

Abstract
α-Diimines are commonly used as supporting ligands for a variety of transition metal-catalyzed processes, most notably in α-olefin polymerization. They are also precursors to valuable synthetic targets, such as chiral 1,2-diamines. Their synthesis is usually performed through acid-catalyzed condensation of amines with α-diketones. Despite the simplicity of this approach, accessing unsymmetrical α-diimines is challenging. Herein, we report the Ti-mediated intermolecular diimination of alkynes to afford a variety of symmetrical and unsymmetrical α-diimines through the reaction of diazatitanacyclohexadiene intermediates with C-nitrosos. These diazatitanacycles can be readily accessed in situ via the multicomponent coupling of Ti NR imidos with alkynes and nitriles. The formation of α-diimines is achieved through formal [4 + 2]-cycloaddition of the C-nitroso to the Ti and γ-carbon of the diazatitanacyclohexadiene followed by two subsequent cycloreversion steps to eliminate nitrile and afford the α-diimine and a Ti oxo.
Alkyne diimination to unsymmetric α-diimines can be achieved via multicomponent reaction of Ti imidos and C-nitrosos. C-nitroso [4 + 2] cycloaddition across a diazatitanacyclohexadiene initiates cascading retrocycloadditions, liberating the α-diimine.
Introduction
α-Diimines (1,4-diaza-1,3-dienes) are widely used as ligands for organometallic complexes,1,2 perhaps most notably in Brookhart-type catalysts for α-olefin polymerization.3–5 The α-diimine ligand scaffold has proven to be remarkably versatile due to its facile stereoelectronic tunability.6–9 For example, modifications to the backbone and N-aryl substituents of α-diimines can exert control over the molecular weight and microstructure of α-olefin polymers by attenuation of chain walking processes.1,10 As ligands, they are often redox non-innocent, which allows for richer and more complex redox processes.11–14 α-Diimines are also precursors to valuable chiral 1,2-diamines through asymmetric hydrogenation,15,16 or to N-heterocyclic carbene (NHC) ligands through cyclization with paraformaldehyde and subsequent deprotonation.17–20
Typically, α-diimines are synthesized through condensation of α-diketones and amines. Despite the apparent simplicity of this approach, accessing unsymmetrical α-diimines through stepwise condensations is synthetically challenging due to poor chemoselectivity.21 Furthermore, since imine formation is reversible, attempts at sequential selective condensations can result in complex mixtures (for example, see Fig. 1A and S95–S98†). Imines are also notoriously difficult to isolate because they are prone to hydrolysis, making methods that generate product mixtures impractical. Highlighting this problem, although amine condensation with glyoxal to afford α-diimines is a common route to symmetric NHCs, examples of unsymmetric NHCs synthesized via condensation with glyoxal are rare.19,22,23
Fig. 1. Challenges and current strategies for unsymmetric α-diimine synthesis.

There are comparatively few examples of unsymmetrical α-diimines, and these are mostly limited to modification of N-aryl substituents, usually involving very sterically encumbered groups,3,9,24 rather than substituents on the backbone (Fig. 1B),11,25 although there are several examples from aldimine cross coupling.26,27 Selective trapping of unsymmetrical diimines via cyclization has also recently been reported.28 Given this methodology gap, developing a route to unsymmetrical α-diimines from simple feedstocks would provide a useful tool for the development of more diverse ligand scaffolds and pharmaceutically relevant building blocks.3,15,16 Our group has reported several examples of Ti-catalyzed oxidative functionalizations of alkynes for the synthesis of multisubstituted N-heterocycles that overcome limitations of classical condensation reactions.29–32 We envisioned that a complementary synthetic route to unsymmetrical α-diimines could be achieved through Ti-mediated oxidative diimination of alkynes. Examples of alkyne diamination or diimination are scarce33–36 despite many reports of alkene diamination.37–42 To the best of our knowledge, there is only a single example of a multicomponent intermolecular alkyne diamination43 and no examples of alkyne diimination. Nevertheless, there are several elegant examples of alkyne difunctionalizations44 using Ti including alkyne carboamination45–47 and iminoamination48 that provide motivation for further exploring Ti-catalyzed or -mediated diamination/diimination.
Previously, we reported that diazatitanacyclohexadienes (prepared from the multicomponent coupling of Ti imidos, alkynes, and nitriles) could undergo oxidation-induced N–N coupling to yield pyrazoles (Fig. 2, top).32 In the interest of further expanding the utility of this unique intermediate, we have begun examining its reactivity with various group transfer reagents. Herein, we report the intermolecular diimination of alkynes by Ti imidos and C-nitrosos to afford unsymmetrical α-diimines (Fig. 2, bottom). This diimination reaction proceeds through a cascading sequence of formal cycloaddition and retrocycloaddition reactions from this key diazatitanacyclohexadiene intermediate. This approach is a useful strategy for synthesizing unsymmetrical α-diimines, which are challenging to access through traditional condensation reactions.
Fig. 2. In situ generated diazatitanacyclohexadiene intermediates. (top) Previous work on oxidative N–N coupling to pyrazoles; (bottom) intermolecular diimination of alkynes (this work).

Results and discussion
Addition of C-nitrosos to diazatitanacyclohexadienes
Addition of PhNO (2a) to diazatitanacycle 1 resulted in rapid, near-quantitative formation of α-diimine 3a (81% by 1H NMR) with the concomitant formation of p-tolunitrile (Fig. 3). It is proposed that this occurs through [4 + 2]-cycloaddition of PhNO to the Ti and γ-carbon of the ligand backbone, followed by [4 + 2]-retrocycloaddition to eliminate p-tolunitrile, and a second cycloreversion to afford 3a and a Ti O species (vide infra).
Fig. 3. Reaction of a C-nitroso with 1 yields an α-diimine 3avia a sequence of cycloaddition/retrocycloaddition steps.

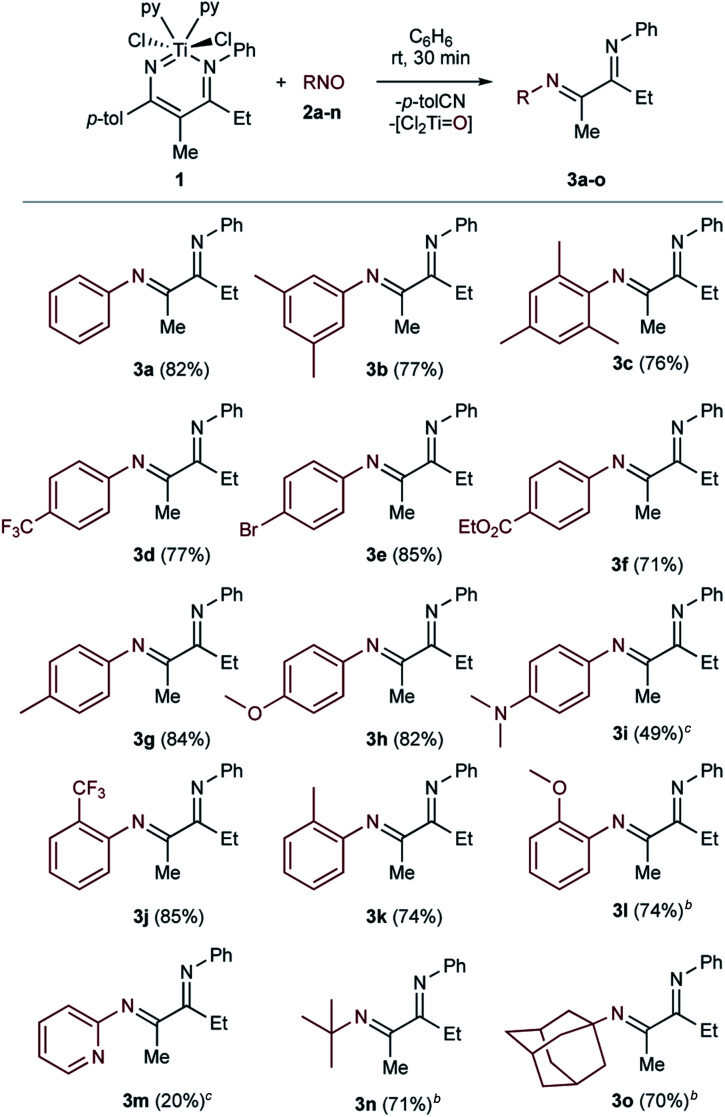
The scope of the C-nitroso reactants examined is shown in Table 1. Conveniently, nitrosoarenes can be readily prepared from the corresponding aniline via Oxone® oxidation.49 In most cases, the α-diimine products were obtained in very good isolated yields (70% to 85%).
Substrate scope of the addition of C-nitrosos to 1a.
|
Conditions: 1 (0.2 mmol), 2a–o (0.2 mmol, 1 equiv.), 3 mL C6H6, N2 atmosphere (glovebox). Isolated yields.
Mixture of imine/enamine tautomers. See ESI for enamine characterization.
1H NMR yield vs. 1,3,5-trimethoxybenzene internal standard = 0.2 M.
Use of sterically demanding nitrosos 2b and 2c resulted in good yields of 3b and 3c (77% and 76%), which are commonly used as bulky α-diimine substituents.3 This method tolerates both electron-poor (3d–3f) and electron-rich (3g–3i) para-substituted (3d–3i) nitrosoarenes, as well as ortho-substituted (3j–3l) nitrosoarenes (3l is formed in a 6 : 1 ratio with its enamine tautomer). The reaction also proceeds cleanly with aliphatic nitrosos: 2-methyl-2-nitrosopropane 2n gives a mixture of tert-butyl substituted α-diimine 3n and its enamine tautomer in a 3.75 : 1 ratio (71%). Similarly, 1-nitrosoadamantane 2o afforded α-diimine 3o and its enamine tautomer in a 3.13 : 1 ratio (70%). While the yield of the reaction was mostly consistent irrespective of nitrosoarene substituent, there were a couple of exceptions. For the reaction with electron-rich 2i, the formation of α-diimine 3i (49% yield by 1H NMR) was accompanied by the formation of unidentified side products that precluded isolation. Also, the reaction with 2-nitrosopyridine 2m yielded only a small amount of 3m by 1H NMR (20%) relative to the amount of p-tolunitrile byproduct (85%).
In situ multicomponent diimine synthesis
Diazatitanacycles such as 1 can be synthesized via the multicomponent coupling of Ti imidos, alkynes, and nitriles.32 With this strategy, one-pot alkyne diiminations were carried out (Fig. 4 and Table 2).
Fig. 4. Comparison of synthesis of 5b using (A) 1 equiv. or (B) 10 equiv. MeCN.

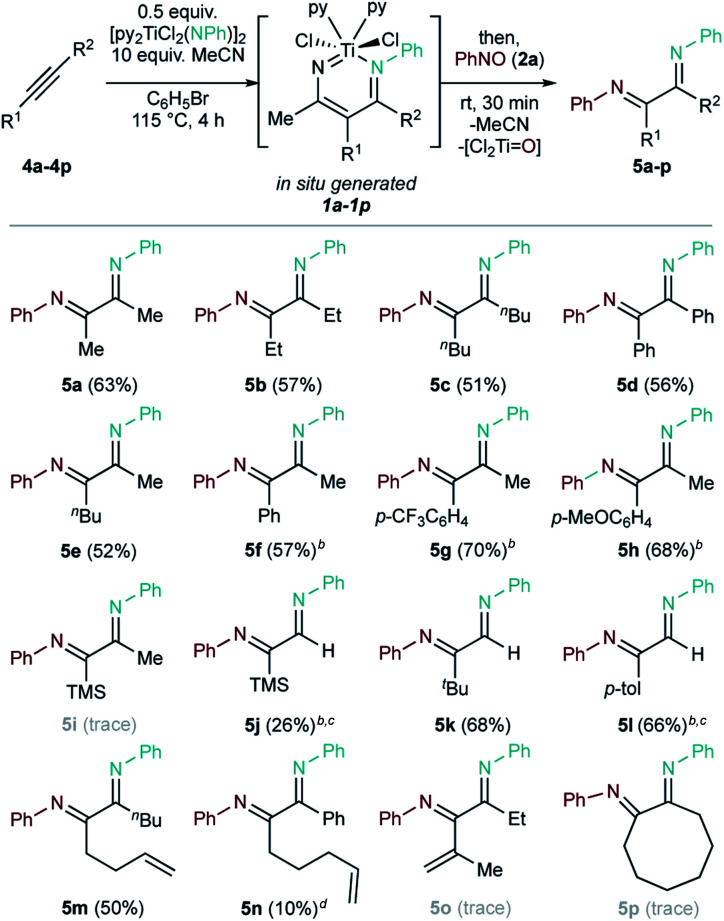
Substrate scope of one-pot in situ diimine synthesis from alkynes, imidos, and C-nitrososa.
|
Conditions: 0.2 mmol (0.5 equiv.) [py2TiCl2(NPh)]2, 0.4 mmol (1 equiv.) alkyne, 4.0 mmol (10 equiv.) MeCN, 4 mL PhBr, 115 °C, 4 h, N2 atmosphere (glovebox); then, 0.4 mmol (1 equiv.) PhNO, rt, 0.5 h. Isolated yields.
Mixture of stereoisomers.
Yield corrected for minor pyrrole impurities.
GC-FID yield (vs. 1,3,5-trimethoxybenzene standard).
In an initial experiment, reactions with 1 equiv. MeCN led to moderate yields of 5b (40%) from reaction of 4b and PhNO (Fig. 4A). MeCN was chosen over p-tolCN (the formal nitrile component from the reactions in Table 1) because of the easier removal of MeCN in vacuo. However, several species from competing side reactions also formed that were difficult to separate from the desired product given the instability of diimines towards hydrolysis: pyrrole (D, 5%) from insertion of a second equivalent of alkyne;29 imine from the hydroamination of 3-hexyne (E, 6%); and azobenzene (F, 12%) from direct metathesis of PhNO with leftover Ti imido.50,51 Using excess MeCN (10 equiv.) suppressed these competing side reactions (Fig. 4B) and led to cleaner formation of the product, with azobenzene as the predominant side-product after removal of volatiles. Conveniently, basic aqueous extraction can remove the Ti byproducts, and azobenzene can be easily removed by sublimation—avoiding the need to perform column chromatography on the sensitive products.
Based on this result, the scope of one-pot oxidative alkyne diimination with [py2TiCl2(NPh)]2 and PhNO was examined (Table 2, 5a–5p). Here, the α-diimine yields are ultimately determined by the yield of in situ formed diazatitanacycle 1a–1p, as the subsequent oxidations are near-quantitative. Symmetrical internal alkynes with both alkyl and aryl substituents formed the respective α-diimines (5a–5d) in good isolated yields (51% to 63%).
Unsymmetrical internal alkyne 4e resulted in the formation of a single α-diimine isomer (5e) in a 52% yield. In contrast, α-diimines 5f–5h give complex 1H and 13C NMR spectra, presumably due to formation of multiple imine stereoisomers. Analysis of 5f by NOESY (Fig. S53 and 54†) showed that chemical exchange occurs between each of these isomers by the presence of EXSY cross-peaks. Additionally, GC-MS revealed only one peak corresponding to the mass of 5f (Fig. S55†).
Imines are well-known to undergo rapid equilibrium between E/Z isomers through inversion (also called the lateral shift mechanism) in nonpolar solvents.52 Confirmation that the complex spectra of 5f–5h were a result of stereoisomer equilibration was obtained through further reaction of the isomer mixtures. For example, reaction of the 5f isomeric mixture with ZnCl2 resulted in 93% yield of α-diimine adduct 6f (Fig. 5). The identities of diimines 5g and 5h were similarly confirmed via ZnCl2 coordination (see ESI†).
Fig. 5. Top: reaction of stereoisomeric mixture 5f with ZnCl2 results in convergence to 6f. Bottom: 13C NMR imine region of (a) 5f (C6D6) and (b) 6f (CDCl3). Inset: crystal structure of 6f showing half of the asymmetric unit with cocrystallized solvent and hydrogens omitted for clarity.

Terminal alkynes 4j–4l were also examined. Trapping Ti NR + terminal alkyne [2 + 2] cycloadducts is challenging: terminal alkynes typically react faster to form pyrrole or alkyne trimerization products.53 Nevertheless, 5k was obtained cleanly in a 68% yield, while 5j and 5l were obtained as a mixture of stereoisomers with small pyrrole impurities (2.4% and 12% pyrrole, respectively).
1,5-Enyne 4m allows for clean formation of 5m (50%), while 1,6-enyne 4n results in formation of 5n (10%) as a mixture with competing carboamination47 products (6%). Here, the shorter linker of 4m compared to 4n prevents the intramolecular alkene insertion that would lead to carboamination. Conjugated enyne 4o yielded only trace 5o, while cyclooctyne 4p predominantly formed alkyne trimer and pyrrole, with only a trace amount of 5p.
5d was also synthesized via a telescoped in situ route from TiCl4(THF)2, azobenzene, and Zn0 powder with only a moderate decrease in yield (Fig. 6). We have previously shown that this strategy is a benchtop-compatible approach to other Ti oxidative amination reactions.54 This one-pot approach facilitates the in situ formation of diazatitanacycle intermediates such as 1 without the need for specialized equipment, making the synthesis of both α-diimines and pyrazoles32 more operationally simple.
Fig. 6. Telescoped one-pot synthesis of 5d from TiCl4(THF)2.

Given that diazatitanacyclohexadiene intermediate formation is regioselective,32,55–57 this method could be used to prepare regioisomeric α-diimines through different combinations of alkynes, imidos, and nitrosos (Fig. 7). For example, reaction of 4k with [py2TiCl2(Np-tol)]2 imido gives 5q (41%), while its regioisomer 5r can be synthesized by using 2g and [py2TiCl2(NPh)]2 (44%). Another regioisomer 5s can be prepared from [py2TiCl2(NtBu)]2 with a 50% yield of diimine stereoisomers, albeit with a small pyrrole impurity (4% yield).
Fig. 7. Modular strategy for the synthesis of α-diimine regioisomers.

Together, these reaction scopes demonstrate that a wide range of both symmetric and unsymmetric α-diimines can be accessed directly through a one-pot multicomponent reaction with a variety of Ti imidos, alkynes, and C-nitrosos.
Proposed mechanism for intermolecular diimination
A plausible mechanism and accompanying DFT calculations for the formation of 5a from 1a (IM1) and PhNO (2a) are shown in Fig. 8. The in situ formation of IM1 from Ti imidos, alkynes, and nitriles through alkyne/imido [2 + 2]-cycloaddition and nitrile insertion has been previously established.32,58–60 The formal [4 + 2] cycloaddition of PhNO to IM1 occurs in a stepwise fashion. First, O-coordination of PhNO to Ti yields IM3. From IM3, the nucleophilic, electron-rich γ-carbon in the metallacycle backbone61 attacks the electrophilic N of coordinated PhNO, generating bicyclic IM4. This process can be further visualized using IBO analysis (Fig. 8a): coordination of PhNO results in a puckering of the γ-C orbital toward the N of coordinated PhNO. Subsequently the new N–C σ-bond is formed in IM4, with simultaneous rearrangement of the Ti–N and N–O π-bonds to a new N–C π-bond and O lone pair, respectively.
Fig. 8. Computed pathway for diimine formation (M06/6-311G(d,p)/SMD, 25 °C, C6H5Br). All free energies are referenced to IM1 = 0.0 kcal mol−1. (a) Intrinsic bond orbitals (IBOs) showing [4 + 2] cycloaddition between NO π-bond (red) orbital of coordinated PhNO and π-backbone (green orbital) of IM3. (b) IBOs showing rearrangement of IM4 to IM5via retro-[4 + 2] cycloaddition.

The tendency of C-nitrosos to undergo Diels–Alder-type cycloaddition reactions is well known,62,63 but examples of this process with metallacycles are scarce.64,65 Titanium β-diketiminate complexes, which differ from diazatitanacyclohexadienes by a degree of unsaturation, have also been observed to undergo [4 + 2]-cycloaddition with ketenes.66,67
IM4 then undergoes rate-determining (12.6 kcal mol−1) retro-[4 + 2] cycloaddition to extrude nitrile, forming azaoxatitanacycle IM5. The retrocycloadditive nature of TS2 can also be visualized by IBO calculations (Fig. 8b), which show the elimination of nitrile by the breaking of C–C and Ti–N σ-bonds in IM4 and the formation of three new N–C, C–C, and Ti–N π-bonds in IM5. Further IBO analysis is provided in Fig. S100.† A similar tandem [4 + 2]-cycloaddition-cycloreversion process to eliminate nitriles has also been proposed in the synthesis of phosphinines.68IM5 then undergoes a haptotropic shift following loss of the coordinated nitrile (IM6) generating η2-(N,O)-bound IM7. Finally, N–O bond cleavage results in exothermic formation of a Ti O species with the bound α-diimine product IM8.69 Cycloreversions of group IV heterometallacycles to yield M = X (M = Ti, Zr; X= O, N) are well-precedented.70–75
Ultimately, nitrile serves as a promoter in the in situ reactions—first forming the key diazatitanacycle intermediate, and then being eliminated prior to product formation. Alternatively, instead of undergoing a [4 + 2]-cycloaddition, a nitroso could directly insert into the [2 + 2] alkyne/Ti NR cycloadduct, bypassing the need for nitrile. Indeed, C-nitrosos undergo insertions with Ti and Zr metallacycles.76,77 However, a nitrile-free control reaction resulted in exclusive formation of azobenzene through metathesis of the Ti imido with PhNO (Fig. 9), making this route unlikely.50,51
Fig. 9. Direct reaction of nitrosos with Ti imidos and alkynes results in formation of azobenzene.

Conclusions
In summary, we have demonstrated a one-pot multicomponent synthesis of α-diimines by the diimination of alkynes by Ti imidos and C-nitrosos. This reaction likely occurs by [4 + 2]-cycloaddition of a nitroso to the Ti and γ-carbon of a diazatitanacyclohexadiene intermediate, followed by two subsequent cycloreversion steps to eliminate nitrile and afford the α-diimine and Ti O. This is an attractive route to complex, unsymmetrical α-diimines that are difficult to obtain through classical condensation reactions, providing new strategies for ligand synthesis,3,18,20 and generally adding to the library of alkyne difunctionalization reactions. Efforts are ongoing to make this cycloaddition–retrocyclization strategy generalizable to other difunctionalizations.
Author contributions
CWF: experimental data collection and analysis, computational analysis; DTE: computational analysis; EK: computational analysis; AJP: experimental data collection and analysis; YC: experimental data collection and analysis; IAT: project direction and all authors contributed to the writing, editing, and revision of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
Financial support was provided by the National Institutes of Health (R35GM119457), and the Alfred P. Sloan Foundation (I. A. T. is a 2017 Sloan Fellow). D. T. E. kindly acknowledges financial support by the ETH Zurich foundation (D. T. E. is a 2019 ESOP scholar). Y. C. acknowledges funding support from the Wayland E. Noland Fellowship (UMN). Instrumentation for the University of Minnesota Chemistry NMR facility was supported from a grant through the National Institutes of Health (S10OD011952). X-ray diffraction experiments were performed with a diffractometer purchased through a grant from NSF/MRI (1229400) and the University of Minnesota. Dr Evan Beaumier, Dr Robin Harkins, and Dr Xuelan Wen are acknowledged for helpful discussions. The Minnesota Supercomputing Institute (MSI) at the University of Minnesota for provided resources that contributed to the results reported within this paper.
Electronic supplementary information (ESI) available: Experimental details and spectroscopic data. CCDC 2072232. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06111a
Notes and references
- Johnson L. K. Killian C. M. Brookhart M. New Pd(ii)- and Ni(ii)-Based Catalysts for Polymerization of Ethylene and α-Olefins. J. Am. Chem. Soc. 1995;117:6414–6415. doi: 10.1021/ja00128a054. [DOI] [Google Scholar]
- Léonard N. G. Chirik P. J. Air-Stable α-Diimine Nickel Precatalysts for the Hydrogenation of Hindered, Unactivated Alkenes. ACS Catal. 2018;8:342–348. doi: 10.1021/acscatal.7b03909. [DOI] [Google Scholar]
- Wang F. Chen C. A continuing legend: the Brookhart-type α-diimine nickel and palladium catalysts. Polym. Chem. 2019;10:2354–2369. doi: 10.1039/C9PY00226J. [DOI] [Google Scholar]
- Chen Z. Brookhart M. Exploring Ethylene/Polar Vinyl Monomer Copolymerizations Using Ni and Pd α-Diimine Catalysts. Acc. Chem. Res. 2018;51:1831–1839. doi: 10.1021/acs.accounts.8b00225. [DOI] [PubMed] [Google Scholar]
- Ittel S. D. Johnson L. K. Brookhart M. Late-Metal Catalysts for Ethylene Homo- and Copolymerization. Chem. Rev. 2000;100:1169–1204. doi: 10.1021/cr9804644. [DOI] [PubMed] [Google Scholar]
- Liu F.-S. Hu H.-B. Xu Y. Guo L.-H. Zai S.-B. Song K.-M. Gao H.-Y. Zhang L. Zhu F.-M. Wu Q. Thermostable α-Diimine Nickel(ii) Catalyst for Ethylene Polymerization: Effects of the Substituted Backbone Structure on Catalytic Properties and Branching Structure of Polyethylene. Macromolecules. 2009;42:7789–7796. doi: 10.1021/ma9013466. [DOI] [Google Scholar]
- Rhinehart J. L. Brown L. A. Long B. K. A Robust Ni(ii) α-Diimine Catalyst for High Temperature Ethylene Polymerization. J. Am. Chem. Soc. 2013;135:16316–16319. doi: 10.1021/ja408905t. [DOI] [PubMed] [Google Scholar]
- Sa S. Jeon M. Kim S. Y. Controlling branch distribution of polyethylenes by steric tuning of Ni α-diimine complexes based on phenanthrenequinone. J. Mol. Catal. A: Chem. 2014;393:263–271. doi: 10.1016/j.molcata.2014.06.020. [DOI] [Google Scholar]
- Dai S. Zhou S. Zhang W. Chen C. Systematic Investigations of Ligand Steric Effects on α-Diimine Palladium Catalyzed Olefin Polymerization and Copolymerization. Macromolecules. 2016;49:8855–8862. doi: 10.1021/acs.macromol.6b02104. [DOI] [Google Scholar]
- Guo L. Dai S. Sui X. Chen C. Palladium and Nickel Catalyzed Chain Walking Olefin Polymerization and Copolymerization. ACS Catal. 2016;6:428–441. doi: 10.1021/acscatal.5b02426. [DOI] [Google Scholar]
- Muresan N. Lu C. C. Ghosh M. Peters J. C. Abe M. Henling L. M. Weyhermöller T. Bill E. Wieghardt K. Bis(α-diimine)iron Complexes: Electronic Structure Determination by Spectroscopy and Broken Symmetry Density Functional Theoretical Calculations. Inorg. Chem. 2008;47:4579–4590. doi: 10.1021/ic7022693. [DOI] [PubMed] [Google Scholar]
- Kreisel K. A. Yap G. P. A. Theopold K. H. Synthesis, Characterization, and Electronic Structure of Diimine Complexes of Chromium. Inorg. Chem. 2008;47:5293–5303. doi: 10.1021/ic800302w. [DOI] [PubMed] [Google Scholar]
- Nishiyama H. Ikeda H. Saito T. Kriegel B. Tsurugi H. Arnold J. Mashima K. Structural and Electronic Noninnocence of α-Diimine Ligands on Niobium for Reductive C–Cl Bond Activation and Catalytic Radical Addition Reactions. J. Am. Chem. Soc. 2017;139:6494–6505. doi: 10.1021/jacs.7b02710. [DOI] [PubMed] [Google Scholar]
- Mashima K. Redox-Active α-Diimine Complexes of Early Transition Metals: From Bonding to Catalysis. Bull. Chem. Soc. Jpn. 2020;93:799–820. doi: 10.1246/bcsj.20200056. [DOI] [Google Scholar]
- Shimizu M. Kamei M. Fujisawa T. Stereocontrol in the reduction of 1,2-diimine with an oxazaborolidine catalyst. Highly stereoselective preparation of (R,R)-1,2-diphenylethylenediamine. Tetrahedron Lett. 1995;36:8607–8610. doi: 10.1016/0040-4039(95)01825-3. [DOI] [Google Scholar]
- Zhu X. Du H. A Highly Stereoselective Metal-Free Hydrogenation of Diimines for the Synthesis of Cis-Vicinal Diamines. Org. Lett. 2015;17:3106–3109. doi: 10.1021/acs.orglett.5b01380. [DOI] [PubMed] [Google Scholar]
- Hopkinson M. N. Richter C. Schedler M. Glorius F. An overview of N-heterocyclic carbenes. Nature. 2014;510:485–496. doi: 10.1038/nature13384. [DOI] [PubMed] [Google Scholar]
- Hintermann L. Expedient syntheses of the N-heterocyclic carbene precursor imidazolium salts IPr·HCl, IMes·HCl and IXy·HCl. Beilstein J. Org. Chem. 2007;3:22. doi: 10.1186/1860-5397-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queval P. Jahier C. Rouen M. Artur I. Legeay J.-C. Falivene L. Toupet L. Crévisy C. Cavallo L. Baslé O. Mauduit M. Multicomponent Synthesis of Unsymmetrical Unsaturated N-Heterocyclic Carbene Precursors and Their Related Transition-Metal Complexes. Angew. Chem., Int. Ed. 2013;52:14103–14107. doi: 10.1002/anie.201308873. [DOI] [PubMed] [Google Scholar]
- Li S. Yang F. Lv T. Lan J. Gao G. You J. Synthesis of unsymmetrical imidazolium salts by direct quaternization of N-substituted imidazoles using arylboronic acids. Chem. Commun. 2014;50:3941–3943. doi: 10.1039/C4CC00474D. [DOI] [PubMed] [Google Scholar]
- Jeon M. Kim S. Y. Ethylene Polymerizations with Unsymmetrical (α-Diimine)nickel(ii) Catalysts. Polym. J. 2008;40:409–413. doi: 10.1295/polymj.PJ2007175. [DOI] [Google Scholar]
- Benhamou L. Chardon E. Lavigne G. Bellemin-Laponnaz S. César V. Synthetic Routes to N-Heterocyclic Carbene Precursors. Chem. Rev. 2011;111:2705–2733. doi: 10.1021/cr100328e. [DOI] [PubMed] [Google Scholar]
- Dumas A. Tarrieu R. Vives T. Roisnel T. Dorcet V. Baslé O. Mauduit M. A Versatile and Highly Z-Selective Olefin Metathesis Ruthenium Catalyst Based on a Readily Accessible N-Heterocyclic Carbene. ACS Catal. 2018;8:3257–3262. doi: 10.1021/acscatal.8b00151. [DOI] [Google Scholar]
- Rosar V. Meduri A. Montini T. Fornasiero P. Zangrando E. Milani B. The contradictory effect of the methoxy-substituent in palladium-catalyzed ethylene/methyl acrylate cooligomerization. Dalton Trans. 2018;47:2778–2790. doi: 10.1039/C7DT04465H. [DOI] [PubMed] [Google Scholar]
- Ellandula S. K. Opoku Amoako C. Mague J. T. Chandrasekaran P. Crystal structure of unsymmetrical [alpha]-diimine palladium(II) complex cis-[{ArN=C(Me)-(Et)C=NAr}PdCl2] [Ar = 2,6-(iPr)2C6H3] Acta Crystallogr., Sect. E: Crystallogr. Commun. 2017;73:1148–1150. doi: 10.1107/S2056989017009616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kison C. Opatz T. Synthesis of Highly Substituted Unsymmetrical 1,2-Diamines, 1,2-Diimines, Imidazolium Salts and Imidazolylidenes by Aldimine Cross-Coupling. Synthesis. 2006;2006:3727–3738. doi: 10.1055/s-2006-950237. [DOI] [Google Scholar]
- Kison C. Meyer N. Opatz T. An Aldimine Cross-Coupling for the Diastereoselective Synthesis of Unsymmetrical 1,2-Diamines. Angew. Chem., Int. Ed. 2005;44:5662–5664. doi: 10.1002/anie.200501705. [DOI] [PubMed] [Google Scholar]
- Wang J. Cheng X. Liu Y. Zhang J. Multicomponent Synthesis of Unsymmetrical 4,5-Disubstituted Imidazolium Salts as N-Heterocyclic Carbene Precursors: Applications in Palladium-Catalyzed Cross-Coupling Reactions. J. Org. Chem. 2021;86:6278–6288. doi: 10.1021/acs.joc.1c00075. [DOI] [PubMed] [Google Scholar]
- Gilbert Z. W. Hue R. J. Tonks I. A. Catalytic formal [2 + 2 + 1] synthesis of pyrroles from alkynes and diazenes via TiII/TiIV redox catalysis. Nat. Chem. 2015;8:63. doi: 10.1038/nchem.2386. [DOI] [PubMed] [Google Scholar]
- Desnoyer A. N. See X. Y. Tonks I. A. Diverse Reactivity of Diazatitanacyclohexenes: Coupling Reactions of 2H-Azirines Mediated by Titanium(ii) Organometallics. 2018;37:4327–4331. doi: 10.1021/acs.organomet.8b00522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumier E. P. Pearce A. J. See X. Y. Tonks I. A. Modern applications of low-valent early transition metals in synthesis and catalysis. Nat. Rev. Chem. 2019;3:15–34. doi: 10.1038/s41570-018-0059-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce A. J. Harkins R. P. Reiner B. R. Wotal A. C. Dunscomb R. J. Tonks I. A. Multicomponent Pyrazole Synthesis from Alkynes, Nitriles, and Titanium Imido Complexes via Oxidatively Induced N–N Bond Coupling. J. Am. Chem. Soc. 2020;142:4390–4399. doi: 10.1021/jacs.9b13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J. Tan Y. J. Leow M. L. Liu X.-W. Copper(ii)/Iron(iii) Co-catalyzed Intermolecular Diamination of Alkynes: Facile Synthesis of Imidazopyridines. Org. Lett. 2012;14:4386–4389. doi: 10.1021/ol301858j. [DOI] [PubMed] [Google Scholar]
- Dwivedi V. Kumar R. Sharma K. Sridhar B. Reddy M. S. Copper-Promoted Regioselective Intermolecular Diamination of Ynamides: Synthesis of Imidazo[1,2-a]pyridines. ACS Omega. 2017;2:2770–2777. doi: 10.1021/acsomega.7b00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C. Hao J. Xu H. Mo Y. Liu H. Han J. Lei A. Heteroaromatic imidazo[1,2-a]pyridines synthesis from C–H/N–H oxidative cross-coupling/cyclization. Chem. Commun. 2012;48:11073–11075. doi: 10.1039/C2CC35927H. [DOI] [PubMed] [Google Scholar]
- Li J. Neuville L. Copper-Catalyzed Oxidative Diamination of Terminal Alkynes by Amidines: Synthesis of 1,2,4-Trisubstituted Imidazoles. Org. Lett. 2013;15:1752–1755. doi: 10.1021/ol400560m. [DOI] [PubMed] [Google Scholar]
- Muñiz K. Martínez C. Development of Intramolecular Vicinal Diamination of Alkenes: From Palladium to Bromine Catalysis. J. Org. Chem. 2013;78:2168–2174. doi: 10.1021/jo302472w. [DOI] [PubMed] [Google Scholar]
- Olson D. E. Su J. Y. Roberts D. A. Du Bois J. Vicinal Diamination of Alkenes under Rh-Catalysis. J. Am. Chem. Soc. 2014;136:13506–13509. doi: 10.1021/ja506532h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y. Cornwall R. G. Du H. Zhao B. Shi Y. Catalytic Diamination of Olefins via N–N Bond Activation. Acc. Chem. Res. 2014;47:3665–3678. doi: 10.1021/ar500344t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñiz K. Barreiro L. Romero R. M. Martínez C. Catalytic Asymmetric Diamination of Styrenes. J. Am. Chem. Soc. 2017;139:4354–4357. doi: 10.1021/jacs.7b01443. [DOI] [PubMed] [Google Scholar]
- Tao Z. Gilbert B. B. Denmark S. E. Catalytic, Enantioselective syn-Diamination of Alkenes. J. Am. Chem. Soc. 2019;141:19161–19170. doi: 10.1021/jacs.9b11261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minakata S. Miwa H. Yamamoto K. Hirayama A. Okumura S. Diastereodivergent Intermolecular 1,2-Diamination of Unactivated Alkenes Enabled by Iodine Catalysis. J. Am. Chem. Soc. 2021;143:4112–4118. doi: 10.1021/jacs.1c00228. [DOI] [PubMed] [Google Scholar]
- Selby J. D. Manley C. D. Feliz M. Schwarz A. D. Clot E. Mountford P. New ligand platforms for developing the chemistry of the Ti N–NR2 functional group and the insertion of alkynes into the N–N bond of a Ti N–NPh2 ligand. Chem. Commun. 2007:4937–4939. doi: 10.1039/B711941K. [DOI] [PubMed] [Google Scholar]
- Manßen M. Schafer L. L. Titanium catalysis for the synthesis of fine chemicals – development and trends. Chem. Soc. Rev. 2020;49:6947–6994. doi: 10.1039/D0CS00229A. [DOI] [PubMed] [Google Scholar]
- Basuli F. Aneetha H. Huffman J. C. Mindiola D. J. A Fluorobenzene Adduct of Ti(iv), and Catalytic Carboamination to Prepare α,β-Unsaturated Imines and Triaryl-Substituted Quinolines. J. Am. Chem. Soc. 2005;127:17992–17993. doi: 10.1021/ja0566026. [DOI] [PubMed] [Google Scholar]
- Basuli F. Wicker B. Huffman J. C. Mindiola D. J. Understanding the role of an easy-to-prepare aldimine–alkyne carboamination catalyst, [Ti(NMe2)3(NHMe2)][B(C6F5)4] J. Organomet. Chem. 2011;696:235–243. doi: 10.1016/j.jorganchem.2010.09.017. [DOI] [Google Scholar]
- Davis-Gilbert Z. W. Yao L. J. Tonks I. A. Ti-Catalyzed Multicomponent Oxidative Carboamination of Alkynes with Alkenes and Diazenes. J. Am. Chem. Soc. 2016;138:14570–14573. doi: 10.1021/jacs.6b09939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C. Shi Y. Odom A. L. A Titanium-Catalyzed Three-Component Coupling To Generate α,β-Unsaturated β-Iminoamines. J. Am. Chem. Soc. 2003;125:2880–2881. doi: 10.1021/ja0284714. [DOI] [PubMed] [Google Scholar]
- Priewisch B. Rück-Braun K. Efficient Preparation of Nitrosoarenes for the Synthesis of Azobenzenes. J. Org. Chem. 2005;70:2350–2352. doi: 10.1021/jo048544x. [DOI] [PubMed] [Google Scholar]
- Dunn S. C. Hazari N. Cowley A. R. Green J. C. Mountford P. Synthesis and Reactions of Group 4 Imido Complexes Supported by Cyclooctatetraene Ligands. Organometallics. 2006;25:1755–1770. doi: 10.1021/om0510526. [DOI] [Google Scholar]
- Blum S. A. Bergman R. G. Nitro and Nitroso Metathesis Reactions with Monomeric Zirconium Imido Complexes. Organometallics. 2004;23:4003–4005. doi: 10.1021/om0495776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin D. Y. Grubbs E. J. McCarty C. G. Uncatalyzed syn-anti Isomerization of Imines, Oxime Ethers, and Haloimines. J. Am. Chem. Soc. 1966;88:2775–2786. doi: 10.1021/ja00964a029. [DOI] [Google Scholar]
- See X. Y. Beaumier E. P. Davis-Gilbert Z. W. Dunn P. L. Larsen J. A. Pearce A. J. Wheeler T. A. Tonks I. A. Generation of TiII Alkyne Trimerization Catalysts in the Absence of Strong Metal Reductants. Organometallics. 2017;36:1383–1390. doi: 10.1021/acs.organomet.7b00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis-Gilbert Z. W. Kawakita K. Blechschmidt D. R. Tsurugi H. Mashima K. Tonks I. A. In situ Catalyst Generation and Benchtop-Compatible Entry Points for TiII/TiIV Redox Catalytic Reactions. Organometallics. 2018;37:4439–4445. doi: 10.1021/acs.organomet.8b00474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman J. I. Effect of conformational change on reactivity in organic chemistry. Evaluations, applications, and extensions of Curtin–Hammett Winstein–Holness kinetics. Chem. Rev. 1983;83:83–134. doi: 10.1021/cr00054a001. [DOI] [Google Scholar]
- Chiu H.-C. See X. Y. Tonks I. A. Dative Directing Group Effects in Ti-Catalyzed [2 + 2 + 1] Pyrrole Synthesis: Chemo- and Regioselective Alkyne Heterocoupling. ACS Catal. 2019;9:216–223. doi: 10.1021/acscatal.8b04669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y. Klein C. K. Tonks I. A. Synthesis of pentasubstituted 2-aryl pyrroles from boryl and stannyl alkynes via one-pot sequential Ti-catalyzed [2 + 2 + 1] pyrrole synthesis/cross coupling reactions. Chem. Sci. 2020;11:10236–10242. doi: 10.1039/D0SC01576H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakita K. Parker B. F. Kakiuchi Y. Tsurugi H. Mashima K. Arnold J. Tonks I. A. Reactivity of terminal imido complexes of group 4–6 metals: Stoichiometric and catalytic reactions involving cycloaddition with unsaturated organic molecules. Coord. Chem. Rev. 2020;407:213118. doi: 10.1016/j.ccr.2019.213118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield A. D. Nova A. Selby J. D. Schwarz A. D. Clot E. Mountford P. Reaction Site Diversity in the Reactions of Titanium Hydrazides with Organic Nitriles, Isonitriles and Isocyanates: Ti Nα Cycloaddition, Ti Nα Insertion and Nα–Nβ Bond Cleavage. Chem.–Eur. J. 2011;17:265–285. doi: 10.1002/chem.201002776. [DOI] [PubMed] [Google Scholar]
- Manßen M. de Graaff S. Meyer M.-F. Schmidtmann M. Beckhaus R. Direct Access to Titanocene Imides via Bis(η5:η1-penta-fulvene)titanium Complexes and Primary Amines. Organometallics. 2018;37:4506–4514. doi: 10.1021/acs.organomet.8b00264. [DOI] [Google Scholar]
- Barluenga J. Pozo C. d. Olano B. Reactions of N-Unsubstituted 4-Amino-1-azadienes Towards Electrophiles. Synthesis. 1996;1996:133–140. doi: 10.1055/s-1996-4160. [DOI] [Google Scholar]
- Carosso S. Miller M. J. Nitroso Diels–Alder (NDA) reaction as an efficient tool for the functionalization of diene-containing natural products. Org. Biomol. Chem. 2014;12:7445–7468. doi: 10.1039/C4OB01033G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maji B. Yamamoto H. Catalytic Enantioselective Nitroso Diels–Alder Reaction. J. Am. Chem. Soc. 2015;137:15957–15963. doi: 10.1021/jacs.5b11273. [DOI] [PubMed] [Google Scholar]
- Bleeke J. R. Hinkle P. V. Rath N. P. Synthesis, Structure, Spectroscopy, and Reactivity of a Metallathiabenzene. Organometallics. 2001;20:1939–1951. doi: 10.1021/om010050s. [DOI] [Google Scholar]
- Holland R. L. O'Connor J. M. Nitroso Compounds Serve as Precursors to Late-Metal η2(N,O)-Hydroxylamido Complexes. Organometallics. 2009;28:394–396. doi: 10.1021/om801105x. [DOI] [Google Scholar]
- Basuli F. Huffman J. C. Mindiola D. J. Reactivity at the β-Diketiminate Ligand Nacnac- on Titanium(iv) (Nacnac- = [Ar]NC(CH3)CHC(CH3)N[Ar], Ar = 2,6-[CH(CH3)2]2C6H3). Diimine-alkoxo and Bis-anilido Ligands Stemming from the Nacnac-Skeleton. Inorg. Chem. 2003;42:8003–8010. doi: 10.1021/ic034853e. [DOI] [PubMed] [Google Scholar]
- Basuli F. Bailey B. C. Watson L. A. Tomaszewski J. Huffman J. C. Mindiola D. J. Four-Coordinate Titanium Alkylidene Complexes:Synthesis ,Reactivity ,and Kinetic Studies Involving the Terminal Neopentylidene Functionality. Organometallics. 2005;24:1886–1906. doi: 10.1021/om049400b. [DOI] [Google Scholar]
- Avarvari N. Le Floch P. Ricard L. Mathey F. 1,3,2-Diazaphosphinines and -Diazaarsinines as Precursors for Polyfunctional Phosphinines and Arsinines. Organometallics. 1997;16:4089–4098. doi: 10.1021/om970414+. [DOI] [Google Scholar]
- Becker L. Strehler F. Korb M. Arndt P. Spannenberg A. Baumann W. Lang H. Rosenthal U. Unusual Nitrile–Nitrile and Nitrile–Alkyne Coupling of Fc–C N and Fc–C C–C N. Chem.–Eur. J. 2014;20:3061–3068. doi: 10.1002/chem.201304478. [DOI] [PubMed] [Google Scholar]
- Kortman G. D. Orr M. J. Hull K. L. Synthesis and Reactivity of Dioxazirconacyclohexenes: Development of a Zirconium–Oxo-Mediated Alkyne–Aldehyde Coupling Reaction. Organometallics. 2015;34:1013–1016. doi: 10.1021/om5008727. [DOI] [Google Scholar]
- Nguyen T. T. Kortman G. D. Hull K. L. Synthesis, Cycloaddition, and Cycloreversion Reactions of Mononuclear Titanocene–oxo Complexes. Organometallics. 2016;35:1713–1725. doi: 10.1021/acs.organomet.6b00111. [DOI] [Google Scholar]
- Doxsee K. M. Mouser J. K. M. Titanium-mediated synthesis of conjugated dienes. Tetrahedron Lett. 1991;32:1687–1690. doi: 10.1016/S0040-4039(00)74304-8. [DOI] [Google Scholar]
- Polse J. L. Andersen R. A. Bergman R. G. Cycloaddition and Cycloreversion Reactions of a Monomeric Ti(iv) Oxo Complex with Terminal and Internal Alkynes. A Reversible Oxametallacyclobutene/Hydroxoacetylide Interconversion. J. Am. Chem. Soc. 1995;117:5393–5394. doi: 10.1021/ja00124a036. [DOI] [Google Scholar]
- Ruck R. T. Zuckerman R. L. Krska S. W. Bergman R. G. Carboamination: Additions of Imine C=N Bonds Across Alkynes Catalyzed by Imidozirconium Complexes. Angew. Chem., Int. Ed. 2004;43:5372–5374. doi: 10.1002/anie.200461063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna T. A. Baranger A. M. Walsh P. J. Bergman R. G. Formation of .alpha,beta.-Unsaturated Imines and Successful Trapping of Oxozirconocene in a [4 + 2] Azaoxametallacyclohexene Retrocycloaddition. J. Am. Chem. Soc. 1995;117:3292–3293. doi: 10.1021/ja00116a042. [DOI] [Google Scholar]
- Doxsee K. M. Juliette J. J. J. Weakley T. J. R. Zientara K. Nitrosoarene and nitrosoalkane insertion reactions of titanacyclobutenes. Inorg. Chim. Acta. 1994;222:305–315. doi: 10.1016/0020-1693(94)03922-4. [DOI] [Google Scholar]
- Nakamoto M. Tilley T. D. Reactions of Zirconacyclopentadienes with Nitrosobenzene. Characterization of Zirconacycle Intermediates and Formation of N-Phenylpyrroles. Organometallics. 2001;20:5515–5517. doi: 10.1021/om010873h. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.