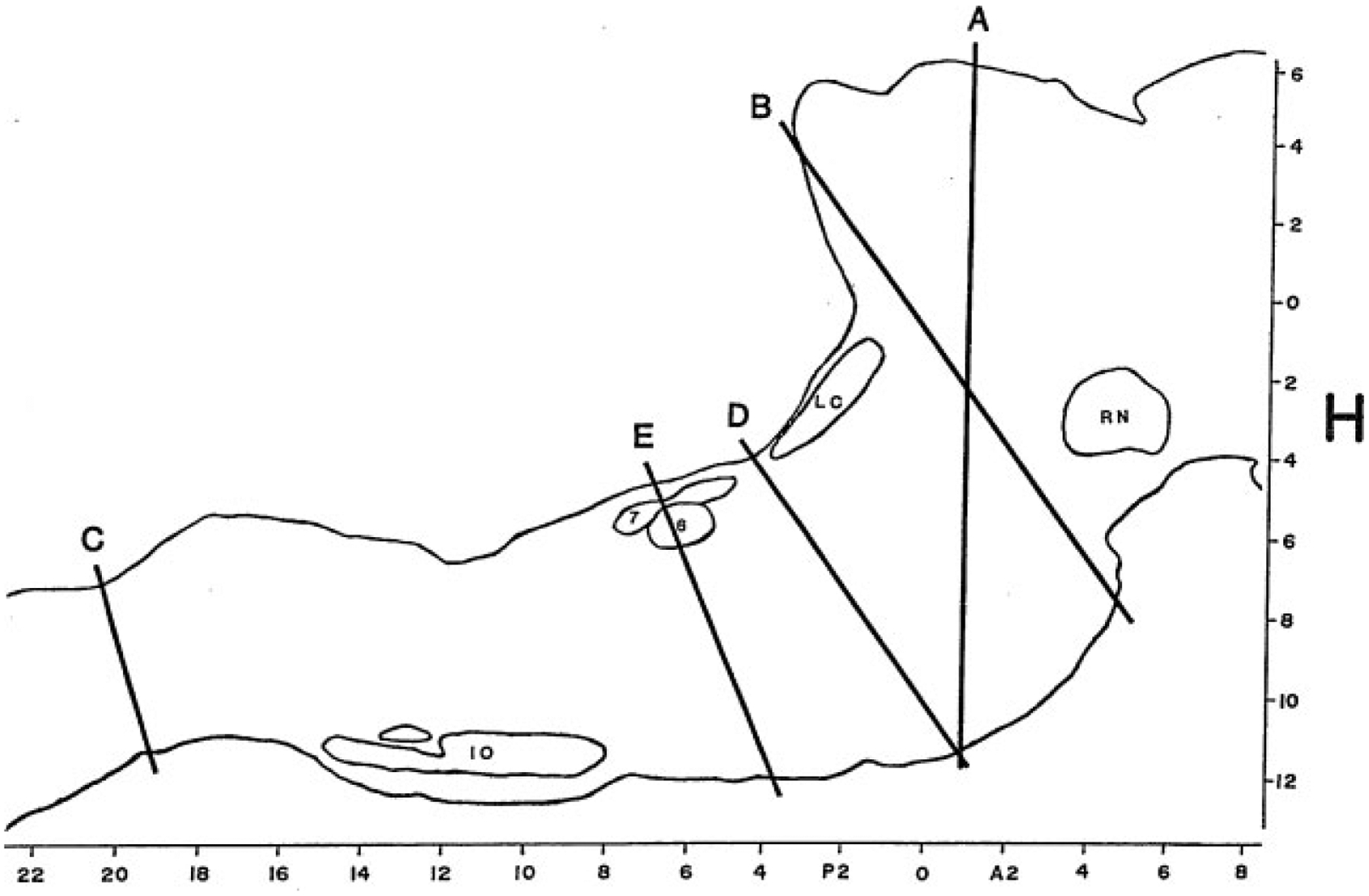
Outline of a sagittal section of the brainstem of the cat drawn from level L = 1.6 of the Berman Atlas indicating the level of key brainstem transection studies. RN, red nucleus; LC, locus coeruleus; 6, abducens nucleus; 7, genu of the facial nerve; IO, inferior olive. H (horizontal) and A-P (anteroposterior) scales are drawn from the atlas.
After the discovery of REM sleep in the cat,45 Jouvet found that this state of EEG desynchrony was normally accompanied by muscle atonia.46 Jouvet then examined the decerebrate cat preparation used by Sherrington and Bard, now adding measures of muscle tone, eye movement, and EEG. When he recorded in the forebrain after separating the forebrain from the brainstem at the midbrain level (Fig. 3, levels A or B), he found no clear evidence of REM sleep. In the first few days after transection, the EEG in the forebrain was always high voltage as in NREM sleep, but when low voltage activity appeared, the PGO spikes that help identify REM sleep in the intact animal were absent from the lateral geniculate where they can be most easily recorded. Thus it appeared that the isolated forebrain had slow wave sleep states and possibly waking, but no clear evidence of REM sleep.
In contrast, the midbrain and brainstem behind the cut showed clear evidence of REM sleep. Muscle atonia appeared with a regular periodicity and duration, similar to that of the intact cat’s REM sleep periods. This atonia was accompanied by PGO spikes with a similar morphology to those seen in the intact animal. The pupils were highly constricted during atonic periods, as in REM sleep in the intact cat.
A further localization of the REM sleep control mechanisms can be achieved by examining the sleep of humans or animals in which the brainstem–spinal cord connection has been severed (Fig. 3, level C). In this case, normal REM sleep in all its manifestations, except for spinally mediated atonia, is present.47 Thus, we can conclude that the region between the caudal medulla and rostral midbrain is sufficient to generate REM sleep.
This approach can be continued by separating the caudal pons from the medulla (Fig. 3, level D or E). In such animals no atonia is present in musculature controlled by the spinal cord, even though electrical or chemical stimulation of the medial medulla in the decerebrate animal suppresses muscle tone.48 Furthermore, neuronal activity in the medulla does not resemble that seen across the REM–NREM sleep cycle, with neuronal discharge very regular for periods of many hours, in contrast to the periodic rate modulation that is linked to the phasic events of REM sleep in the intact animal (Fig. 4).49 This demonstrates that the medulla and spinal cord together, although they may contain circuitry whose activation can suppress muscle tone, are not sufficient to generate this aspect of REM sleep when disconnected from more rostral brainstem structures, and they are also not sufficient to generate the phasic bursts of activity that characterize REM sleep.
Figure 4.

States seen in the chronic medullary cat. Note the absence of periods of atonia. EMG, electromyogram; EKG, electrocardiogram; RESP, thoracic strain gauge. Calibration, 50 μV. (From Siegel et al.49)
In contrast, the regions rostral to this cut show aspects of REM sleep (Fig. 2 bottom, Fig. 5).50 In these regions we can see the progression from isolated to grouped PGO spikes and the accompanying reduction in PGO spike amplitude that occurs in the pre-REM sleep period and the REM sleep periods in the intact animal. We also see increased forebrain unit activity, with unit spike bursts in conjunction with PGO spikes, just as in REM sleep.49,51
Figure 5.
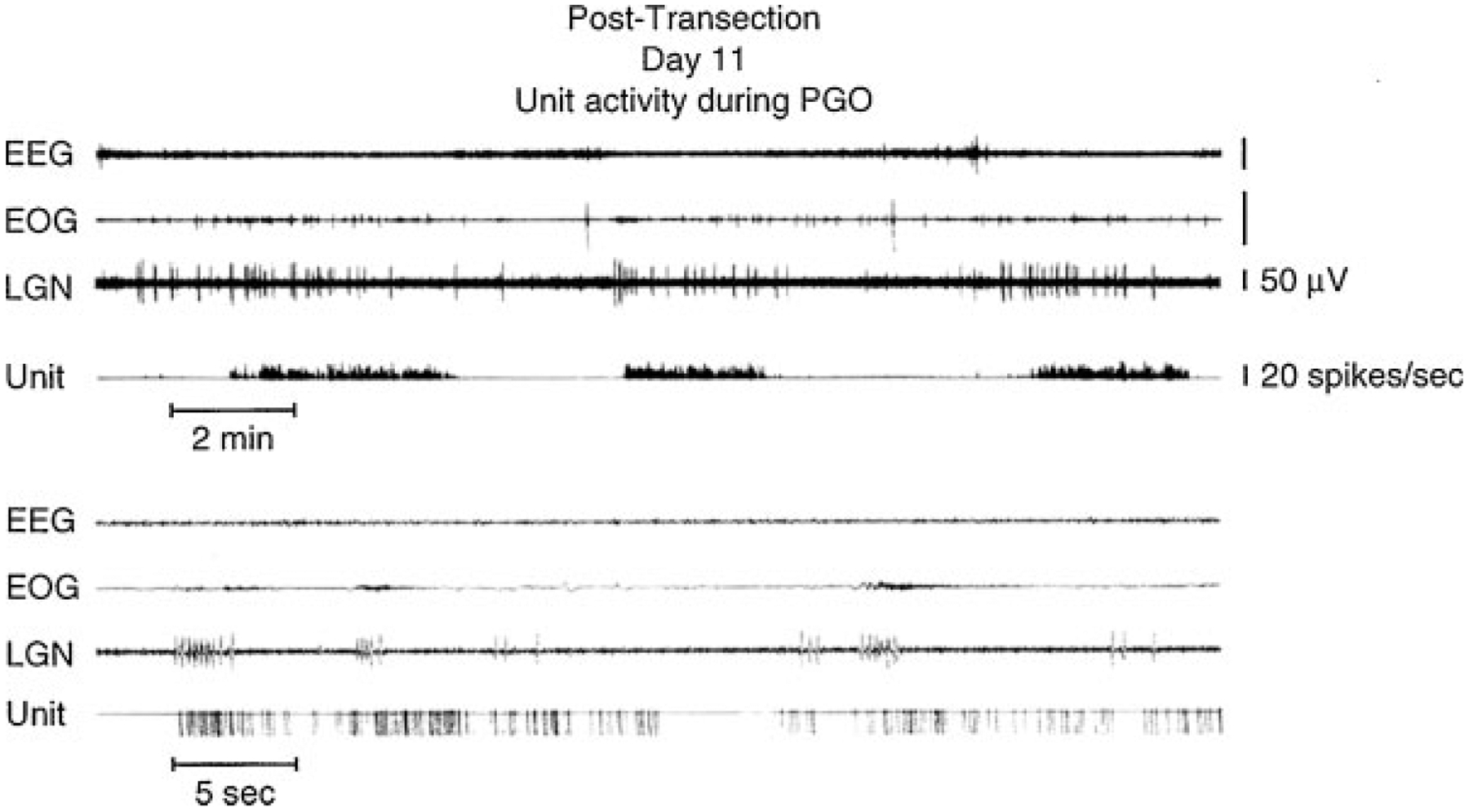
Midbrain unit: electroencephalographic (EEG), electro-oculographic (EOG), and lateral geniculate nucleus (LGN) activity rostral to chronic transections at the pontomedullary junction. In the upper portion of the figure, the unit channel displays the output of an integrating digital counter resetting at 1-second intervals. In the lower portion, one pulse is produced for each spike by a window discriminator. (From Siegel.51)
To summarize, this work shows that when pontine regions are connected to the medulla, atonia, rapid eye movements and the associated unit activity of REM sleep occur, whereas the medulla and spinal cord together, disconnected from the pons are not sufficient to generate these local aspects of REM sleep. When the pons is connected to the forebrain, forebrain aspects of REM sleep are seen, but the forebrain without attached pons does not generate these aspects of REM sleep. Further confirmation of the importance of the pons and caudal midbrain comes from the studies of Matsuzaki et al.52 They found that when two cuts were placed, one at the junction of the midbrain and pons and the other at the junction of the pons and medulla, one could see periods of PGO spikes in the isolated pons, but no signs of REM sleep in structures rostral or caudal to the “pontine island.”
These transection studies demonstrate, by positive evidence, that the pons is sufficient to generate the pontine signs of REM sleep, i.e., the periodic pattern of PGO spikes and irregular neuronal activity that characterizes REM sleep. One can conclude that the pons is the crucial region for the generation of REM sleep. Below, we will consider in more detail the structures within this region that synthesize the core elements of REM sleep.
However, it is also clear that the pons alone does not generate all the phenomena of REM sleep. Atonia requires the inactivation of brainstem systems facilitating muscle tone and the activation of motor inhibitory systems in the medulla.24,53,54 In the intact animal, forebrain mechanisms interact with pontine mechanisms to regulate the amplitude and periodicity of PGO spikes,55 which, in turn, is linked to the twitches and rapid eye movements of REM sleep. We know from cases of human REM sleep behavior disorder that the motor activity expressed in dreams is tightly linked to the imagery of the dream.56 Extrapolating to dream imagery in normal humans, one can hypothesize that since the structure of REM sleep results from an interaction of forebrain and brainstem mechanisms, the dream itself is not just passively driven from the brainstem, but rather represents the result of a dynamic interaction between forebrain and brainstem structures.
Localized Lesion Studies
The transection studies point to a relatively small portion of the brainstem, the pons, and caudal midbrain, as critical for REM sleep generation. Further specification of the core regions can be achieved by destroying portions of the pons in an otherwise intact animal and seeing which areas are necessary and which are unnecessary for REM sleep generation. It was shown that neurons in medial pontine regions, including the giant cell region, were not important in REM sleep control53,57,58 because near total destruction of these cells was followed by normal amounts of REM sleep as soon as anesthesia dissipated.59,60 However, lesions of the subcoeruleus and adjacent regions with cytotoxins did cause a prolonged reduction in the amount of REM sleep. According to one study, the extent of this loss was proportional to the percentage of cholinergic cells lost in subcoeruleus and adjacent regions of the brainstem of the cat.61 In rats, lesion or inactivation of the same region below the locus coeruleus (called the sub-laterodorsal nucleus in the terminology of Swanson62) has been found to reduce REM sleep.34
Although large lesions may eliminate all aspects of REM sleep, small bilaterally symmetrical lesions within the pons can eliminate specific aspects of REM sleep. Lesions of lateral pontine structures allow muscle atonia during REM sleep. However, PGO spikes and the associated rapid eye movements are absent when lesions include the region surrounding the superior cerebellar peduncle of the cat (Fig. 6, top).63 This points to a role for this lateral region in the generation of PGO waves and the associated phasic activity of REM sleep.
Figure 6.
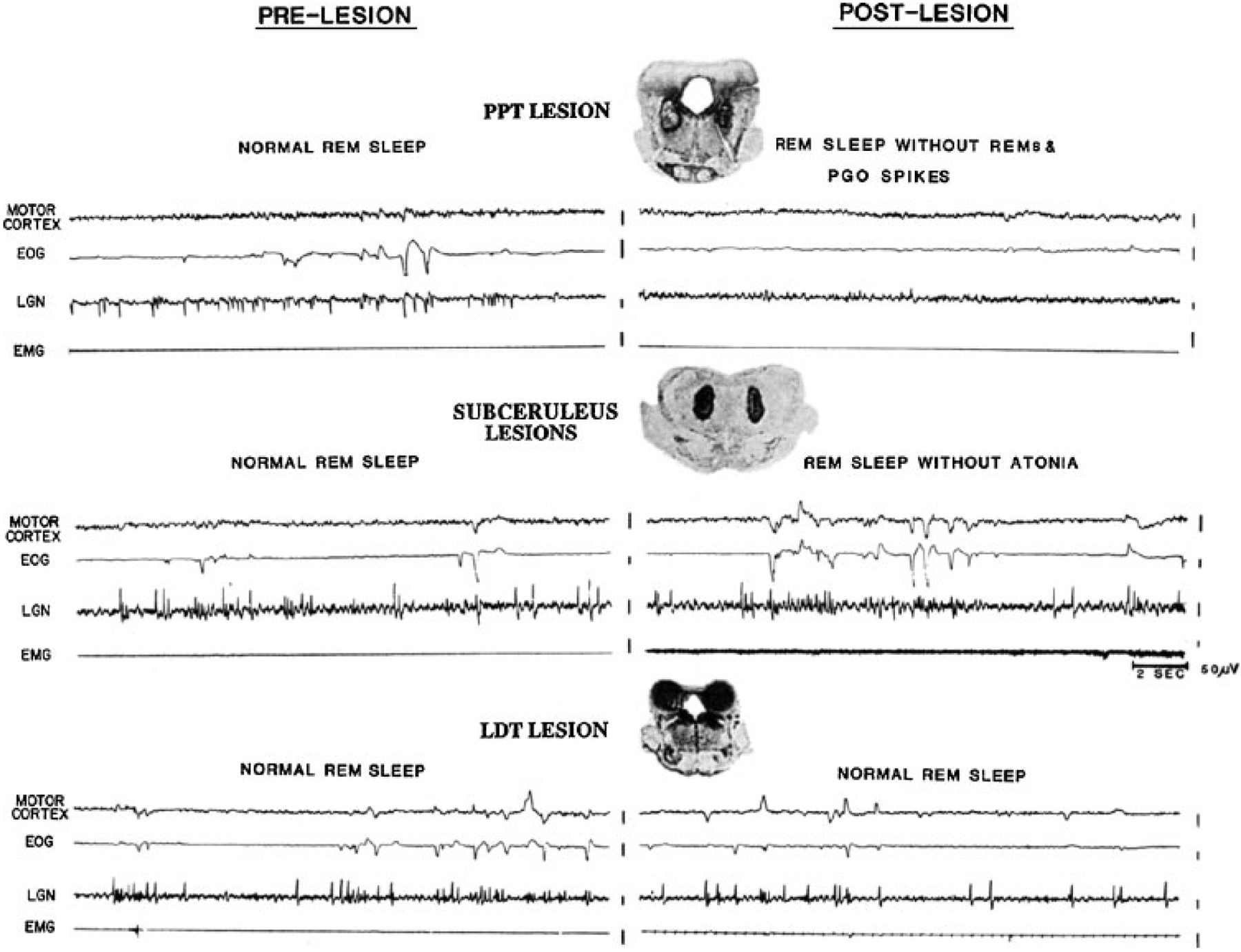
Twenty-second polygraph tracings of rapid eye movement (REM) sleep before and after lesions, together with a coronal section through the center of the pontine lesions. Electroencephalogram (EEG) voltage reduction of REM sleep (recorded from motor cortex) was present after both lesions. Top, radiofrequency lesions of the pedunculopontine region diminished ponto-geniculo-occipital (PGO) spikes and eye movement bursts during REM sleep. Bottom, lesions in the region ventral to the locus coeruleus produced REM sleep without atonia without any diminution of PGO spike or REM frequency. (Reprinted from Shouse and Siegel,63 with permission from Elsevier Science.)
Small lesions confined to portions of the subcoeruleus regions result in a very unusual syndrome. After NREM sleep, these animals enter REM sleep as indicated by lack of responsiveness to the environment, PGO spikes, EEG desynchrony, and pupil constriction. However, they lack the muscle atonia that normally characterizes this state (Fig. 6, bottom).64,65 During “REM sleep without atonia” these animals appear to act out dreams, attacking objects that are not visible, exhibiting unusual affective behaviors and ataxic locomotion. When they are awakened, normal behavior resumes. More recent studies have demonstrated that lesions of a system extending from the ventral midbrain to the medial medulla can cause REM sleep without atonia and that activation of this system can suppress muscle tone.53,66–69
This subcoeruleus region is under the control of midbrain regions. A midbrain region located just beneath and lateral to the periaqueductal gray (and called the dorsocaudal central tegmental field in the cat), appears to inhibit REM sleep by inhibiting the critical “REM-on” subcoeruleus neurons. Muscimol, a GABAA receptor agonist, injected into this midbrain region silences these cells and increases REM sleep, presumably by blocking the inhibition.70 The same phenomena have been observed when muscimol is injected into the corresponding region of guinea pig71 and the rat.35 In the rat, this midbrain region has been called the deep mesencephalic nucleus.
Increasing the levels of GABA in the subcoeruleus region (also called the pontine oralis nucleus in the rat and cat) produces an increase in waking, rather than the increase in REM sleep seen with GABA injection into the midbrain regions indicated above.72,73 This is another reminder that, despite the sleep-inducing effect of systemic administration of GABA receptor activating hypnotic medications, local manipulation shows that the effect of GABA on sleep and waking states varies across brain regions. Blocking GABA in the subcoeruleus has been reported to increase REM sleep in the cat.74
Stimulation Studies
The first study showing that chemical stimulation could elicit REM sleep was done by George et al.75 They found that application of the acetylcholine agonist carbachol to specific regions of the pons ventral to the locus coeruleus could elicit REM sleep in the cat. An impressive proof that a unique REM sleep generation mechanism was being activated was the long duration of the elicited REM sleep periods with aspects of REM sleep persisting for hours or even days after injection. Microinjection of acetylcholine into this region in the decerebrate cat produces an immediate suppression of decerebrate rigidity. Later studies showed that depending on the exact site, either REM sleep or just atonia in a waking state could be triggered by cholinergic stimulation.76–78 When stimulation was applied to the lateral regions whose lesion blocked PGO waves, continuous PGO spikes were generated even though the animal was not always behaviorally asleep. Increased REM sleep has been reported in the rat after microinjection of cholinergic agonists into the subcoeruleus region,79–81 although this effect is certainly not as robust as it is in the cat.82
The first study demonstrating a role for glutamate in the control of REM sleep was done in the cat. It was found that a profound suppression of muscle tone could be elicited by the injection of glutamate into the subcoeruleus region or into the ventral medullary region.31,48,83 Further work has demonstrated that the pontine cells in this inhibitory region receiving this cholinergic input use glutamate as their transmitter and project directly to glutamate responsive regions of the medial medulla.31,84–86
Work in the rat has emphasized the strong triggering of REM sleep by glutamatergic excitation of this region.34,87 However, glutamatergic excitation of this region in the cat also increases REM sleep,88 suggesting that the difference in response in the two species does not indicate a fundamental difference in control features, although it does suggest species differences in the relative potency of these transmitters or perhaps in the pattern of distribution of receptors for them.
Neuronal Activity, Transmitter Release
The transection, lesion, and stimulation studies all point to the same regions of the pons and caudal midbrain as the critical region for the generation of the state of REM sleep as a whole, and smaller subregions in the brainstem and forebrain in the control of its individual components. The pons contains a complex variety of cells differing in their neurotransmitter, receptors and axonal projections. Unit recording techniques allow an analysis of the interplay between these cell groups and their targets to further refine our identification of REM sleep mechanisms.
Most cells within the medial brainstem reticular formation are maximally active in waking, greatly reduce discharge rate in NREM sleep and increase discharge rate back to waking levels in REM sleep.41,42,58,89,90 Discharge is most regular in NREM sleep and is relatively irregular in both waking and REM sleep. The similarity of the waking and REM sleep discharge pattern suggests a similar role of these cells in both states. Indeed, most of these cells have been shown to be active in waking in relation to specific lateralized movements of the head, neck, tongue, face, or limbs. For example, a cell may discharge only with extension of the ipsilateral forelimb or abduction of the tongue. The twitches that are normally visible in facial and limb musculature during REM sleep and the phenomenon of REM sleep without atonia suggest that these cells command movements that are blocked by the muscle tone suppression of REM sleep. Lesion of these cells has little or no effect on REM sleep duration or periodicity,59,91 but does dramatically prevent movements of the head and neck92 that can normally be observed in waking.
Microinjection of cholinergic agonists into the pons triggers REM sleep. Microdialysis studies show that pontine acetylcholine release is greatly increased during natural REM sleep when compared with either NREM sleep or waking.93 Recordings of neuronal activity within the cholinergic cell population demonstrate the substrates of this release. Certain cholinergic cells are maximally active in REM sleep (REM-on cells). Others are active in both waking and REM sleep.94 Presumably the REM sleep-on cholinergic cells project to the acetylcholine responsive region in the subcoeruleus area.95
Cells with activity selective for REM sleep can be identified within the subcoeruleus area in both cats96 and rats.35 Anatomic studies using Fos labeling and tract tracing suggest that these neurons are glutamatergic and that some of them project to the ventral medullary region involved in the triggering of the muscle atonia of REM sleep.31,34,35,48,84–86,97
Monoamine-containing cells have a very different discharge profile. Most if not all noradrenergic98,99 and serotonergic100 cells of the midbrain and pontine brainstem, and histaminergic25 cells of the posterior hypothalamus are continuously active during waking, decrease their activity during NREM sleep, and further reduce or cease activity during REM sleep (Fig. 7). As was pointed out above, these cell groups are not critical for REM sleep generation, but it is likely that they modulate the expression of REM sleep. The cessation of discharge in monoaminergic cells during REM sleep appears to be caused by the release of GABA onto these cells,27–29,101 presumably by REM sleep-active GABAergic brainstem neurons.102,103 Administration of a GABA agonist to the raphe cell group increases REM sleep duration,28 demonstrating a modulatory role for this cell group in REM sleep control.
Figure 7.

Activity of a “REM sleep-off” cell recorded in the locus coeruleus. EEG, electroencephalogram; EOG, electro-oculogram; LGN, lateral geniculate nucleus’ EMG, electromyogram.
Other cholinergic cells in lateral pontine regions discharge in bursts before each ipsilateral PGO wave.104,105 These cells may therefore participate in the triggering of these waves. We know from other studies that PGO waves are tonically inhibited in waking by serotonin input.106–108 Therefore, it is likely that certain groups of cholinergic cells receive direct or perhaps indirect serotonergic inhibition in waking, and that the decrease of this inhibition in NREM sleep and REM sleep facilitates PGO wave and REM sleep generation.
A more global mapping of neurons active in REM sleep can be achieved by using the Fos labeling to identify neurons active within the 20- or more minute period before sacrifice. Quattrochi et al demonstrated that microinjections of the cholinergic agonist carbachol that triggered episodes of continuous PGO waves in waking activated neurons within the laterodorsal and pedunculopontine nuclei of the cat. Destruction of these nuclei blocks these waves.108–110
More extensive Fos mapping has been done to identify neurons activated during REM sleep in the rat. Verret et al111 found that only a few cholinergic neurons from the laterodorsal and pedunculopontine tegmental nuclei were Fos-labeled after REM sleep. In contrast, a large number of noncholinergic Fos-labeled cells was observed in the laterodorsal tegmental nucleus; subcoeruleus region; and the lateral, ventrolateral, and dorsal periaqueductal gray of the midbrain. In addition, cells in other regions outside of the brainstem regions critical for REM sleep control were labeled. These included neurons in the α and ventral gigantocellular reticular nuclei of the medulla, dorsal and lateral paragigantocellular reticular112 nuclei, and the nucleus raphe obscurus. In a second study, an effort was made to identify the source of the GABAergic input thought to cause the cessation of discharge in locus coeruleus cells during REM sleep.29 Verret et al83 found that the dorsal and lateral paragigantocellular reticular nuclei of the medulla and regions of the periaqueductal gray of the midbrain, regions with large percentages of GABAergic cells, are active in REM sleep. Maloney et al102 found GABAergic cells adjacent to the locus coeruleus that expressed Fos during periods of REM sleep. Because the critical phenomena of REM sleep do not appear to require the medulla, it seems likely that the periaqueductal gray GABAergic neurons and GABAergic neurons adjacent to locus coeruleus and raphe nuclei are sufficient to suppress the activity of noradrenergic and serotonergic neurons,28,113 although medullary neurons may participate in the intact animal.
Fos mapping has also been used to identify forebrain regions likely to control REM sleep. The preoptic region, important in NREM sleep control contains neurons that express Fos maximally in REM-sleep-deprived animals, suggesting that these neurons may be related to the triggering or maintenance of REM sleep by brainstem systems, perhaps in coordination with the triggering of NREM sleep by this region.8 Fos studies also indicate that melanin concentrating hormone neurons, which are located in the hypothalamus, express Fos during periods with large amounts of REM sleep and that intracerebroventricular administration of melanin-concentrating hormone increases the amount of subsequent REM sleep.114,115 These results suggest that melanin concentrating hormone neurons are an additional source of forebrain modulation of REM sleep.
CONTROL OF MUSCLE TONE IN SLEEP
Abnormalities of muscle tone control underlie many sleep disorders. During NREM sleep muscle tone is greatly reduced. During REM sleep, central motor systems are highly active, whereas motoneurons are hyperpolarized producing a further reduction of muscle tone.116 The normal suppression of tone in the tongue and laryngeal muscles in NREM sleep and their further suppression in REM sleep is a major contributing factor in sleep apnea. The failure of muscle tone suppression in REM sleep causes REM sleep behavior disorder. Triggering of the REM sleep muscle tone control mechanism in waking is responsible for cataplexy.
Early work using intracellular recording and microiontophoresis has shown that motoneuron hyperpolarization during REM sleep was accompanied by the release of glycine onto motoneurons.116,117 Microdialysis sampling showed that both GABA and glycine are released onto motoneurons during atonia induced by carbachol in the cat.118 This release occurs in ventral horn motoneurons as well as in hypoglossal motoneurons. The glycinergic inhibition during a carbachol-elicited REM sleep-like state was investigated with immunohistochemistry and found to be due to the activation of glycinergic neurons in the nucleus reticularis gigantocellularis and nucleus magnocellularis in the rostro-ventral medulla and the ventral portion of the nucleus paramedianus reticularis,117 regions whose activation has been shown to suppress muscle tone in the unanesthetized decerebrate animal.31 A second population was located in the caudal medulla adjacent to the nucleus ambiguus; these neurons may be responsible for the REM-sleep-related inhibition of motoneurons that innervate the muscles of the larynx and pharynx.
In related work, it has been shown that norepinephrine and serotonin release onto motoneurons is decreased during atonia.119 Because these monoamines are known to excite motoneurons and GABA and glycine are known to inhibit them, it appears that the coordinated activity of these cell groups produces motoneuron hyperpolarization and hence atonia in REM sleep by a combination of inhibition and disfacilitation.
The inhibitory and facilitatory systems are strongly and reciprocally linked. Electrical stimulation of the pontine inhibitory area (PIA), located in the subcoeruleus region,31 produces muscle tone suppression. Even though the pontine inhibitory area is within a few millimeters of the noradrenergic locus coeruleus, electrical stimulation in the pontine inhibitory area that suppresses muscle tone will always cause a cessation of activity in the noradrenergic neurons of the locus coeruleus and other facilitatory cell groups.120 This indicates that the process responsible for the suppression of muscle tone causes the linked inhibition and disfacilitation. Cells that are maximally active in REM sleep (REM-on cells) are present in the pontine inhibitory area and also in the region of the medial medulla, which receives pontine inhibitory area projections (Fig. 8).
Figure 8.
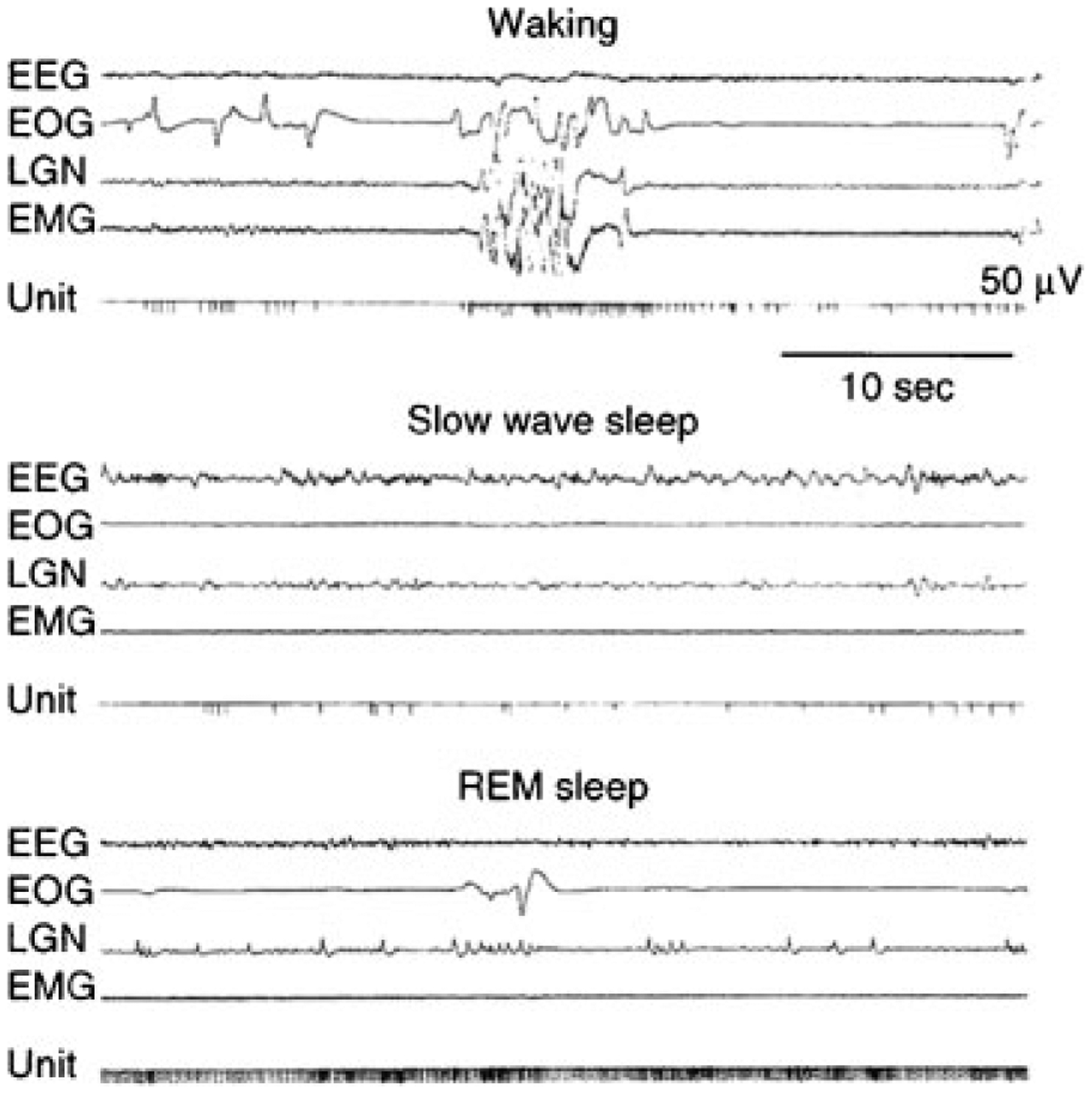
Activity of medullary “REM sleep-on” cell. Note the tonic activity during rapid eye movement (REM) sleep. In waking, activity is generally absent even during vigorous movement. However, some activity is seen during movements involving head lowering and postural relaxation. EEG, electroencephalogram; EOG, electro-oculogram; LGN, lateral geniculate nucleus; EMG, electromyogram.
The release of GABA and glycine onto motoneurons during REM sleep atonia is most likely mediated by a pathway from the pontine inhibitory area to the medial medulla.85,86 The pontine region triggering this release is not only sensitive to acetylcholine, but also responds to glutamate (Fig. 9).31,84 The medullary region with descending projections to motoneurons can be subdivided into a rostral portion responding to glutamate and a caudal portion responding to acetylcholine (Fig. 9).48,121 The medullary interaction with pontine structures is critical for muscle tone suppression because inactivation of pontine regions greatly reduces the suppressive effects of medullary stimulation on muscle tone.122,123 An ascending pathway from the medulla to the pons may mediate the inhibition of locus coeruleus during atonia and may also help recruit other active inhibitory mechanisms. Thus damage anywhere in the medial pontomedullary region can block muscle atonia (perhaps causing REM sleep behavior disorder) by interrupting ascending and descending portions of the pontomedullary inhibitory system, as can muscimol injection into the pontine inhibitory region.122
Figure 9.

Sagittal map of pontomedullary inhibitory areas. Electrical stimulation produced atonia at all the points mapped. All electrically defined inhibitory sites were microinjected with glutamate or cholinergic agonists. Filled symbols represent points at which microinjections decreased muscle tone (to <30% of baseline values or to complete atonia). Open circles indicate points at which injections increased or produced no change in baseline values. Glutamate injections are shown at the top, acetylcholine (ACh) and carbachol (Carb) injections at the bottom. At the bottom, circles and triangles represent ACh and Carb injections, respectively. 4V, fourth ventricle; 5ME, mesencephalic trigeminal tract; 6, abducens nucleus; 7G, genu of the facial nerve; IO, inferior olivary nucleus; LC, locus coeruleus nucleus; NGC, nucleus gigantocellularis; NMC, nucleus magnocellularis; NPM, nucleus paramedianus; PG, pontine gray; PT, pyramid tract; SO, superior olivary nucleus; T, nucleus of the trapezoid body; TB, trapezoid body. (From Lai and Siegel.48)
The success of jaw appliances indicates that reduced jaw muscle activity can contribute to closure of the airway in sleep apnea. Jaw muscle relaxation is a common initial sign of cataplexy and tonic muscle activation underlies bruxism. Investigation of the control of masseter motor neurons allows analysis of the regulation of muscle tone on one side of the face, while using the other side as a control for changes in behavioral state caused by application of neurotransmitter agonist and antagonists.124 Using this model, it was determined that tonic glycine release reduces muscle tone in both waking and NREM sleep. However, blockade of glycine receptors did not prevent the suppression of muscle tone in REM sleep. In a similar manner, blockade of GABA receptors alone or in combination with glycine receptors increased tone in waking and NREM sleep, but did not prevent the suppression of masseter tone125 or of genioglossus tone in REM sleep.126 However, both of these manipulations increased phasic masseter muscle activity in REM sleep.
Further studies showed that a blockade of glutamate receptors reduces the normal enhancement of muscle tone in waking relative to the level in NREM sleep. Glutamate also contributes to the phasic motor activity during REM sleep. However, reduction in glutamate alone is not sufficient to account for the suppression of muscle tone in REM sleep because stimulation of N-methyl-d-aspartate (NMDA) and non-NMDA glutamate receptors does not appear to restore muscle tone in REM sleep.127
A study in the anesthetized rat suggested that activation of norepinephrine receptors, in combination with the activation of glutamate receptors, was sufficient to potently increase muscle tone in the masseter muscles in REM sleep. A study of the hypoglossal motor nucleus in the unanesthetized rat concluded that the suppression of muscle tone in REM sleep was mediated to a large extent by a reduction in norepinephrine release.128 Thus, this work in the context of prior microdialysis analysis of transmitter release suggests that the reduction of norepinephrine release may be a key factor regulating muscle tone, along with the above-described changes in amino acid release. These conclusions are consistent with prior work indicating that cataplexy was linked to a reduction in the activity of noradrenergic neurons (see below).24 Although the current literature suggests that trigeminal, hypoglossal, and ventral horn motoneurons are subjected to similar neurochemical control across the sleep cycle, direct quantitative comparison of the neurochemical control of these systems has not been made, and it is likely that some aspects of control may differ across systems as well as species.
The role of reduced serotonin release in the suppression of muscle tone has been investigated in the hypoglossal nucleus of the rat. It was found that the modulation of genioglossus activity across natural sleep-wake states was not greatly affected by endogenous input from serotonergic neurons, although prior studies in vagotomized and anesthetized rats had shown an effect of serotonin on muscle tone under these aphysiologic conditions.129–131
Recent work suggests that inhibition of motor output is accompanied by a neurochemically similar inhibition of sensory relays during REM sleep.132 Such sensory inhibition may be important in preserving sleep, especially by blocking the sensory input produced by twitches breaking through the motor inhibition of REM sleep. The failure of this inhibition may contribute to sleep disruption and increased motor activity in sleep in pathologic states.
In contrast to norepinephrine, serotonin, and histamine cell groups, it was reported that mesencephalic dopaminergic neurons do not appear to alter their discharge rate across the sleep cycle.133 Dopamine release in the amygdala measured by dialysis does not significantly vary across the sleep cycle.134,135 In disagreement with this finding, a Fos study indicated that dopaminergic neurons within the ventral portion of the mesencephalic tegmentum were activated during periods of increased REM sleep.136 A unit recording study indicated that dopaminergic neurons in the ventral tegmental area of the midbrain show maximal burst firing in both waking and REM sleep.137 Other work using the Fos labeling technique identified a wake active dopaminergic cell population in the ventral periaqueductal gray.138 In dialysis measurements of dopamine release, we have seen reduced dopamine release in the dorsal horn of the spinal cord during the REM sleep-like state triggered by carbachol. We did not see such a decrease in the ventral horn or hypoglossal nucleus.119 These data suggest either heterogeneity in the behavior of sleep cycle activity of dopaminergic neurons, presynaptic control of dopamine release independent of action potentials in the cell somas, or variable vesicle size allowing greater release of transmitters per action potential.
Fig. 10 illustrates some of the anatomic and neurochemical substrates of the brainstem generation of REM sleep.
Figure 10.
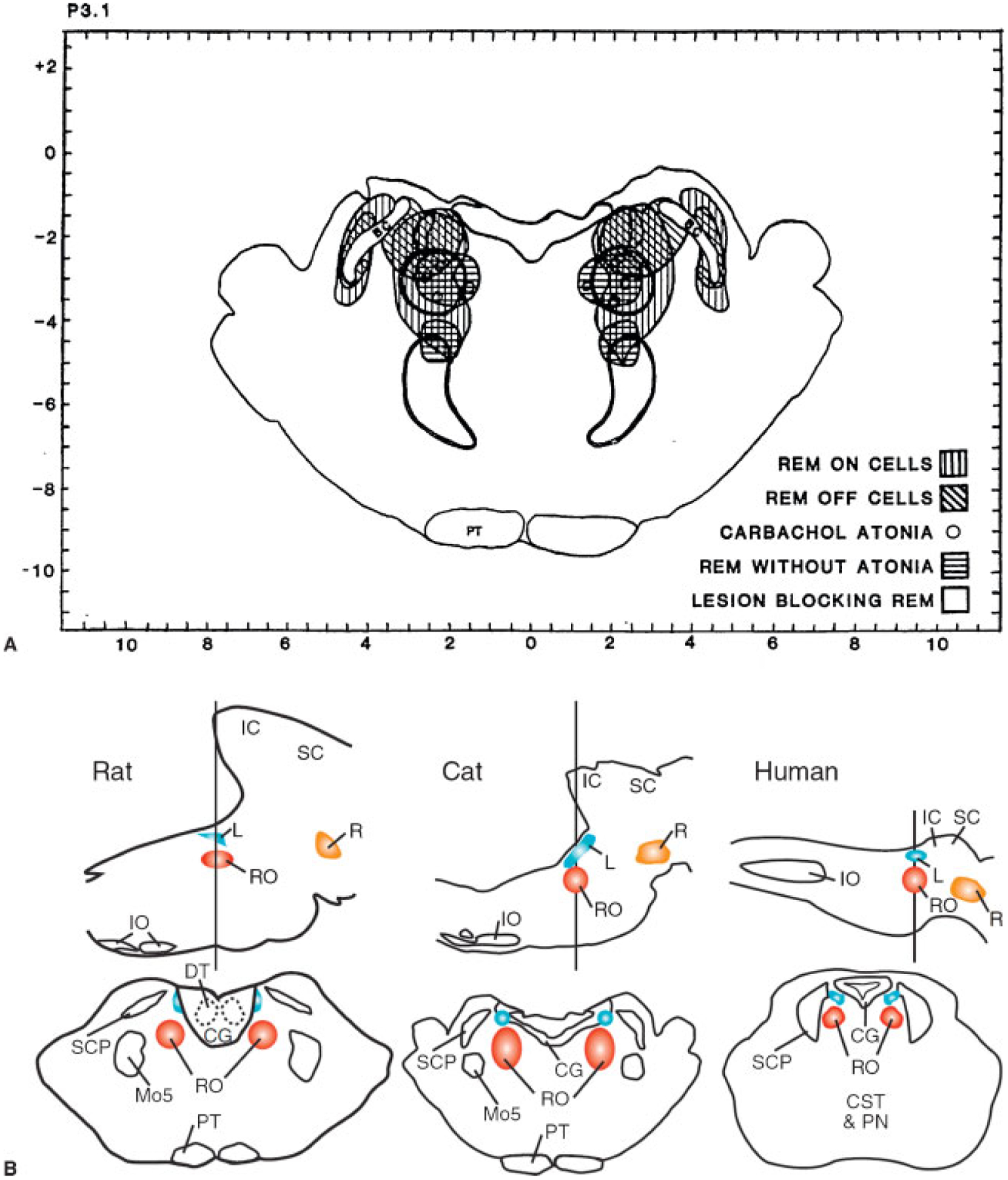
(A and B) Anatomic relation of “REM sleep-on” and “sleep-off” cells, carbachol-induced atonia sites, lesions blocking atonia but not preventing rapid eye movement (REM) sleep, and lesions completely blocking REM sleep. The inhibitory regions shown in Figs. 9–12 are not plotted. (From Siegel JM, Rogawski MA. A function for REM sleep: regulation of noradrenergic receptor sensitivity. Brain Res 1988;13:213–233.) Bottom of figure shows anatomical locations of REM on areas in cats, rat, and projected location in human in sagittal and coronal views. (From Siegel JM. The stuff dreams are made of: anatomical substrates of REM sleep. Nat Neurosci 2006;9:721–722.)
NARCOLEPSY AND HYPOCRETIN
The persistent sleepiness of narcolepsy appears to be related to activation of sleep active neurons or disfacilitation of wake active neurons. Narcolepsy has also been characterized as a disease of the REM sleep mechanism. Narcoleptics often have REM sleep within 5 minutes of sleep onset, in contrast to normal individuals who rarely show such “sleep- onset REM sleep.” Most narcoleptics experience cataplexy, a sudden loss of muscle tone with the same reflex suppression that is seen in REM sleep. High amplitude theta activity in the hippocampus, characteristic of REM sleep, is also prominent in cataplexy as observed in dogs.24,139 Further evidence for links between narcolepsy and REM sleep comes from studies of neuronal activity during cataplexy. Many of the same cell populations in the pons and medulla that are tonically active only during REM sleep in normals, become active during cataplexy in narcoleptics.43,140 Likewise, cells in the locus coeruleus, which cease discharge only in REM sleep in normal animals, invariably cease discharge in cataplexy.141 However, just as cataplexy differs behaviorally from REM sleep in its maintenance of consciousness, not all neuronal aspects of REM sleep are present during cataplexy. As was noted above, in the normal animal, noradrenergic, serotonergic, and histaminergic cells are all tonically active in waking, reduce discharge in NREM sleep, and cease discharge in REM sleep.24,141 However, unlike noradrenergic cells, serotonergic cells do not cease discharge during cataplexy, only reducing discharge to quiet waking levels. Histaminergic cells actually increase discharge in cataplexy relative to quiet waking levels (Fig. 11).142 These findings allow us to identify some of the cellular substrates of cataplexy. Medullary inhibition and noradrenergic disfacilitation are linked to cataplexy’s loss of muscle tone. In contrast, the maintained activity of histamine neurons is a likely substrate for the maintenance of consciousness during cataplexy that distinguishes cataplexy from REM sleep. Thus, the study of neuronal activity in the narcoleptic animal provides an insight into both narcolepsy and the normal role of these cell groups across the sleep cycle.
Figure 11.
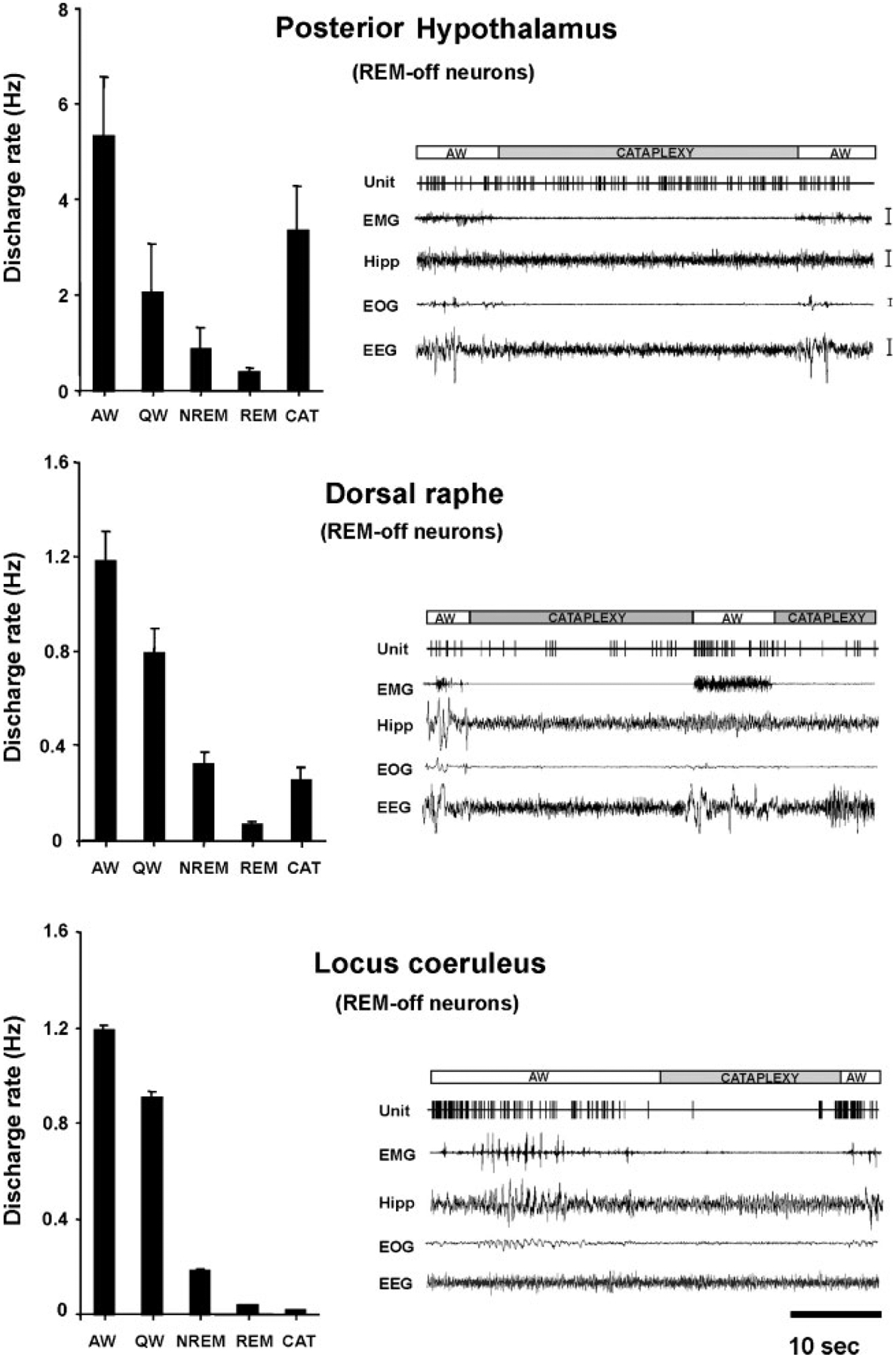
Comparison of mean discharge rates in sleep-waking states and cataplexy of rapid eye movement (REM-) off cells recorded from three brain regions. Posterior hypothalamic histaminergic neurons remain active, whereas dorsal raphe serotonergic neurons reduced discharge, and locus coeruleus noradrenergic neurons cease discharge during cataplexy. All of these cell types were active in waking, reduced discharge in non-REM (NREM) sleep, and were silent or nearly silent in REM sleep. EMG, electromyogram; HIPP, hippocampus; EOG, electro-oculogram; EEG, electroencephalogram; AW, quiet waking; QW, quiet waking. (From John et al.24)
In 2001, it was discovered that most human narcolepsy was caused by a loss of hypothalamic cells containing the peptide hypocretin (Fig. 12).143–145 On average 90% of these cells are lost in narcolepsy. Subsequently, it was discovered that a lesser reduction in the number of hypocretin cells was seen in Parkinson’s disease, with a loss of up to 60% of hypocretin cells.146,147 It was found that administration of the peptide to genetically narcoleptic dogs reversed symptoms of the disorder,148 and that nasal administration reversed sleepiness in monkeys,149 suggesting that similar treatment could be uniquely effective for narcolepsy and perhaps for other disorders characterized by sleepiness.
Figure 12.

Loss of hypocretin cells in human narcolepsy. Distribution of cells in perifornical and dorsomedial hypothalamic regions of normal and narcoleptic humans. (From Thannickal et al.144)
In further work in normal animals, it was determined that identified hypocretin neurons fire maximally during active waking (Fig. 13).26 This discharge was reduced or absent during aversive waking situations, even if the EEG indicated high levels of alertness. This is consistent with the hypothesis that release of hypocretin facilitates motor activity during emotionally charged activities of the sort that trigger cataplexy in narcoleptics, such as laughter.150–152 Even normal individuals experience weakness at these times, seen in the “doubling over” that often accompanies laughter or the weakness that can result from other sudden onset, strong emotions. Studies of hypocretin release in the cat,153 and preliminary studies in humans are also consistent with this hypothesis.154 In the absence of the hypocretin-mediated motor facilitation, muscle tone is lost at these times. Hypocretin cells also send ascending projections to cortical and basal forebrain regions. In the absence of hypocretin mediated facilitation of forebrain arousal centers, waking periods are truncated, resulting in the sleepiness of narcolepsy.151
Figure 13.
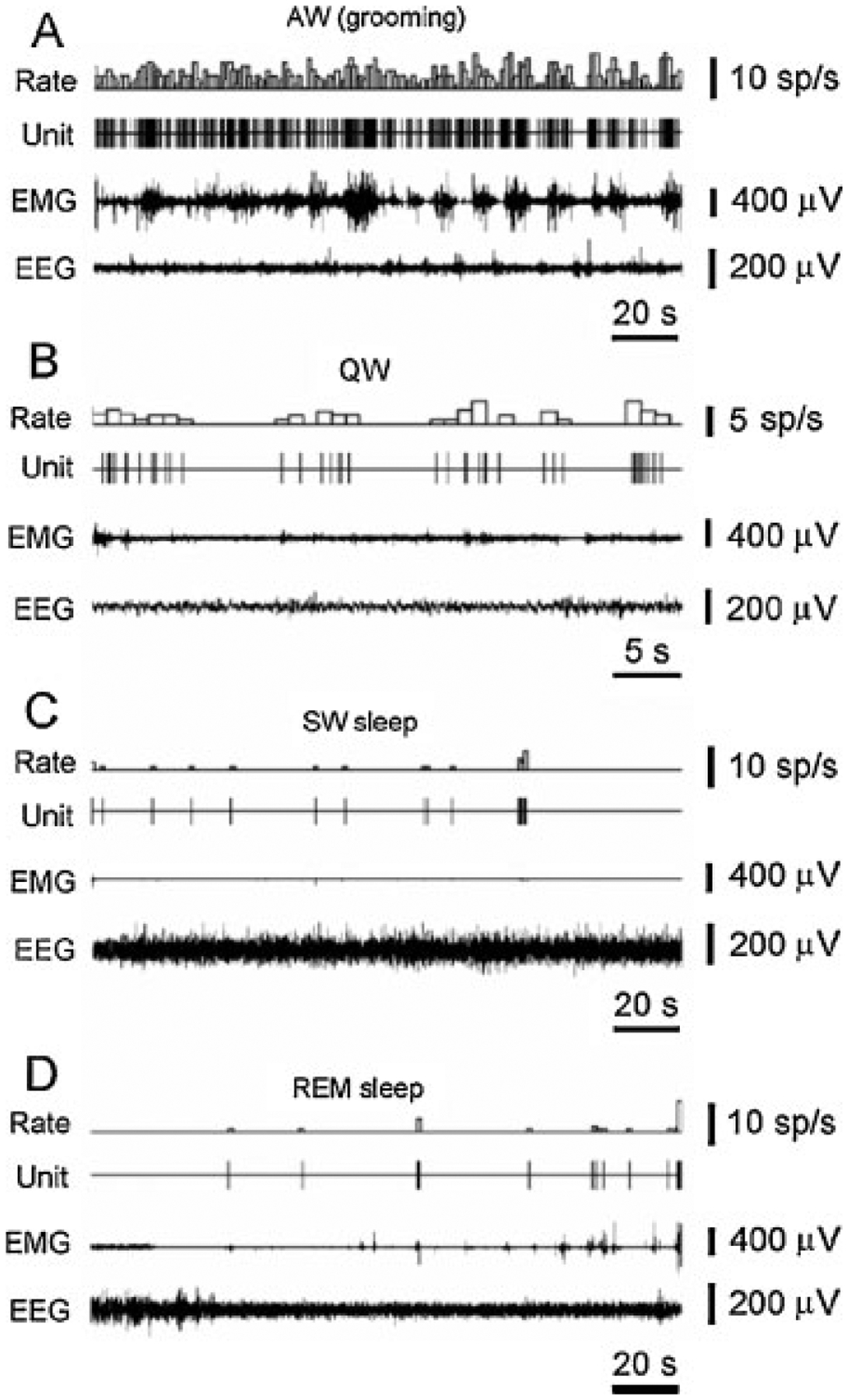
Firing rate of hypocretin cells in waking and sleep behaviors in freely moving rats. The discharge pattern of a representative hypocretin neuron across the sleep–waking cycle in the freely moving rat. (A) High firing rates are seen during AW (active waking – grooming). (B) Reduced firing rate or cessation of activity is seen in QW (quiet waking) and drowsiness. (C) A further decrease or cessation of firing is seen during SW sleep. (D) Minimal firing rate is seen during the tonic phase of rapid eye movement (REM) sleep. Brief Hcrt cell discharge bursts are correlated with muscle twitches during the phasic events of REM sleep. EMG, electromyogram; EEG, electroencephalogram. (From Mileykovskiy et al.26)
The functions of hypocretin have been investigated in knockout animals that do not have the peptide using operant reinforcement tasks. Hypocretin knockout mice were deficient in the performance of bar presses to secure food or water reinforcement. However, they did not differ from their normal littermates in their performance when trained to bar press to avoid foot shock. Periods of poor performance on the positive reinforcement tasks were characterized by EEG deactivation. Fos labeling of normal mice showed that the positive reinforcement task used in this study was characterized by activation of hypocretin neurons. However, hypocretin neurons were not activated in the negative reinforcement task despite high levels of EEG activation, indicating that nonhypocretin systems mediate arousal during this behavior. This study led to the conclusion that hypocretin neurons are critically involved in arousal linked to positive reinforcement, and that in their absence such behaviors are impaired. However, they are not required to maintain arousal in conditions of negative reinforcement, indicating that other brain systems subserve this role.
Hypocretin appears to act largely by modulating the release of amino acid neurotransmitters.155 Systemic injection of hypocretin causes a release of glutamate in certain hypocretin innervated regions producing a potent postsynaptic excitation.124,156 In other regions it facilitates GABA release, producing postsynaptic inhibition.153,157 The loss of these competing inhibitory and facilitatory influences in narcolepsy appears to leave brain motor regulatory and arousal systems less stable than the tightly regulated balance that can be maintained in the presence of hypocretin (Fig. 14). According to this hypothesis, this loss of stability is the underlying cause of narcolepsy, with the result being inappropriate loss of muscle tone in waking and inappropriate increases of muscle tone during sleep resulting in a striking increased incidence of REM sleep behavior disorder in narcoleptics. In the same manner, although a principal symptom of narcolepsy is intrusions of sleep into the waking period, narcoleptics sleep poorly at night with frequent awakenings.158–160 In other words, narcoleptics are not simply weaker and sleepier than normal. Rather, their muscle tone and sleep–waking state regulation is less stable than that in normal individuals as a result of the loss of hypocretin function.
Figure 14.
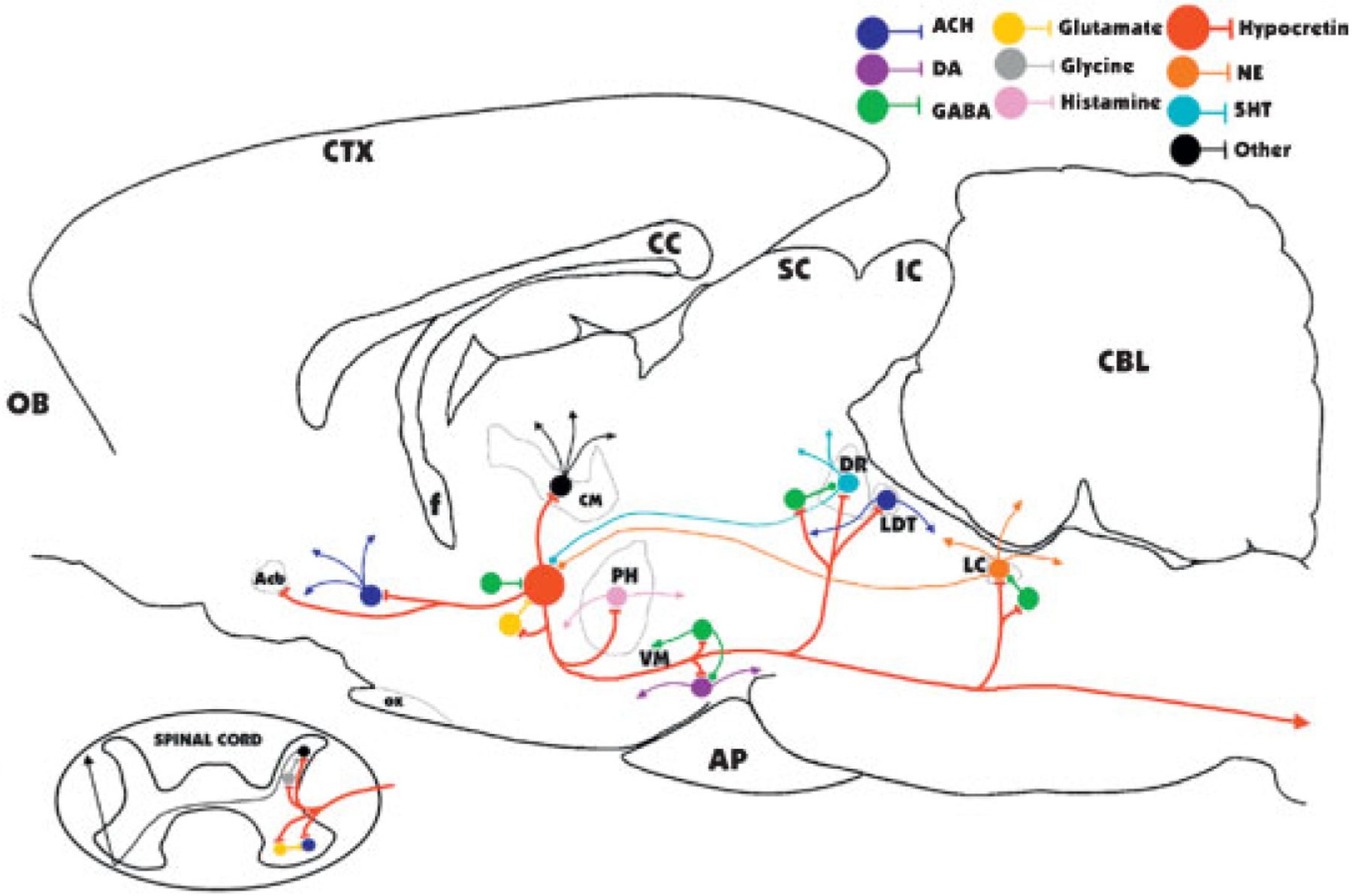
Major identified synaptic interactions of hypocretin neurons. Lines terminated by perpendicular lines denote excitation; circular terminations indicate inhibition. ACH, acetylcholine; DA, dopamine; NE, norepinephrine; 5HT, serotonin; OB, olfactory bulb; Acb, nucleus accumbens; f, fornix; OX, optic chiasm; CM, centromedian nucleus of the thalamus; PH, posterior hypothalamus; VM, ventral midbrain; AP, anterior pituitary; SC, superior colliculus; IC, inferior colliculus; DR, dorsal raphe; LDT, laterodorsal tegmental and pedunculopontine; LC, locus coeruleus; CBL, cerebellum.
THE FUNCTIONS OF SLEEP
A discussion of the function(s) of REM and NREM sleep is beyond the scope of this article. However, phylogenetic data and a critical consideration of physiologic data suggests that a universal function of sleep is to conserve energy and time behavior for conditions optimal for acquiring food and escaping predators.161–163 Other functions that can be fulfilled in some species in waking may have “migrated” into sleep in some species. Genetic success is best served by maximizing sleep time when vital needs have been met. This contrasts with the demands and attractions of human society, which values waking activity for entertainment and professional advancement. But, as in other animals, increased wake time in humans does not necessarily produce a genetic advantage in terms of maximizing offspring.164
Further relevant literature can be found at http://www.npi.ucla.edu/sleepresearch.
REFERENCES
- 1.von-Economo C Sleep as a problem of localization. J Nerv Ment Dis 1930;71:249–259 [Google Scholar]
- 2.von-Economo C Die encephalitis lethargica. Wien Deuticke; 1918 [Google Scholar]
- 3.Nauta WJH. Hypothalamic regulation of sleep in rats: an experimental study. J Neurophysiol 1946;9:285–316 [DOI] [PubMed] [Google Scholar]
- 4.Szymusiak R, McGinty D. Sleep suppression following kainic acid-induced lesions of the basal forebrain. Exp Neurol 1986;94(3):598–614 [DOI] [PubMed] [Google Scholar]
- 5.Sterman MB, Clemente CD. Forebrain inhibitory mechanisms: sleep patterns induced by basal forebrain stimulation in the behaving cat. Exp Neurol 1962;6:103–117 [DOI] [PubMed] [Google Scholar]
- 6.Sagar SM, Sharp FR, Curran T. Expression of c-fos protein in brain: metabolic mapping at the cellular level. Science 1988;240(4857):1328–1331 [DOI] [PubMed] [Google Scholar]
- 7.Basheer R, Sherin JE, Saper CB, et al. Effects of sleep on wake-induced c-fos expression. J Neurosci 1997;17:9746–9750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gvilia I, Xu F, McGinty D, Szymusiak R. Homeostatic regulation of sleep: a role for preoptic area neurons. J Neurosci 2006;26(37):9426–9433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sherin JE, Shiromani PJ, McCarley RW, Saper CB. Activation of ventrolateral preoptic neurons during sleep. Science 1996;271(5246):216–219 [DOI] [PubMed] [Google Scholar]
- 10.Suntsova N, Szymusiak R, Alam MN, Guzman-Marin R, McGinty D. Sleep-waking discharge patterns of median preoptic nucleus neurons in rats. J Physiol 2002;543(Pt 2): 665–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Findlay ALR, Hayward JN. Spontaneous activity of single neurones in the hypothalamus of rabbits during sleep and waking. J Physiol 1969;201(1):237–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaitin KI. Preoptic area unit activity during sleep and wakefulness in the cat. Exp Neurol 1984;83(2):347–357 [DOI] [PubMed] [Google Scholar]
- 13.Szymusiak R, McGinty DJ. Sleep-related neuronal discharge in the basal forebrain of cats. Brain Res 1986;370(1):82–92 [DOI] [PubMed] [Google Scholar]
- 14.Alam MN, McGinty D, Szymusiak R. Thermosensitive neurons of the diagonal band in rats: relation to wakefulness and non-rapid eye movement sleep. Brain Res 1997;752(1–2): 81–89 [DOI] [PubMed] [Google Scholar]
- 15.Szymusiak R, Gvilia I, McGinty D. Hypothalamic control of sleep. Sleep Med 2007;8(4):291–301 [DOI] [PubMed] [Google Scholar]
- 16.Szymusiak R, McGinty D. Hypothalamic regulation of sleep and arousal. Ann N Y Acad Sci 2008;1129(1):275–286 [DOI] [PubMed] [Google Scholar]
- 17.McCormick DA, Pape HC. Properties of a hyperpolarization-activated cation current and its role in rhythmic oscillation in thalamic relay neurones. J Physiol 1990;431: 291–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCormick DA. Cholinergic and noradrenergic modulation of thalamocortical processing. Trends Neurosci 1989; 12(6):215–221 [DOI] [PubMed] [Google Scholar]
- 19.Steriade M, Gloor P, Llinás RR, Lopes de Silva FH, Mesulam MM. Report of IFCN Committee on Basic Mechanisms. Basic mechanisms of cerebral rhythmic activities. Electroencephalogr Clin Neurophysiol 1990;76(6):481–508 [DOI] [PubMed] [Google Scholar]
- 20.Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science 1993;262(5134):679–685 [DOI] [PubMed] [Google Scholar]
- 21.Steriade M Brain electrical activity and sensory processing during waking and sleep. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier Saunders; 2005:101–119 [Google Scholar]
- 22.Steriade M, Sleep, epilepsy and thalamic reticular inhibitory neurons. Trends Neurosci 2005;28(6):317–324 [DOI] [PubMed] [Google Scholar]
- 23.Jacobs BL, Fornal CA. Activity of serotonergic neurons in behaving animals. Neuropsychopharmacology 1999;21 (2, Suppl):9S–15S [DOI] [PubMed] [Google Scholar]
- 24.John J, Wu MF, Boehmer LN, Siegel JM. Cataplexy-active neurons in the posterior hypothalamus: implications for the role of histamine in sleep and waking behavior. Neuron 2004;42(4): 619–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steininger TL, Alam MN, Gong H, Szymusiak R, McGinty D. Sleep-waking discharge of neurons in the posterior lateral hypothalamus of the albino rat. Brain Res 1999;840(1–2):138–147 [DOI] [PubMed] [Google Scholar]
- 26.Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron 2005;46(5):787–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nitz D, Siegel JM. GABA release in the posterior hypothalamus of the cat as a function of sleep/wake state. Am J Physiol 1996;271:R1707–R1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nitz D, Siegel JM. GABA release in the dorsal raphe nucleus: role in the control of REM sleep. Am J Physiol 1997;273(1 Pt 2):R451–R455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nitz D, Siegel JM. GABA release in the locus coeruleus as a function of sleep/wake state. Neuroscience 1997;78:795–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blanco-Centurion C, Gerashchenko D, Shiromani PJ. Effects of saporin-induced lesions of three arousal populations on daily levels of sleep and wake. J Neurosci 2007; 27(51):14041–14048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai YY, Siegel JM. Pontomedullary glutamate receptors mediating locomotion and muscle tone suppression. J Neurosci 1991;11(9):2931–2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siegel JM. The neurotransmitters of sleep. J Clin Psychiatry 2004;65(Suppl 16):4–7 [PMC free article] [PubMed] [Google Scholar]
- 33.Siegel JM. REM sleep. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia: Elsevier Saunders; 2005:120–135 [Google Scholar]
- 34.Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature 2006;441(7093): 589–594 [DOI] [PubMed] [Google Scholar]
- 35.Luppi PH, Gervasoni D, Verret L, et al. Paradoxical (REM) sleep genesis: the switch from an aminergic-cholinergic to a GABAergic-glutamatergic hypothesis. J Physiol (Paris) 2006;100(5–6):271–283 [DOI] [PubMed] [Google Scholar]
- 36.Hirshkowitz M, Schmidt MH. Sleep-related erections: clinical perspectives and neural mechanisms. Sleep Med Rev 2005;9(4):311–329 [DOI] [PubMed] [Google Scholar]
- 37.Parmeggiani PL. Thermoregulation and sleep. Front Biosci 2003;8:s557–s567 [DOI] [PubMed] [Google Scholar]
- 38.Villablanca J. Behavioral and polygraphic study of “sleep” and “wakefulness” in chronic decerebrate cats. Electroencephalogr Clin Neurophysiol 1966;21(6):562–577 [DOI] [PubMed] [Google Scholar]
- 39.Morrison AR, Bowker RM. The biological significance of PGO spikes in the sleeping cat. Acta Neurobiol Exp (Warsz) 1975;35(5–6):821–840 [PubMed] [Google Scholar]
- 40.De Gennaro L, Ferrara M. Sleep deprivation and phasic activity of REM sleep: independence of middle-ear muscle activity from rapid eye movements. Sleep 2000;23(1):81–85 [PubMed] [Google Scholar]
- 41.Siegel JM, Tomaszewski KS. Behavioral organization of reticular formation: studies in the unrestrained cat. I. Cells related to axial, limb, eye, and other movements. J Neurophysiol 1983;50(3):696–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siegel JM, Tomaszewski KS, Wheeler RL. Behavioral organization of reticular formation: studies in the unrestrained cat. II. Cells related to facial movements. J Neurophysiol 1983;50(3):717–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siegel JM, Nienhuis R, Fahringer HM, et al. Activity of medial mesopontine units during cataplexy and sleep-waking states in the narcoleptic dog. J Neurosci 1992; 12(5):1640–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bard P, Macht MB. The behavior of chronically decerebrate cats. In: Wolstenholme GEW, O’Conner CMO, eds. Neurological Basis of Behavior. London: Churchill; 1958: 55–75 [Google Scholar]
- 45.Dement WC. The occurrence of low voltage, fast, electroencephalogram patterns during behavioral sleep in the cat. Electroencephalogr Clin Neurophysiol 1958;10(2):291–296 [DOI] [PubMed] [Google Scholar]
- 46.Jouvet M Research on the neural structures and responsible mechanisms in different phases of physiological sleep. Arch Ital Biol 1962;100:125–206 [PubMed] [Google Scholar]
- 47.Adey WR, Bors E, Porter RW. EEG sleep patterns after high cervical lesions in man. Arch Neurol 1968;19(4):377–383 [DOI] [PubMed] [Google Scholar]
- 48.Lai YY, Siegel JM. Medullary regions mediating atonia. J Neurosci 1988;8(12):4790–4796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siegel JM, Tomaszewski KS, Nienhuis R. Behavioral states in the chronic medullary and midpontine cat. Electroencephalogr Clin Neurophysiol 1986;63(3):274–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Siegel JM, Nienhuis R, Tomaszewski KS. REM sleep signs rostral to chronic transections at the pontomedullary junction. Neurosci Lett 1984;45(3):241–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Siegel JM. Pontomedullary interactions in the generation of REM sleep. In: McGinty DJ, Drucker-Colin R, Morrison A, Parmeggiani PL, eds. Brain Mechanisms of Sleep. New York: Raven Press; 1985:157–174 [Google Scholar]
- 52.Matsuzaki M Differential effects of sodium butyrate and physostigmine upon the activities of para-sleep in acute brain stem preparations. Brain Res 1969;13(2):247–265 [DOI] [PubMed] [Google Scholar]
- 53.Schenkel E, Siegel JM. REM sleep without atonia after lesions of the medial medulla. Neurosci Lett 1989;98(2): 159–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mileykovskiy BY, Kiyashchenko LI, Kodama T, Lai YY, Siegel JM. Activation of pontine and medullary motor inhibitory regions reduces discharge in neurons located in the locus coeruleus and the anatomical equivalent of the midbrain locomotor region. J Neurosci 2000;20(22):8551–8558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gadea-Ciria M Tele-encephalic versus cerebellar control upon ponto-geniculo-occipital waves during paradoxical sleep in the cat. Experientia 1976;32(7):889–890 [DOI] [PubMed] [Google Scholar]
- 56.Mahowald MW, Schenck CH, Bornemann MA. Pathophysiologic mechanisms in REM sleep behavior disorder. Curr Neurol Neurosci Rep 2007;7(2):167–172 [DOI] [PubMed] [Google Scholar]
- 57.Siegel JM, McGinty DJ. Pontine reticular formation neurons: relationship of discharge to motor activity. Science 1977;196(4290):678–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Siegel JM, McGinty DJ, Breedlove SM. Sleep and waking activity of pontine gigantocellular field neurons. Exp Neurol 1977;56(3):553–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sastre JP, Sakai K, Jouvet M. Are the gigantocellular tegmental field neurons responsible for paradoxical sleep? Brain Res 1981;229(1):147–161 [DOI] [PubMed] [Google Scholar]
- 60.Drucker-Colín R, Pedraza JGB. Kainic acid lesions of gigantocellular tegmental field (FTG) neurons does not abolish REM sleep. Brain Res 1983;272(2):387–391 [DOI] [PubMed] [Google Scholar]
- 61.Webster HH, Jones BE. Neurotoxic lesions of the dorsolateral pontomesencephalic tegmentum-cholinergic cell area in the cat. II. Effects upon sleep-waking states. Brain Res 1988;458(2):285–302 [DOI] [PubMed] [Google Scholar]
- 62.Swanson LW. Brain Maps: Structure of the Rat Brain. Elsevier; 1992 [Google Scholar]
- 63.Shouse MN, Siegel JM. Pontine regulation of REM sleep components in cats: integrity of the pedunculopontine tegmentum (PPT) is important for phasic events but unnecessary for atonia during REM sleep. Brain Res 1992; 571(1):50–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hendricks JC, Morrison AR, Mann GL. Different behaviors during paradoxical sleep without atonia depend on pontine lesion site. Brain Res 1982;239(1):81–105 [DOI] [PubMed] [Google Scholar]
- 65.Jouvet M, Delorme F. Locus coeruleus et sommeil paradoxal. Comptes Rendus de l’ Academie des Sciences 1965; 159:895–899 [Google Scholar]
- 66.Lai YY, Shalita T, Hajnik T, et al. Neurotoxic N-methyl-D-aspartate lesion of the ventral midbrain and mesopontine junction alters sleep-wake organization. Neuroscience 1999; 90(2):469–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lai YY, Hsieh KC, Nguyen D, Shalita T, Peever J, Siegel JM. Neurotoxic lesions at the ventral mesopontine junction change in sleep time and muscle activity during sleep: an animal model of motor disorders in sleep. Neuroscience 2008;154(2):431–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Holmes CJ, Mainville LS, Jones BE. Distribution of cholinergic, GABAergic and serotonergic neurons in the medial medullary reticular formation and their projections studied by cytotoxic lesions in the cat. Neuroscience 1994; 62(4):1155–1178 [DOI] [PubMed] [Google Scholar]
- 69.Lai YY, Siegel JM. Cardiovascular and muscle tone changes produced by microinjection of cholinergic and glutamatergic agonists in dorsolateral pons and medial medulla. Brain Res 1990;514(1):27–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Crochet S, Onoe H, Sakai K. A potent non-monoaminergic paradoxical sleep inhibitory system: a reverse microdialysis and single-unit recording study. Eur J Neurosci 2006;24(5): 1404–1412 [DOI] [PubMed] [Google Scholar]
- 71.Vanini G, Torterolo P, McGregor R, Chase MH, Morales FR. GABAergic processes in the mesencephalic tegmentum modulate the occurrence of active (rapid eye movement) sleep in guinea pigs. Neuroscience 2007;145(3):1157–1167 [DOI] [PubMed] [Google Scholar]
- 72.Xi MC, Morales FR, Chase MH. Interactions between GABAergic and cholinergic processes in the nucleus pontis oralis: neuronal mechanisms controlling active (rapid eye movement) sleep and wakefulness. J Neurosci 2004;24(47): 10670–10678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Watson CJ, Soto-Calderon H, Lydic R, Baghdoyan HA. Pontine reticular formation (PnO) administration of hypocretin-1 increases PnO GABA levels and wakefulness. Sleep 2008;31(4):453–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xi MC, Morales FR, Chase MH. Interactions between GABAergic and cholinergic processes in the nucleus pontis oralis: neuronal mechanisms controlling active (rapid eye movement) sleep and wakefulness. J Neurosci 2004;24(47): 10670–10678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.George R, Haslett WL, Jenden DJ. A cholinergic mechanism in the brainstem reticular formation: induction of paradoxical sleep. Int J Neuropharmacol 1964;3:541–552 [DOI] [PubMed] [Google Scholar]
- 76.Vanni-Mercier G, Sakai K, Lin JS, Jouvet M. Mapping of cholinoceptive brainstem structures responsible for the generation of paradoxical sleep in the cat. Arch Ital Biol 1989;127(3):133–164 [PubMed] [Google Scholar]
- 77.Katayama Y, DeWitt DS, Becker DP, Hayes RL. Behavioral evidence for a cholinoceptive pontine inhibitory area: descending control of spinal motor output and sensory input. Brain Res 1984;296(2):241–262 [DOI] [PubMed] [Google Scholar]
- 78.Mitler MM, Dement WC. Cataplectic-like behavior in cats after micro-injections of carbachol in pontine reticular formation. Brain Res 1974;68(2):335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gnadt JW, Pegram GV. Cholinergic brainstem mechanisms of REM sleep in the rat. Brain Res 1986;384(1):29–41 [DOI] [PubMed] [Google Scholar]
- 80.Shiromani PJ, Fishbein W. Continuous pontine cholinergic microinfusion via mini-pump induces sustained alterations in rapid eye movement (REM) sleep. Pharmacol Biochem Behav 1986;25(6):1253–1261 [DOI] [PubMed] [Google Scholar]
- 81.Velazquez-Moctezuma J, Gillin JC, Shiromani PJ. Effect of specific M1, M2 muscarinic receptor agonists on REM sleep generation. Brain Res 1989;503(1):128–131 [DOI] [PubMed] [Google Scholar]
- 82.Deurveilher S, Hars B, Hennevin E. Pontine microinjection of carbachol does not reliably enhance paradoxical sleep in rats. Sleep 1997;20(8):593–607 [PubMed] [Google Scholar]
- 83.Verret L, Fort P, Gervasoni D, Léger L, Luppi PH. Localization of the neurons active during paradoxical (REM) sleep and projecting to the locus coeruleus noradrenergic neurons in the rat. J Comp Neurol 2006;495(5): 573–586 [DOI] [PubMed] [Google Scholar]
- 84.Lai YY, Siegel JM. Muscle tone suppression and stepping produced by stimulation of midbrain and rostral pontine reticular formation. J Neurosci 1990;10(8):2727–2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lai YY, Clements JR, Siegel JM. Glutamatergic and cholinergic projections to the pontine inhibitory area identified with horseradish peroxidase retrograde transport and immunohistochemistry. J Comp Neurol 1993;336(3):321–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lai YY, Clements JR, Wu XY, et al. Brainstem projections to the ventromedial medulla in cat: retrograde transport horseradish peroxidase and immunohistochemical studies. J Comp Neurol 1999;408(3):419–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Boissard R, Gervasoni D, Schmidt MH, Barbagli B, Fort P, Luppi PH. The rat ponto-medullary network responsible for paradoxical sleep onset and maintenance: a combined microinjection and functional neuroanatomical study. Eur J Neurosci 2002;16(10):1959–1973 [DOI] [PubMed] [Google Scholar]
- 88.Onoe H, Sakai K. Kainate receptors: a novel mechanism in paradoxical (REM) sleep generation. Neuroreport 1995; 6(2):353–356 [PubMed] [Google Scholar]
- 89.Siegel JM. Behavioral functions of the reticular formation. Brain Res 1979;180:69–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Siegel JM, Wheeler RL, McGinty DJ. Activity of medullary reticular formation neurons in the unrestrained cat during waking and sleep. Brain Res 1979;179(1):49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Drucker-Colin RR, Pedraza JGB. Kainic acid lesions of gigantocellular tegmental field (FTG) neurons does not abolish REM sleep. Brain Res 1983;272:387–391 [DOI] [PubMed] [Google Scholar]
- 92.Suzuki SS, Siegel JM, Wu MF. Role of pontomedullary reticular formation neurons in horizontal head movements: an ibotenic acid lesion study in the cat. Brain Res 1989; 484(1–2):78–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kodama T, Takahashi Y, Honda Y. Enhancement of acetylcholine release during paradoxical sleep in the dorsal tegmental field of the cat brain stem. Neurosci Lett 1990; 114(3):277–282 [DOI] [PubMed] [Google Scholar]
- 94.Steriade M, Datta S, Paré D, Oakson G, Curró Dossi RC. Neuronal activities in brain-stem cholinergic nuclei related to tonic activation processes in thalamocortical systems. J Neurosci 1990;10(8):2541–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Greene RW, Gerber U, McCarley RW. Cholinergic activation of medial pontine reticular formation neurons in vitro. Brain Res 1989;476(1):154–159 [DOI] [PubMed] [Google Scholar]
- 96.Sakai K, Crochet S, Onoe H. Pontine structures and mechanisms involved in the generation of paradoxical (REM) sleep. Arch Ital Biol 2001;139(1–2):93–107 [PubMed] [Google Scholar]
- 97.Shiromani PJ, Lai YY, Siegel JM. Descending projections from the dorsolateral pontine tegmentum to the paramedian reticular nucleus of the caudal medulla in the cat. Brain Res 1990;517(1–2):224–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hobson JA, McCarley RW, Wyzinski PW. Sleep cycle oscillation: reciprocal discharge by two brainstem neuronal groups. Science 1975;189(4196):55–58 [DOI] [PubMed] [Google Scholar]
- 99.Fenik V, Marchenko V, Janssen P, Davies RO, Kubin L. A5 cells are silenced when REM sleep-like signs are elicited by pontine carbachol. J Appl Physiol 2002;93(4):1448–1456 [DOI] [PubMed] [Google Scholar]
- 100.McGinty DJ, Harper RM. Dorsal raphe neurons: depression of firing during sleep in cats. Brain Res 1976;101(3):569–575 [DOI] [PubMed] [Google Scholar]
- 101.Gervasoni D, Darracq L, Fort P, Soulière F, Chouvet G, Luppi PH. Electrophysiological evidence that noradrenergic neurons of the rat locus coeruleus are tonically inhibited by GABA during sleep. Eur J Neurosci 1998;10(3):964–970 [DOI] [PubMed] [Google Scholar]
- 102.Maloney KJ, Mainville L, Jones BE. Differential c-Fos expression in cholinergic, monoaminergic, and GABAergic cell groups of the pontomesencephalic tegmentum after paradoxical sleep deprivation and recovery. J Neurosci 1999;19(8):3057–3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Torterolo P, Yamuy J, Sampogna S, Morales FR, Chase MH. GABAergic neurons of the cat dorsal raphe nucleus express c-fos during carbachol-induced active sleep. Brain Res 2000;884(1–2):68–76 [DOI] [PubMed] [Google Scholar]
- 104.Datta S, Siwek DF. Single cell activity patterns of pedunculopontine tegmentum neurons across the sleep-wake cycle in the freely moving rats. J Neurosci Res 2002; 70(4):611–621 [DOI] [PubMed] [Google Scholar]
- 105.Steriade M, Paré D, Datta S, Oakson G, Curró Dossi R. Different cellular types in mesopontine cholinergic nuclei related to ponto-geniculo-occipital waves. J Neurosci 1990; 10(8):2560–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ruch-Monachon MA, Jalfre M, Haefely W. Drugs and PGO waves in the lateral geniculate body of the curarized cat. IV. The effects of acetylcholine, GABA and benzodiazepines on PGO wave activity. Arch Int Pharmacodyn Ther 1976;219(2):308–325 [PubMed] [Google Scholar]
- 107.Wu MF, Siegel JM. Facilitation of the acoustic startle reflex by ponto-geniculo-occipital waves: effects of PCPA. Brain Res 1990;532(1–2):237–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brooks DC, Gershon MD. An analysis of the effect of reserpine upon ponto- geniculo-occipital wave activity in the cat. Neuropharmacology 1972;11:499–510 [DOI] [PubMed] [Google Scholar]
- 109.Quattrochi JJ, Bazalakova M, Hobson JA. From synapse to gene product: prolonged expression of c-fos induced by a single microinjection of carbachol in the pontomesencephalic tegmentum. Brain Res Mol Brain Res 2005;136(1–2): 164–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Datta S, Li G, Auerbach S. Activation of phasic pontine-wave generator in the rat: a mechanism for expression of plasticity-related genes and proteins in the dorsal hippocampus and amygdala. Eur J Neurosci 2008;27(7):1876–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Verret L, Léger L, Fort P, Luppi PH. Cholinergic and noncholinergic brainstem neurons expressing Fos after paradoxical (REM) sleep deprivation and recovery. Eur J Neurosci 2005;21(9):2488–2504 [DOI] [PubMed] [Google Scholar]
- 112.Goutagny R, Luppi PH, Salvert D, Lapray D, Gervasoni D, Fort P. Role of the dorsal paragigantocellular reticular nucleus in paradoxical (rapid eye movement) sleep generation: a combined electrophysiological and anatomical study in the rat. Neuroscience 2008;152(3):849–857 [DOI] [PubMed] [Google Scholar]
- 113.Pompeiano O, Hoshino K. Tonic inhibition of dorsal pontine neurons during the postural atonia produced by an anticholinesterase in the decerebrate cat. Arch Ital Biol 1976;114(3):310–340 [PubMed] [Google Scholar]
- 114.Hanriot L, Camargo N, Courau AC, Leger L, Luppi PH, Peyron C. Characterization of the melanin-concentrating hormone neurons activated during paradoxical sleep hypersomnia in rats. J Comp Neurol 2007;505(2):147–157 [DOI] [PubMed] [Google Scholar]
- 115.Verret L, Goutagny R, Fort P, et al. A role of melanin-concentrating hormone producing neurons in the central regulation of paradoxical sleep. BMC Neurosci 2003;4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chase MH, Morales FR. Control of motoneurons during sleep. In: Kryger MH, Roth T, Dement WC, eds. Principles of Sleep Medicine. 4th ed. Philadelphia: Elsevier Saunders; 2005:154–168 [Google Scholar]
- 117.Morales FR, Sampogna S, Rampon C, Luppi PH, Chase MH. Brainstem glycinergic neurons and their activation during active (rapid eye movement) sleep in the cat. Neuroscience 2006;142(1):37–47 [DOI] [PubMed] [Google Scholar]
- 118.Kodama T, Lai YY, Siegel JM. Changes in inhibitory amino acid release linked to pontine-induced atonia: an in vivo microdialysis study. J Neurosci 2003;23(4):1548–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lai YY, Kodama T, Siegel JM. Changes in monoamine release in the ventral horn and hypoglossal nucleus linked to pontine inhibition of muscle tone: an in vivo microdialysis study. J Neurosci 2001;21(18):7384–7391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Cessation of activity in red nucleus neurons during stimulation of the medial medulla in decerebrate rats. J Physiol 2002;545(Pt 3): 997–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kodama T, Lai YY, Siegel JM. Enhancement of acetylcholine release during REM sleep in the caudomedial medulla as measured by in vivo microdialysis. Brain Res 1992;580 (1–2):348–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kohyama J, Lai YY, Siegel JM. Inactivation of the pons blocks medullary-induced muscle tone suppression in the decerebrate cat. Sleep 1998;21(7):695–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Siegel JM, Nienhuis R, Tomaszewski KS. Rostral brainstem contributes to medullary inhibition of muscle tone. Brain Res 1983;268(2):344–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Peever JH, Lai YY, Siegel JM. Excitatory effects of hypocretin-1 (orexin-A) in the trigeminal motor nucleus are reversed by NMDA antagonism. J Neurophysiol 2003; 89(5):2591–2600 [DOI] [PubMed] [Google Scholar]
- 125.Brooks PL, Peever JH. Glycinergic and GABA(A)-mediated inhibition of somatic motoneurons does not mediate rapid eye movement sleep motor atonia. J Neurosci 2008;28(14):3535–3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Morrison JL, Sood S, Liu H, Park E, Nolan P, Horner RL. GABAA receptor antagonism at the hypoglossal motor nucleus increases genioglossus muscle activity in NREM but not REM sleep. J Physiol 2003;548(Pt 2):569–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Burgess C, Lai D, Siegel J, Peever J. An endogenous glutamatergic drive onto somatic motoneurons contributes to the stereotypical pattern of muscle tone across the sleep-wake cycle. J Neurosci 2008;28(18):4649–4660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chan E, Steenland HW, Liu H, Horner RL. Endogenous excitatory drive modulating respiratory muscle activity across sleep-wake states. Am J Respir Crit Care Med 2006;174(11): 1264–1273 [DOI] [PubMed] [Google Scholar]
- 129.Fenik VB, Davies RO, Kubin L. Noradrenergic, serotonergic and GABAergic antagonists injected together into the XII nucleus abolish the REM sleep-like depression of hypoglossal motoneuronal activity. J Sleep Res 2005;14(4):419–429 [DOI] [PubMed] [Google Scholar]
- 130.Jelev A, Sood S, Liu H, Nolan P, Horner RL. Microdialysis perfusion of 5-HT into hypoglossal motor nucleus differentially modulates genioglossus activity across natural sleep-wake states in rats. J Physiol 2001;532(Pt 2):467–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sood S, Raddatz E, Liu X, Liu H, Horner RL. Inhibition of serotonergic medullary raphe obscurus neurons suppresses genioglossus and diaphragm activities in anesthetized but not conscious rats. J Appl Physiol 2006;100(6):1807–1821 [DOI] [PubMed] [Google Scholar]
- 132.Taepavarapruk N, Taepavarapruk P, John J, et al. State-dependent release of glycine in Clarke’s column of the upper lumbar spinal cord. Sleep 2003;26:A11–A12 [Google Scholar]
- 133.Miller JD, Farber J, Gatz P, Roffwarg H, German DC. Activity of mesencephalic dopamine and non-dopamine neurons across stages of sleep and walking in the rat. Brain Res 1983;273(1):133–141 [DOI] [PubMed] [Google Scholar]
- 134.Shouse MN, Staba RJ, Saquib SF, Farber PR. Monoamines and sleep: microdialysis findings in pons and amygdala. Brain Res 2000;860(1–2):181–189 [DOI] [PubMed] [Google Scholar]
- 135.Shouse MN, Siegel JM, Wu MF, Szymusiak R, Morrison AR. Mechanisms of seizure suppression during rapid-eye-movement (REM) sleep in cats. Brain Res 1989;505(2): 271–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Maloney KJ, Mainville L, Jones BE. c-Fos expression in dopaminergic and GABAergic neurons of the ventral mesencephalic tegmentum after paradoxical sleep deprivation and recovery. Eur J Neurosci 2002;15(4):774–778 [DOI] [PubMed] [Google Scholar]
- 137.Dahan L, Astier B, Vautrelle N, Urbain N, Kocsis B, Chouvet G. Prominent burst firing of dopaminergic neurons in the ventral tegmental area during paradoxical sleep. Neuropsychopharmacology 2007;32(6):1232–1241 [DOI] [PubMed] [Google Scholar]
- 138.Lu J, Jhou TC, Saper CB. Identification of wake-active dopaminergic neurons in the ventral periaqueductal gray matter. J Neurosci 2006;26(1):193–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mignot E, Nishino S, Sharp LH, et al. Heterozygosity at the canarc-1 locus can confer susceptibility for narcolepsy: induction of cataplexy in heterozygous asymptomatic dogs after administration of a combination of drugs acting on monoaminergic and cholinergic systems. J Neurosci 1993; 13(3):1057–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Siegel JM, Nienhuis R, Fahringer HM, et al. Neuronal activity in narcolepsy: identification of cataplexy-related cells in the medial medulla. Science 1991;252(5010):1315–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wu MF, Gulyani SA, Yau E, Mignot E, Phan B, Siegel JM. Locus coeruleus neurons: cessation of activity during cataplexy. Neuroscience 1999;91(4):1389–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.John J, Wu MF, Boehmer LN, Siegel JM. Cataplexy-active neurons in the posterior hypothalamus: implications for the role of histamine in sleep and waking behavior. Neuron 2004;42:619–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Peyron C, Faraco J, Rogers W, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med 2000;6(9): 991–997 [DOI] [PubMed] [Google Scholar]
- 144.Thannickal TC, Moore RY, Nienhuis R, et al. Reduced number of hypocretin neurons in human narcolepsy. Neuron 2000;27(3):469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Thannickal TC, Siegel JM, Nienhuis R, Moore RY. Pattern of hypocretin (orexin) soma and axon loss, and gliosis, in human narcolepsy. Brain Pathol 2003;13(3):340–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fronczek R, Overeem S, Lee SY, et al. Hypocretin (orexin) loss in Parkinson’s disease. Brain 2007;130(Pt 6):1577–1585 [DOI] [PubMed] [Google Scholar]
- 147.Thannickal TC, Lai YY, Siegel JM. Hypocretin (orexin) cell loss in Parkinson’s disease. Brain 2007;130(Pt 6):1586–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.John J, Wu MF, Siegel JM. Systemic administration of hypocretin-1 reduces cataplexy and normalizes sleep and waking durations in narcoleptic dogs. Sleep Res Online 2000;3(1):23–28 [PMC free article] [PubMed] [Google Scholar]
- 149.Deadwyler SA, Porrino L, Siegel JM, Hampson RE. Systemic and nasal delivery of orexin-A (Hypocretin-1) reduces the effects of sleep deprivation on cognitive performance in nonhuman primates. J Neurosci 2007;27(52):14239–14247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gulyani S, Wu M-F, Nienhuis R, John J, Siegel JM. Cataplexy-related neurons in the amygdala of the narcoleptic dog. Neuroscience 2002;112(2):355–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Siegel JM. Hypocretin (orexin): role in normal behavior and neuropathology. Annu Rev Psychol 2004;55:125–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Siegel JM, Boehmer LN. Narcolepsy and the hypocretin system—where motion meets emotion. Nat Clin Pract Neurol 2006;2(10):548–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kiyashchenko LI, Mileykovskiy BY, Maidment N, et al. Release of hypocretin (orexin) during waking and sleep states. J Neurosci 2002;22(13):5282–5286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Blouin AM, Fried I, Staba RJ, et al. Hypocretin release during wake and sleep in the human brain. Abstract Viewer/Itinerary Planner. Washington, DC: Society for Neuroscience; 2007 [Google Scholar]
- 155.van den Pol AN, Gao XB, Obrietan K, Kilduff TS, Belousov AB. Presynaptic and postsynaptic actions and modulation of neuroendocrine neurons by a new hypothalamic peptide, hypocretin/orexin. J Neurosci 1998;18(19):7962–7971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.John J, Wu M-F, Kodama T, Siegel JM. Intravenously administered hypocretin-1 alters brain amino acid release: an in vivo microdialysis study in rats. J Physiol 2003;548 (Pt 2):557–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Liu RJ, van den Pol AN, Aghajanian GK. Hypocretins (orexins) regulate serotonin neurons in the dorsal raphe nucleus by excitatory direct and inhibitory indirect actions. J Neurosci 2002;22(21):9453–9464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Siegel JM. Narcolepsy: a key role for hypocretins (orexins). Cell 1999;98(4):409–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Guilleminault C, Anognos A. Narcolepsy. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practice of Sleep Medicine. 3rd ed. Philadelphia: WB Saunders; 2000:676–686 [Google Scholar]
- 160.de Waal FB, Aureli F, Judge PG. Coping with crowding. Sci Am 2000;282(5):76–81 [DOI] [PubMed] [Google Scholar]
- 161.Siegel JM. The REM sleep-memory consolidation hypothesis. Science 2001;294(5544):1058–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Siegel JM. Clues to the functions of mammalian sleep. Nature 2005;437(7063):1264–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Siegel JM. Do all animals sleep? Trends Neurosci 2008; 31(4):208–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kripke DF. Sleep and mortality. Psychosom Med 2003; 65(1):74. [DOI] [PubMed] [Google Scholar]