

Abstract
Phosphoinositide-3- kinase (PI3K) signaling regulates cellular proliferation, survival and metabolism, and its aberrant activation is one of the most frequent oncogenic events across human cancers. In the last few decades, research focused on the development of PI3K inhibitors, from preclinical tool compounds to the highly specific medicines approved to treat patients with cancer. Herein we discuss current paradigms for PI3K inhibitors in cancer therapy, focusing on clinical data and mechanisms of action. We also discuss current limitations in the use of PI3K inhibitors including toxicities and mechanisms of resistance, with specific emphasis on approaches aimed to improve their efficacy.
Introduction
The PI3K pathway is a key regulatory hub for cell growth, survival, and metabolism. Activation of PI3K is a frequent hallmark of cancer, highlighted by the prevalence of somatic mutations in genes encoding key components of this pathway. Over the last two decades, the clinical development of PI3K inhibitors has evolved considerably; from the first-in-class tool compounds that lacked potency, specificity, and appropriate drug-like properties, all the way to medicines that have been approved by several regulatory agencies for the treatment of different types of cancer such as leukemia and breast cancer. These most recent agents present improved potency, selectivity, and pharmacological profiles, and can be safely administered to patients as monotherapies or in combination with other anti-cancer agents. Despite the extraordinary progress in the field, there are still may questions surrounding the clinical use of PI3K inhibitors. In this review, we summarize the current clinical investigations of compounds targeting PI3K. We also discuss relevant mechanisms of action behind the clinical efficacy and current limitations of these therapeutic approaches, including drug resistance. Finally, we discuss potential strategies aimed to improve the therapeutic index of these inhibitors and how novel chemical entities are required to unveil the full potential of PI3K inhibition in cancer therapy.
PI3K activation in cancer
Given its essential cellular functions, PI3K is evolutionary conserved across metazoans and, in humans, it has undergone successive gene duplication events giving rise to different isoforms. There are four catalytic subunits encoded in our genomes: p110α and p110β (encoded by PIK3CA and PIK3CB, respectively) are ubiquitously expressed, while p110δ and p110γ (encoded by PIK3CD and PIK3CG, respectively) are restricted to immune lineages (reviewed by 1–3) (Figure 1A).
Figure 1. The PI3K-AKT pathway and the most common PIK3CA mutations in cancer.

a. The gene expression profile of PI3K isoforms in normal human tissues shown in log2 (TPM+1) scale based on http://gepia.cancer-pku.cn (visited on April 2021). b. Receptor tyrosine kinase (RTK) activation by insulin or growth factors mediates tyrosine phosphorylation which allows the recruitment of the lipid kinase PI3K to the plasma membrane through the p85 regulatory subunit. PI3K phosphorylates the lipid phosphatidylinositol 4,5-bisphosphate (PIP2) to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3). The lipid phosphatase PTEN dephosphorylates PIP3 back to PIP2. PIP3 recruits the serine/threonine kinase AKT to the plasma membrane, where it gets phosphorylated and activated through phosphorylation at T308 by the PDK1 kinase and S473 by mTORC2 kinase complex. AKT then phosphorylates a numerous of substrates promoting glucose, metabolism, cell cycle arrest, cell growth, proliferation, and translation. P denotes protein phosphorylation events. c. The most frequent mutations of the PI3KCA gene by MSK-IMPACT. d. The percentages of the alteration frequency of the PIK3CA gene across human cancers by MSK-IMPACT. Amino-terminal adaptor-binding domain (ABD); Ras-binding domain (RBD); Protein-kinase-C homology-2 (C2).
PI3K is an enzyme that catalyzes the synthesis of the second messenger phosphatidylinositol (3,4,5)-trisphosphate (PIP3) by phosphorylating phosphatidylinositol 4,5-bisphosphate (PIP2), an abundant lipid found at the plasma membrane. This biochemical reaction is mediated by a catalytic subunit, p110, which forms a heterodimer with a regulatory subunit, p85 (or p84/p101 for the p110γ isoform). The interaction with the regulatory subunit controls the appropriate localization, regulation, and stability necessary for the activity of the catalytic subunit. The p85 regulatory subunit has two Src Homology 2 (SH2) domains that are critical for the interaction with phospho-tyrosine (pTyr) residues found in activated receptor tyrosine kinases (RTKs) and/or adaptor proteins (e.g., IRS-1 and ERBB3). However, when the SH2 domains are not binding pTyr residues, they mediate inhibitory intermolecular contacts with p110, maintaining the catalytic subunit in an inactive conformation4. Detailed structural studies have been undertaken in this context and revealed a complex network of interactions between catalytic and regulatory subunits required for PI3K activation5. The RAS GTPases directly interact with the p110 subunit to regulate PI3K activity6. Whether this interaction contributes directly to PI3K catalytic activity or regulates PI3K stability or localization remains less understood. Regardless, the interaction between RAS GTPases and PI3K appears to be necessary for RAS-driven lung tumorigenesis in murine models7.
PI3K is commonly activated by receptor tyrosine kinase (RTK) stimulation. Some RTKs are particularly efficient at activating this enzyme, including IR, PDGFR, and HER2. The increased concentration of PIP3 at the plasma membrane triggers the recruitment of proteins containing the PIP3-binding pleckstrin homology (PH) domain and activation of downstream pathways2. Among the proteins that contain PH domains, PDK1 and AKT kinases are the key canonical downstream effectors of PI3K activation8,9. Activated AKT phosphorylates an array of effector proteins that control fundamental cellular processes, including mTORC1, an important downstream effector complex that regulates cell growth, translation, and metabolic fitness10. AKT activates mTORC1 by phosphorylating and inhibiting TSC2 and PRAS40, two negative regulators of mTORC111. AKT also directly phosphorylates and inhibits the FOXO transcription factors impacting cell survival and cell cycle progression, as well as many other processes regulated downstream of FOXO target genes12 (Figure 1B). Despite the rapid activation of PI3K upon growth factor stimulation, PIP3 is short-lived. Downstream signaling is rapidly attenuated due to the activity of phosphatase and tensin homolog (PTEN) and other lipid phosphatases that antagonizes the enzymatic activity of PI3K by directly de-phosphorylating PIP313 (Figure 1B).
A frequent mechanism of PI3K activation in cancers is the presence of mutations in the PIK3CA gene. Such gain-of-function mutations generally affect two different domains of p110α, the kinase and the helical domains, where the mutational hotspots affect the H1047 and the E542/E545 codons, respectively14. Although mutations at these sites account for most PIK3CA mutations, other variants can be found in the p85-binding (ABD) and C2 domains (Figure 1C). Structural insights into how these mutants lead to hyperactivation of PI3K suggest that the helical and kinase domain mutations activate the kinase via distinct molecular mechanisms. Helical domain mutations, which lead to a lysine replacing a glutamate (i.e. E542K and E545K), abrogates an intramolecular inhibitory interaction between p110α and the N-terminal SH2 domain of its regulatory subunit, p85, leading to constitutive activity that mimics pTyr stimulation15,16. On the other hand, the kinase domain mutations appear to increase the specific activity of p110α by affecting binding lipid membranes and its substrate, PIP216.
PIK3CA mutations are most frequently found in gynecological malignancies, breast adenocarcinomas, and head and neck cancers (Figure 1D). However, other tumor types including lung, bladder, and colorectal adenocarcinomas, as well as overgrowth syndromes, often carry PIK3CA activating mutations17–19. Mutation bias has also been observed in tumors, similar to other driver oncogenes. For instance, head and neck cancers are more likely to carry helical E542/545 hotspot mutations20. PIK3CA amplifications are also frequent across cancer types and have been suggested to increase p110α activity. Mutations in other genes can also lead to hyperactivation of the PI3K pathway. Deletions, non-sense, and loss-of-function missense mutations in PTEN are frequent in many cancers, including prostate and breast cancer, glioblastoma, and melanoma, among others. Although loss of PTEN results in accumulation of PIP3 and elevated PI3K activity, it is important to highlight that this effect appears to be mediated to a greater extent by p110β21. Mutations in PIK3R1, the gene encoding for the p85 regulatory subunit, lead to activation of PI3K signaling. Although infrequent, they are found in uterine, ovarian, and prostate cancers22.
Mechanism of action of PI3K inhibitors
To date, most PI3K inhibitors that have entered clinical development are reversible, ATP-competitive inhibitors. The few covalent inhibitors that irreversibly inhibit PI3K are mostly natural products and have not been tested in late phase clinical trials.
PI3K inhibitors can induce a diverse set of cellular responses including apoptosis and/or cell cycle arrest (Figure 2). These effects can be achieved by different mechanisms that are regulated by the PI3K/AKT pathway. For instance, both alpelisib, a p110α inhibitor, and copanlisib, a pan-p110 inhibitor, caused apoptosis and cell cycle arrest in a subset of breast cancer cells23–25. Although some cell lines undergo apoptosis when treated with these inhibitors, the mechanism by which cells die is not entirely clear and may involve dynamic regulation of BH3 proteins (i.e. BAX/BAD are substrates of AKT) and transcriptional programs mediated by FOXO transcription factors among other mechanisms26,27. In addition, PI3K inhibitors often lead to suppression of mTORC1 in sensitive cells, and inhibition of mTORC1 can promote apoptosis by impairing translation, especially of pro-survival and growth proteins and increasing autophagy10,28–31.
Figure 2. The non-cell autonomous and the cell-autonomous effects of PI3K inhibitors.

PI3K inhibitors have well-studied cell-autonomous effects in cancer cells that lead to a cytostatic and cytotoxic response. These include the activation of intrinsic apoptosis, the reduction of glucose metabolism, translation inhibition, and changes in transcriptional regulation through the FOXO transcription factors among others. However, in addition to these effects, PI3K inhibitors can also exert non-cell autonomous affects in the organism and tumor microenvironment that can significantly contribute to the antitumoral effect. For example, administration of PI3K inhibitors lead to short and long-term metabolic responses that affect tumor nutrient availability and glycemic and insulinemic response. These compounds also have remarkable anti-angiogenic properties that are mediated by the alpha isoform. Inhibitors that target the delta and gamma isoform of PI3K inhibitors have been shown to affect macrophage activation (including polarization and phagocytosis), inhibition of regulatory T-cells, and inhibition of suppressive myeloid cells and neutrophils. Most of these changes result in activation of cytotoxic T-cells that promote cancer cell killing. The predominant PI3K isoform mediating each of these cellular effects have been highlighted in red (alpha), green (delta), and blue (gamma).
The anti-tumoral effects induced by PI3K inhibitors in vivo are not necessarily the result of cancer cell-intrinsic mechanisms, as these drugs have also been shown to affect other compartments within the tumor microenvironment (Figure 2). Angiogenesis is highly dependent on PI3K signaling, both during embryonic development and tumorigenesis32. Inhibitors that specifically target p110α have been shown to impair functional angiogenesis, which can also contribute to tumor shrinkage33.
In addition, PI3K is an important signaling node necessary for the maturation and activation of different immune cells. Therefore, inhibition of this enzyme can lead to anti-tumoral effects that are partially mediated by the immune system. For example, it has been shown that macrophages require PI3Kγ for polarization and suppression of T-cell activation in syngeneic mouse models of cancer; hence, inhibition of this isoform with selective inhibitors has been shown to enhance the immune recognition of a solid tumor model 34. Similarly, myeloid suppressive cells can be inhibited with duvelisib, a p110δ/γ inhibitor that enhance T-cell-mediated cytotoxicity and tumor regression35. In both experiments, inhibition of PI3K increased the efficacy of anti-PD-L1 therapies.
In lymphocytes, PI3Kδ is required for proper antigen receptor signaling and both inactivation and hyperactivation of this isoform has important functional consequences36,37. In tumors, genetic or pharmacologic blockade of PI3Kδ leads to the inhibition of suppressive T-regulatory lymphocytes, hence promoting the activation of cytotoxic lymphocytes38. While PI3Kδ/γ inhibitors have been mostly developed and approved in the context of hematologic malignancies, there is increasing preclinical evidence for using these inhibitors in solid tumors38,35. Additional mechanisms regarding the impact of PI3K inhibitors in the tumor stroma have been previously discussed in detail39.
Another effect of PI3K inhibition is the systemic metabolic adaptation. Acute treatment with these inhibitors induces hyperinsulinemia, which in turn can activate pro-survival pathways in the cancer cell40. However, long-term treatment with PI3K inhibitors has been shown to reduce adiposity, hyperglycemia, and increase survival in mice and monkeys41.
Clinical development of PI3K inhibitors
The first inhibitors of the PI3K pathway, isolated and characterized more than 25 years ago, were used to study the role of PI3K in lymphocyte activation and trafficking42–45, insulin receptor signaling46 and DNA synthesis and repair47–49 among other biological processes. Both wortmannin and LY294002 were used as PI3K inhibitors, albeit these are in fact promiscuous agents that target also other members of the phosphatidylinositol 3-kinase-related kinases (PIKK) family such as mTOR and DNA-PK. This broad activity against a number of different kinases prevented their clinical implementation and development; of note, wortmannin and LY294002 have been extensively used as tool compounds for more than a decade.
More specific inhibitors of PI3K were later developed and began to be tested in preclinical models across several cancer types50–53. The rationale behind the development of these pan-PI3K inhibitors (small molecules that inhibit all of the p110 isoforms) was the attempt of widening their therapeutic window by limiting their activity specifically to PI3K. Buparlisib is a pan-PI3K inhibitor that exemplifies the initial excitement for these compounds, and from which we have learned much in terms of tolerability and specificity. The combination of buparlisib and the ER degrader fulvestrant showed convincing efficacy in preclinical models of ER-positive breast cancer54,55. After the results from the first-in-human phase I clinical trial in solid tumors56, buparlisib was tested in combination with either aromatase inhibitors57 or fulvestrant58 in ER-positive breast cancer patients, with some encouraging clinical responses but also important tolerability issues. In addition to the common side effects also observed with other PI3K inhibitors (rash, hyperglycemia, fatigue and diarrhea as discussed more below), buparlisib treatment induced neurological symptoms (e.g., depression) in a substantial proportion of patients, likely due to its impact blood brain barrier permeability59. In HER2-positive breast cancer, buparlisib was tested in combination with trastuzumab, both in the laboratory60,61 and in the clinical setting62–64 where promising clinical benefits were again accompanied by serious adverse events that rendered this therapeutic strategy impractical. Buparlisib was also tested in combination with olaparib in both breast and ovarian cancers65,66. However, the lack of biomarkers of sensitivity (beyond BRCA1/2 mutations) and the dose-limiting adverse effects observed in a significant proportion of patients precluded the design of larger trials testing the effectiveness of this combination. Another PI3K inhibitor with predominant and potent activity against PI3Kα and PI3Kδ that underwent substantial clinical development is copanlisib67. Copanlisib demonstrated impressive efficacy and encouraging tolerability leading to the FDA approval for the treatment of follicular lymphoma68 and it is now being tested with a number of combinatory treatments in patients with non-Hodgkin lymphoma (NCT04263584, NCT03474744, NCT03877055), including the anti-CD20 antibody rituximab69. Although this compound is considered a pan-PI3K inhibitor, its clinical efficacy in lymphoma is likely the result of the potent PI3Kδ inhibition. In addition, copanlisib is now being tested in clinical trials in patients with thyroid cancers (NCT04462471), with advanced solid cancers that have alterations in PIK3CA and PTEN (NCT04317105), and patients with HER2+ breast cancers (NCT04108858) among others. However, one of the clinical challenges of copanlisib is its administration, which is an intravenous infusion that has to be conducted at the hospital.
Despite the disappointing results obtained with buparlisib, clinical trials showed for the first time that PIK3CA mutant breast cancers might be particularly sensitive to PI3K inhibition and paved the way for the development of isoform-specific p110α inhibitors, which were predicted to have a better therapeutic window for cancers bearing this frequent genomic alteration. The first clinical PI3Kα-specific inhibitor, alpelisib, was developed with the aim of targeting the PI3K pathway (when activated by the p110α isoform), but limiting the emergence of toxicities attributed to inhibition of other isoforms of p11025,70. The phase I first-in-human trial of alpelisib in solid cancers with PIK3CA mutations demonstrated single-agent activity71, and several clinical studies were launched to test its activity in combination with targeted therapies, including anti-hormonal agents, in breast cancer patients. The combination of fulvestrant and alpelisib was tested in a phase I trial enrolling patients with ER-positive metastatic breast and showed promising clinical activity in the PIK3CA mutant subpopulation72. This early observation was confirmed in a large Phase III clinical trial (SOLAR1) where the progression-free survival (PFS) of PIK3CA mutant breast cancer patients treated with alpelisib plus fulvestrant was 11 months in comparison to 5.7 months of the patients treated with fulvestrant monotherapy73. These results granted FDA approval of this regime in ER-positive PIK3CA mutant metastatic breast cancer patients.
Another p110α inhibitor that was developed for solid tumors is taselisib. This potent inhibitor showed remarkable efficacy in preclinical studies74. During clinical trials, taselisib, in combination with endocrine therapy, demonstrated significant an increased response rate and duration of response compared to the placebo arms75. Nevertheless, the clinical activity was negated because of intractable toxicity, such as diarrhea, hyperglycemia, colitis, and stomatitis. The main difference between alpelisib and taselisib is the specificity for the p110 isoforms; taselisib is not only a potent p110α inhibitor but is also very active against p110δ. In addition, taselisib half-life is around 39 hours76, which could potentially result in drug accumulation with daily treatment regimes. Although PIK3CA mutant breast cancer has been the focus for both drugs, they have been tested in many different preclinical models, spanning across several cancer types. For example, taselisib showed interesting activity in head and neck cancer, another malignancy where genomic alterations of PIK3CA are frequently observed77. Preclinical studies suggested cooperativity with radiation therapy78 and in clinical trials, responses to taselisib monotherapy were also observed in heavily pre-treated patients with PIK3CA mutant head and neck cancer77.
More recently a potent and highly selective p110α inhibitor (GDC-0077) has been developed for clinical use. Mechanism of action studies indicate that GDC-0077 selectively degrades PI3Kα in a proteasome-dependent manner79. Although the completed results from the Phase I clinical trial have not yet been published, GDC-0077 is being tested in combination with endocrine agents and palbociclib in advanced ER+ breast cancer patients that harbor PIK3CA mutations (NCT04191499, NCT03006172).
Despite the efforts in targeting PI3K in solid tumors, the developments in hematological malignancies have also been significant. In this regard, specific inhibitors targeting the p110δ/γ isoforms have been developed and approved for the treatment of different forms of leukemia and lymphomas. For example, the PI3Kδ inhibitor idelalisib is approved by the FDA for the treatment of relapsed chronic lymphocytic leukemia, follicular non-Hodgkin lymphoma and small lymphocytic lymphoma80. Similarly, duvelisib, a dual inhibitor of the p110δ/γ isoforms, was approved by the FDA for the treatment of relapsed chronic lymphocytic leukemia, follicular lymphoma, and small lymphocytic lymphoma81. Another compound that has been recently approved for the treatment of follicular and marginal zone lymphoma is the dual PI3Kδ and casein kinase-1ε inhibitor umbralisib82.
A snapshot of the PI3K inhibitors that have been tested in advanced clinical phases or that are currently approved by the FDA can be found in Table 1.
Table 1. Pharmacological properties of clinical PI3K inhibitors.
PI3K inhibitors that have reached late phases of clinical development and/or have been approved by the FDA for the treatment of cancer are listed.
| In vitro IC50 (nM) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PI3K inhibitor | Company | Structure | PI3Kα | PI3Kβ | PI3K | PI3Kγ | Clinical | Dose | t1/2 (h) | Registration trial | Ref |
| Alpelisib | Novartis |
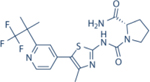
|
4.6 | 1156 | 290 | 250 | Approved for HR+/Her2- mBC in combination with Fulvestrant | Oral; 300 mg daily | 8 | NCT02437318 | 71–73 |
| Buparlisib | Novartis |
|
52 | 166 | 116 | 262 | Discontinued during Phase III | Oral; 100 mg daily | 40 | NCT01633060 | 58,121 |
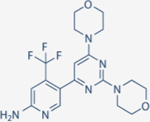
| Copanlisib | Bayer |
|
0.5 | 3.7 | 0.7 | 6.4 | Approved for relapsed follicular lymphoma | IV; 60 mg three times a month | 39.1 | NCT01660451 | 68,69 |
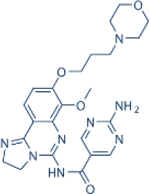
| Duvelisib | Verastem |
|
1602 | 85 | 2.5 | 27 | Approved for relapsed follicular lymphoma, Chronic lymphocytic leukemia; small lymphocytic leukemia | Oral; 25 mg twice daily | 4.7 | NCT02004522 | 81 |
| Idelalisib | Gilead |
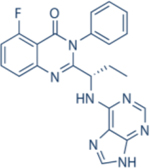
|
8600 | 4000 | 19 | 110 | Approved for relapsed follicular lymphoma, Chronic lymphocytic leukemia; small lymphocytic leukemia | Oral; 150 mg twice daily | 8.2 | NCT01282424 | 122,123 |
| Inavolisib | Genentech |
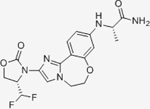
|
0.038 | N/A | N/A | N/A | Ongoing Phase III in combination with Fulvestrant and palbociclib for HR+/Her2- mBC | Oral; 9 mg daily | 18 | NCT04191499 | 124,125 |
| Taselisib | Genentech |
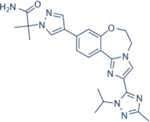
|
0.029 | 8 | 0.12 | 0.97 | Discontinued during Phase III | Oral; 4 mg daily | 39.3 | NCT02340221 | 76,77,94 |
| Umbrasilib | TG Therapeutics |
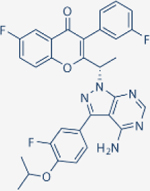
|
>10000 | >10000 | 6.2 | 1400 | Approved for relapsed follicular lymphoma and marginal zone lymphoma | Oral; 800 mg daily | 91 | NCT02793583 | 82 |
Finally, another emerging use of p110α inhibitors worth noting is in the treatment of the non-cancerous PIK3CA-related overgrowth spectrum (PROS), a group of heterogeneous disorders characterized by overgrowth and vascular anomalies. Preclinical and early clinical work has shown that alpelisib could be beneficial for the treatment of these disorders83,84 and clinical trials aimed at testing efficacy are currently underway.
Improving the therapeutic index of PI3K inhibitors
PI3K inhibitors have demonstrated clinically meaningful benefit in breast cancer, and additional indications and combinations are being investigated. It is likely that we have yet to realize the full benefit of completely inhibiting PI3K signaling in cancers. At a minimum, the therapeutic window of PI3K inhibitors is limited by on-target, off tumor toxicity. In particular, the induction of hyperglycemia and hyperinsulinemia are observed as major dose limiting toxicities for p110α inhibitors73. Insulin signaling in muscle and liver requires PI3K signaling, and inhibition of PI3K in these tissues impairs insulin signaling, leading to insulin resistance. Thus, it is likely that induction of hyperinsulinemia and hyperglycemia prevents dosing high enough to fully suppress PI3K signaling in the tumor.
With the recent approval of alpelisib, there is increased need to find methods to mitigate hyperglycemia. In the clinical trial of alpelisib that led to its approval for breast cancer, 37% of patients developed grade 3–4 hyperglycemia, and this toxicity was one of the main causes of dose interruptions and reductions73. Although the progression-free survival (PFS) was 11 months for the alpelisib treatment arm (approximately double the PFS of the control arm), the majority of patients required dose interruptions and/or dose reductions. Moreover, recent laboratory studies reveal that the higher insulin levels may activate P3K signaling in cancer cells, thereby decreasing the degree of the PI3K inhibition in the cancer cells40. More effective mitigation of hyperinsulinemia and hyperglycemia may lead to more uninterrupted dosing, which in turn might result in even superior clinical efficacy. Although metformin was employed to treat most patients in early trials, newer and more potent therapeutic approaches to manage hyperglycemia may have a significant impact on the patients’ quality of life. One such class of agents are the SGLT2 inhibitors, and in preclinical studies, their concomitant administration with PI3K inhibitors abrogated induction of insulin and led to better tumor growth control in preclinical studies 40. Notably, ketogenic diet also yielded promising preclinical results as well. Future clinical studies will reveal the impact of more effective management of hyperglycemia on this class of agents85.
To improve the therapeutic index of PI3K inhibitors careful consideration should also be given to patient stratification (Figure 3). In fact, alpelisib was only approved for PIK3CA-mutated advanced breast cancer as detected by an FDA-approved companion diagnostic PCR test to select for patients who have PIK3CA mutations in tumor tissue and/or in circulating tumor DNA (ctDNA) isolated from plasma. As expected, in SOLAR-1, a meaningful benefit was not observed for alpelisib-fulvestrant in the cohort without PIK3CA-mutated cancer73. Apart from PIK3CA mutations being a powerful biomarker for alpelisib sensitivity, the SOLAR-1 study and a phase I/II study of alpelisib plus anti-estrogen therapy (NCT01870505) showed a wide range of clinical benefit. These results prompted additional investigations for additional molecular stratifiers86, which identified PTEN loss in a subset of cancers with primary resistance. Accordingly, excluding tumors with PTEN loss might yield meaningful improvements in the response rates of PI3K inhibitors.
Figure 3. Proposed approaches to increase the therapeutic index of PI3K inhibitors.

Several factors that may increase the therapeutic index of PI3K inhibitors are required to improve the clinical application of these agents. These include pharmacological strategies, such as careful consideration of dose and schedule; patient stratification allowing the identification of biomarkers and mediators of drug resistance; the effect of metabolism and diet, and the successful management of common toxicities such as hyperglycemia, GI problems, and rash among others. Similar to many other therapies, there are novel modalities to inhibit PI3K that need to be considered, which should overcome the current limitations of PI3K inhibitors in the clinic.
As highlighted above, multiple PI3K inhibitors failed to provide a clinical meaningful outcome (i.e buparlisib, pictilisib), in part because of substantial toxicities, highlighting the importance of dose and scheduling as another key solution in increasing their therapeutic index (Figure 3). Through the lens of targeted therapies, the attention to classical clinical pharmacology characteristics (such as dose, regimen, bioaccumulation, and others) has often been suboptimally investigated, mostly because targeted agents were anticipated to have a positive safety profile and because continuous dosing has been favored with the assumption of prolonged target inhibition87. In order to deliver the full potential of PI3K inhibitors, new scheduling and dosing may be less toxic while preserving efficacy. For instance, in preclinical models both metronomic (daily) and intermittent (weekly) inhibition of PI3K using different doses of pictilisib has been studied88. The different treatment doses and schedules examined in this study elicited similar tumor responses in combination with fulvestrant88. Moreover, a first-in-human, Phase I dose-escalation study of the p110α inhibitor serabelisib (TAK-117) found that intermitted dosing had an acceptable safety profile and enabled higher doses and total weekly exposures in comparison to once-daily dosing89, suggesting that alternative dosing and scheduling strategies might expand the therapeutic window of PI3K inhibitors.
In order to overcome the current limitations of PI3K inhibitors, novel strategies to target this enzyme have been envisioned. For instance, a potential strategy to mitigate toxicity is to develop selective inhibitors for mutant PI3K, targeting either the helical domain, E545K, or the kinase domain, H1047R, mutations. Selective compounds that specifically inhibit these mutant proteins but spare wildtype p110α have the promise of fully suppressing cancer signaling while having minimal effect on PI3K signaling in normal tissues, which in turn would ameliorate the metabolic toxicities associated to non-selective PI3K signaling inhibition. Since the helical and kinase domain mutations reside in distinct regions of p110α, likely increasing the enzyme’s activity via distinct molecular mechanisms, it seems unlikely that a single molecular entity would be able to inhibit both mutants while sparing the wildtype isoform. Thus, it is quite conceivable that there will be different mutation-specific selective inhibitors for the helical and kinase domain mutants, respectively. The impact of inhibiting wildtype p110α in cancers that harbor an oncogenic variant remain largely unknown. Although mutant-selective inhibitors hold great promise, there may be indications in which targeting wild-type PI3K may have important anti-tumor activity, and in these indications, a pan p110α or AKT inhibitor compound administered concomitantly with a potent anti-hyperglycemia regimen may be the preferred approach.
An emerging chemical technology that could be of interest for targeting PI3K is the use of proteolysis targeting chimeras (PROTAC) to promote specific degradation of proteins. These heterobifunctional molecules have the ability to bind a target and recruit ubiquitin ligase complexes (namely CRL2VHL or CRL4CRBN)90. Using the large collection of PI3K kinase inhibitors available to date, it is in principle possible to create such molecules; however, the advantage of these compounds in terms of therapeutic efficacy and toxicity over the classic kinase inhibitors remains to be addressed. For instance, GDC0077 was reported to induce degradation of mutant p110α, although the mechanism that leads to proteolysis has not yet been discovered79. If mutant p110α is more prone to degradation, this could represent a promising starting point for mutant-specific inhibitors. Bifunctional molecules that enforce protein-protein interactions, such as the so-called small-molecule protein ligands interface stabilizers (SPLINTs), could be engineered to re-establish and/or stabilize the inhibitory interaction with p8591. This would be particularly interesting in PIK3CA helical mutant tumors, in which such interaction is compromised.
PI3K inhibitors with clinical experience are orthosteric antagonists and only interact within the catalytic pocket of p110α. However, the mechanism of activation of PI3K involves many steps that could be potentially targeted with allosteric inhibitors. For instance, structural and biochemical studies have shown that pTyr peptides and RAS proteins promote dynamic changes in p85-p110α dimers that result in increased kinase activity16,92. Therefore, allosteric inhibitors that leverage these mechanisms could lead to novel targeting strategies that are selective for cancers with specific mechanisms of activating the PI3K pathway.
Therapeutic combinations
It has long been speculated that PI3K inhibitors would be most effective as part of combination strategies, especially when used in earlier lines of therapy. Currently, alpelisib is approved in combination with anti-estrogens in breast cancer, where a few additional combinations are generating a great interest. Preclinical studies demonstrate that combined CDK4/6 and p110α inhibition is highly effective in PIK3CA mutant breast cancers93, and clinical trials of the combination are currently underway. Indeed, recent data demonstrate that the triplet combination of palbociclib, taselisib, and fulvestrant has promising efficacy in heavily pretreated PIK3CA-mutant ER+ breast cancer94. There is also interest in developing PI3K inhibitors in ERBB2 amplified breast cancers. HER2 provides a strong signal to PI3K (via HER3), and preclinical studies demonstrate that ERBB2 amplified breast cancers are among the most sensitive cancers to single-agent PI3K inhibitors. This has led to the clinical development of PI3K inhibitors and HER2-targeting agents in this subset of breast cancers. In addition, and as mentioned before, PI3K inhibitors have shown intriguing activity with PARP inhibitors in BRCA1 mutant breast and ovarian cancers65,66, and trials assessing this combo are underway. There is also substantial preclinical data examining other combinations with conventional chemotherapy and radiation therapy, and clinical trials have begun to assess their efficacies. For example, a trial of buparlisib and paclitaxel for head and neck cancer had demonstrated an overall survival benefit compared to paclitaxel95.
It has been long-appreciated that concomitant inhibition of the PI3K-AKT and MEK-ERK pathways is effective at killing cancer cells96. However, clinical trials of these combos have been disappointing, likely because toxicity prevents full suppression of both pathways. Indeed, the challenges with respect to therapeutic index of single agent p110α inhibitors underscores the difficulties combining these agents with MEK and/or ERK inhibitors, each of which have their own challenges with therapeutic index. Thus, these combinations are ripe for mutant selective PI3K inhibitors in PIK3CA mutant cancers. For example, a combination of such PI3K inhibitor with a MEK inhibitor would suppress MEK/ERK systemically, but concomitant inhibition of PI3K and MEK/ERK pathways would occur exclusively in cancer cells. Furthermore, the advent of mutant KRAS selective inhibitors97 holds promise for combinations with PI3K inhibitors. Indeed, preclinical experiments have demonstrated PI3K inhibitors may be quite powerful combination partners for KRAS G12C inhibitors98. Furthermore, some KRAS mutant cancers (e.g. KRAS G12D GI cancers) have significant flux through the PI3K pathway in lung cancers50, and concomitant inhibition of KRAS and PI3K might have significant anti-tumor efficacy. It is also well-known that several colorectal cancers harbor both KRAS and PIK3CA mutations, suggesting that combined KRAS and PI3K inhibition might be especially effective in these malignancies. In KRAS mutant cancers, a mutant selective KRAS inhibitor might combine well with a PI3K inhibitor because both pathways will be suppressed specifically in cancer cells, but the MEK/ERK pathway will remain intact in the normal cells of the body, increasing significantly the tolerability of the combination.
Resistance mechanisms
Drug resistance continues to be a major challenge in cancer therapy, and is determined by the pharmacological properties of the drugs in concert with the cell-intrinsic and extrinsic properties of the heterogeneous tumor environment99. Similar to the majority of cancer drugs, resistance to PI3K inhibitors is a key factor limiting the impact of these agents in the clinic. Resistance mechanisms can be divided into de novo, where no initial clinical responses are observed due to intrinsic refractoriness of the tumors (or very rapid adaptation to PI3K inhibition), and acquired, when prolonged clinical response is followed by therapy escape. There are several well-characterized mechanisms of resistance to PI3K inhibitors, encompassing the reactivation of the PI3K pathway and the activation of compensatory parallel signaling cascades, which in turn restore cell homeostasis following PI3K suppression.
Initial studies conducted over a decade ago identified the expression and activation of RTKs as a way that can fuel downstream signaling pathways limiting the response to PI3K inhibitors100–103. RTKs such as HER3 and others can stimulate PI3K by binding of pTyr to the SH2 domains of p85, thereby relieving its inhibition of p110α. The relevance of this cellular adaptation to PI3K inhibitors is underscored by the observation that combinatorial treatments targeting RTKs and PI3K led to superior antitumor activity in the preclinical setting in PI3K-driven tumors61,104,105.
A clinically validated mechanism of acquired resistance to p110α specific inhibitors is the loss of function mutations in the tumor suppressor PTEN, which leads to increased p110β signaling106. Loss of PTEN was first found to limit the effectiveness of p110α inhibition by analyzing different metastases of a patient that showed an impressive initial response to alpelisib monotherapy followed by widespread progression106. In addition, analyses of plasma and tumors from a recent clinical trial attesting the combination of alpelisib and aromatase inhibitors (NCT01870505) uncovered loss of function PTEN mutations in 25% of patients with de novo resistance86. Mechanistically, inhibition of the p110β isoform was required to restore antitumor activity in breast cancer cells that lost PTEN expression and became resistant to alpelisib106. Likewise, initial efficacy of p110α inhibition was mitigated by rapid accumulation of PIP3 produced by the p110β isoform which re-activates PI3K107. Consistently, the addition of a p110β inhibitor to p110α prevented PIP3 rebound and led to greater anti-tumor effects compared to single agent treatment in breast cancer and in PTEN deficient preclinical models of prostate cancer108. The biological determinants of the increased dependency on PI3K p110β in PTEN loss cancer cells remains to be delineated.
Resistance to PI3K inhibitors can also been mediated by parallel activation of related kinases that feed into the PI3K pathway. For instance, activation of mTORC1 can drive resistance to PI3K inhibitors and everolimus can re-sensitize breast and head and neck cancers to PI3Kα blockade23,109–111. Mechanistically, we and others have shown that in breast cancer cells resistant to p110α inhibition, the PDK1-SGK axis can overcome AKT inhibition by activating mTORC1 signaling110,111. More recently, we identified and validated several negative regulators of mTORC1 including TSC1, TSC2, ITFG2, TBC1D7, AKT1S1, STK11, NPRL2, NPRL3 among others, whose loss reduced the sensitivity to p110α inhibition112. Other kinases that have been proposed to confer resistance to alpelisib include the overexpression and/or overactivation of PIM and PKC kinases, which maintain downstream pathway activation in an AKT-independent manner109,113. Moreover, inhibition of other kinases such as cyclin-dependent kinase 4 and 6 (CDK4/6) can restore sensitivity to alpelisib in breast cancer cells in cancers that maintain RB phosphorylation despite inhibition of PI3K93.
A more recent mechanism of resistance to PI3K inhibitors involving a cancer-cell extrinsic mechanism was described in murine models of breast cancer, showing that an increase in blood glucose and insulin following treatment with PI3K inhibitors was sufficient to activate PI3K signaling, even in the presence of PI3K inhibitors, through systematic glucose-insulin feedback40. As mentioned above, this feedback could be inhibited by sodium glucose cotransporter 2 (SGLT2) inhibition or ketogenic diet that prevent hyperinsulinemia40. Another study found that expression of the FOXM1 transcription factor upon PI3K inhibition is a biomarker of resistance due to metabolic changes driven by the expression of lactate dehydrogenase (LDH)114. Thus, changes in the metabolomic milieu could predict response to PI3K inhibitors and rapid assessment of these alterations may help identify patients who would benefit from PI3K inhibitors. In addition, pharmacological targeting of specific metabolic enzymes, which are inherently druggable, may offer effective new therapeutic alternatives9.
A critical example of a highly uniform adaptive mechanism is the activation of ER signaling upon PI3Kα inhibition, which drives resistance to PI3K inhibitors115. The importance of the PI3K pathway in ER+ breast cancer is underscored by the high frequency of activating mutations in PIK3CA (~40%) in this cancer subtype116. In this regard, PI3Kα inhibition elicited an increased dependency of PIK3CA-mutant breast cancer cells on ER signaling, which limited their sensitivity and was reversed by the addition of anti-ER therapies115. Mechanistically, PI3Kα inhibition regulated ER activity by the phosphorylation of the epigenetic regulator KMT2D by the PI3K effectors AKT/SGK117,118. The clinical utility of combined inhibition of ER and PI3Kα was established in the SOLAR-1 clinical trial, where the addition of alpelisib to anti-ER therapy led to a substantial increase in progression-free survival73.
In the context of prostate cancer, which is characterized by its dependence on androgen receptor (AR) and PI3K signaling through loss of PTEN, inhibition of PI3K paradoxically activates AR-mediated transcription as a survival feedforward mechanism119. Similar to ER in breast cancer, the inhibition of PI3K (with pan-PI3K inhibitors) or AKT, together with anti-AR therapy, led to tumor shrinkage. This has raised hopes for more durable disease control through combination of PI3K inhibitors and anti-AR therapy in prostate cancer with loss of PTEN. Currently, AKT inhibitors in combination with anti-AR therapy are showing promising results in phase II clinical trials120. Altogether, it is notable that the targets whose inhibition elicits powerful adaptive responses seem to belong to cellular growth pathways that are important for the growth of both normal and cancer cells and that are regulated by complex feedback loops.
Conclusion
Although PIK3CA is among the most common oncogenes in cancer and several studies have demonstrated its validity as a biomarker for PI3K inhibition, skepticism remains in considering this pathway a suitable and satisfactory target for cancer therapy. This could be for a number of reasons. First, PIK3CA mutations do not necessarily translate to poor prognosis or aggressive phenotypes, as seen in other oncogenes such as EGFR in lung cancer. Second, the relatively narrow therapeutic window of these agents, due to the relevance of the PI3K pathway for the homeostasis of normal cells, can limit its therapeutic window. Even in tumors that are highly addicted to this signaling cascade for proliferation and survival, dose-limiting toxicities result in short and incomplete target engagement. Third, inhibition of the pathway often causes a fast and tissue-dependent activation of molecular feedbacks that limit the long-term efficacy of these agents. However, this can also be an opportunity to design rationale-based combinatorial strategies and new drug discovery approaches that increase the efficacy and safety of PI3K inhibitors.
In our opinion, and considering the new and exciting body of evidence which led to the FDA approval of PI3K inhibitors, these agents should not be limited to very specific cancer subtypes and lines of therapy, but rather tested in other cancer types to broaden their application. This implies identifying other tumor types, or subtypes, that are likely to respond to these medicines, developing appropriate combinatorial therapies that maximize their benefit, reducing toxicities that lead to treatment discontinuation, and developing novel chemical modalities that would allow cancer-specific inhibition of PI3K.
Acknowledgements
We would like to dedicate this review to the memory of Jose Baselga, who was one of the pioneers in the clinical development of PI3K inhibitors. PC work is supported by the NCI K99/R00 Pathway to Independence Award (K99CA245122). ET is supported by grants from JKTG foundation, Breast Cancer Research Alliance, Institute of Cancer Informatics, and NCI K22 Transition to Independence award. MS is supported by the NIH grants P30 CA008748.
Footnotes
Conflicts of interest
PC is founder and advisory board member of Venthera, Inc. ET has received honorarium for invited lectures at AstraZeneca and Oric Pharmaceuticals. MS is an Astrazeneca employee, holds AstraZeneca equity and is a co-founder of medendi.org. In the past two years, MS received research funding from Daiichi-Sankio, AstraZeneca, Menarini Ricerche, Puma Biotechnologies and Targimmune. JAE was a Novartis employee and holds Novartis equity. JAE is a co-founder of Treeline Biosciences.
References
- 1.Bilanges B, Posor Y. & Vanhaesebroeck B. PI3K isoforms in cell signalling and vesicle trafficking. Nat Rev Mol Cell Biol 20, 515–534 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Thorpe LM, Yuzugullu H. & Zhao JJ PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 15, 7–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fruman DA et al. The PI3K Pathway in Human Disease. Cell 170, 605–635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burke JE Structural Basis for Regulation of Phosphoinositide Kinases and Their Involvement in Human Disease. Mol Cell 71, 653–673 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Vadas O, Burke JE, Zhang X, Berndt A. & Williams RL Structural basis for activation and inhibition of class I phosphoinositide 3-kinases. Sci Signal 4, re2 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez-Viciana P. et al. Phosphatidylinositol-3-OH kinase direct target of Ras. Nature 370, 527–532 (1994). [DOI] [PubMed] [Google Scholar]
- 7.Gupta S. et al. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell 129, 957–968 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Park WS et al. Comprehensive identification of PIP3-regulated PH domains from C. elegans to H. sapiens by model prediction and live imaging. Mol Cell 30, 381–392 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoxhaj G. & Manning BD The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer 20, 74–88 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu GY & Sabatini DM mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol 21, 183–203 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saxton RA & Sabatini DM mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eijkelenboom A. & Burgering BMT FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol 14, 83–97 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Worby CA & Dixon JE PTEN. Annu Rev Biochem 83, 641–669 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Samuels Y. et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 304, 554 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Miled N. et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science 317, 239–242 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Burke JE, Perisic O, Masson GR, Vadas O. & Williams RL Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110α (PIK3CA). PNAS 109, 15259–15264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailey MH et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–385.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Madsen RR, Vanhaesebroeck B. & Semple RK Cancer-Associated PIK3CA Mutations in Overgrowth Disorders. Trends in Molecular Medicine 24, 856–870 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Castel P, Rauen KA & McCormick F. The duality of human oncoproteins: drivers of cancer and congenital disorders. Nat Rev Cancer 20, 383–397 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lui VWY et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov 3, 761–769 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jia S. et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 454, 776–779 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheung LWT et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov 1, 170–185 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elkabets M. et al. mTORC1 inhibition is required for sensitivity to PI3K p110α inhibitors in PIK3CA-mutant breast cancer. Sci Transl Med 5, 196ra99 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Will M. et al. Rapid induction of apoptosis by PI3K inhibitors is dependent upon their transient inhibition of RAS-ERK signaling. Cancer Discov 4, 334–347 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fritsch C. et al. Characterization of the novel and specific PI3Kα inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol. Cancer Ther 13, 1117–1129 (2014). [DOI] [PubMed] [Google Scholar]
- 26.She Q-B et al. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell 8, 287–297 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salih DAM & Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol 20, 126–136 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang W. et al. Autophagy promotes escape from phosphatidylinositol 3-kinase inhibition in estrogen receptor-positive breast cancer. FASEB J 32, 1222–1235 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takuwa N, Fukui Y. & Takuwa Y. Cyclin D1 expression mediated by phosphatidylinositol 3-kinase through mTOR-p70(S6K)-independent signaling in growth factor-stimulated NIH 3T3 fibroblasts. Mol Cell Biol 19, 1346–1358 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Faber AC et al. mTOR inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1. Cancer Discov 4, 42–52 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Liu L, Chang H, Zou Z. & Xing D. Downregulation of MCL-1 and upregulation of PUMA using mTOR inhibitors enhance antitumor efficacy of BH3 mimetics in triple-negative breast cancer. Cell Death Dis 9, 137 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soler A, Angulo-Urarte A. & Graupera M. PI3K at the crossroads of tumor angiogenesis signaling pathways. Mol Cell Oncol 2, e975624 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soler A. et al. Inhibition of the p110α isoform of PI 3-kinase stimulates nonfunctional tumor angiogenesis. J Exp Med 210, 1937–1945 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaneda MM et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 539, 437–442 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis RJ et al. Anti-PD-L1 Efficacy Can Be Enhanced by Inhibition of Myeloid-Derived Suppressor Cells with a Selective Inhibitor of PI3Kδ/γ. Cancer Res 77, 2607–2619 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okkenhaug K. et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science 297, 1031–1034 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Angulo I. et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science 342, 866–871 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ali K. et al. Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 510, 407–411 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okkenhaug K, Graupera M. & Vanhaesebroeck B. Targeting PI3K in Cancer: Impact on Tumor Cells, Their Protective Stroma, Angiogenesis, and Immunotherapy. Cancer Discov 6, 1090–1105 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hopkins BD et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560, 499–503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ortega-Molina A. et al. Pharmacological inhibition of PI3K reduces adiposity and metabolic syndrome in obese mice and rhesus monkeys. Cell Metab 21, 558–570 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei Z. et al. Death-associated protein kinase 1 (DAPK1) controls CD8+ T cell activation, trafficking, and antitumor activity. FASEB J 35, e21138 (2021). [DOI] [PubMed] [Google Scholar]
- 43.Ding J, Vlahos CJ, Liu R, Brown RF & Badwey JA Antagonists of phosphatidylinositol 3-kinase block activation of several novel protein kinases in neutrophils. J Biol Chem 270, 11684–11691 (1995). [DOI] [PubMed] [Google Scholar]
- 44.Anel A. et al. Two signaling pathways can lead to Fas ligand expression in CD8+ cytotoxic T lymphocyte clones. Eur J Immunol 25, 3381–3387 (1995). [DOI] [PubMed] [Google Scholar]
- 45.Fruman DA et al. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85alpha. Science 283, 393–397 (1999). [DOI] [PubMed] [Google Scholar]
- 46.Sanchez-Margálet V, Goldfine ID, Vlahos CJ & Sung CK Role of phosphatidylinositol-3-kinase in insulin receptor signaling: studies with inhibitor, LY294002. Biochem Biophys Res Commun 204, 446–452 (1994). [DOI] [PubMed] [Google Scholar]
- 47.Hayakawa J. et al. Inhibition of BAD phosphorylation either at serine 112 via extracellular signal-regulated protein kinase cascade or at serine 136 via Akt cascade sensitizes human ovarian cancer cells to cisplatin. Cancer Res 60, 5988–5994 (2000). [PubMed] [Google Scholar]
- 48.Misra UK & Pizzo SV Binding of receptor-recognized forms of alpha2-macroglobulin to the alpha2-macroglobulin signaling receptor activates phosphatidylinositol 3-kinase. J Biol Chem 273, 13399–13402 (1998). [DOI] [PubMed] [Google Scholar]
- 49.Graness A, Adomeit A, Heinze R, Wetzker R. & Liebmann C. A novel mitogenic signaling pathway of bradykinin in the human colon carcinoma cell line SW-480 involves sequential activation of a Gq/11 protein, phosphatidylinositol 3-kinase beta, and protein kinase Cepsilon. J Biol Chem 273, 32016–32022 (1998). [DOI] [PubMed] [Google Scholar]
- 50.Ihle NT et al. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther 3, 763–772 (2004). [PubMed] [Google Scholar]
- 51.Folkes AJ et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem 51, 5522–5532 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Junttila TT et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 15, 429–440 (2009). [DOI] [PubMed] [Google Scholar]
- 53.Maira S-M et al. Identification and Characterization of NVP-BKM120, an Orally Available Pan-Class I PI3-Kinase Inhibitor. Mol Cancer Ther 11, 317–328 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Sanchez CG et al. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res 13, R21 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller TW et al. ERα-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov 1, 338–351 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bendell JC et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol 30, 282–290 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Mayer IA et al. Stand up to cancer phase Ib study of pan-phosphoinositide-3-kinase inhibitor buparlisib with letrozole in estrogen receptor-positive/human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol 32, 1202–1209 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baselga J. et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet Oncology 18, 904–916 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Geuna E. et al. Buparlisib, an oral pan-PI3K inhibitor for the treatment of breast cancer. Expert Opin Investig Drugs 24, 421–431 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Serra V. et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 68, 8022–8030 (2008). [DOI] [PubMed] [Google Scholar]
- 61.Rexer BN, Chanthaphaychith S, Dahlman K. & Arteaga CL Direct inhibition of PI3K in combination with dual HER2 inhibitors is required for optimal antitumor activity in HER2+ breast cancer cells. Breast Cancer Res 16, R9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saura C. et al. Phase Ib study of Buparlisib plus Trastuzumab in patients with HER2-positive advanced or metastatic breast cancer that has progressed on Trastuzumab-based therapy. Clin Cancer Res 20, 1935–1945 (2014). [DOI] [PubMed] [Google Scholar]
- 63.Loibl S. et al. Neoadjuvant buparlisib plus trastuzumab and paclitaxel for women with HER2+ primary breast cancer: A randomised, double-blind, placebo-controlled phase II trial (NeoPHOEBE). Eur J Cancer 85, 133–145 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pistilli B. et al. Phase II study of buparlisib (BKM120) and trastuzumab in patients with HER2+ locally advanced or metastatic breast cancer resistant to trastuzumab-based therapy. Breast Cancer Res Treat 168, 357–364 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ibrahim YH et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov 2, 1036–1047 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Juvekar A. et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov 2, 1048–1063 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu N. et al. BAY 80–6946 is a highly selective intravenous PI3K inhibitor with potent p110α and p110δ activities in tumor cell lines and xenograft models. Mol Cancer Ther 12, 2319–2330 (2013). [DOI] [PubMed] [Google Scholar]
- 68.Dreyling M. et al. Phosphatidylinositol 3-Kinase Inhibition by Copanlisib in Relapsed or Refractory Indolent Lymphoma. J Clin Oncol 35, 3898–3905 (2017). [DOI] [PubMed] [Google Scholar]
- 69.Matasar MJ et al. Copanlisib plus rituximab versus placebo plus rituximab in patients with relapsed indolent non-Hodgkin lymphoma (CHRONOS-3): a double-blind, randomised, placebo-controlled, phase 3 trial. The Lancet Oncology S1470204521001455 (2021) doi: 10.1016/S1470-2045(21)00145-5. [DOI] [PubMed] [Google Scholar]
- 70.Furet P. et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg Med Chem Lett 23, 3741–3748 (2013). [DOI] [PubMed] [Google Scholar]
- 71.Juric D. et al. Phosphatidylinositol 3-Kinase α-Selective Inhibition With Alpelisib (BYL719) in PIK3CA-Altered Solid Tumors: Results From the First-in-Human Study. J Clin Oncol 36, 1291–1299 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Juric D. et al. Alpelisib Plus Fulvestrant in PIK3CA-Altered and PIK3CA-Wild-Type Estrogen Receptor-Positive Advanced Breast Cancer: A Phase 1b Clinical Trial. JAMA Oncol 5, e184475 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.André F. et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor–Positive Advanced Breast Cancer. New England Journal of Medicine 380, 1929–1940 (2019). [DOI] [PubMed] [Google Scholar]
- 74.Ndubaku CO et al. Discovery of 2-{3-[2-(1-isopropyl-3-methyl-1H-1,2–4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}−2-methylpropanamide (GDC-0032): a β-sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor activity. J Med Chem 56, 4597–4610 (2013). [DOI] [PubMed] [Google Scholar]
- 75.Saura C. et al. Neoadjuvant letrozole plus taselisib versus letrozole plus placebo in postmenopausal women with oestrogen receptor-positive, HER2-negative, early-stage breast cancer (LORELEI): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol 20, 1226–1238 (2019). [DOI] [PubMed] [Google Scholar]
- 76.Juric D. et al. Phase I Dose-Escalation Study of Taselisib, an Oral PI3K Inhibitor, in Patients with Advanced Solid Tumors. Cancer Discov 7, 704–715 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jhaveri K. et al. Phase I Basket Study of Taselisib, an Isoform-Selective PI3K Inhibitor, in Patients with PIK3CA-Mutant Cancers. Clin Cancer Res 27, 447–459 (2021). [DOI] [PubMed] [Google Scholar]
- 78.Zumsteg ZS et al. Taselisib (GDC-0032), a Potent β-Sparing Small Molecule Inhibitor of PI3K, Radiosensitizes Head and Neck Squamous Carcinomas Containing Activating PIK3CA Alterations. Clin Cancer Res 22, 2009–2019 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hong R. et al. Abstract PD4–14: GDC-0077 is a selective PI3Kalpha inhibitor that demonstrates robust efficacy in PIK3CA mutant breast cancer models as a single agent and in combination with standard of care therapies. in Poster Discussion Abstracts PD4–14-PD4–14 (American Association for Cancer Research, 2018). doi: 10.1158/1538-7445.SABCS17-PD4-14. [DOI] [Google Scholar]
- 80.Furman RR et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med 370, 997–1007 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Flinn IW et al. The phase 3 DUO trial: duvelisib vs ofatumumab in relapsed and refractory CLL/SLL. Blood 132, 2446–2455 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fowler NH et al. Umbralisib, a Dual PI3Kδ/CK1ε Inhibitor in Patients With Relapsed or Refractory Indolent Lymphoma. J Clin Oncol JCO2003433 (2021) doi: 10.1200/JCO.20.03433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Castel P. et al. Somatic PIK3CA mutations as a driver of sporadic venous malformations. Sci Transl Med 8, 332ra42 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Venot Q. et al. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature 558, 540–546 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hanker AB, Kaklamani V. & Arteaga CL Challenges for the Clinical Development of PI3K Inhibitors: Strategies to Improve Their Impact in Solid Tumors. Cancer Discov 9, 482–491 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Razavi P. et al. Alterations in PTEN and ESR1 promote clinical resistance to alpelisib plus aromatase inhibitors. Nat Cancer 1, 382–393 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Toska E. & Baselga J. Pharmacology in the Era of Targeted Therapies: The Case of PI3K Inhibitors. Clin Cancer Res 22, 2099–2101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang W. et al. Strategically Timing Inhibition of Phosphatidylinositol 3-Kinase to Maximize Therapeutic Index in Estrogen Receptor Alpha-Positive, PIK3CA-Mutant Breast Cancer. Clin Cancer Res 22, 2250–2260 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Juric D. et al. A First-in-Human, Phase I, Dose-Escalation Study of TAK-117, a Selective PI3Kα Isoform Inhibitor, in Patients with Advanced Solid Malignancies. Clin Cancer Res 23, 5015–5023 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lai AC & Crews CM Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov 16, 101–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fischer ES, Park E, Eck MJ & Thomä NH SPLINTS: small-molecule protein ligand interface stabilizers. Curr Opin Struct Biol 37, 115–122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Siempelkamp BD, Rathinaswamy MK, Jenkins ML & Burke JE Molecular mechanism of activation of class IA phosphoinositide 3-kinases (PI3Ks) by membrane-localized HRas. J Biol Chem 292, 12256–12266 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vora SR et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 26, 136–149 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pascual J. et al. Triplet Therapy with Palbociclib, Taselisib, and Fulvestrant in PIK3CA-Mutant Breast Cancer and Doublet Palbociclib and Taselisib in Pathway-Mutant Solid Cancers. Cancer Discov (2020) doi: 10.1158/2159-8290.CD-20-0553. [DOI] [PubMed] [Google Scholar]
- 95.Soulières D. et al. Buparlisib and paclitaxel in patients with platinum-pretreated recurrent or metastatic squamous cell carcinoma of the head and neck (BERIL-1): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Oncol 18, 323–335 (2017). [DOI] [PubMed] [Google Scholar]
- 96.Engelman JA et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med 14, 1351–1356 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ostrem JM, Peters U, Sos ML, Wells JA & Shokat KM K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Misale S. et al. KRAS G12C NSCLC Models Are Sensitive to Direct Targeting of KRAS in Combination with PI3K Inhibition. Clin Cancer Res 25, 796–807 (2019). [DOI] [PubMed] [Google Scholar]
- 99.Vasan N, Baselga J. & Hyman DM A view on drug resistance in cancer. Nature 575, 299–309 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.O’Reilly KE et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 66, 1500–1508 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chandarlapaty S. et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 19, 58–71 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Serra V. et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene 30, 2547–2557 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chakrabarty A, Sánchez V, Kuba MG, Rinehart C. & Arteaga CL Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc Natl Acad Sci U S A 109, 2718–2723 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tao JJ et al. Antagonism of EGFR and HER3 enhances the response to inhibitors of the PI3K-Akt pathway in triple-negative breast cancer. Sci Signal 7, ra29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wong MH et al. Cotargeting of epidermal growth factor receptor and PI3K overcomes PI3K-Akt oncogenic dependence in pancreatic ductal adenocarcinoma. Clin Cancer Res 20, 4047–4058 (2014). [DOI] [PubMed] [Google Scholar]
- 106.Juric D. et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature 518, 240–244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Costa C. et al. Measurement of PIP3 levels reveals an unexpected role for p110β in early adaptive responses to p110α-specific inhibitors in luminal breast cancer. Cancer Cell 27, 97–108 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schwartz S. et al. Feedback suppression of PI3Kα signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kβ. Cancer Cell 27, 109–122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Elkabets M. et al. AXL mediates resistance to PI3Kα inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 27, 533–546 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Castel P. et al. PDK1-SGK1 Signaling Sustains AKT-Independent mTORC1 Activation and Confers Resistance to PI3Kα Inhibition. Cancer Cell 30, 229–242 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bago R. et al. The hVps34-SGK3 pathway alleviates sustained PI3K/Akt inhibition by stimulating mTORC1 and tumour growth. EMBO J 35, 1902–1922 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cai Y. et al. Genomic alterations in PIK3CA-mutated breast cancer result in mTORC1 activation and limit sensitivity to PI3Kα inhibitors. Cancer Res (2021) doi: 10.1158/0008-5472.CAN-20-3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Le X. et al. Systematic Functional Characterization of Resistance to PI3K Inhibition in Breast Cancer. Cancer Discov 6, 1134–1147 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ros S. et al. Metabolic Imaging Detects Resistance to PI3Kα Inhibition Mediated by Persistent FOXM1 Expression in ER+ Breast Cancer. Cancer Cell 38, 516–533.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bosch A. et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci Transl Med 7, 283ra51 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ciriello G. et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 163, 506–519 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Toska E. et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science 355, 1324–1330 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Toska E. et al. PI3K Inhibition Activates SGK1 via a Feedback Loop to Promote Chromatin-Based Regulation of ER-Dependent Gene Expression. Cell Rep 27, 294–306.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Carver BS et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 19, 575–586 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.de Bono JS et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin Cancer Res 25, 928–936 (2019). [DOI] [PubMed] [Google Scholar]
- 121.Di Leo A. et al. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet Oncology 19, 87–100 (2018). [DOI] [PubMed] [Google Scholar]
- 122.Gopal AK et al. PI3Kδ Inhibition by Idelalisib in Patients with Relapsed Indolent Lymphoma. N Engl J Med 370, 1008–1018 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Salles G. et al. Efficacy and safety of idelalisib in patients with relapsed, rituximab- and alkylating agent-refractory follicular lymphoma: a subgroup analysis of a phase 2 study. Haematologica 102, e156–e159 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Juric D. et al. Abstract OT1–08-04: A first-in-human phase Ia dose escalation study of GDC-0077, a p110a-selective and mutant-degrading PI3K inhibitor, in patients with PIK3CA -mutant solid tumors. in Ongoing Clinical Trials OT1–08-04-OT1–08–04 (American Association for Cancer Research, 2020). doi: 10.1158/1538-7445.SABCS19-OT1-08-04. [DOI] [Google Scholar]
- 125.Kalinsky K. et al. Abstract CT109: A phase I/Ib study evaluating GDC-0077 plus fulvestrant in patients with PIK3CA -mutant, hormone receptor-positive/HER2-negative breast cancer. in Tumor Biology CT109–CT109 (American Association for Cancer Research, 2020). doi: 10.1158/1538-7445.AM2020-CT109. [DOI] [Google Scholar]