

Graphical abstract
Keywords: Pyrazolopyrimidines, Structure-activity relationships, Synthesis, Tuberculosis, Antimicrobial resistance
Abstract
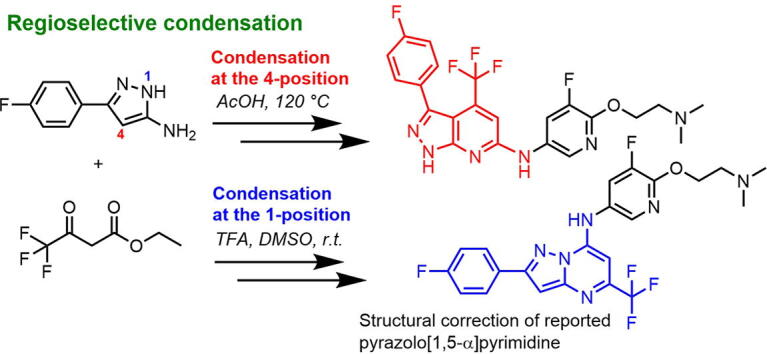
During our studies into preparing analogues of pyrazolopyrimidine as ATP synthesis inhibitors of Mycobacterium tuberculosis, a regiospecific condensation reaction between ethyl 4,4,4-trifluoroacetoacetate and 3-(4-fluorophenyl)-1H-pyrazol-5-amine was observed which was dependent on the specific reaction conditions employed. This work identifies optimized reaction conditions to access either the pyrazolo[3,4-β]pyridine or the pyrazolo[1,5-α]pyrimidine scaffold. This has led to the structural confirmation of the previously reported pyrazolopyrimidine 17b which was reported as pyrazolo[1,5-α]pyrimidine structure 2 which was corrected to pyrazolo[3,4-β]-pyrimidine 19.
Antimicrobial resistance (AMR) is a global health threat, and the World Health Organisation has declared that AMR is one of the top ten global public health threats facing humanity [1]. AMR occurs when bacteria no longer respond to medicines due to genetic changes, and is often driven by the misuse of antimicrobial agents. Tuberculosis (TB) is one of the leading causes of death from infectious disease globally. In 2019, there were 10 million new cases of active TB, with 1.4 million deaths. Drug resistance against the current standard treatment for Mycobacterium tuberculosis (M.tb), the causative agent of TB, is on the rise with increases in rifampicin-resistant TB (RR-TB) and multi-drug resistant TB (MDR-TB) being detected. In 2019, treatment success rates of 85% were observed for drug-susceptible TB, while this rate dropped to 56% for MDR-TB [2]. These resistant strains of M.tb require longer treatment courses (including injectables), which are more expensive and more difficult to access. The development of novel TB drugs to treat these resistant strains of M.tb is urgently needed.
There has been steady progress in the past fifteen years to develop novel drugs for treating TB, with many of these agents currently in clinical trials as part of combination therapy regimens [3]. Novel ATP synthase inhibitors include bedaquiline, which was first drug to be conditionally approved by the FDA in 2012, and its second generation analogues TBAJ-587 and TBAJ-876, which have recently entered phase I clinical trial [4], [5], [6], [7], [8], [9], [10]. There have been recent reports of squaramides [11], tetrahydroisoquinolines [12], 2,4-diaminoquinolines and pyrazolo[1,5-α]pyrimidines [13], [14] as potential ATP synthesis pathway inhibitors of M.tb. In particular, the pyrazolo[1,5-α]pyrimidine scaffold has attracted interest from our research group, with previous compounds having shown potent activity against M.tb [13], [14]. Derivatives with this scaffold have also been shown to be potential antianxiety agents [15] and the structurally similar triazolopyrimidines were reported to have antimalarial [16], [17], [18] and antileishmaniasis [19] properties.
Recent work from Tantry and co-workers has described pyrazolo[1,5-α]pyrimidines as potent ATP synthesis pathway inhibitors for the treatment of tuberculosis [13]. Our group has also been focused on elucidating structural activity relationships of novel pyrazolo[1,5-α]pyrimidines as potential inhibitors of M.tb. During the initial screening of a series of pyrazolo[1,5-α]pyrimidines for inhibition of ATP synthesis, we attempted to resynthesize compounds 1 and 2 (compound 17a and 17b, respectively, from Tantry and co-workers) as starting points for our drug discovery campaign (Fig. 1).
Fig. 1.

Pyrazolo[1,5-α]pyrimidines synthesized by Tantry and co-workers as potent ATP synthesis pathway inhibitors for the treatment of tuberculosis.
Synthesis of 1 was achieved following the reported reaction conditions (Scheme 1). Condensation of 3-(4-fluorophenyl)-3-oxopropanenitrile 3 with hydrazine hydrate gave 3-(4-fluorophenyl)-1H-pyrazol-5-amine 4. Reaction of aminopyrazole 4 with β-keto ester 5 gave pyrazolopyrimidinone intermediate 6 exclusively. Treatment of pyrazolopyrimidinone 6 with phosphorous oxychloride provided 7, which underwent nucleophilic substitution with 6-[2-(dimethylamino)ethoxy]-5-fluoropyridin-3-amine 8 to give 1 in poor yield. This reaction could be improved using Buchwald-Hartwig amination reaction conditions to afford compound 1.
Scheme 1.

Synthesis of pyrazolo[1,5-α]pyrimidine 1.
Synthesis of target compound 2 was more challenging. Following the reported condensation reaction conditions [13], reaction of aminopyrazole 4 and ethyl 4,4,4-trifluoroacetate 9 in acetic acid at 120 °C for 4 h gave the alternative cyclized pyrazolo[3,4-β]pyridinone 11, instead of the desired pyrazolo[1,5-α]pyrimidinone 10 (Scheme 2).
Scheme 2.

Synthesis of pyrazolo[3,4-β]pyridinone 12.
This could be explained if the condensation reaction had occurred at the 4-position of aminopyrazole 4, instead of at the usual 1-position which was observed in the previous formation of pyrazolo[1,5-α]pyrimidinone 6 (Scheme 1). Aminopyrazoles typically undergo condensation reactions at the 1-position with symmetrical β-diketones or β-keto esters to yield the pyrazolo[1,5-α]pyrimidinone scaffold [14]. However in rare cases, this condensation reaction undergoes condensation at the 4-position of aminopyrazoles to yield pyrazolo[3,4-β]pyridinones. This was observed when the β-keto ester ethyl 4,4,4-trifluoroacetate 9 was used in the condensation reaction with various aminopyrazoles [20], [21]. The electron withdrawing CF3 group in ethyl 4,4,4-trifluoroacetate 9, makes it sufficiently activated for electrophilic attack at the reactive 4-position of the aminopyrazole ring to take place.
With the condensation reaction forming the undesired pyrazolo[3,4-β]pyridinone core 11, alternative methods to access pyrazolo[1,5-α]pyrimidinone core 10 were investigated. There have been reports of similar reactions between ethyl 4,4,4-trifluoroacetate 9 and aminopyrazoles where the 4-position of the pyrazole ring was blocked with a methyl group to prevent the condensation reaction occurring at this position. This led to exclusive formation of pyrazolo[1,5-α]pyrimidinones [22]. However this method was not feasible as our aminopyrazole 4 is required to be unsubstituted at the 4-position.
Similar reactions between the related aminopyrazole 13 and hexafluoroacetylacetone 14 have been reported in the literature (Scheme 3). This reaction was reported first by Nam and co-workers which outlines exclusive formation of pyrazolo[3,4-β]pyridine 16 when the reaction was heated to 140 °C without solvent (condition 1) [23]. However, Petrov and co-workers report the same reaction could yield the other regioisomer pyrazolo[1,5-α]-pyrimidine 15 by altering the reaction conditions [24]. They found that carrying out the reaction at lower temperatures (20 °C) in DMSO gave exclusive formation of pyrazolo[1,5-α]-pyrimidine 15 (condition 2).
Scheme 3.

Formation of pyrazolo[3,4-β]pyridine 16 and pyrazolo[1,5-α]-pyrimidine 15 scaffold.
Encouraged by these results, aminopyrazole 4 and ethyl 4,4,4-trifluoroacetate 9 were dissolved in DMSO and stirred at r.t for 24 h (Scheme 4). However, no reaction occurred with both starting materials still present after 48 h. The reaction was repeated with catalytic amounts of trifluoroacetic acid as an acid catalyst. After 24 h, desired pyrazolo[1,5-α]-pyrimidinone 10 was isolated in 59% yield as the sole product. Trifluoroacetic acid was the catalyst of choice, as acetic acid readily reacted with the NH2 group of aminopyrazole 4 to form the unwanted acetamide byproduct.
Scheme 4.

Formation of pyrazolo[1,5-α]pyrimidinone 10.
The structure of 10 was confirmed unambiguously by single-crystal X-ray crystallography (Fig. 2) [25]. A single crystal of 10 was obtained by slow recrystallization by diffusing n-hexanes into a dichloromethane solution of 10. The X-ray crystal structure confirms the desired pyrazolo[1,5-α]-pyrimidinone core.
Fig. 2.

X-ray crystal structure of 10. Data for crystal structure 10 is available from CCDC 2105641.
Pyrazolo[3,4-β]-pyrimidinone 11 and pyrazolo[1,5-α]-pyrimidinone 10 were converted to the corresponding chlorides 17 and 18 respectively, using phosphorous oxychloride (Scheme 5). Buchwald-Hartwig amination with 8 gave the final products pyrazolo[3,4-β]-pyrimidine 19 and pyrazolo[1,5-α]-pyrimidine 2 respectively.
Scheme 5.

Synthesis of pyrazolo[3,4-β]-pyrimidine 19 and pyrazolo[1,5-α]-pyrimidine 2.
From the analytical data generated through this work, it was now clear that the structure of compound 17b reported by Tantry and co-workers [13] is actually pyrazolo[3,4-β]-pyrimidine 19. The 1H NMR spectra of 17b and 19 matched, except a NH peak at 13.7 ppm was missing in 17b and it also had an extra peak at 6.68 ppm (Table 1). For 1H NMR spectrum of 2 and 19, please refer to the ESI) [26]. This could be explained if the peak at 6.68 ppm is a ‘folded in’ or ‘aliased’ peak of the true peak at 13.7 ppm. This can be caused by the narrow spectral width setting which was insufficient to encompass all the peaks in the spectrum [27]. The 1H NMR spectrum of 19 showed it clearly had two NH’s present (confirmed by D2O exchange), while the 1H NMR spectrum of pyrazolo[1,5-α]-pyrimidine 2 clearly indicates it contains only one NH.
Table 1.
1H NMR comparison of reported 17b and pyrazolo[3,4-β]-pyrimidine 19 in DMSO, 400 MHz.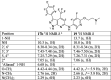
| Position | 17b1H NMR δ a | 191H NMR δ |
|---|---|---|
| 1-NH | – | 13.7 (s, 1H) |
| NH | 10.3 (s, 1H) | 10.0 (s, 1H) |
| 2′, 6′ | 8.30–8.34 (m, 2H) | 8.31–8.34 (m, 2H) |
| 3′, 5′ | 7.45–7.48 (m, 2H) | 7.46–7.50 (m, 2H) |
| 5, 4″ | 7.25–7.29 (m, 2H) | 7.26–7.31 (m, 2H) |
| 2″ | 7.05 (s, 1H) | 7.06 (s, 1H) |
| ‘Aliased’ 1-NH | 6.68 (s, 1H) | – |
| O-CH2 | 4.42–4.44 (m, 2H) | 4.42 (t, J = 5.9 Hz, 2H) |
| N-CH2 | 2.76 (m, 2H) | 2.66 (t, J = 5.8 Hz, 2H) |
| N(CH3)2 | 2.29 (s, 6H) | 2.23 (s, 6H) |
a Data published by Tantry and co-workers [13].
Minimum inhibitory concentration values for pyrazolo[1,5-α]-pyrimidines 2 and pyrazolo[3,4-β]-pyrimidine 19 were determined against both aerobic (replicating; MABA) and anaerobic (non-replicating; LORA) cultures of M.tb (Table 2). Results indicate both analogues possess moderate MIC90 values of ∼8 µg/mL against MABA bacterial cultures and ∼11 µg/mL against LORA cultures. The compounds were additionally tested for mammalian cell toxicity (with Vero green monkey-derived epithelial kidney cells), where both compounds had IC50s 11 ∼ 12 μg/mL. In comparison, the value for bedaquiline in repeat assays was between 4 and 16 μg/mL.
Table 2.
Inhibitory properties of pyrazolo[1,5-α]-pyrimidines 2 and pyrazolo[3,4-β]-pyrimidine 19.
| Compound | MABA MIC90a (µg/mL) | LORA MIC90a (µg/mL) | VERO IC50b(µg/mL) |
|---|---|---|---|
| Bedaquilinec | 0.04 | 0.08 | 4 ∼ 16 in repeat assays |
| 2 | 7.9 | 10.9 | 12.0 |
| 19 | 7.70 | 10.95 | 11.43 |
aMIC90 (µg/mL); minimum inhibitory concentration for 90% inhibition of growth of M.tb strain H37Rv, determined under aerobic (replicating; MABA) [28] or non-replicating (LORA) [29] conditions, determined at the Institute for Tuberculosis Research, University of Illinois at Chicago. Each value is the mean of at least two independent determinations; bIC50 values (µg/mL) in green monkey kidney epithelial (VERO) cells as a measure of mammalian cell toxicity [30]; cBedquiline is a known ATP synthase inhibitor for treatment of multi-drug-resistant tuberculosis.
A unique condensation reaction between ethyl 4,4,4-trifluoroacetate 9 and 3-(4-fluorophenyl)-1H-pyrazol-5-amine 4 has resulted in two distinct regioisomers depending on the reaction conditions. Condensation reaction conditions using ethanol with acetic acid at high temperatures resulted in the formation of pyrazolo[3,4-β]-pyrimidinone 11, while the same reaction carried out in DMSO at room temperature with catalytic TFA resulted in the exclusive formation of the pyrazolo[1,5-α]-pyrimidinone 10. This provides a unique regioselective method for accessing both regioisomers and highlights that care must be taken when assigning structures of products derived from condensation reactions with CF3-substituted β-keto esters. This chemistry was successfully utilized in synthesizing the analogues pyrazolo[1,5-α]-pyrimidines 2 and pyrazolo[3,4-β]-pyrimidine 19 as potential ATP synthesis inhibitors of M.tb. The reported structure of 17b has been confirmed and corrected with X-ray crystallography and 1H NMR spectroscopy as pyrazolo[3,4-β]-pyrimidine 19.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgments
The authors thank Tanya Groutso for the X-ray crystallography and the financial support from the following donors:
This work was supported by the Bill & Melinda Gates Foundation (#OPP1017459), the U.S. Agency for International Development (GHS-A-00-08-00012-00), the U.K. Department for International Development (DFID), and Irish Aid, 23-27 Henry Street, Limerick, Eire.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.tetlet.2021.153611.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.World Health Organization, Antimicrobial resistance, 2020.
- 2.World Health Organization, Global tuberculosis report 2020, 2020.
- 3.Tweed C.D., Dawson R., Burger D.A., Conradie A., Crook A.M., Mendel C.M., Conradie F., Diacon A.H., Ntinginya N.E., Everitt D.E., Haraka F., Li M., van Niekerk C.H., Okwera A., Rassool M.S., Reither K., Sebe M.A., Staples S., Variava E., Spigelman M. Bedaquiline, moxifloxacin, pretomanid, and pyrazinamide during the first 8 weeks of treatment of patients with drug-susceptible or drug-resistant pulmonary tuberculosis: a multicentre, open-label, partially randomised, phase 2b trial. Lancet Respiratory Med. 2019;7(12):1048–1058. doi: 10.1016/S2213-2600(19)30366-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blaser A., Sutherland H.S., Tong A.S.T., Choi P.J., Conole D., Franzblau S.G., Cooper C.B., Upton A.M., Lotlikar M., Denny W.A., Palmer B.D. Structure-activity relationships for unit C pyridyl analogues of the tuberculosis drug bedaquiline. Biorg. Med. Chem. 2019;27(7):1283–1291. doi: 10.1016/j.bmc.2019.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi P.J., Conole D., Sutherland H.S., Blaser A., Tong A.S.T., Cooper C.B., Upton A.M., Palmer B.D., Denny W.A. Synthetic Studies to Help Elucidate the Metabolism of the Preclinical Candidate TBAJ-876-A Less Toxic and More Potent Analogue of Bedaquiline. Molecules. 2020;25(6):1423. doi: 10.3390/molecules25061423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi P.J., Sutherland H.S., Tong A.S.T., Blaser A., Franzblau S.G., Cooper C.B., Lotlikar M.U., Upton A.M., Guillemont J., Motte M., Queguiner L., Andries K., Van den Broeck W., Denny W.A., Palmer B.D. Synthesis and evaluation of analogues of the tuberculosis drug bedaquiline containing heterocyclic B-ring units. Bioorg. Med. Chem. Lett. 2017;27(23):5190–5196. doi: 10.1016/j.bmcl.2017.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sutherland H.S., Tong A.S.T., Choi P.J., Blaser A., Conole D., Franzblau S.G., Lotlikar M.U., Cooper C.B., Upton A.M., Denny W.A., Palmer B.D. 3,5-Dialkoxypyridine analogues of bedaquiline are potent antituberculosis agents with minimal inhibition of the hERG channel. Biorg. Med. Chem. 2019;27(7):1292–1307. doi: 10.1016/j.bmc.2019.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sutherland H.S., Tong A.S.T., Choi P.J., Blaser A., Franzblau S.G., Cooper C.B., Upton A.M., Lotlikar M., Denny W.A., Palmer B.D. Variations in the C-unit of bedaquiline provides analogues with improved biology and pharmacology. Biorg. Med. Chem. 2020;28(1) doi: 10.1016/j.bmc.2019.115213. [DOI] [PubMed] [Google Scholar]
- 9.Sutherland H.S., Tong A.S.T., Choi P.J., Conole D., Blaser A., Franzblau S.G., Cooper C.B., Upton A.M., Lotlikar M.U., Denny W.A., Palmer B.D. Structure-activity relationships for analogs of the tuberculosis drug bedaquiline with the naphthalene unit replaced by bicyclic heterocycles. Biorg. Med. Chem. 2018;26(8):1797–1809. doi: 10.1016/j.bmc.2018.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tong A.S.T., Choi P.J., Blaser A., Sutherland H.S., Tsang S.K.Y., Guillemont J., Motte M., Cooper C.B., Andries K., Van den Broeck W., Franzblau S.G., Upton A.M., Denny W.A., Palmer B.D., Conole D. 6-Cyano Analogues of Bedaquiline as Less Lipophilic and Potentially Safer Diarylquinolines for Tuberculosis. ACS Med. Chem. Lett. 2017;8(10):1019–1024. doi: 10.1021/acsmedchemlett.7b00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tantry S.J., Markad S.D., Shinde V., Bhat J., Balakrishnan G., Gupta A.K., Ambady A., Raichurkar A., Kedari C., Sharma S., Mudugal N.V., Narayan A., Naveen Kumar C.N., Nanduri R., Bharath S., Reddy J., Panduga V., Prabhakar K.R., Kandaswamy K., Saralaya R., Kaur P., Dinesh N., Guptha S., Rich K., Murray D., Plant H., Preston M., Ashton H., Plant D., Walsh J., Alcock P., Naylor K., Collier M., Whiteaker J., McLaughlin R.E., Mallya M., Panda M., Rudrapatna S., Ramachandran V., Shandil R., Sambandamurthy V.K., Mdluli K., Cooper C.B., Rubin H., Yano T., Iyer P., Narayanan S., Kavanagh S., Mukherjee K., Balasubramanian V., Hosagrahara V.P., Solapure S., Ravishankar S., Hameed P.S. Discovery of Imidazo[1,2-a]pyridine Ethers and Squaramides as Selective and Potent Inhibitors of Mycobacterial Adenosine Triphosphate (ATP) Synthesis. J. Med. Chem. 2017;60(4):1379–1399. doi: 10.1021/acs.jmedchem.6b01358. [DOI] [PubMed] [Google Scholar]
- 12.Lu G.L., Tong A.S.T., Conole D., Sutherland H.S., Choi P.J., Franzblau S.G., Upton A.M., Lotlikar M.U., Cooper C.B., Denny W.A., Palmer B.D. Synthesis and structure-activity relationships for tetrahydroisoquinoline-based inhibitors of Mycobacterium tuberculosis. Bioorg. Med. Chem. 2020;28(22) doi: 10.1016/j.bmc.2020.115784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tantry S.J., Shinde V., Balakrishnan G., Markad S.D., Gupta A.K., Bhat J., Narayan A., Raichurkar A., Jena L.K., Sharma S., Kumar N., Nanduri R., Bharath S., Reddy J., Panduga V., Prabhakar K.R., Kandaswamy K., Kaur P., Dinesh N., Guptha S., Saralaya R., Panda M., Rudrapatna S., Mallya M., Rubin H., Yano T., Mdluili K., Cooper C.B., Balasubramanian V., Sambandamurthy V.K., Ramachandran V., Shandil R., Kavanagh S., Narayanan S., Iyer P., Mukherjee K., Hosagrahara V.P., Solapure S., Hameed P.S., Ravishankar S. Scaffold morphing leading to evolution of 2,4-diaminoquinolines and aminopyrazolopyrimidines as inhibitors of the ATP synthesis pathway. Med. Chem. Comm. 2016;7(5):1022–1032. [Google Scholar]
- 14.Soares de Melo C., Feng T.S., van der Westhuyzen R., Gessner R.K., Street L.J., Morgans G.L., Warner D.F., Moosa A., Naran K., Lawrence N., Boshoff H.I., Barry C.E., 3rd, Harris C.J., Gordon R., Chibale K. Aminopyrazolo[1,5-a]pyrimidines as potential inhibitors of Mycobacterium tuberculosis: Structure activity relationships and ADME characterization. Bioorg. Med. Chem. 2015;23(22):7240–7250. doi: 10.1016/j.bmc.2015.10.021. [DOI] [PubMed] [Google Scholar]
- 15.Kirkpatrick W.E., Okabe T., Hillyard I.W., Robins R.K., Dren A.T., Novinson T. 3-Halo-5,7-dimethylpyrazolo [1,5-a]pyrimidines, a nonbenzodiazepinoid class of antianxiety agents devoid of potentiation of central nervous system depressant effects of ethanol or barbiturates. J. Med. Chem. 1977;20(3):386–393. doi: 10.1021/jm00213a014. [DOI] [PubMed] [Google Scholar]
- 16.Gujjar R., El Mazouni F., White K.L., White J., Creason S., Shackleford D.M., Deng X., Charman W.N., Bathurst I., Burrows J., Floyd D.M., Matthews D., Buckner F.S., Charman S.A., Phillips M.A., Rathod P.K. Lead optimization of aryl and aralkyl amine-based triazolopyrimidine inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with antimalarial activity in mice. J. Med. Chem. 2011;54(11):3935–3949. doi: 10.1021/jm200265b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kokkonda S., Deng X., White K.L., Coteron J.M., Marco M., de Las Heras L., White J., El Mazouni F., Tomchick D.R., Manjalanagara K., Rudra K.R., Chen G., Morizzi J., Ryan E., Kaminsky W., Leroy D., Martinez-Martinez M.S., Jimenez-Diaz M.B., Bazaga S.F., Angulo-Barturen I., Waterson D., Burrows J.N., Matthews D., Charman S.A., Phillips M.A., Rathod P.K. Tetrahydro-2-naphthyl and 2-Indanyl Triazolopyrimidines Targeting Plasmodium falciparum Dihydroorotate Dehydrogenase Display Potent and Selective Antimalarial Activity. J. Med. Chem. 2016;59(11):5416–5431. doi: 10.1021/acs.jmedchem.6b00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coteron J.M., Marco M., Esquivias J., Deng X., White K.L., White J., Koltun M., El Mazouni F., Kokkonda S., Katneni K., Bhamidipati R., Shackleford D.M., Angulo-Barturen I., Ferrer S.B., Jimenez-Diaz M.B., Gamo F.J., Goldsmith E.J., Charman W.N., Bathurst I., Floyd D., Matthews D., Burrows J.N., Rathod P.K., Charman S.A., Phillips M.A. Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J. Med. Chem. 2011;54(15):5540–5561. doi: 10.1021/jm200592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.da Silva Edson R., Boechat Nubia, Pinheiro Luiz C.S., Bastos Monica M., Costa Carolina C.P., Bartholomeu Juliana C., da Costa Talita H. Novel selective inhibitor of Leishmania (Leishmania) amazonensis arginase. Chem. Biol. Drug Des. 2015;86(5):969–978. doi: 10.1111/cbdd.12566. [DOI] [PubMed] [Google Scholar]
- 20.Balicki R. Studies in the Field of Nitrogen Heterocyclic Compounds. Part XI. Abnormal Cyclocondensation of Ethyl 4,4,4-trifluoroacetoacetate with Aminopyrazoles. Pol. J. Chem. 1983;57(7–9):789–797. [Google Scholar]
- 21.Hansen B.B., Jepsen T.H., Larsen M., Sindet R., Vifian T., Burhardt M.N., Larsen J., Seitzberg J.G., Carnerup M.A., Jerre A., Molck C., Lovato P., Rai S., Nasipireddy V.R., Ritzen A. Fragment-Based Discovery of Pyrazolopyridones as JAK1 Inhibitors with Excellent Subtype Selectivity. J. Med. Chem. 2020;63(13):7008–7032. doi: 10.1021/acs.jmedchem.0c00359. [DOI] [PubMed] [Google Scholar]
- 22.Nam N.L., Grandberg I.I., Sorokin V.I. Condensation of 5-Aminopyrazoles Unsubstituted in Position 1 with OI-Keto Esters. Chem. Heterocyclic Compd. 2003;39(9):1210–1212. [Google Scholar]
- 23.Nam N.L., Grandberg I.I., Sorokin V.I. Pyrazolopyrimidines Based on 5-Aminopyrazoles Unsubstituted at the Position 1. Chem. Heterocyclic Compd. 2002;38(11):1371–1374. [Google Scholar]
- 24.Petrov A.A., Emelina E.E., Selivanov S.I. α-aminoazoles in synthesis of heterocycles: IV. Regiodirection of 3(5)-amino-5(3)-methylpyrazole reaction with hexafluoroacetylacetone. Russ. J. Org. Chem. 2011;44(2):263–269. [Google Scholar]
- 25.Data for crystal structure 10 is available from CCDC; CCDC 2105641 contains the supplementary crystallographic data for this paper. This data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 26.Reported 1H NMR spectrum of 17b: (400 MHz, DMSO-d6): δ 2.29 (s, 6H), 2.76 (m, 2H), 4.42-4.44 (m, 2H), 6.68 (s, 1H), 7.05 (s, 1H), 7.25-7.29 (m, 2H), 7.45-7.48 (m, 2H), 8.30-8.34 (m, 2H), 10.03 (s, 1H).
- 27.Torres Allan M., Price William S. Common problems and artifacts encountered in solution-state NMR experiments. Concepts Magn. Resonance Part A. 2016;45A(2):e21387. doi: 10.1002/cmr.a.2016.45A.issue-210.1002/cmr.a.21387. [DOI] [Google Scholar]
- 28.Collins L., Franzblau S.G. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 1997;41(5):1004–1009. doi: 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cho S.H., Warit S., Wan B., Hwang C.H., Pauli G.F., Franzblau S.G. Low-Oxygen-Recovery Assay for High-Throughput Screening of Compounds against Nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2007;51(4):1380–1385. doi: 10.1128/AAC.00055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Falzari K., Zhu Z., Pan D., Liu H., Hongmanee P., Franzblau S.G. In vitro and in vivo activities of macrolide derivatives against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2005;49(4):1447–1454. doi: 10.1128/AAC.49.4.1447-1454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.