

Abstract
Background
Vancomycin-resistant Enterococcus faecium (VREfm) cause a wide range of hospital infections. Ireland has had one of the highest invasive VREfm infection rates in Europe over the last decade, yet little is known about Irish VREfm.
Objectives
To investigate the population structure of Irish VREfm, explore diversity by analysing the vanA transposon region and compare Irish, Danish and global isolates.
Methods
E. faecium (n = 648) from five Irish hospitals were investigated, including VREfm [547 rectal screening and 53 bloodstream infection (BSI)] isolates and 48 vancomycin-susceptible (VSEfm) BSI isolates recovered between June 2017 and December 2019. WGS and core-genome MLST (cgMLST) were used to assess population structure. Genetic environments surrounding vanA were resolved by hybrid assembly of short-read (Illumina) and long-read (Oxford Nanopore Technologies) sequences.
Results
All isolates belonged to hospital-adapted clade A1 and the majority (435/648) belonged to MLST ST80. The population structure was highly polyclonal; cgMLST segregated 603/648 isolates into 51 clusters containing mixtures of screening and BSI isolates, isolates from different hospitals, and VREfm and VSEfm. Isolates within clusters were closely related (mean average ≤16 allelic differences). The majority (96.5%) of VREfm harboured highly similar vanA regions located on circular or linear plasmids with multiple IS1216E insertions, variable organization of vanA operon genes and 78.6% harboured a truncated tnpA transposase. Comparison of 648 Irish isolates with 846 global E. faecium from 30 countries using cgMLST revealed little overlap.
Conclusions
Irish VREfm are polyclonal, yet harbour a characteristic plasmid-located vanA region with multiple IS1216E insertions that may facilitate spread.
Introduction
Enterococcus faecium, a resident of the normal human gastrointestinal (GI) flora has emerged as an important nosocomial pathogen responsible for bloodstream-, abdominal-, urinary tract- and IV catheter-related infections.1 Acquired resistance to ampicillin, gentamicin (high level), vancomycin and linezolid has increased worldwide among hospital-associated E. faecium, narrowing treatment options.1–3 Patients colonized in the GI tract with vancomycin-resistant E. faecium (VREfm) act as VREfm reservoirs and disseminators into the hospital environment.4 Previous studies identified 2–10 asymptomatic VREfm carriers for each VREfm-infected patient.4 For over a decade, the Republic of Ireland has reported one of the highest rates of invasive VREfm infections in Europe, ranging between 32.5% and 45.8% (2006–2019).2
Molecular typing of E. faecium requires specific approaches due to its highly recombinant genome. Typing methods that are effective for discriminating isolates of other nosocomial pathogens (e.g. MRSA) including MLST and PFGE are poorly discriminatory for E. faecium compared with WGS approaches, such as core-genome MLST (cgMLST).5–7 The dissemination of plasmids encoding vancomycin resistance genes (e.g. vanA) through diverse lineages presents an additional challenge in VREfm surveillance.6–8
Previous E. faecium WGS studies revealed a well-defined hospital population, termed clade A1,9,10 distinct from animal and community isolates.9 Clade A1 isolates tend to be enriched with mobile genetic elements (MGEs), ISs and pathogenicity islands.9,11 VREfm can arise de novo from vancomycin-susceptible E. faecium (VSEfm) following the acquisition of a vanA-encoding MGE.7,8 WGS-based VREfm studies revealed a diverse array of genetically distinct clones (polyclonal population) circulating in hospitals, with evidence of both intra- and inter-hospital spread of particular clones, and in some instances the circulation of predominant vanA plasmids.6,8 A previous study of VREfm from Irish hospitals provided evidence of a unique vanA transposon with a truncated transposase gene in 21 isolates investigated, distinct from UK isolates.7
The purpose of this study was to investigate the population structure of VREfm from Irish hospitals using WGS, to explore diversity by investigating the vanA region and to identify potential characteristic features associated with Irish VREfm. To place Irish isolates in a global context, the isolates were compared for relatedness with a large collection of international VREfm and VSEfm.
Materials and methods
Isolates
In total, 648 E. faecium from separate patients in five Irish hospitals were investigated. A large acute Dublin hospital (H1) was the main site for the study. H1 isolates were recovered between June 2017 and December 2019 including VREfm from rectal screening (n = 480) and all E. faecium bloodstream infection (BSI) isolates (n = 72). Isolates from four other hospitals included all VREfm screening and E. faecium BSI isolates recovered between October 2019 and December 2019; H2 isolates were from Dublin (n = 26), H3 from West (n = 26), H4 from South-East (n = 26) and H5 from South (n = 4) regions of Ireland. In total, 547 VREfm screening, 53 VREfm BSI and 48 VSEfm BSI isolates were investigated (Table S1, available as Supplementary data at JAC Online).
Species identification and susceptibility testing
Species identification and vanA/vanB vancomycin resistance gene detection was undertaken by PCR.12–14 Vancomycin and teicoplanin susceptibility were determined using the VITEK®2 system (bioMérieux, Marcy-l’Étoile, France) using the EUCAST (v11.0) interpretative criteria.15
Conjugation
Conjugative vanA-encoding plasmid transfer was undertaken by filter mating using the plasmid-free rifampicin- and fusidic acid-resistant recipient E. faecium strain 64/3 as described previously.16 Putative transconjugants were confirmed as E. faecium and tested for vanA/vanB by PCR.12–14
WGS
All 648 E. faecium, the E. faecium 64/3 recipient strain and several E. faecium 64/3 transconjugants underwent WGS using MiSeq (Illumina, The Netherlands) short-read sequencing as described previously.14 Briefly, libraries scaled to yield ≥50× coverage were prepared using the Nextera DNA Flex Library Preparation Kit (Illumina) and underwent paired-end sequencing with the 600 cycle MiSeq Reagent Kit v3 (Illumina).
For isolates/transconjugants selected for hybrid assembly, DNA was extracted using the MagAttract HMW kit (QIAGEN, UK) and long-read sequencing performed using the MinION platform (Oxford Nanopore Technologies, UK), using MinKNOW software v1.7.10 (Oxford Nanopore), as described previously.14,17
Analysis of WGS data
Quality trimming, de novo assembly using Velvet (v1.1.04) and neighbour-joining phylogenetic tree construction was undertaken in SeqSphere+ (v7.0.4, Ridom GmbH, Germany) using default settings except inclusion of the optimize coverage cut-off parameter with the E. faecium cgMLST scheme (1423 loci).5 Neighbour-joining trees were based on allelic differences using gene-by-gene allele calling against the defined core genes in the SeqSphere+ E. faecium cgMLST scheme.5 Using this approach, alleles are used instead of SNPs or concatenated sequences to mitigate the effects of recombination and to enable a global and public nomenclature (cgMLST.org Nomenclature Server; https://www.cgmlst.org/ncs). Visualization and annotation of trees was undertaken using Interactive Tree Of Life (iTOL) v5.18 Traditional MLST STs were extracted in silico. Clade A1 isolates were confirmed by comparison with reference clade A1 E. faecium genomes (Aus0004, DO and E8377; GenBank accession numbers: CP003351.1, CP003583.1 and LR135401.1, respectively) by k-mer distance estimation using MASH (v2.2; https://github.com/marbl/Mash).
To place Irish isolates in a global context, Irish isolates (n = 648) were compared for relatedness with 846 global clade A1 isolates [547 VREfm (304 vanA, 243 vanB) and 299 VSEfm] originating from 30 countries, spanning a 30 year period (1986–2016) from the study of van Hal et al.19 (Table S2) using cgMLST in SeqSphere+. Irish isolates (n = 640) were also compared with a secondary comparator group consisting of 989 VREfm [862/989 (87%) rectal screening and 27/989 (13%) infection isolates] from patients in four hospitals in Copenhagen, Denmark between 2017 and 2018. The majority (983/989; 99%) were vanA VREfm, while the remaining 6 (1%) were vanB VREfm. Danish VREfm were sequenced as previously described.20 Read files from both Irish and Danish isolates were trimmed with BBDuk (https://sourceforge.net/projects/bbmap/) with the parameters ktrim = r, k = 23, mink = 11, hdist = 1, tbo, qtrim = r and minlength = 30. De novo assembly was performed using SKESA21 v2.3.1 with default settings except inclusion of the parameter –allow_snps.
Assembly and analysis of vanA-encoding regions and plasmids
Long-read MinION and paired-end short-read MiSeq FASTQ files were used for hybrid assemblies using Unicycler (v0.4.8).22 The genetic organization of vanA-encoding plasmids from Irish isolates was determined following hybrid assembly and these were used as reference sequences for further analysis. MiSeq reads were mapped against reference plasmid sequences and percentage depth and breadth of coverage calculated using Burrows–Wheeler Aligner, SAMtools and BedTools.23–25 Alignment quality was assessed using Tablet.26 vanA-encoding genomes resolved by hybrid assembly were annotated using RAST v2.0 (http://rast.nmpdr.org/)27 and visualized using SnapGene (GSL Biotech; https://snapgene.com). Irish VREfm and VSEfm sequence reads and sequences resolved by hybrid assembly have been deposited in GenBank under BioProject PRJNA734127. Danish VREfm sequence reads have been deposited in GenBank under BioProjects PRJNA573568, PRJNA686881, PRJNA691722, PRJNA702038 and PRJNA740173.
Ethics
Ethical approval for this project was provided by the School of Dental Science & Dublin Dental University Hospital Research Ethics Committee (Reference DSREC2020-01-02).
Results
Genomic epidemiology of Irish hospital E. faecium
In total, 648 E. faecium isolates (547 screening and 101 BSI) from patients in five Irish hospitals recovered between June 2017 and December 2019 were investigated. The majority (600/648; 92.5%) were vancomycin resistant, harbouring vanA (vancomycin MIC > 4 mg/L; 547 screening and 53 BSI), while the remaining 48 (7.4%) (all BSI) were vancomycin susceptible (vancomycin MIC ≤ 4 mg/L). The majority of isolates were from H1 (n = 552), a large Dublin hospital, whereas the remainder were from four other hospitals (H2, H3, H4 and H5; n = 96).
All 648 isolates belonged to clade A1; the majority were identified as ST80 (435/648; 67.1%) by conventional MLST, with the remainder belonging to ST117 (n = 23), ST203 (n = 61), ST612 (n = 34) and ST787 (n = 28). Four isolates were non-typeable due to a previously described pstS housekeeping gene deletion.28 Nine ‘novel’ isolates were submitted to pubMLST.org for ST assignment. Using cgMLST, the 648 isolates segregated into 51 clusters (≤24 allelic differences) and 43 singletons, with an inter-cluster allelic difference range of 22–1201. Clusters contained mixtures of screening and BSI isolates, isolates from different hospitals, and VREfm and VSEfm (Figure 1, Table S1).
Figure 1.

Neighbour-joining tree (NJT) based on cgMLST of 648 clade A1 E. faecium isolates, recovered between June 2017 and December 2019 from five Irish hospitals. The 648 isolates divided into 51 clusters and 43 singletons, with an inter-cluster allelic difference range of 22–1201. Clusters are shaded in grey and predominant CTs labelled accordingly. Isolates within clusters were closely related; the allelic differences between isolates in the three largest clusters were as follows: CT1916, range 0–24, mean 8.8 (71 isolates); CT1598, range 0–16, mean 5.2 (55 isolates); CT2933, range 0–22, mean 5.5 (64 isolates clustering, two not assigned a CT). Scale bar represents the phylogenetic distance between isolates based on cgMLST. Metadata are represented encircling the NJT as denoted by the colour legends, data represented are (a) STs, (b) vancomycin phenotype, (c) source of isolate, and (d) origin hospital location of isolates.*Indicates isolates SJ47 (CT2023) and SJ266 (CT3175) within ST80 CT1916 cluster.
The two largest clusters consisted of ST80 isolates and were defined by their predominant cgMLST complex types (CTs). Cluster CT1916 [70 VREfm, 1 VSEfm (BSI)] had an allelic difference range of 0–24 (mean 8.8) and contained screening and BSI isolates from four hospitals recovered between December 2017 and November 2019, of which 65/71 were from H1 (Table S1). Two isolates within CT1916 were identified as CT2023 (SJ47) and CT3175 (SJ266). Cluster CT2933 (59 VREfm and 5 BSI VSEfm) had an average allelic difference range of 0–22 (mean 5.5) and contained screening and BSI isolates recovered from four hospitals between May 2019 and November 2019. Two isolates (SJ55, SJ64) within CT2933 were not assigned a CT.
The majority of ST80 isolates (412/435; 94.7%) divided into 27 clusters of ≥2 isolates by cgMLST (Figure S1), but clusters differed widely, with an inter-cluster allelic difference range of 26–268. Within clusters, close relatedness between screening and BSI isolates, isolates from different hospitals and between VREfm and VSEfm was evident; for example, ST80 CT1598 [intra-cluster allelic difference range 0–16 (mean 5.2)] contained 54 isolates from 14 H1 wards and three H3 isolates. CT1598 also contained one VSEfm (BSI_SJ72) with zero allelic differences to VREfm within the cluster (Figure S1).
Irish, global and Danish E. faecium comparison
In order to place Irish isolates in a global context, 648 Irish isolates were compared with 846 global E. faecium from 30 countries, which revealed very little overlap between the two groups of isolates (Figure 2). None of the Irish isolates clustered with any of the 101 isolates investigated from the UK, the closest geographic neighbour of Ireland. A small number of Irish isolates (10 in total) were identified in four clusters of related isolates consisting of both Irish and global isolates (Figure 2). Cluster 1 consisted of two isolates separated by 20 allelic differences including one Irish ST18 CT3387 VREfm and one ST18 CT173 VREfm (vanA) from Slovenia. Cluster 2 consisted of three isolates including two Irish ST262 CT3166 VREfm and one German ST262 CT1016 VREfm (vanA), which was separated by 20 allelic differences from the nearest Irish isolate. Cluster 3 consisted of 19 isolates including one Irish ST1478 CT24 VREfm and 18 global isolates from five countries with a maximum allelic difference between the isolates of 30. The global isolates in cluster 3 included four Norwegian ST117 CT24 VSEfm, one German ST117 CT24 VREfm (vanA), one German ST117 CT1020 VREfm (vanA), one German ST1486 CT178 VREfm (vanB), five Spanish ST117 CT24 isolates (two VREfm and three VSEfm), two Danish ST117 CT24 VREfm (both vanA), one ST1201 CT24 Danish VREfm (vanA), one Belgian ST117 CT24 VREfm (vanB) and two Dutch ST117 CT24 VREfm (both vanB). The closest global isolate to the Irish isolate in cluster 3 was a Norwegian isolate, which differed by seven allelic differences. Cluster 4 consisted of 22 ST203 CT20 isolates including 6 Irish isolates (5 VREfm and 1 VSEfm) and 16 global isolates from six countries. The global isolates in cluster 4 included one Norwegian VSEfm, two German VREfm (both vanA), one Danish VREfm (vanA), one Belgian VREfm (vanA), three Dutch VREfm (all vanA) and eight Australian VREfm (all vanA). The closest global isolate to an Irish isolate in cluster 4 was the Belgian isolate, which differed by four allelic differences.
Figure 2.

Minimum spanning tree of Irish Clade A1 E. faecium (n = 648) (600 VREfm and 48 VSEfm) isolated between June 2017 and December 2019 and global Clade A1 E. faecium (n = 846) [547 VREfm (304 vanA, 243 vanB) and 299 VSEfm] originating from 30 countries, spanning a 30 year period (1986–2016) from the study of van Hal et al.19 using cgMLST in SeqSphere+. Isolates are coloured by country of origin as indicated in the legend. A small number of Irish isolates (10 in total) were identified in four clusters of related isolates consisting of both Irish and global isolates. The four clusters of related isolates are encircled and numbered 1–4. The ST and CT of the isolates within the clusters are indicated.
Irish E. faecium (n = 640) were also compared with 989 Danish VREfm recovered from four hospitals during 2017–18. Denmark was chosen as a direct comparator due to its increase in VREfm prevalence, caused by the introduction of an ST80 clone.8 The minimum spanning tree of Danish VREfm revealed 30 clusters and 37 singletons. The largest clusters were ST1421, ST203, ST80 and ST117 containing 420, 294, 107 and 56 isolates, respectively. The comparison of Irish and Danish isolates revealed 64 clusters and 69 singletons. Clusters were predominantly country-specific, with only three containing isolates from both countries (Figure S2).
Structural organization of the vanA region in Irish VREfm
The extrachromosomal vanA-harbouring regions of four VREfm, SJ10 (ST789, CT1601), SJ11 (non-typeable by conventional MLST, CT24), BSI_SJ40 (ST80, CT1598) and RC_19_023 (ST80, CT1916) were resolved using hybrid assembly. The vanA regions varied in size (SJ10vanA, 11 022 bp; SJ11vanA, 13 252 bp; BSI_SJ40vanA, 13 269 bp; RC_19_023vanA, 12 883 bp) and differed from the Tn1546 vanA operon prototype from VREfm BM4147 (GenBank accession number: M97297). All four vanA regions harboured large deletions in the tnpA transposase gene, mainly in the 3′ end, the extent of which varied (Figures 3 and 4). Other differences included multiple IS1216E insertions, different orientations of the vanA operon genes and in the case of SJ11vanA, BSI_SJ40vanA and RC-19_023vanA, a cadmium efflux accessory protein gene insertion (Figure 3). RC_19_23vanA lacked vanY but remained phenotypically resistant to vancomycin and teicoplanin. Both SJ11 and BSI_SJ40 exhibited 100% sequence identity to SJ10vanA. The vanA region was compared across all VREfm investigated and 96.6% (580/600) of isolates harboured a vanA region with >90% sequence identity to the comparative SJ10vanA reference used.
Figure 3.

Schematic diagram showing the structural organization of vanA transposon regions closed by hybrid assembly from VREfm isolates from Irish hospitals. (a) SJ10 (SJ10vanA, 11 022 bp); (b) SJ11 (SJ11vanA, 13 252 bp); (c) BSI_SJ40 (BSI_SJ40vanA, 13 269 bp); (d) SJ82 (10 823 bp vanA region from plasmid pSJ82vanA); (e) SJ245 (14 168 bp vanA region from plasmid pSJ245vanA); (f) RC_19_039 (13 601 bp vanA region from plasmid pRC_19_039); (g) RC_19_023 (RC_19_023vanA, 12 883 bp); and (h) the structural organization of the prototype vanA transposon Tn1546 from VREfm BM4147 (GenBank accession number: M97297). VREfm isolates were recovered from H1 (a–e), H3 (f) and H4 (g). Genes and their orientation are denoted by directional arrows and labelled with corresponding gene names. A reference size scale bar is shown at the bottom of the diagram. The extent of the transposase tnpA gene sequences deleted (Δ) in the various assemblies is shown in Figure 4.
Figure 4.
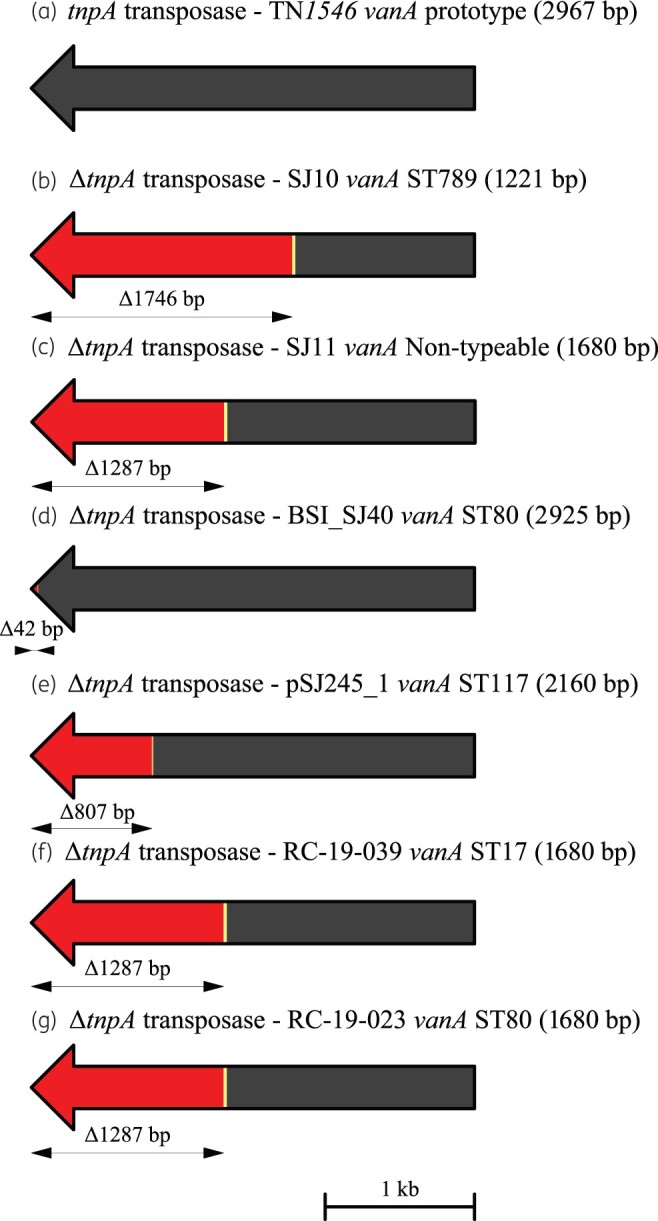
Schematic diagram showing the extent of deletions and other mutations in the tnpA transposase gene within the vanA region closed by hybrid assembly from VREfm isolates from Irish hospitals. The arrows indicate the direction of transcription. Deletions are shown in red, bp substitutions are indicated in yellow and bp insertions are indicated in green. The extent of sequences deleted (Δ) is shown beneath each tnpA gene in bp. (a) WT tnpA (transposase) gene (2967 bp) from the prototype vanA transposon Tn1546;29 (b) ΔtnpA gene from isolate SJ10 (SJ10vanA, 1221 bp); (c) ΔtnpA gene from isolate SJ11 (SJ11vanA, 1680 bp); (d) ΔtnpA gene from isolate BSI_SJ40 (BSI_SJ40vanA, 2925 bp); (e) ΔtnpA gene from plasmid pSJ245vanA in isolate SJ245 (pSJ245vanA, 2160 bp); (f) ΔtnpA gene from isolate RC_19_039 (RC_19_039vanA, 1680 bp); and (g) ΔtnpA gene from isolate RC_19_023 (RC_19_023vanA, 1680 bp). VREfm isolates were recovered from H1 (b–e), H3 (f) and H4 (g). A reference size scale bar is shown at the bottom of the diagram.
SJ10vanA harboured multiple insertions of IS1216E within and around the vanA region, with 96.6% (580/600) of VREfm harbouring a highly similar vanA region, as demonstrated by >90% sequence identity, indicating enrichment of IS1216E around vanA in Irish VREfm (Figure 3).
Terminal 5′ and 3′ 1000 bp sequences from the TN1546 prototype tnpA transposase gene (2967 bp)29 were used as references to compare the tnpA region across all VREfm. The majority (578/600; 96.3%) harboured a tnpA region with >99% sequence identity to the 5′ tnpA reference. In contrast, the majority (472/600; 78.6%) lacked the 3′ tnpA region with <40% sequence identity to the 3′ tnpA reference. These findings demonstrated the majority of VREfm harboured a tnpA gene with large 3′-end deletions.
Plasmids encoding vanA
Attempts to close plasmids harbouring SJ10vanA, SJ11vanA, BSI_SJ40vanA and RC_19_023vanA by hybrid assembly were unsuccessful but in each case yielded vanA on a single large contig that failed to circularize [89 310 bp (SJ10), 116 749 bp (SJ11), 131 341 bp (BSI_SJ40) and 109 124 bp (RC_19_023)]. These contigs are likely to be plasmids that exhibit linear topology as reported recently.30–32 Using the SJ10vanA contig as a reference plasmid-like sequence, it was clear this element was highly similar throughout all VREfm, with 95.8% (575/600) exhibiting >85% sequence identity. The SJ10vanA plasmid-like contig exhibited 99.9% and 99.8% sequence similarity to the vanA contigs from SJ11 and BSI_SJ40, respectively. Plasmids encoding vanA were successfully transferred from SJ11 (116 749 bp plasmid) and BSI_SJ40 (131 341 bp plasmid) to recipient strain E. faecium 63/4 by conjugation, but not from SJ10 or RC_19_023vanA (see below).
Three vanA-encoding plasmids were successfully resolved by hybrid assembly from VREfm SJ82 (ST203, CT20) and SJ245 (ST117, CT2929) from H1, and RC_19_039 (ST17, CT294) from H3. Plasmids were named pSJ82vanA (48 934 bp), pSJ245vanA (40 559 bp) and pRC_19_039vanA (66 751 bp), respectively (Figure 3). All plasmids encoded additional resistance genes (Figures S3–S5). Plasmid pSJ82vanA lacked a tnpA transposase gene, whereas plasmids pSJ245vanA and pRC_19_039vanA each harboured a truncated tnpA (Figures 3 and 4).
Plasmid pSJ82vanA was identified in five screening VREfm (SJ3, SJ12, SJ71, SJ77 and SJ82), all with 100% sequence coverage identity to pSJ82vanA. All five were indistinguishable by cgMLST (ST203, CT20) and were recovered from five H1 wards over 14 months (Table S1), indicating inter-ward spread and persistence over time. The vanA region of pSJ245vanA exhibited 95.5% similarity to the SJ10vanA reference, but there was much rearrangement in the vanA region in SJ245, including the insertion of ISEfa5 (Figure 3). Plasmid pSJ245vanA was identified in five ST117 isolates (SJ220, SJ273, SJ274, JH085 and SJ24) with >99% sequence identity, four of which were recovered within 7 days in two H1 wards and one 4 months later on another ward (Table S1). All five isolates were indistinguishable by cgMLST and formed a phylogenetic cluster (ST117, CT2929; Figure 1). These findings indicate the spread of a single clone in H1 and highlight the usefulness of pairing cgMLST and plasmid analysis. Plasmid pRC_19_039vanA was identified in five ST17 CT2934 isolates (JH010, JH022, BSI_SJ49, RC_19_033 and RC_19_039) by >99% sequence identity. Three originated from H1 with evidence of persistence on Ward 6 from September 2018 to October 2019; the remaining two isolates were from H3 (Table S1).
To investigate whether the large transferable vanA-encoding contigs identified in isolates SJ11 and BSI_SJ40 by hybrid assembly were linear plasmids, the sequences of the contigs were compared with the corresponding sequences of linear plasmids pELF1 (143 316 bp), pELF2 (108 102 bp) and pELF_USZ (101 837 bp) recently described in VREfm isolates from Japan30,31 and Switzerland, respectively.32 Comparing the vanA contigs from SJ11 (116 749 bp) and BSI_SJ40 (131 341 bp) revealed a clear homology (Figure S6). Each plasmid and contig harboured genes encoding putative replication initiation proteins and proteins with a putative role in DNA partitioning and transfer. These findings strongly indicated that the vanA-encoding contigs identified in isolates SJ11 and BSI_SJ40 were linear plasmids, which were named pELF_SJ11 and pELF_BSI_SJ40, respectively (Figure S6). Details of pELF_SJ11 and pELF_BSI_JS40 genes and putative proteins encoded are provided in Tables S3 and S4. An alignment of pELF_SJ11 and pELF_BSI_SJ40 with the vanA-encoding contig from SJ10 is shown in Figure S7.
Transmission of vanA via IS1216E translocation and plasmid conjugation
Conjugation of vanA to the E. faecium 64/3 recipient was successful for four VREfm screening isolates investigated [SJ11 (non-typeable, CT2), SJ82 (ST203, CT20), SJ245 (ST117, CT2929) and SJ267 (ST18, CT1898)] and the BSI isolate BSI_SJ40 (ST80, CT1598). Transconjugants were phenotypically resistant to vancomycin and teicoplanin and showed the identical sequence coverage to the SJ10vanA reference as their corresponding donor isolate (81.2%–100%) (Table S5). The donor isolates SJ11, SJ267 and BSI_SJ40 and their corresponding E. faecium 64/3 transconjugant derivatives showed near identical coverage to the SJ10vanA plasmid-like contig (Table S5). Linear plasmids pELF_SJ11 and pELF_BSI_SJ40, identified in donor isolates SJ11 and BSI_SJ40, respectively, were identified in their corresponding E. faecium 64/3 transconjugant derivatives and exhibited 99.9% and 100% coverage to the respective parental linear plasmids. Plasmid pSJ245 in isolate SJ245 was also shown to be conjugative, with the corresponding E. faecium 64/3 transconjugant derivatives SJ245:Efm 64/3 TC1 and SJ245:Efm 64/3 TC2 exhibiting 100% sequence coverage to pSJ245. Both transconjugants showed 95.5% sequence coverage to the reference SJ10vanA, identical to the coverage in the donor isolate SJ245 (Table S5).
The transconjugant SJ82:Efm 64/3 TC1 underwent hybrid assembly and the vanA plasmid identified (pSJ82vanA_TC) was much larger (175 921 bp) than plasmid pSJ82vanA (48 934 bp) in donor isolate SJ82 (Figure 5). Examination of the hybrid assembly of SJ82 revealed another large plasmid, termed pSJ82_B (232 026 bp), in addition to pSJ82vanA. Analysis of pSJ82_TC revealed that it was composed of the vanA operon from pSJ82vanA flanked on either side by IS1216E (9826 bp) inserted within a section of pSJ82_B (166 095 bp) (Figures 3d and 5).
Figure 5.

Schematic diagrams of the structural organization of (a) plasmids pSJ82_B and pSJ82vanA from VREfm donor strain SJ82, and (b) the vancomycin resistance plasmid pSJ82_TC from the transconjugant SJ82:Efm 64/3 TC1. Diagrams are partially annotated for clarity. Genes of interest and their orientation are represented by arrows: red indicates antibiotic resistance genes, orange indicates ISs/transposases and blue indicates genes encoding known proteins. The regions from plasmids resolved by hybrid assembly of paired-end Illumina MiSeq short reads with Oxford Nanopore Technologies long reads present in the donor isolate SJ82 (i.e. pSJ82_B and pSJ82vanA) that are present in the hybrid transconjugant plasmid pSJ82_TC are marked as follows: pSJ82_B region highlighted in blue and pSJ82vanA region highlighted in red. Plasmid regions in pSJ82vanA and pSJ82_B shaded in black were not present in pSJ82_TC.
Mobilization of the vanA region from pSJ82vanA into pSJ82_B was evident by the identical sequence coverage of the parent and the transconjugant to the SJ10vanA reference, with both having 81.2% sequence coverage (Table S4). The pSJ82vanA vanA region likely moved via IS1216E-mediated translocation to pSJ82_B and there was loss of 65 931 bp of pSJ82_B, giving rise to the hybrid plasmid pSJ82_TC (Figure 5).
Discussion
Ireland has consistently reported one of the highest invasive VREfm infection rates in Europe over the last decade.2 This study used WGS analysis to determine that the population structure of E. faecium from Irish hospitals consisted of diverse clonal lineages, correlating with previous WGS-based European studies of clade A1 E. faecium,6,7,9,33 with ST80 accounting for the majority of isolates (61.7%). The predominance of ST80 in hospital H1 (66.5%) was reflected in the four other hospitals (H2, H3, H4 and H5) (75%). Isolates belonging to the same ST determined by conventional MLST were distantly related by cgMLST (Figure 1).
A threshold of ≤20 cgMLST allelic differences has been previously proposed to describe E. faecium isolates as indistinguishable or highly related.5,6 Clusters of related isolates could be defined by the CT to which the majority of isolates in that cluster belonged. The majority of VREfm harboured identical or highly related vanA regions, with 96.6% exhibiting >90% sequence identity to the SJ10vanA reference (Table S1, Figure 3). This finding of identical/closely related vanA regions in genetically highly related and unrelated isolates provides a challenge for surveillance. Along with expansion of existing VREfm clones in the hospital environment, the transfer of a vanA MGE could give rise to new VREfm clones from VSEfm. The circulation of a common vanA element among diverse VREfm lineages was also observed previously in Denmark, where 81% of isolates investigated harboured an identical vanA plasmid.8 In comparison with this Danish vanA plasmid (pV24-5), the Irish isolates had a median sequence coverage of 69.3% (range 10.8%–91.8%), indicating this plasmid has not spread widely among Irish VREfm.
Another notable feature of Irish VREfm was the persistence of clones in hospitals over extended periods, e.g. the ST80 CT1598 cluster contained highly related isolates recovered from 14 wards in H1 and three isolates from H3, recovered over 25 months (Table S1). In H1 there were only three allelic differences between the earliest isolate SJ6 (24 July 2017) from Ward 2 and the latest isolate SJ291 (28 June 2019) from Ward 12. Isolates from H3 (RC_19_30, RC_19_052, RC_19_053) exhibited ≤3 allelic differences from isolates from H1 (SJ1, SJ6, SJ17, BSI_SJ37, BSI_SJ72) (Table S1). These findings indicate widespread transmission throughout H1 and inter-hospital spread.
Conjugation experiments with selected isolates demonstrated the transmissibility of vanA via conjugative plasmids, such as the circular plasmid pSJ245 and the linear plasmids pELF_SJ11 and pELF_BSI_SJ40 (Table S5, Figures S5 and S7). The ability of vanA to translocate from one plasmid to another in a clinical isolate and then to transfer to a new recipient by conjugative plasmid transfer was also demonstrated (Figure 5).
ST80 predominated among VREfm in Irish hospitals, accounting for 67.1% (435/648) of isolates. Studies from Copenhagen (11 hospitals) revealed a substantial increase in VREfm during 2011–15 resulting from the introduction, expansion and spread of a single vanA-encoding ST80 clone.6,8,34 The significant variation within the ST80 population correlates with the diversity of clade A1 E. faecium isolates in Ireland. A comparison of Irish, global and Danish isolates showed little overlap between isolates from different countries (Figure 2 and Figure S2). This finding suggests that local evolution plays a significant role in the epidemiology of VREfm, with no evidence for widespread inter-country spread of strains, which can occur with more clonal organisms such as MRSA.2 A recent study of VREfm from 31 countries, which showed localized VREfm evolution along with evidence of localized acquisition of van genes,19 supports this suggestion.
This study reports the first vanA-harbouring VREfm with multiple insertions of an identical IS element, IS1216E (Figure 3). Previous reports of different vanA iterations have shown a single IS element insertion into the middle of the vanA operon.6 To date, no previous studies have described this multiple division of the vanA operon by IS1216E. The vanA operon organization determined for VREfm isolate SJ10 demonstrates van operon rearrangement, with the transposase, resolvase, VanR and VanS being encoded following an insertion of IS1216E downstream of vanY and vanZ at the end of the operon (Figure 3). Previous research indicates that clade A1 E. faecium harbouring the linezolid resistance gene poxtA flanked by IS1216E can loop out and form small transposable elements (TEs).35 Multiple IS1216E insertions throughout the vanA operon could lead to instability and/or aid its dissemination. A recent study reported that IS1216 can form cointegrates by both a copy-in mechanism, involving one copy of IS1216 located on a plasmid and a target sequence, and by a targeted conservative mechanism involving two copies of IS1216, one located on a plasmid and the second in the target sequence.36 The latter mechanism occurred at a significantly higher frequency. It is likely that a targeted conservative mechanism involving IS1216 contributed to the rearrangements of the vanA region of VRE containing several copies of IS1216. The high prevalence of IS1216E in Irish VREfm may facilitate mobility of the vanA operon in the absence of a functional tnpA transposase gene.
Conclusions
Irish VREfm are diverse yet harbour a characteristic vanA region with a truncated transposase and multiple insertions of IS1216E that may facilitate spread. The vanA region was identified on several closed conjugative plasmids and on conjugative plasmids with linear topology. A comparison of Irish, global and Danish VREfm/VSEfm showed little overlap indicating that local evolution is important in the epidemiology of VREfm. Current Irish guidelines only recommend VREfm screening of patients upon hospital admission to high-risk areas.37 Implementing universal VRE admission screening would very likely help to mitigate the spread.
Supplementary Material
Acknowledgements
D.C.C., S.A.E., N.L.K., B.A.McM. and A.C.S. wish to acknowledge the support of the staff of the Irish National MRSA Reference Laboratory at St. James’s Hospital, Dublin, Ireland and the staff of the clinical microbiology laboratories of the five Irish hospitals that contributed isolates for the study. We thank Peter Kinnevey for technical support.
Funding
This work was supported by Irish Health Research Board grant number ILP-POR-2019-010.
Transparency declarations
None of the authors have any conflicts of interest to declare.
Supplementary data
Tables S1 to S5 and Figures S1 to S7 are available as Supplementary data at JAC Online.
Contributor Information
Sarah A. Egan, Microbiology Research Unit, Division of Oral Biosciences, Dublin Dental University Hospital, University of Dublin, Trinity College, Lincoln Place, Dublin 2, Ireland
Nicole L. Kavanagh, Microbiology Research Unit, Division of Oral Biosciences, Dublin Dental University Hospital, University of Dublin, Trinity College, Lincoln Place, Dublin 2, Ireland
Anna C. Shore, Microbiology Research Unit, Division of Oral Biosciences, Dublin Dental University Hospital, University of Dublin, Trinity College, Lincoln Place, Dublin 2, Ireland
Sarah Mollerup, Department of Clinical Microbiology, Hvidovre University Hospital, Hvidovre, Denmark.
José A. Samaniego Castruita, Department of Clinical Microbiology, Hvidovre University Hospital, Hvidovre, Denmark
Brian O’Connell, Department of Clinical Microbiology, School of Medicine, University of Dublin, Trinity College Dublin, St. James’s Hospital, Dublin 8, Ireland; National MRSA Reference Laboratory, St. James’s Hospital, James’s Street, Dublin 8, Ireland.
Brenda A. McManus, Microbiology Research Unit, Division of Oral Biosciences, Dublin Dental University Hospital, University of Dublin, Trinity College, Lincoln Place, Dublin 2, Ireland
Grainne I. Brennan, National MRSA Reference Laboratory, St. James’s Hospital, James’s Street, Dublin 8, Ireland
Mette Pinholt, Department of Clinical Microbiology, Hvidovre University Hospital, Hvidovre, Denmark.
Henrik Westh, Department of Clinical Microbiology, Hvidovre University Hospital, Hvidovre, Denmark.
David C. Coleman, Microbiology Research Unit, Division of Oral Biosciences, Dublin Dental University Hospital, University of Dublin, Trinity College, Lincoln Place, Dublin 2, Ireland
References
- 1. Arias CA, Murray BE. The rise of the Enterococcus: beyond vancomycin resistance. Nat Rev Microbiol 2012; 10: 266–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. ECDC. Antimicrobial resistance in the EU/EAA (EARS-Net). Annual Epidemiological Report for 2019. 2020. https://www.ecdc.europa.eu/en/publications-data/surveillance-antimicrobial-resistance-europe-2019.
- 3. Miller WR, Munita JM, Arias CA. Mechanisms of antibiotic resistance in enterococci. Expert Rev Anti Infect Ther 2014; 12: 1221–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cattoir V, Leclercq R. Twenty-five years of shared life with vancomycin-resistant enterococci: is it time to divorce? J Antimicrob Chemother 2013; 68: 731–42. [DOI] [PubMed] [Google Scholar]
- 5. de Been M, Pinholt M, Top J et al. Core genome multilocus sequence typing scheme for high-resolution typing of Enterococcus faecium. J Clin Microbiol 2015; 53: 3788–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pinholt M, Gumpert H, Bayliss S et al. Genomic analysis of 495 vancomycin-resistant Enterococcus faecium reveals broad dissemination of a vanA plasmid in more than 19 clones from Copenhagen, Denmark. J Antimicrob Chemother 2017; 72: 40–7. [DOI] [PubMed] [Google Scholar]
- 7. Raven KE, Reuter S, Reynolds R et al. A decade of genomic history for healthcare-associated Enterococcus faecium in the United Kingdom and Ireland. Genome Res 2016; 26: 1388–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pinholt M, Bayliss SC, Gumpert H et al. WGS of 1058 Enterococcus faecium from Copenhagen, Denmark, reveals rapid clonal expansion of vancomycin-resistant clone ST80 combined with widespread dissemination of a vanA-containing plasmid and acquisition of a heterogeneous accessory genome. J Antimicrob Chemother 2019; 74: 1776–85. [DOI] [PubMed] [Google Scholar]
- 9. Lebreton F, van Schaik W, Manson McGuire A et al. Emergence of epidemic multidrug-resistant Enterococcus faecium from animal and commensal strains. mBio 2013; 4: e00534-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Raven KE, Gouliouris T, Brodrick H et al. Complex routes of nosocomial vancomycin-resistant Enterococcus faecium transmission revealed by genome sequencing. Clin Infect Dis 2017; 64: 886–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gouliouris T, Raven KE, Ludden C et al. Genomic surveillance of Enterococcus faecium reveals limited sharing of strains and resistance genes between livestock and humans in the United Kingdom. mBio 2018; 9: e01780-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dutka-Malen S, Evers S, Courvalin P. Detection of glycopeptide resistance genotypes and identification to the species level of clinically relevant enterococci by PCR. J Clin Microbiol 1995; 33: 24–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dutka-Malen S, Evers S, Courvalin P. Detection of glycopeptide resistance genotypes and identification to the species level of clinically relevant enterococci by PCR. Erratum in: J Clin Microbiol 1995; 33: 1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Egan SA, Shore AC, O'Connell B et al. Linezolid resistance in Enterococcus faecium and Enterococcus faecalis from hospitalized patients in Ireland: high prevalence of the MDR genes optrA and poxtA in isolates with diverse genetic backgrounds. J Antimicrob Chemother 2020; 75: 1704–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. EUCAST. Breakpoint tables for interpretation of MICs and zone diameters. Version 11.0. 2021. https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_11.0_Breakpoint_Tables.pdf.
- 16. Bender JK, Fleige C, Lange D et al. Rapid emergence of highly variable and transferable oxazolidinone and phenicol resistance gene optrA in German Enterococcus spp. clinical isolates. Int J Antimicrob Agents 2018; 52: 819–27. [DOI] [PubMed] [Google Scholar]
- 17. Egan SA, Corcoran S, McDermott H et al. Hospital outbreak of linezolid-resistant and vancomycin-resistant ST80 Enterococcus faecium harbouring an optrA-encoding conjugative plasmid investigated by whole-genome sequencing. J Hosp Infect 2020; 105: 726–35. [DOI] [PubMed] [Google Scholar]
- 18. Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 2021; 49: W293–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Hal SJ, Willems RJL, Gouliouris T et al. The global dissemination of hospital clones of Enterococcus faecium. Genome Med 2021; 13: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pinholt M, Mollerup S, Boye K et al. Investigation of the introduction and dissemination of vanB Enterococcus faecium in the Capital Region of Denmark – and development of a rapid and accurate clone specific vanB E. faecium PCR. J Antimicrob Chemother 2021; 76: 2260–7. [DOI] [PubMed] [Google Scholar]
- 21. Souvorov A, Agarwala R, Lipman DJ. SKESA: strategic k-mer extension for scrupulous assemblies. Genome Biol 2018; 19: 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wick RR, Judd LM, Gorrie CL et al. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol 2017; 13: e1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2009; 25: 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H, Handsaker B, Wysoker A et al. The Sequence Alignment/Map (SAM) format and SAMtools. Bioinformatics 2009; 25: 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010; 26: 841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Milne I, Stephen G, Bayer M et al. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform 2013; 14: 193–202. [DOI] [PubMed] [Google Scholar]
- 27. Overbeek R, Olson R, Pusch GD et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res 2014; 42: D206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carter GP, Buultjens AH, Ballard SA et al. Emergence of endemic MLST non-typeable vancomycin-resistant Enterococcus faecium. J Antimicrob Chemother 2016; 71: 3367–71. [DOI] [PubMed] [Google Scholar]
- 29. Arthur M, Molinas C, Depardieu F et al. Characterization of Tn1546, a Tn3-related transposon conferring glycopeptide resistance by synthesis of depsipeptide peptidoglycan precursors in Enterococcus faecium BM4147. J Bacteriol 1993; 175: 117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hashimoto Y, Kita I, Suzuki M et al. First report of the local spread of vancomycin-resistant enterococci ascribed to the interspecies transmission of a vanA gene cluster-carrying linear plasmid. mSphere 2020; 5: e00102-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hashimoto Y, Taniguchi M, Uesaka K et al. Novel multidrug-resistant enterococcal mobile linear plasmid pELF1 encoding vanA and vanM gene clusters from a Japanese vancomycin-resistant enterococci isolate. Front Microbiol 2019; 10: 2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boumasmoud M, Haunreiter VD, Schweizer TA et al. Genomic surveillance of vancomycin-resistant Enterococcus faecium reveals spread of a linear plasmid conferring a nutrient utilization advantage. bioRxiv 2021; 10.1101/2021.05.07.442932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gorrie C, Higgs C, Carter G et al. Genomics of vancomycin-resistant Enterococcus faecium. Microb Genom 2019; 5: e000283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pinholt M, Larner-Svensson H, Littauer P et al. Multiple hospital outbreaks of vanA Enterococcus faecium in Denmark, 2012–13, investigated by WGS, MLST and PFGE. J Antimicrob Chemother 2015; 70: 2474–82. [DOI] [PubMed] [Google Scholar]
- 35. Shan X, Li XS, Wang N et al. Studies on the role of IS1216E in the formation and dissemination of poxtA-carrying plasmids in an Enterococcus faecium clade A1 isolate. J Antimicrob Chemother 2020; 75: 3126–30. [DOI] [PubMed] [Google Scholar]
- 36. Harmer CJ, Hall RM. IS26 family members IS257 and IS1216 also form cointegrates by copy-in and targeted conservative routes. mSphere 2020; 5: e00811-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Royal College of Physicians of Ireland (RCPI) Clinical Advisory Group on Healthcare Associated Infections. Guidelines for the Prevention and Control of Multi-drug resistant organisms (MDRO) excluding MRSA in the healthcare setting. 2012. https://www.hpsc.ie/a-z/microbiologyantimicrobialresistance/infectioncontrolandhai/guidelines/Guidelines%20for%20the%20Prevention%20and%20Control%20of%20MDRO_Final%20Revised_July%202014.pdf.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.