

Abstract
Immune checkpoint inhibitors (ICIs) induce a durable response in a wide range of tumor types, but only a minority of patients outside these ‘responsive’ tumor types respond, with some totally resistant. The primary predictor of intrinsic immune resistance to ICIs is the complete or near‐complete absence of lymphocytes from the tumor, so‐called immunologically cold tumors. Here, we propose two broad approaches to convert ‘cold’ tumors into ‘hot’ tumors. The first is to induce immunogenic tumor cell death, through the use of oncolytic viruses or bacteria, conventional cancer therapies (e.g. chemotherapy or radiation therapy) or small molecule drugs. The second approach is to target the tumor microenvironment, and covers diverse options such as depleting immune suppressive cells; inhibiting transforming growth factor‐beta; remodelling the tumor vasculature or hypoxic environment; strengthening the infiltration and activation of antigen‐presenting cells and/or effector T cells in the tumor microenvironment with immune modulators; and enhancing immunogenicity through personalised cancer vaccines. Strategies that successfully modify cold tumors to overcome their resistance to ICIs represent mechanistically driven approaches that will ultimately result in rational combination therapies to extend the clinical benefits of immunotherapy to a broader cancer cohort.
Keywords: immune checkpoint inhibitor, tumor microenvironment, immune surveillance and resistance, cold tumor, therapeutic strategy, cancer immunotherapy
In this review, we propose two broad approaches to convert ‘cold’ tumours into ‘hot’ tumours: targeting tumour cells, and remodelling the tumour microenvironment. Strategies that successfully modify cold tumours will ultimately result in rational combination therapies to extend the clinical benefits of immunotherapy to a broader cancer cohort.

Introduction
Solid tumors that lack or have few tumor‐infiltrating lymphocytes (TILs) are considered immunologically ‘cold’ (Figure 1a and b). 1 , 2 , 3 Cold tumors usually result from a low burden of tumor neoantigens or their total absence because of gene silencing. 4 Other causes include defective antigen presentation because of loss or mutation of β2‐microglobulin, 5 deletion of specific major histocompatibility complex (MHC) alleles, 6 defects in interferon (IFN)‐γ signalling 7 and defects of differentiation, migration and antigen processing by dendritic cells (DCs) 3 in response to soluble, tumor‐derived immune suppressive factors, such as interleukin‐10 (IL‐10) and transforming growth factor‐β (TGF‐β). TIL ‘desertification’ is also caused by the loss or low expression of specific chemokines and their corresponding receptors, such as CXCR3 and its chemokine ligands CXCL9 and CXCL10, 8 as well as CCR5 9 and its ligands CCL3, CCL4 and CCL5. 10 Finally, physical barriers such as a dense extracellular matrix, defective tumor vasculature and hypoxia can all be contributors. 3 In contrast, immunologically ‘hot’ tumors contain many infiltrating T cells (Figure 1b) whose implied antitumor activity is being held in check by immune checkpoints invoked either by the tumor or by the immunosuppressive tumor microenvironment (TME).
Figure 1.
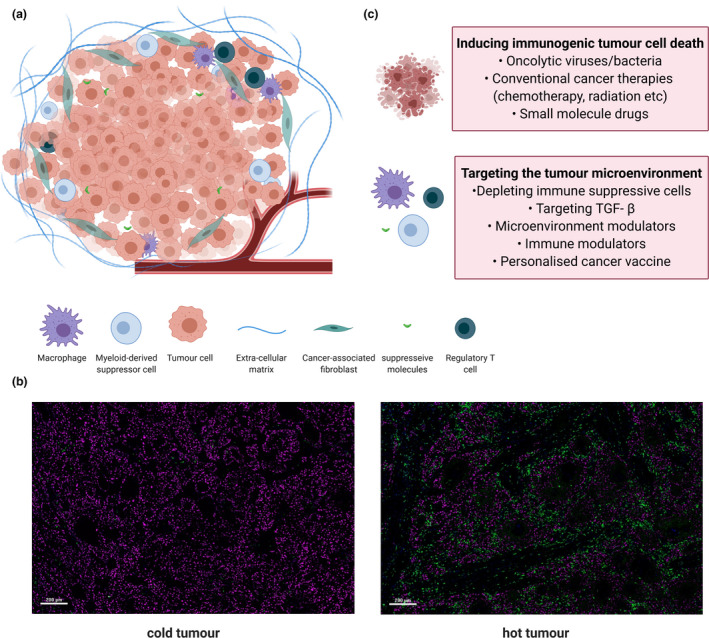
Therapeutic strategies to remodel immunogenically cold tumors. (a) Features of an immunologically cold tumor. (b) Representative fluorescent images showing the cold and hot head and neck tumors with T infiltration visualised by CD3+ T cells (green) and a tumor marker (magenta). (c) Two therapeutic strategies that focus on inducing immunogenic tumor cell death and targeting the tumor microenvironment to convert ‘cold’ tumors into ‘hot’ ones. TGF‐β: Transforming growth factor‐beta.
For decades, practitioners of immunotherapy operated under the presumption that the best way to unleash the power of the immune system on cancer was to make cancer cells more immunogenic, provoking an enormous number of studies that typically combined a putative tumor antigen and an adjuvant powerful enough to generate a T‐cell effector response, including cytokines, 11 synthetic virus‐like preparations such as Iscomatrix, 12 and varieties of DCs. 13 Remarkably, about a decade ago, studies began to elicit some significant clinical responses in advanced cancers, such as disseminated melanoma and non‐small‐cell lung cancer, even without the administration of a tumor antigen. These successful studies utilised immune checkpoint inhibitors (ICIs), which are typically blocking antibodies directed against cytotoxic T‐lymphocyte‐associated protein 4 (CTLA4), programmed cell death protein 1 (PD‐1) or programmed cell death 1 ligand 1 (PD‐L1). This approach proved successful because it de‐repressed and greatly amplified a pre‐existing T‐cell response to the tumor. To support this concept, responses to ICIs have mostly been observed in immunologically ‘hot’ tumors: those with high pre‐existing immune response. 14 , 15 Frequently, responsive tumors also carried a high mutational burden that implied tumor ‘neoantigens’ were being generated, processed and presented as potential T‐cell targets; and increased PD‐L1 expression in cancer cells and/or antigen‐presenting cells (APCs), indicative of suppression of an otherwise potentially effective T‐cell response. ICIs have a broad impact on the immune system, many treated patients experience a discrete spectrum of adverse events known as immune‐related adverse events (irAEs), 16 which represent systemic or organ‐specific immune damage at sites unrelated to their tumor – whether a therapeutic response to the cancer is achieved or not.
Where a therapeutic response does occur, it can be rapid and potent; however, the therapeutic impact of ICIs in immunologically 'cold' tumors, where the tumor has not been as immunogenic, is generally poor. 17 Notwithstanding the often‐sizeable clinical problem of treating autoimmune manifestations of ICI, the most pressing challenge for cancer therapy has now understandably refocused on next‐generation therapeutic strategies to remodel 'cold' tumors into ‘hot’ ones, either prior to or coincident with ICI being administered. Attempts at improving immunogenicity have focused on improving immunogenicity in the tumor, reducing immunosuppression in the TME and enhancing T‐cell activation through utilising various approaches to: (1) target tumor cells, (2) target the TME or (3) augment immunity with presensitised adoptively transferred immune cells. 18 Here, we review and compare the progress and challenges of strategies that focus on tumor cells and their surrounding TME to transform ‘cold’ tumor into ‘hot’ ones (Figure 1c).
Inducing immunogenic tumor cell death (ICD)
Promoting a tumor’s immunogenicity means improving its ability to induce an immune response. Immunogenicity can be influenced by many factors, but two with paramount importance are antigenicity and adjuvanticity. 19 Antigenicity means that the tumor can generate antigen targets to which the host has not been tolerised. Given the genetic instability of many forms of cancer, antigenicity can be achieved via the expression of neoantigens, viral oncoproteins and overexpressed self‐antigens. 20 , 21 Neoantigens have been widely investigated in personalised cancer vaccine approaches as they result from mutated or ectopic proteins and peptides, and are often specific to that individual’s tumor. 22 Adjuvanticity results from the presence of danger‐associated molecular patterns (DAMPs) that initiate tumor antigen recognition by APCs and the subsequent activation of the innate and adaptive immune systems. 23 Tumor cell death that is associated with the release of DAMPs that bind to specific receptors on the surface of APCs to activate the antitumor immune response is referred to as ICD. Until recent years, ICD was often referred to as immunogenic apoptosis as the majority of ICD occur via apoptosis. 24 Recently, many other forms of cell death, including necroptosis, pyroptosis and ferroptosis, have been studied for their capacity to induce a durable immune response. 25 Enormous efforts have been made to induce the tumor itself to undergo ICD and thus activate antitumor immunity (Figure 2). These approaches have included (1) oncolytic viruses (OVs) or bacteria that selectively replicate in tumor cells, releasing soluble antigens and generating danger signals; (2) conventional cancer therapies (radiotherapy, chemotherapy and phototherapy) that induce DNA damage and generate a ‘vaccine pool’ of neoantigen in situ to initiate the immune response; and (3) small molecule drugs that target a cancer’s oncogenic pathways and/or positively influencing the TME.
Figure 2.
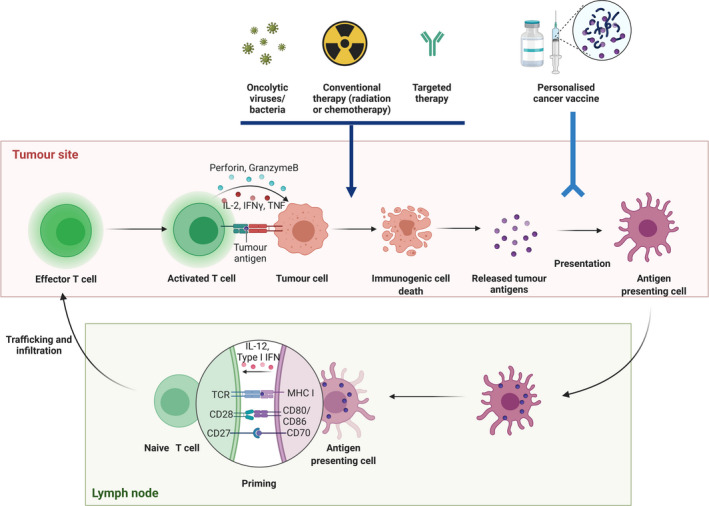
The cancer immunity cycle and selective methods to increase tumor immunogenicity. Tumor antigens are released as cancer cells die and are captured by the antigen‐presenting dendritic cells in the tumor site, and further presented to T cells in the lymph node. T cells are activated and proliferate, and effector T cells traffic and infiltrate into the tumor site. Activated effector T cells recognise and kill the cancer cells. Oncolytic viruses/bacteria, conventional therapy and targeted therapies are able to induce immunogenic cell death. Personalised cancer vaccines typically comprise tumor‐associated antigens or tumor‐specific antigens in a vaccine vector (RNA, DNA, viral, bacteria, protein, peptide and antigen‐presenting cells) with an immune adjuvant. IFN, interferon; IL, interleukin; MHC, major histocompatibility complex; TCR, T‐cell receptor; TNF, tumor necrosis factor.
Oncolytic viruses/bacteria
Oncolytic viruses play important roles in different steps in the cancer immunity cycle 26 , 27 : (1) inducing ICD through selective replication in tumor cells; and (2) triggering innate and subsequent adaptive immune responses through the release of soluble antigens and danger signals 28 (Figure 2). To date, both DNA and RNA viruses have been used to generate OVs. 29 Advantages of DNA viruses include a large genome that can be edited without compromising replication, and high‐fidelity DNA polymerases that ensure the integrity of the viral genome. Conversely, the small genome of RNA viruses has limited ability to encode large transgenes. However, the smaller RNA viruses can go through the blood–brain barrier and potentially target the tumors in the central nervous system. Pre‐existing immune response to some RNA viruses is also less common, making them more suitable for systemic delivery. 28
Currently, two oncolytic immunotherapies have been approved by the US Food and Drug Administration (FDA): one is intravascular injection of bacillus Calmette–Guérin (BCG, live attenuated tuberculosis vaccine) for the treatment of bladder cancer 30 ; and the other is talimogene laherparepvec (T‐VEC), an attenuated herpes simplex virus type 1 (HSV‐1) that is engineered and approved for advanced, refractory melanoma. T‐VEC is engineered to improve antigen presentation and T‐cell priming by inserting the gene for human granulocyte–macrophage colony‐stimulating factor (GM‐CSF). 31 While intradermal BCG has been used for bladder cancer for several decades, more recent approval for T‐VEC was based on data from the pivotal phase III OPTiM trial, which demonstrated tumor control at certain anatomical sites and favorable tolerability. 32 However, T‐VEC was not associated with prolonged survival as visceral metastatic sites respond poorly, so its broader uptake as a single agent in patients with metastatic disease has been low, and thus far limited to melanoma. 33 , 34
Despite this, the initial proof‐of‐concept studies with T‐VEC have proved attractive for combination therapies. Several studies have shown that the adjuvant‐like activities of the OVs could turn melanoma, 35 , 36 triple‐negative breast cancer (TNBC) 37 and brain tumors 38 ‘hot’, effectively priming them for subsequent ICI treatment. Ribas et al. 35 investigated the effect of T‐VEC on immune cell infiltration and subsequent treatment efficacy of anti‐PD‐1 antibody in a phase 1b clinical trial. In 21 patients with advanced melanoma, tumor size decreased by 62%, while 33% experienced a complete response. Another virus to show promise in preclinical mouse models is the Maraba rhabdovirus, shown by Bourgeois‐Daigneault et al. 37 to provide long‐term protection against TNBC and to sensitise 4T1 tumors to ICI. 37 A recent report on a phase I trial using coxsackievirus A21 against non‐muscle‐invasive bladder cancer demonstrated acceptable safety profile and virus‐induced ‘immunological heat’ within the tumor microenvironment. 39 Samson et al. 38 showed that reovirus could up‐regulate the expression of IFN‐regulated genes and PD‐1/PD‐L1 axis expression in a preclinical glioma model. Addition of anti‐PD‐1 to reovirus enhanced systemic response in these tumors. 38 In addition, studies that augment the OV's function via cytokine and chemokine coexpression have been reported. Ge et al. 40 showed that OV delivering tethered IL‐12 was able to avoid systemic toxicity and cure all mice with late‐stage colon cancer, by facilitating T‐cell infiltration, increasing IFN‐γ and decreasing suppressive factor TGF‐β in the tumor.
Other approaches utilise rotavirus, influenza or the yellow fever vaccines to elicit antitumor immunity and may potentially be quickly approved, given their extensively understood safety profile. For instance, when administered via intratumoral injection, the FDA‐approved unadjuvanted seasonal influenza vaccine could inhibit tumor growth by increasing the number of antitumor TILs and decreasing regulatory B cells in a mouse model. 41 This process creates systemic immune responses and sensitises tumors to subsequent ICIs. Importantly, intratumoral vaccination also generates protection against subsequent influenza virus lung infection. A recent study reported that pyroptosis‐induced inflammation initiates rigorous antitumor immunity and can synergise with ICIs. 42 A murine study demonstrated that by implanting Gasdermin E‐positive B16 melanoma cells, mice were protected from subsequent challenge with wild‐type B16 cells. This was because of the pyroptosis of Gasdermin E‐positive B16 melanoma cells, which enhanced antitumor immunity through tumor‐associated macrophages, tumor‐infiltrating natural killer (NK) and cytotoxic T‐lymphocyte (CTL) cells. 43
Conventional cancer therapies
Radiotherapy is a major treatment option for many cancer patients. In addition to mediating DNA damage‐induced cancer cell death, radiotherapy can modulate tumor immunogenicity by activating the cyclic guanosine monophosphate–adenosine monophosphate synthase (cyclic GMP‐AMP synthase) – stimulator of interferon gene (cGAS‐STING) pathway. 44 , 45 Several studies have demonstrated activation of the cGAS‐STING pathway and radiation‐induced type I interferon induction, 46 with increased MHC‐I expression 47 , 48 and antigen processing and presentation within the tumor. 49 These events result in the activation of innate and, subsequently, adaptive antitumor immunity, and increase the tumor’s immune responsiveness. 50 , 51 Indeed, the immunogenic effects of radiation can result in systemic immunity, reflected in shrinkage of non‐irradiated distal lesions, referred to as the abscopal effect, 52 , 53 , 54 a feature that can be further harnessed by combination ICI. Recent studies reported the induction of systemic antitumor T‐cell responses in chemorefractory metastatic lung cancer following irradiation and CTLA4 blockade. 55 However, overall radiotherapy dose and fraction must be cautiously tuned, as high doses of radiation can attenuate STING activation and even damage TILs recruited to the irradiated organ. Preclinical data suggest that moderately hypofractionated radiotherapy offers the best chance of favorable immunomodulation. 56 Unfortunately, these positive impacts of radiotherapy remain relatively rare and are counterbalanced by activation of immune suppression, via increased TGF‐β secretion by cancer‐associated fibroblasts (CAFs). 57 Hence, radiotherapy combined with TGF‐β inhibition may potentially enhance systemic antitumor responses. Cross‐presentation by specialised DCs is critical for priming tumor‐specific T‐cell responses against solid tumors, 58 and this process could be induced through radiotherapy. 52 A recent report on utility of a in situ vaccine, combining Fms‐like tyrosine kinase 3 ligand (Flt3L), radiotherapy and a Toll‐like receptors (TLR)‐3 agonist, demonstrated systemic clinical tumor regression through recruitment, antigen loading and activation of intratumoral, cross‐presenting DCs. 59
Ultimately, the interplay between multiple variables including radiotherapy dose, fractionation, schedule and choice of systemic immune mediator will all need to be considered, and carefully evaluated in preclinical and translationally focused clinical trials.
It has long been appreciated that some, but not all, chemotherapy can induce antitumor immune responses. 60 , 61 Chemotherapy may elicit antitumor immunity via three main mechanisms: the 'on‐target' or direct impact on tumor cells that induce their immunogenicity; the 'off‐target' effects on different immune cell subsets; and impact on whole‐body physiology that reshape antitumor immunosurveillance. 62 Like radiotherapy, chemotherapy‐induced DNA damage may also trigger the cGAS‐STING pathway to increase tumor immunogenicity. 63 There is evidence that chemotherapy can activate immune cells, such as M1‐like macrophages, 64 APCs 65 and T cells. 66 However, chemotherapy also has limitations 67 : chemotherapeutic agents are usually strongly cytotoxic and eliminate tumor cells that might otherwise present neoantigens generated by DNA damage. In any event, new chemotherapy‐induced mutations tend to be limited as treatment generally applied to established cancers, potential exceptions being chemotherapy given in an adjuvant setting. Interestingly, ICI combined with chemotherapy is associated with a high risk of adverse events, 68 as ICIs are known to have a distinct toxicity profile from that caused by conventional chemotherapy. ICI‐related adverse events are identified as irAEs and occur commonly in skin, gut, lung, skeletal muscle and various endocrine organs including the pituitary gland. 69 While advert events are increased in patients treated with chemotherapy and ICI, to date there are no in vivo mechanistic studies exploring this issue. It is possible that this combination treatment is particularly effective at breaking peripheral immune tolerance; however, this remains to be proven. Despite this, combined chemotherapy and ICI has become the standard of care and is under continual refinement for patients with breast, non‐small‐cell lung cancer, colorectal, gastric, gastro‐oesophageal carcinoma or some lymphomas. 62
Photothermal therapy (PTT), a form of hyperthermia, can also induce ICD. 70 By focusing precisely controlled near‐infrared (NIR) laser light onto a tumor, tumor‐associated antigens (TAAs) can be released in situ. 71 Huang et al. 72 reported a strategy for controlled release of anti‐PD‐L1 via controlled NIR, which increased the recruitment of TILs and boosted the immune activity against tumors. Zhang et al. 73 further investigated the combination of PTT agent IR820 and indoleamine 2,3‐dioxygenase (IDO) inhibitor 1‐methyl‐tryptophan (1MT), which evoked a helpful immune response against tumor metastasis and recurrence. These data provide a promising alternative to transform a 'cold' tumor into a 'hot' one. High‐intensity focused ultrasound (HIFU) is a non‐invasive therapy that is used in the clinic to thermally ablate solid tumors with high spatial precision, 74 and to mechanically disrupt tumors. 75 HIFU‐mediated tumor fractionation may cause ICD and increase inflammation 76 and potentially lead to immune sensitisation. 77 Eranki et al. 78 recently demonstrated a combination of HIFU and ICI significantly enhanced systemic antitumor responses and cured the majority of mice with large, established unilateral and bilateral neuroblastoma tumors. Most interestingly, a significant abscopal effect was observed using HIFU, suggesting a long‐term memory response. These studies provide proof‐of‐concept data for using HIFU to stimulate systemic immunity that may act as an adjuvant to ICI therapy.
Small molecule drugs
Tumor‐intrinsic oncogenic signalling pathways can also play a role in driving or blunting an immune response; hence, targeted therapies could potentially exert direct effects on antitumor immunity (Figure 2). For example, Deng J et al. 79 discovered that drugs inhibiting cyclin‐dependent kinases 4 and 6 (CDK4/6) could increase T‐cell activation and facilitate their penetration into tumors. Goel et al. 80 demonstrated CDK4/6 inhibitors enhanced tumor antigen presentation through stimulation of type III IFN production, as well as suppressed regulatory T‐cell (Treg) proliferation. These data indicate CDK4/6 inhibitors are able to boost tumor immunogenicity 81 and might have a synergistic effect with ICIs. As CDK4/6 inhibitors have already been approved for breast cancer, and they act synergistically with anti‐PD‐1 therapy in mouse models, the findings demonstrate the potential of combining CDK4/6 and ICIs in clinical trials.
The mitogen‐activated protein kinase (MAPK) kinase (MEK) pathway is frequently up‐regulated in various human solid tumors. A recent study showed that MAPK activity could suppress the expression of MHC‐I and MHC‐II, and tumor cells can evade antigen presentation through activating the MAPK pathway in TNBC. 82 Preclinical models have demonstrated that MEK inhibitors may increase T‐cell infiltration into the tumor site and improve survival of TILs. 83 Similar results were observed with BRAF inhibitors in melanoma. 84 , 85 BRAF inhibitors can abrogate an immune suppressive TME and augment effector T‐cell infiltration and function. 86 , 87 , 88 These results offer a rational therapeutic strategy linking inhibitors targeting the BRAF‐MAPK pathway with cancer immunotherapy. A recent phase 1b clinical trial demonstrated MEK inhibitor, cobimetinib, in combination with anti‐PD‐L1, atezolizumab, had manageable safety and clinical activity in cold tumors, irrespective of KRAS or BRAF status. 89 However, a phase III (IMblaze370) clinical trial investigating this combination in previously treated metastatic colorectal cancers did not meet the primary endpoint of improved overall survival. 90 Data suggest that BRAF/MEK inhibitors favorably alter the TME within two weeks; however, this effect is lost by several weeks after treatment. 91 These results emphasise the importance of considering the sequencing and ideal timing of combination therapies.
A third example comes in the form of agents that block the DNA damage response, whose normal function is to ensure genome integrity, but which can act to preserve cancer cell viability. Therapeutic inhibition of DNA damage response with agents such as poly (ADP‐ribose) polymerase (PARP) inhibitors has improved clinical benefits for BRCA‐mutated tumors. 92 Consequently, PARP inhibitors generate high levels of DNA damage, and increase T‐cell infiltration, IFN‐γ production and PD‐L1 expression. 93 Combining DNA damage inhibitors such as PARP inhibitor with ICIs may increase response rates to ICIs, as is under investigation in multiple clinical trials. 94
Targeting the TME
The second major strategy to amplify antitumor immunity is to target the TME, so as to facilitate an immune response to the cancer cells embedded within it. As the TME is almost always suppressive for T‐cell responses, such efforts typically include inhibiting immune suppressive cells, such as Tregs and myeloid‐derived suppressive cells (MDSCs); decreasing the expression or the effects of intensely immune suppressive molecules such as TGF‐β; using immune modulators to enhance the recruitment, trafficking and activity of APCs and TILs; and reversing adverse metabolic factors such as hypoxia by remodelling the tumor vasculature or other means.
Depleting immune suppressive cells
Cancers very frequently accumulate immune suppressive cells, such as Tregs and MDSCs, in the TME. Tregs promote tumor growth through inhibiting antitumor immune responses. 95 MDSCs are a heterogeneous group of immature myeloid cells with strong immune suppressive functions 96 , 97 that impede the local immune response. Certain molecular signalling pathways were shared between Tregs, MDSCs and immune effector cells; hence, it poses a significant challenge to specifically target Tregs or MDSCs without compromising a variety of immune effector cells in the TME. 98 With recent advances in technology, immune suppressive cells with specific biological properties such as Th1‐like Tregs (T‐bet+IFN‐γ+Foxp3+) have been defined. 99 Increased expression of IFN‐γ by Tregs drives the fragility of surrounding wild‐type Tregs, boosts antitumor immunity and, most importantly, is required for responsiveness to anti‐PD1. 100 In addition, the body is capable of developing different strategies against antitumor immunity. Recent studies demonstrated depletion of Tregs in large tumors induced the generation of MDSCs 101 and reprogrammed fibroblast differentiation, 102 thus promoting tumor growth. Undoubtedly, immune suppressive cells are responsible for maintaining peripheral tolerance, so depleting these cells other than in cancerous tissues increases the risk of systemic irAEs.
Targeting TGF‐β
Depleting or functionally blocking immune suppressive molecules such as TGF‐β holds greater promise. TGF‐β tends to suppress tumor growth at early stages of tumorigenesis. 103 However, during the tumor’s progression, TGF‐β instead promotes tumor growth by facilitating immune evasion and epithelial‐to‐mesenchymal transition. 104 , 105 Three forms of inhibitor might in principle block the TGF‐β pathway – small molecule inhibitors, antibodies and receptor‐based TGF‐β traps, 106 all of which have shown promising effects in preclinical studies and progressed to clinical trials. Galunisertib is the most promising small molecule inhibitor, targeting the TGF‐βRI kinase. The most extensively studied antibody, fresolimumab (GC1008), which sequesters all isoforms of TGF‐β, has also progressed to clinical trials. 107 However, both small molecules and antibodies have had little clinical impact. By contrast, receptor‐based TGF‐β traps appear to hold greater promise. Bintrafusp alfa (M7824) is an innovative first‐in‐class bifunctional fusion protein composed of an IgG1 PD‐L1 antibody and the extracellular domain of the human TGF‐β receptor II (TGF‐βRII or TGF‐β trap). 106 Preclinical studies manifested that bintrafusp alfa inhibited tumor growth and spontaneous metastasis more effectively than either the TGF‐β trap or anti‐PD‐L1 alone. 108 In patients with heavily pretreated advanced solid tumors, bintrafusp alfa has demonstrated manageable safety with durable clinical efficacy, 109 , 110 , 111 and encouraging long‐term survival in a two‐year follow‐up study. 112 Clinical studies of bintrafusp alfa in combination with radiotherapy or chemotherapy are currently proceeding.
TME modulators
Stromal cells within the TME also play a key role in inhibiting immune responses to support tumor growth. The extracellular matrix produced by CAFs hinders the immune response via blocking TIL infiltration, compressing tumor blood vessels and inducing hypoxia. 113 Hence, combining ICIs with antiangiogenic therapies, including vascular endothelial growth factor (VEGF) inhibitor – bevacizumab 114 ; anti‐VEGFR‐2 antibody – ramucirumab 115 ; and small molecule antiangiogenic multi‐kinase inhibitors such as axitinib, 116 offer a rational therapeutic strategy and have shown meaningful activity in clinical trials. A phase I study that combined CTLA4 blockade (ipilimumab) and bevacizumab 117 found that, compared with patients receiving ipilimumab alone, those who received combination therapy had substantially higher infiltration of effector T cells and macrophages in their tumor, and increased circulating memory T cells. The combination of pembrolizumab and axitinib for advanced renal cell carcinoma was assessed in a phase III study. 116 The pembrolizumab–axitinib group resulted in prolonged overall‐ and progression‐free survival, as well as a 59.3% objective response rate. 116 The same strategy is now under investigation in multiple clinical trials to refine the optimal patient characteristics for this approach.
Studies have shown that hypoxia drives recruitment of MDSCs and tumor‐associated macrophage. 118 , 119 Jayaprakash et al. 120 reported that the hypoxia‐activated prodrug TH‐302 caused an influx of T cells into hypoxic zones and significantly reduced MDSC density. Consistent with these observations, combining the hypoxia/prodrug and ICIs cured more than 80% of mice with experimental prostate cancer. 120 These findings suggest that targeted hypoxia reduction may restore antitumor immune responses and resensitise hypoxic tumors to ICIs. A further adverse effect of tumor hypoxia is that cancer cell consumption of adenosine triphosphate (ATP) is accelerated, significantly increasing the production of adenosine diphosphate (ADP) in the TME. 121 In turn, ADP is reduced to potently immunosuppressive free adenosine through the action of cancer cell surface exoenzymes such as CD39 and CD73 induced by the hypoxia, in effect, a ‘vicious cycle’ of events. Adenosine binding (through receptors with various isoforms) adversely affects virtually every facet of humoral and cellular immunity, and binding to the A2A isoform expressed on TILs dampens IFN‐γ secretion and CTL/NK cytotoxicity. Beavis et al. 122 have recently shown that pharmacological inhibition of adenosine binding can greatly amplify ICI, particularly when the two agents are coadministered with CAR T cells.
Immune modulators
CD40 is broadly expressed on a wide range of immune cells, such as DCs, B cells and macrophages. It is a cell surface member of the TNF receptor superfamily and plays a major role in activating and licensing DCs to prime effective T cells and re‐educate macrophages. 123 CD40 agonists have been developed and evaluated as a novel cancer immunotherapy, 123 and manifested T‐cell‐mediated 124 or macrophage‐dependent 125 antitumor activity. CD40 agonists, in conjunction with chemotherapy, have shown a significant effect on suppressing tumor growth in pancreatic cancer, 126 mesothelioma 127 and other advanced solid tumors. 128 These results suggest CD40 activation could be a critical mechanism to convert ‘cold’ tumors into ‘hot’ and in particular sensitise them to chemotherapy or ICIs.
Pattern recognition receptors (PRRs) are a diverse family of receptors and are widely expressed on innate immune cells. 129 Five families of PRRs have been reported: Toll‐like receptors (TLRs), nucleotide‐binding oligomerisation domain (NOD)‐like receptors (NLRs), C‐type lectin receptors (CLRs), RIG‐I‐like receptors (RLRs) and cytosolic DNA sensors (CDSs). The immunostimulatory characteristics of PRRs can alter the immune suppressive TME and prompt the activation of APCs, driving tumor‐specific T‐cell responses. Studies have shown that PRRs agonists can activate antigen presentation through resident myeloid cells in the TME. 130 Toll‐like receptor 9 (TLR9) activation can foster innate and adaptive immune responses, subsequently improving immune‐mediated tumor control. Kapp et al. 131 showed lefitolimod, a potent TLR9 agonist, could activate immune cells and facilitate their differentiation into antitumor effector cells and their trafficking into the TME. Zanker et al. 132 recently demonstrated intratumoral administration of TLR 7/8 agonist 3M052 was able to induce a T‐cell‐inflamed TME and suppress lung metastasis in preclinical TNBC. However, depending on the TLR and the tumor type studied, TLR agonists showed apparently contradictory results by either promoting or suppressing tumor progression in preclinical studies. 133 These inconsistent results make it difficult to ensure safety or efficacy when moving into human studies.
The STING pathway is activated when DNA is detected within the cell cytoplasm and binds to cGAS, which then generates cGAMP. 134 Downstream STING signalling leads to APC activation and cytokine production, and subsequently promotes the priming and recruitment of effector immune cells. 135 Thus, STING agonists may potentially boost de novo innate and subsequent adaptive immune responses, and have the potential to transform the therapeutic landscape if optimised. The first STING agonist investigated in the immunotherapy field was the molecule DMXAA. DMXAA showed antitumor activity in preclinical models but was subsequently demonstrated to activate mouse, but not human, STING. 136 The first generation of human STING agonists includes MIW815 (ADU‐S100) and MK‐1454. However, because of ubiquitous STING expression in tumor and normal tissue, 137 these agents induce inflammatory cytokines with little specificity when administered systemically in mouse cancer models. This feature restricts their application in clinical trials to direct intratumoral injection and, as a result, limits their potential use to a narrow set of tumors. Thus, STING agonists that are suitable for systemic administration are needed. A small number of intravenously administered STING agonists have recently begun evaluation in clinical trials (NCT03843359, NCT04420884 and NCT04096638). Undoubtedly, non‐nucleotide small molecule STING agonist that can be administrated systemically is the next focus of STING agonist development, providing this can be done safely at a therapeutic dose, given the expected immune activation with such an approach. Recent studies reported by Pan et al. 138 and Chin et al. 139 have demonstrated promising results in preclinical models.
Indoleamine‐pyrrole 2,3‐dioxygenase (IDO)‐1 catalyses the conversion of amino acid tryptophan to kynurenine, which then suppresses immune effector function and activates suppressive immune cells. Preclinical studies demonstrated that IDO1 inhibitors alleviate the suppression of CTL and NK cells. 140 It has been hypothesised that as immune‐metabolic adjuvants, IDO‐1 inhibitors may exert little effect on their own, but may extend the efficacy of ICIs. Unfortunately, combining anti‐PD1 with epacadostat, a selective IDO‐1 inhibitor, did not improve progression‐free or overall survival over anti‐PD1 monotherapy in advanced melanoma. 141 The use of IDO‐1 inhibition to sensitise tumors to ICIs thus remains uncertain, although it must be noted that combined ICI/IDO trials with other IDO inhibitors remain in development.
Among cytokines, interferons are considered the most potent for inducing cellular immune responses. 142 Continuous successful discoveries have been made since 1969, when Ion Gresser reported the antitumor effect of purified murine interferons in mice. 143 Clinical trials soon followed using partially purified IFN‐α from healthy human donor blood leucocytes. 144 Leucocyte‐derived IFN‐α was used to treat 38 patients with malignant lymphoma, multiple myeloma and metastatic breast cancer, and objective tumor regression occurred in 50%. 145 Recombinant IFN‐α2 thus became the first approved human immunotherapeutic for cancer treatment. Subsequently, IFN‐α demonstrated antitumor effects in various tumor types including melanoma, Kaposi sarcoma, bladder and renal cell carcinomas. 142 Furthermore, IFN‐α2 proved to be clinically effective in viral infections, 146 and IFN‐β in relapsing and remitting multiple sclerosis. 147 However, further clinical interest and use were reduced as IFN‐α2 and IFN‐β have been associated with systemic adverse effects, which are often dose‐limiting. 148
IFN‐γ has also been evaluated as a single‐agent cancer treatment in several prospective randomised trials 149 before the modern era of cancer immunotherapy. Limited clinical benefit has been achieved, although Zhang et al. 150 observed that tumor MHC‐I expression was increased, along with T‐cell infiltration and PD‐L1 expression when systemic IFN‐γ was given at weekly intervals. These data suggest that systemic IFN‐γ may potentially convert cold tumors into ‘hot’ tumors and work in concert with ICI therapy.
Other immune cytokines, such as IL‐2, IL‐12, IL‐7, IFN‐γ and tumor necrosis factor (TNF), are able to mediate the recruitment and expansion of lymphocytes within the tumor. The strategy of activating these cytokines specifically at the tumor site to maximise their effects while reducing systematic toxicity has been challenging, high‐dose IL‐2 having been administered to patients for close to 40 years. 151 Some of these approaches combine an antibody targeting a tumor antigen with a modified cytokine. Cergutuzumab amunaleukin is a CEA‐targeted immune cytokine, which comprises a single IL‐2 variant moiety with abolished CD25 binding and a carcinoembryonic antigen (CEA)‐specific antibody (CEA‐IL2v). 152 Superior efficacy was observed with CEA‐IL‐2v plus anti‐PD‐L1 in CEA‐positive preclinical tumor models compared with anti‐PD‐L1 monotherapy. 152 Kim et al. 153 demonstrated systemic delivery of recombinant human IL‐7 increased both tumor‐reactive and bystander CD8 TILs in tumors.
One major challenge facing immune modulators, especially cytokines and immune agonists, is unacceptable systemic toxicity, which may in part be avoided by intratumoral administration. STING or TLR agonists are currently being evaluated in this way, although systemic toxicity is not completely eliminated. An alternative is to use a prodrug approach. NKTR‐214 is a recombinant human IL‐2 preparation that is masked with polyethylene glycol (PEG). It becomes active only when the PEG chains are hydrolysed 154 and is now being investigated in a phase I/II clinical trial. 155
Personalised cancer vaccines
As many cancers accumulate a range of genetic alterations, it ought in principle be possible to raise a robust immune response against antigens unique to the tumor. 156 Cancer vaccines usually comprise soluble TAAs or tumor‐specific antigens (e.g. oncogenic viral or neoantigens) in a vaccine vector (e.g. protein or peptide‐based, nucleic acid‐based, viral or bacterial vector‐based or cell‐based) combined with immune adjuvants (e.g. TLR agonist, CD40 agonist, STING agonist or GM‐CSF) (Figure 2). 157 They have proven immunogenic in many preclinical models and resulted in some encouraging results in early clinical trials. 157 The typical workflow for neoepitope selection and vaccine manufacture involves three steps 157 : first, identify the available mutations through whole‐exome sequencing (WES) and narrow the spectrum to expressed genes via RNA sequencing (RNA‐seq); second, DNA from normal tissue is used to determine the human leucocyte antigen (HLA), and neoepitopes are ranked for binding to the patient’s HLA alloforms; and third,, validated epitopes are incorporated into a personalised cancer vaccine with an immune adjuvant and administered to patients.
Three different platforms of personalised neoantigen vaccines have been tested in melanoma patients: (1) DC vaccines (ClinicalTrials.gov identifier: NCT00683670) 158 ; (2) synthetic long peptide vaccine (NeoVax, NCT01970358) 159 ; and (3) RNA vaccine (IVAC MUTANOME, NCT02035956). 160 These studies demonstrated that the approach is feasible, is safe and induces strong neoepitope‐specific T‐cell‐dependent immune responses, encouraging accelerated development of this approach. Hepatocellular cancer (HCC) is also being evaluated in a randomised European multi‐centre phase I/II clinical trial. 161 However, recent data demonstrated exome‐derived mutated HLA ligands to be rarely presented in HCC. 162 In addition, generating effective immunity against tumors with a relatively low mutational load, such as glioblastoma (GBM) and virtually all paediatric cancers, is another challenge. Despite this, a phase I clinical trial testing multi‐epitope, personalised neoantigen vaccination in GBM recently reported increased TIL infiltration with enriched memory phenotype. 163
As suggested above, HLA epitope prediction is a critical challenge for cancer vaccine development, as current algorithms have limited predictive power. Recently, performance improvement has been achieved in this field. To predict HLA class I peptide presentation across a large fraction of the population, Sarkizova et al. 164 eluted 95 HLA‐A, HLA‐B, HLA‐C and HLA‐G mono‐allelic cell lines and profiled more than 185 000 peptides using mass spectrometry. They identified canonical peptides, and unique and shared binding submotifs for each HLA alloform, and other motifs whose presentation varied with peptide length. 164 This algorithm then correctly predicted endogenous peptide presentation for more than 75% of peptides generated in 11 patient‐derived tumor cell lines. 164
Of course, the immunosuppressive TME undoubtedly reduces the efficacy of neoantigen vaccines. 156 Combining these vaccines with other approaches such as inhibiting immune suppressive molecules, depleting immune suppressive cells and blocking immune checkpoints is also proceeding. Recent studies have shown that synergistic or additive effects can be observed between ICIs and cancer vaccines. 165 , 166 Having moved beyond the first critical hurdle of clinical development, future studies on personalised neoantigen vaccines will focus on neoantigen prediction; manufacturing efficiencies, turnaround time and affordability; and identifying the most suitable clinical settings for this approach.
The idea of targeting the non‐mutated TAAs is fascinating as certain solid tumor types have low mutational burden. And even in melanoma, which is considered to have a high mutation rate, a significant proportion of patients harbour only a moderate‐to‐low mutational burden. 167 Nonetheless, the clinical studies in TAA‐based cancer vaccine have been mostly disappointing over the past twenty years, largely because of low immunogenicity. 168 Nonetheless, an RNA vaccine targeting multiple non‐mutated TAA prevalent in melanoma recently induced durable objective response in ICI‐experienced melanoma patients in a phase I clinical trial. 169 Most importantly, the antigen‐specific cytotoxic T‐cell responses are durable at a level that are generally reported for adoptive T‐cell therapy in some responders. 169 These results shed light on the feasibility of using conventional tumor‐associated antigens as targets for cancer vaccination in patients with relatively low mutational burden.
Conclusion
Investigators and clinicians in the field of immuno‐oncology are now armed with a considerable range of approaches to address the intrinsic immune resistance of cold tumors. The main challenge now is how to rationally combine and dose these many options to maximise therapeutic impact and understand what is being achieved pharmacodynamically while minimising off‐target effects. A necessary prerequisite for success is to more comprehensively understand tumor–immune interactions across the gamut of clinical cancer development – not only is each case unique at the time of presentation, but the interplay between cancer and host immunity is bound to have been shaped by the immunomodulatory mutations cancer accumulates over time. Understanding the dynamic changes in cancer immunity as a result of previous therapies is also of utmost importance. High‐resolution analyses, such as single‐cell sequencing and high‐dimensional cell phenotyping, are now enabling the capture of various immune cells in the TME and determination of their molecular and functional status. To capture the dynamic changes in cellular composition and functional status, both baseline and on‐treatment biopsies are in need.
In this review, we have outlined many possible strategies to remodel immunologically cold tumors and sensitise them to ICIs; the potential promise of each is counterbalanced by limitations for clinical translation. Given the countless possibilities, we believe a reasonable starting point is to focus on extending sometimes remarkable benefits currently enjoyed by a minority of patients treated with ICIs to a broader patient population. One reasonable way to assess progress and track potential success is to build approaches that make ‘cold’ tumors progressively ‘warmer’. We trust this review opens new possibilities and encourages the rational design of such therapeutic interventions.
Apart from the strategies discussed here, some other options have also been demonstrated to improve tumor immunogenicity, such as epigenetic modulators. 170 Strategies that involve the adoptive transfer of immune cells generated, stimulated and/or genetically modified (prime among them, CAR T cells), also hold great promise. 171 As a result of constraints in length and scope, these topics are not discussed in detail here.
Conflicts of interest
PJN received research grants from Roche/Genentech, BMS, Allergan, Compugen and Juno/Celgene, outside the submitted work. JD reports research support from Roche Genentech, Lilly, Astra Zeneca, BeiGene, Novartis, Bristol‐Myers Squibb and GlaxoSmithKline, and consulting fees from Amgen, Eisai and Pierre‐Fabre. The authors have no other conflicts of interest to declare.
Author contributions
Minyu Wang: Visualization; Writing‐original draft. Sen Wang: Writing‐original draft. Jayesh Desai: Writing‐review & editing. Joseph Trapani: Supervision; Writing‐review & editing. Paul Neeson: Supervision; Writing‐review & editing.
Acknowledgments
The authors acknowledge long‐term funding support from the National Health and Medical Research Council (NHMRC) of Australia and Cancer Council Victoria, in the form of many Program, Project, Venture and Fellowship grants over the past 20 years. JAT and PJN are also very grateful to the Peter MacCallum Cancer Foundation for generous philanthropic support. In particular, JAT is a Rosie Lew Fellow of the Foundation. SW thanks the Hudson Institute of Medical Research for the support. The figures included in this paper were created with BioRender.com.
Contributor Information
Joseph A Trapani, Email: joe.trapani@petermac.org.
Paul J Neeson, Email: paul.neeson@petermac.org.
References
- 1. Halse H, Colebatch AJ, Petrone P et al. Multiplex immunohistochemistry accurately defines the immune context of metastatic melanoma. Sci Rep 2018; 8: 11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang M, Huang YK, Kong JC et al. High‐dimensional analyses reveal a distinct role of T‐cell subsets in the immune microenvironment of gastric cancer. Clin Transl Immunology 2020; 9: e1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov 2019; 18: 197–218. [DOI] [PubMed] [Google Scholar]
- 4. Trujillo JA, Sweis RF, Bao R, Luke JJ. T cell‐inflamed versus non‐T cell‐inflamed tumors: a conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol Res 2018; 6: 990–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zaretsky JM, Garcia‐Diaz A, Shin DS et al. Mutations associated with acquired resistance to PD‐1 blockade in melanoma. N Engl J Med 2016; 375: 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z, Swanton C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med 2015; 7: 283ra254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sharma P, Hu‐Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017; 168: 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mikucki ME, Fisher DT, Matsuzaki J et al. Non‐redundant requirement for CXCR3 signalling during tumoricidal T‐cell trafficking across tumour vascular checkpoints. Nat Commun 2015; 6: 7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dangaj D, Bruand M, Grimm AJ et al. Cooperation between constitutive and inducible chemokines enables T cell engraftment and immune attack in solid tumors. Cancer Cell 2019; 35: 885–900 e810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gonzalez‐Martin A, Gomez L, Lustgarten J, Mira E, Manes S. Maximal T cell‐mediated antitumor responses rely upon CCR5 expression in both CD4+ and CD8+ T cells. Cancer Res 2011; 71: 5455–5466. [DOI] [PubMed] [Google Scholar]
- 11. Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer 2004; 4: 11–22. [DOI] [PubMed] [Google Scholar]
- 12. Drane D, Gittleson C, Boyle J, Maraskovsky E. ISCOMATRIX adjuvant for prophylactic and therapeutic vaccines. Expert Rev Vaccines 2007; 6: 761–772. [DOI] [PubMed] [Google Scholar]
- 13. Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol 2014; 15: e257–e267. [DOI] [PubMed] [Google Scholar]
- 14. Tumeh PC, Harview CL, Yearley JH et al. PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515: 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang M, Wang S, Trapani JA, Neeson PJ. Challenges of PD‐L1 testing in non‐small cell lung cancer and beyond. J Thorac Dis 2020; 12: 4541–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martins F, Sofiya L, Sykiotis GP et al. Adverse effects of immune‐checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol 2019; 16: 563–580. [DOI] [PubMed] [Google Scholar]
- 17. Ochoa de Olza M, Navarro Rodrigo B, Zimmermann S, Coukos G. Turning up the heat on non‐immunoreactive tumours: opportunities for clinical development. Lancet Oncol 2020; 21: e419–e430. [DOI] [PubMed] [Google Scholar]
- 18. Shi Y, Lammers T. Combining nanomedicine and immunotherapy. Acc Chem Res 2019; 52: 1543–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 2017; 17: 97–111. [DOI] [PubMed] [Google Scholar]
- 20. Vigneron N. Human tumor antigens and cancer immunotherapy. Biomed Res Int 2015; 2015: 948501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Andersen RS, Thrue CA, Junker N et al. Dissection of T‐cell antigen specificity in human melanoma. Cancer Res 2012; 72: 1642–1650. [DOI] [PubMed] [Google Scholar]
- 22. Keenan TE, Burke KP, Van Allen EM. Genomic correlates of response to immune checkpoint blockade. Nat Med 2019; 25: 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 2012; 12: 860–875. [DOI] [PubMed] [Google Scholar]
- 24. Messmer MN, Snyder AG, Oberst A. Comparing the effects of different cell death programs in tumor progression and immunotherapy. Cell Death Differ 2019; 26: 115–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tang R, Xu J, Zhang B et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol 2020; 13: 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harrington K, Freeman DJ, Kelly B, Harper J, Soria JC. Optimizing oncolytic virotherapy in cancer treatment. Nat Rev Drug Discov 2019; 18: 689–706. [DOI] [PubMed] [Google Scholar]
- 27. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov 2015; 14: 642–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol 2018; 18: 498–513. [DOI] [PubMed] [Google Scholar]
- 29. Sivanandam V, LaRocca CJ, Chen NG, Fong Y, Warner SG. Oncolytic viruses and immune checkpoint inhibition: the best of both worlds. Mol Ther Oncolytics 2019; 13: 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Biot C, Rentsch CA, Gsponer JR et al. Preexisting BCG‐specific T cells improve intravesical immunotherapy for bladder cancer. Sci Transl Med 2012; 4: 137ra172. [DOI] [PubMed] [Google Scholar]
- 31. Kohlhapp FJ, Kaufman HL. Molecular pathways: mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clin Cancer Res 2016; 22: 1048–1054. [DOI] [PubMed] [Google Scholar]
- 32. Andtbacka RH, Kaufman HL, Collichio F et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol 2015; 33: 2780–2788. [DOI] [PubMed] [Google Scholar]
- 33. Kaufman HL, Amatruda T, Reid T et al. Systemic versus local responses in melanoma patients treated with talimogene laherparepvec from a multi‐institutional phase II study. J Immunother Cancer 2016; 4: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Andtbacka RH, Ross M, Puzanov I et al. Patterns of clinical response with talimogene laherparepvec (T‐VEC) in patients with melanoma treated in the OPTiM phase III clinical trial. Ann Surg Oncol 2016; 23: 4169–4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ribas A, Dummer R, Puzanov I et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti‐PD‐1 immunotherapy. Cell 2017; 170: 1109–1119 e1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Puzanov I, Milhem MM, Minor D et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB‐IV melanoma. J Clin Oncol 2016; 34: 2619–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bourgeois‐Daigneault MC, Roy DG, Aitken AS et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple‐negative breast cancer to immune checkpoint therapy. Sci Transl Med 2018; 10: eaao1641. [DOI] [PubMed] [Google Scholar]
- 38. Samson A, Scott KJ, Taggart D et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med 2018; 10: eaam7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Annels NE, Mansfield D, Arif M et al. Phase I trial of an ICAM‐1‐targeted immunotherapeutic‐coxsackievirus A21 (CVA21) as an oncolytic agent against non muscle‐invasive bladder cancer. Clin Cancer Res 2019; 25: 5818–5831. [DOI] [PubMed] [Google Scholar]
- 40. Ge Y, Wang H, Ren J et al. Oncolytic vaccinia virus delivering tethered IL‐12 enhances antitumor effects with improved safety. J Immunother Cancer 2020; 8: e000710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Newman JH, Chesson CB, Herzog NL et al. Intratumoral injection of the seasonal flu shot converts immunologically cold tumors to hot and serves as an immunotherapy for cancer. Proc Natl Acad Sci USA 2020; 117: 1119–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang Q, Wang Y, Ding J et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature 2020; 579: 421–426. [DOI] [PubMed] [Google Scholar]
- 43. Zhang Z, Zhang Y, Xia S et al. Gasdermin E suppresses tumour growth by activating anti‐tumour immunity. Nature 2020; 579: 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017; 548: 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mackenzie KJ, Carroll P, Martin CA et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017; 548: 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dillon MT, Bergerhoff KF, Pedersen M et al. ATR inhibition potentiates the radiation‐induced inflammatory tumor microenvironment. Clin Cancer Res 2019; 25: 3392–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Reits EA, Hodge JW, Herberts CA et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med 2006; 203: 1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sharabi AB, Nirschl CJ, Kochel CM et al. Stereotactic radiation therapy augments antigen‐specific PD‐1‐mediated antitumor immune responses via cross‐presentation of tumor antigen. Cancer Immunol Res 2015; 3: 345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chang MC, Chen YL, Lin HW et al. Irradiation enhances abscopal anti‐tumor effects of antigen‐specific immunotherapy through regulating tumor microenvironment. Mol Ther 2018; 26: 404–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Matsumura S, Wang B, Kawashima N et al. Radiation‐induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol 2008; 181: 3099–3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Keam SP, Halse H, Nguyen T et al. High dose‐rate brachytherapy of localized prostate cancer converts tumors from cold to hot. J Immunother Cancer 2020; 8: e000792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hutchinson L. Radiotherapy: abscopal responses: pro‐immunogenic effects of radiotherapy. Nat Rev Clin Oncol 2015; 12: 504. [DOI] [PubMed] [Google Scholar]
- 53. Golden EB, Demaria S, Schiff PB, Chachoua A, Formenti SC. An abscopal response to radiation and ipilimumab in a patient with metastatic non‐small cell lung cancer. Cancer Immunol Res 2013; 1: 365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Keam S, Gill S, Ebert MA, Nowak AK, Cook AM. Enhancing the efficacy of immunotherapy using radiotherapy. Clin Transl Immunology 2020; 9: e1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Formenti SC, Rudqvist NP, Golden E et al. Radiotherapy induces responses of lung cancer to CTLA‐4 blockade. Nat Med 2018; 24: 1845–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dovedi SJ, Cheadle EJ, Popple AL et al. Fractionated radiation therapy stimulates antitumor immunity mediated by both resident and infiltrating polyclonal T‐cell populations when combined with PD‐1 blockade. Clin Cancer Res 2017; 23: 5514–5526. [DOI] [PubMed] [Google Scholar]
- 57. McLaughlin M, Patin EC, Pedersen M et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat Rev Cancer 2020; 20: 203–217. [DOI] [PubMed] [Google Scholar]
- 58. Hildner K, Edelson BT, Purtha WE et al. Batf3 deficiency reveals a critical role for CD8α+ dendritic cells in cytotoxic T cell immunity. Science 2008; 322: 1097–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hammerich L, Marron TU, Upadhyay R et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat Med 2019; 25: 814–824. [DOI] [PubMed] [Google Scholar]
- 60. Pfirschke C, Engblom C, Rickelt S et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 2016; 44: 343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nowak AK, Robinson BWS, Lake RA. Gemcitabine exerts a selective effect on the humoral immune response: implications for combination chemo‐immunotherapy. Cancer Res 2002; 62: 2353–2358. [PubMed] [Google Scholar]
- 62. Galluzzi L, Humeau J, Buque A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol 2020; 17: 725–741. [DOI] [PubMed] [Google Scholar]
- 63. Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS‐STING pathway of cytosolic DNA sensing. Nat Immunol 2016; 17: 1142–1149. [DOI] [PubMed] [Google Scholar]
- 64. Wanderley CW, Colon DF, Luiz JPM et al. Paclitaxel reduces tumor growth by reprogramming tumor‐associated macrophages to an M1 profile in a TLR4‐dependent manner. Cancer Res 2018; 78: 5891–5900. [DOI] [PubMed] [Google Scholar]
- 65. Ma Y, Mattarollo SR, Adjemian S et al. CCL2/CCR2‐dependent recruitment of functional antigen‐presenting cells into tumors upon chemotherapy. Cancer Res 2014; 74: 436–445. [DOI] [PubMed] [Google Scholar]
- 66. Schaer DA, Geeganage S, Amaladas N et al. The folate pathway inhibitor pemetrexed pleiotropically enhances effects of cancer immunotherapy. Clin Cancer Res 2019; 25: 7175–7188. [DOI] [PubMed] [Google Scholar]
- 67. Helleday T. Making immunotherapy 'cold' tumours 'hot' by chemotherapy‐induced mutations‐a misconception. Ann Oncol 2019; 30: 360–361. [DOI] [PubMed] [Google Scholar]
- 68. Yang W, Men P, Xue H, Jiang M, Luo Q. Risk of gastrointestinal adverse events in cancer patients treated with immune checkpoint inhibitor plus chemotherapy: a systematic review and meta‐analysis. Front Oncol 2020; 10: 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Puzanov I, Diab A, Abdallah K et al. Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer 2017; 5: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Duan X, Chan C, Lin W. Nanoparticle‐mediated immunogenic cell death enables and potentiates cancer immunotherapy. Angew Chem Int Ed Engl 2019; 58: 670–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ng CW, Li J, Pu K. Recent progresses in phototherapy‐synergized cancer immunotherapy. Adv Funct Mater 2018; 28: 1804688–1804708. [Google Scholar]
- 72. Huang L, Li Y, Du Y et al. Mild photothermal therapy potentiates anti‐PD‐L1 treatment for immunologically cold tumors via an all‐in‐one and all‐in‐control strategy. Nat Commun 2019; 10: 4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang D, Zhang J, Li Q, Song A, Li Z, Luan Y. Cold to hot: rational design of a minimalist multifunctional photo‐immunotherapy nanoplatform toward boosting immunotherapy capability. ACS Appl Mater Interfaces 2019; 11: 32633–32646. [DOI] [PubMed] [Google Scholar]
- 74. Chapman A, ter Haar G. Thermal ablation of uterine fibroids using MR‐guided focused ultrasound‐a truly non‐invasive treatment modality. Eur Radiol 2007; 17: 2505–2511. [DOI] [PubMed] [Google Scholar]
- 75. Eranki A, Farr N, Partanen A et al. Mechanical fractionation of tissues using microsecond‐long HIFU pulses on a clinical MR‐HIFU system. Int J Hyperthermia 2018; 34: 1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hu Z, Yang XY, Liu Y et al. Investigation of HIFU‐induced anti‐tumor immunity in a murine tumor model. J Transl Med 2007; 5: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. van den Bijgaart RJ, Eikelenboom DC, Hoogenboom M, Futterer JJ, den Brok MH, Adema GJ. Thermal and mechanical high‐intensity focused ultrasound: perspectives on tumor ablation, immune effects and combination strategies. Cancer Immunol Immunother 2017; 66: 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Eranki A, Srinivasan P, Ries M et al. High‐intensity focused ultrasound (HIFU) triggers immune sensitization of refractory murine neuroblastoma to checkpoint inhibitor therapy. Clin Cancer Res 2020; 26: 1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Deng J, Wang ES, Jenkins RW et al. CDK4/6 inhibition augments antitumor immunity by enhancing T‐cell activation. Cancer Discov 2018; 8: 216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Goel S, DeCristo MJ, Watt AC et al. CDK4/6 inhibition triggers anti‐tumour immunity. Nature 2017; 548: 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Minton K. Cancer immunotherapy: cell cycle inhibitors boost tumour immunogenicity. Nat Rev Drug Discov 2017; 16: 679. [DOI] [PubMed] [Google Scholar]
- 82. Loi S, Dushyanthen S, Beavis PA et al. RAS/MAPK activation is associated with reduced tumor‐infiltrating lymphocytes in triple‐negative breast cancer: therapeutic cooperation between MEK and PD‐1/PD‐L1 immune checkpoint inhibitors. Clin Cancer Res 2016; 22: 1499–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ebert PJR, Cheung J, Yang Y et al. MAP kinase inhibition promotes T cell and anti‐tumor activity in combination with PD‐L1 checkpoint blockade. Immunity 2016; 44: 609–621. [DOI] [PubMed] [Google Scholar]
- 84. Hu‐Lieskovan S, Mok S, Homet Moreno B et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci Transl Med 2015; 7: 279ra241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kakavand H, Wilmott JS, Menzies AM et al. PD‐L1 expression and tumor‐infiltrating lymphocytes define different subsets of MAPK inhibitor‐treated melanoma patients. Clin Cancer Res 2015; 21: 3140–3148. [DOI] [PubMed] [Google Scholar]
- 86. Sapkota B, Hill CE, Pollack BP. Vemurafenib enhances MHC induction in BRAF(V600E) homozygous melanoma cells. Oncoimmunology 2013; 2: e22890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Boni A, Cogdill AP, Dang P et al. Selective BRAFV600E inhibition enhances T‐cell recognition of melanoma without affecting lymphocyte function. Cancer Res 2010; 70: 5213–5219. [DOI] [PubMed] [Google Scholar]
- 88. Proietti I, Skroza N, Michelini S et al. BRAF inhibitors: molecular targeting and immunomodulatory actions. Cancers (Basel) 2020; 12: 1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hellmann MD, Kim TW, Lee CB et al. Phase Ib study of atezolizumab combined with cobimetinib in patients with solid tumors. Ann Oncol 2019; 30: 1134–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Eng C, Kim TW, Bendell J et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): a multicentre, open‐label, phase 3, randomised, controlled trial. Lancet Oncol 2019; 20: 849–861. [DOI] [PubMed] [Google Scholar]
- 91. Frederick DT, Piris A, Cogdill AP et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res 2013; 19: 1225–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Minchom A, Aversa C, Lopez J. Dancing with the DNA damage response: next‐generation anti‐cancer therapeutic strategies. Ther Adv Med Oncol 2018; 10: 1758835918786658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Jiao S, Xia W, Yamaguchi H et al. PARP inhibitor upregulates PD‐L1 expression and enhances cancer‐associated immunosuppression. Clin Cancer Res 2017; 23: 3711–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Brown JS, O'Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov 2017; 7: 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sharabi A, Tsokos MG, Ding Y, Malek TR, Klatzmann D, Tsokos GC. Regulatory T cells in the treatment of disease. Nat Rev Drug Discov 2018; 17: 823–844. [DOI] [PubMed] [Google Scholar]
- 96. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid‐derived suppressor cells in the tumor microenvironment. Trends Immunol 2016; 37: 208–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ostrand‐Rosenberg S, Fenselau C. Myeloid‐derived suppressor cells: immune‐suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol 2018; 200: 422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer 2020; 19: 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Di Pilato M, Kim EY, Cadilha BL et al. Targeting the CBM complex causes Treg cells to prime tumours for immune checkpoint therapy. Nature 2019; 570: 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Overacre‐Delgoffe AE, Chikina M, Dadey RE et al. Interferon‐γ drives Treg fragility to promote anti‐tumor immunity. Cell 2017; 169: 1130–1141 e1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhang B, Jia H, Liu J et al. Depletion of regulatory T cells facilitates growth of established tumors: a mechanism involving the regulation of myeloid‐derived suppressor cells by lipoxin A4. J Immunol 2010; 185: 7199–7206. [DOI] [PubMed] [Google Scholar]
- 102. Zhang Y, Lazarus J, Steele NG et al. Regulatory T‐cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis. Cancer Discov 2020; 10: 422–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Seoane J, Gomis RR. TGF‐β family signaling in tumor suppression and cancer progression. Cold Spring Harb Perspect Biol 2017; 9: a022277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. David JM, Dominguez C, McCampbell KK, Gulley JL, Schlom J, Palena C. A novel bifunctional anti‐PD‐L1/TGF‐β trap fusion protein (M7824) efficiently reverts mesenchymalization of human lung cancer cells. Oncoimmunology 2017; 6: e1349589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Trapani JA. The dual adverse effects of TGF‐β secretion on tumor progression. Cancer Cell 2005; 8: 349–350. [DOI] [PubMed] [Google Scholar]
- 106. Lind H, Gameiro SR, Jochems C et al. Dual targeting of TGF‐β and PD‐L1 via a bifunctional anti‐PD‐L1/TGF‐β RII agent: status of preclinical and clinical advances. J Immunother Cancer 2020; 8: e000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Morris JC, Tan AR, Olencki TE et al. Phase I study of GC1008 (fresolimumab): a human anti‐transforming growth factor‐beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS One 2014; 9: e90353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Lan Y, Zhang D, Xu C et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD‐L1 and TGF‐β. Sci Transl Med 2018; 10: eaan5488. [DOI] [PubMed] [Google Scholar]
- 109. Strauss J, Heery CR, Schlom J et al. Phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD‐L1 and TGFβ, in advanced solid tumors. Clin Cancer Res 2018; 24: 1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Cho BC, Daste A, Ravaud A et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF‐β and PD‐L1, in advanced squamous cell carcinoma of the head and neck: results from a phase I cohort. J Immunother Cancer 2020; 8: e000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yoo C, Oh DY, Choi HJ et al. Phase I study of bintrafusp alfa, a bifunctional fusion protein targeting TGF‐β and PD‐L1, in patients with pretreated biliary tract cancer. J Immunother Cancer 2020; 8: e000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Cho BC, Kim TM, Vicente D et al. Two‐year follow‐up of bintrafusp alfa, a bifunctional fusion protein targeting TGF‐β and PD‐L1, for second‐line (2L) treatment of non‐small cell lung cancer (NSCLC). J Clin Oncol 2020; 38: 9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ott PA, Hodi FS, Buchbinder EI. Inhibition of immune checkpoints and vascular endothelial growth factor as combination therapy for metastatic melanoma: an overview of rationale, preclinical evidence, and initial clinical data. Front Oncol 2015; 5: 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. McDermott DF, Huseni MA, Atkins MB et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med 2018; 24: 749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Herbst RS, Arkenau H‐T, Santana‐Davila R et al. Ramucirumab plus pembrolizumab in patients with previously treated advanced non‐small‐cell lung cancer, gastro‐oesophageal cancer, or urothelial carcinomas (JVDF): a multicohort, non‐randomised, open‐label, phase 1a/b trial. Lancet Oncol 2019; 20: 1109–1123. [DOI] [PubMed] [Google Scholar]
- 116. Rini BI, Plimack ER, Stus V et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal‐cell carcinoma. N Engl J Med 2019; 380: 1116–1127. [DOI] [PubMed] [Google Scholar]
- 117. Hodi FS, Lawrence D, Lezcano C et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res 2014; 2: 632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Noman MZ, Desantis G, Janji B et al. PD‐L1 is a novel direct target of HIF‐1α, and its blockade under hypoxia enhanced MDSC‐mediated T cell activation. J Exp Med 2014; 211: 781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Corzo CA, Condamine T, Lu L et al. HIF‐1α regulates function and differentiation of myeloid‐derived suppressor cells in the tumor microenvironment. J Exp Med 2010; 207: 2439–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Jayaprakash P, Ai M, Liu A et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J Clin Invest 2018; 128: 5137–5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst 2001; 93: 266–276. [DOI] [PubMed] [Google Scholar]
- 122. Beavis PA, Henderson MA, Giuffrida L et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest 2017; 127: 929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Vonderheide RH. CD40 agonist antibodies in cancer immunotherapy. Annu Rev Med 2020; 71: 47–58. [DOI] [PubMed] [Google Scholar]
- 124. Bajor DL, Mick R, Riese MJ et al. Long‐term outcomes of a phase I study of agonist CD40 antibody and CTLA‐4 blockade in patients with metastatic melanoma. Oncoimmunology 2018; 7: e1468956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Beatty GL, Chiorean EG, Fishman MP et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011; 331: 1612–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Beatty GL, Torigian DA, Chiorean EG et al. A phase I study of an agonist CD40 monoclonal antibody (CP‐870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res 2013; 19: 6286–6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Nowak AK, Cook AM, McDonnell AM et al. A phase 1b clinical trial of the CD40‐activating antibody CP‐870,893 in combination with cisplatin and pemetrexed in malignant pleural mesothelioma. Ann Oncol 2015; 26: 2483–2490. [DOI] [PubMed] [Google Scholar]
- 128. Vonderheide RH, Burg JM, Mick R et al. Phase I study of the CD40 agonist antibody CP‐870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology 2013; 2: e23033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Aleynick M, Svensson‐Arvelund J, Flowers CR, Marabelle A, Brody JD. Pathogen Molecular Pattern Receptor Agonists: Treating Cancer by Mimicking Infection. Clin Cancer Res 2019; 25: 6283–6294. [DOI] [PubMed] [Google Scholar]
- 130. Smits EL, Cools N, Lion E et al. The Toll‐like receptor 7/8 agonist resiquimod greatly increases the immunostimulatory capacity of human acute myeloid leukemia cells. Cancer Immunol Immunother 2010; 59: 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Kapp K, Volz B, Oswald D, Wittig B, Baumann M, Schmidt M. Beneficial modulation of the tumor microenvironment and generation of anti‐tumor responses by TLR9 agonist lefitolimod alone and in combination with checkpoint inhibitors. Oncoimmunology 2019; 8: e1659096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Zanker DJ, Spurling AJ, Brockwell NK et al. Intratumoral administration of the Toll‐like receptor 7/8 agonist 3M–052 enhances interferon‐driven tumor immunogenicity and suppresses metastatic spread in preclinical triple‐negative breast cancer. Clin Transl Immunology 2020; 9: e1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Pradere JP, Dapito DH, Schwabe RF. The Yin and Yang of Toll‐like receptors in cancer. Oncogene 2014; 33: 3485–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP‐AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013; 339: 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Gajewski TF, Higgs EF. Immunotherapy with a sting. Science 2020; 369: 921–922. [DOI] [PubMed] [Google Scholar]
- 136. Conlon J, Burdette DL, Sharma S et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6‐dimethylxanthenone‐4‐acetic acid. J Immunol 2013; 190: 5216–5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Walker MM, Crute BW, Cambier JC, Getahun A. B cell‐intrinsic STING signaling triggers cell activation, synergizes with B cell receptor signals, and promotes antibody responses. J Immunol 2018; 201: 2641–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Pan BS, Perera SA, Piesvaux JA et al. An orally available non‐nucleotide STING agonist with antitumor activity. Science 2020; 369: eaba6098. [DOI] [PubMed] [Google Scholar]
- 139. Chin EN, Yu C, Vartabedian VF et al. Antitumor activity of a systemic STING‐activating non‐nucleotide cGAMP mimetic. Science 2020; 369: 993–999. [DOI] [PubMed] [Google Scholar]
- 140. Prendergast GC, Mondal A, Dey S, Laury‐Kleintop LD, Muller AJ. Inflammatory reprogramming with IDO1 inhibitors: turning immunologically unresponsive 'cold' tumors 'hot'. Trends Cancer 2018; 4: 38–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Long GV, Dummer R, Hamid O et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO‐301/KEYNOTE‐252): a phase 3, randomised, double‐blind study. Lancet Oncol 2019; 20: 1083–1097. [DOI] [PubMed] [Google Scholar]
- 142. Borden EC, Sen GC, Uze G et al. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov 2007; 6: 975–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Gresser I, Bourali C, Levy JP, Fontaine‐Brouty‐Boye D, Thomas MT. Increased survival in mice inoculated with tumor cells and treated with interferon preparations. Proc Natl Acad Sci USA 1969; 63: 51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Strander H, Cantell K, Carlstrom G, Jakobsson PA. Clinical and laboratory investigations on man: systemic administration of potent interferon to man. J Natl Cancer Inst 1973; 51: 733–742. [DOI] [PubMed] [Google Scholar]
- 145. Gutterman JU, Blumenschein GR, Alexanian R et al. Leukocyte interferon‐induced tumor regression in human metastatic breast cancer, multiple myeloma, and malignant lymphoma. Ann Intern Med 1980; 93: 399–406. [DOI] [PubMed] [Google Scholar]
- 146. Lin FC, Young HA. Interferons: Success in anti‐viral immunotherapy. Cytokine Growth Factor Rev 2014; 25: 369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Rudick RA, Goelz SE. Beta‐interferon for multiple sclerosis. Exp Cell Res 2011; 317: 1301–1311. [DOI] [PubMed] [Google Scholar]
- 148. Borden EC, Parkinson D. A perspective on the clinical effectiveness and tolerance of interferon‐alpha. Semin Oncol 1998; 25: 3–8. [PubMed] [Google Scholar]
- 149. Alberts DS, Marth C, Alvarez RD et al. Randomized phase 3 trial of interferon γ‐1b plus standard carboplatin/paclitaxel versus carboplatin/paclitaxel alone for first‐line treatment of advanced ovarian and primary peritoneal carcinomas: results from a prospectively designed analysis of progression‐free survival. Gynecol Oncol 2008; 109: 174–181. [DOI] [PubMed] [Google Scholar]
- 150. Zhang S, Kohli K, Black RG et al. Systemic interferon‐γ increases MHC class I expression and T‐cell infiltration in cold tumors: results of a phase 0 clinical trial. Cancer Immunol Res 2019; 7: 1237–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Rosenberg SA. IL‐2: the first effective immunotherapy for human cancer. J Immunol 2014; 192: 5451–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Klein C, Waldhauer I, Nicolini VG et al. Cergutuzumab amunaleukin (CEA‐IL2v), a CEA‐targeted IL‐2 variant‐based immunocytokine for combination cancer immunotherapy: overcoming limitations of aldesleukin and conventional IL‐2‐based immunocytokines. Oncoimmunology 2017; 6: e1277306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Kim JH, Kim YM, Choi D et al. Hybrid Fc‐fused interleukin‐7 induces an inflamed tumor microenvironment and improves the efficacy of cancer immunotherapy. Clin Transl Immunology 2020; 9: e1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Charych DH, Hoch U, Langowski JL et al. NKTR‐214, an engineered cytokine with biased IL2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clin Cancer Res 2016; 22: 680–690. [DOI] [PubMed] [Google Scholar]
- 155. Bentebibel SE, Hurwitz ME, Bernatchez C et al. A first‐in‐human study and biomarker analysis of NKTR‐214, a novel IL2R βγ‐biased cytokine, in patients with advanced or metastatic solid tumors. Cancer Discov 2019; 9: 711–721. [DOI] [PubMed] [Google Scholar]
- 156. Sahin U, Tureci O. Personalized vaccines for cancer immunotherapy. Science 2018; 359: 1355–1360. [DOI] [PubMed] [Google Scholar]
- 157. Hu Z, Ott PA, Wu CJ. Towards personalized, tumour‐specific, therapeutic vaccines for cancer. Nat Rev Immunol 2018; 18: 168–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Carreno BM, Magrini V, Becker‐Hapak M et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen‐specific T cells. Science 2015; 348: 803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Ott PA, Hu Z, Keskin DB et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017; 547: 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Sahin U, Derhovanessian E, Miller M et al. Personalized RNA mutanome vaccines mobilize poly‐specific therapeutic immunity against cancer. Nature 2017; 547: 222–226. [DOI] [PubMed] [Google Scholar]
- 161. Buonaguro L, Consortium H . Developments in cancer vaccines for hepatocellular carcinoma. Cancer Immunol Immunother 2016; 65: 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Loffler MW, Mohr C, Bichmann L et al. Multi‐omics discovery of exome‐derived neoantigens in hepatocellular carcinoma. Genome Med 2019; 11: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Keskin DB, Anandappa AJ, Sun J et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019; 565: 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Sarkizova S, Klaeger S, Le PM et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat Biotechnol 2020; 38: 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Fu J, Kanne DB, Leong M et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD‐1 blockade. Sci Transl Med 2015; 7: 283ra252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Ali OA, Lewin SA, Dranoff G, Mooney DJ. Vaccines combined with immune checkpoint antibodies promote cytotoxic T‐cell activity and tumor eradication. Cancer Immunol Res 2016; 4: 95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Hugo W, Zaretsky JM, Sun L et al. Genomic and transcriptomic features of response to anti‐PD‐1 therapy in metastatic melanoma. Cell 2017; 168: 542. [DOI] [PubMed] [Google Scholar]
- 168. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004; 10: 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Sahin U, Oehm P, Derhovanessian E et al. An RNA vaccine drives immunity in checkpoint‐inhibitor‐treated melanoma. Nature 2020; 585: 107–112. [DOI] [PubMed] [Google Scholar]
- 170. Loo Yau H, Ettayebi I, De Carvalho DD. The cancer epigenome: exploiting its vulnerabilities for immunotherapy. Trends Cell Biol 2019; 29: 31–43. [DOI] [PubMed] [Google Scholar]
- 171. Slaney CY, Wang P, Darcy PK, Kershaw MH. CARs versus BiTEs: a comparison between T cell‐redirection strategies for cancer treatment. Cancer Discov 2018; 8: 924–934. [DOI] [PubMed] [Google Scholar]