

ABSTRACT
Sepsis-induced myocardial dysfunction (SIMD) is ubiquitous in septic shock patients and is associated with high morbidity and mortality rates. Heat shock protein 22 (Hsp22), which belongs to the small HSP family of proteins, is involved in several biological functions. However, the function of Hsp22 in lipopolysaccharide (LPS)-induced myocardial injury is not yet established. This study was aimed at investigating the underlying mechanistic aspects of Hsp22 in myocardial injury induced by LPS. In this study, following the random assignment of male C57BL/6 mice into control, LPS-treated, and LPS + Hsp22 treated groups, relevant echocardiograms and staining were performed to scrutinize the cardiac pathology. Plausible mechanisms were proposed based on the findings of the enzyme-linked immunosorbent assay and Western blotting assay. A protective role of Hsp22 against LPS-induced myocardial injury emerged, as evidenced from decreased levels of creatinine kinase-MB (CK-MB), lactate dehydrogenase (LDH), and enhanced cardiac function. The post-LPS administration-caused spike in inflammatory cytokines (IL-1β, IL-6, TNF-α and NLRP3) was attenuated by the Hsp22 pre-treatment. In addition, superoxide dismutase (SOD) activity and B-cell lymphoma-2 (Bcl2) levels were augmented by Hsp22 treatment resulting in lowering of LPS-induced oxidative stress and cardiomyocyte apoptosis. In summary, the suppression of LPS-induced myocardial injury by Hsp22 overexpression via targeting of inflammation, oxidative stress, and apoptosis in cardiomyocytes paves the way for this protein to be employed in the therapy of SIMD.
KEYWORDS: Sepsis-induced myocardial dysfunction, heat shock protein 22, lipopolysaccharide, inflammatory, oxidative stress, apoptosis
Introduction
Inflammation is vitally involved in protection against tissue damage and/or infection to safeguard the wellbeing of an organism [1,2]. Sepsis is a systemic inflammatory response syndrome following an infection, causing high mortality rates in such patients [3,4]. Sepsis-induced myocardial dysfunction (SIMD), characterized by adverse outcomes and increased mortality, is an indicator of poor prognosis in patients with sepsis [5]. Epidemiological studies have documented myocardial injury or cardiac dysfunction in 25–50% of patients with sepsis [6]. Earlier work has revealed the involvement of inflammation, apoptosis, mitochondrial damage, and oxidative stress in myocardial damage in these patients [7,8].
Lipopolysaccharide (LPS) is a constituent of the outer membrane of gram-negative bacteria and is an important risk factor for sepsis. The stimulation of many pro-inflammatory markers, including IL-1β, IL-6, and TNF-α, by this molecule to trigger several immune cascades has been documented [9,10]. Inflammatory pathways mediated by oxidative stress are also known to be stimulated by LPS [11]. Furthermore, the abrogation of LPS-induced cardiac dysfunction by the suppression of apoptosis has also been documented [12].
The vital role played by the ubiquitous protein heat shock protein 22 (Hsp22, also known as HspB8) in chaperone activity and anti-apoptotic function is well known [13]. Hsp22 is highly expressed in a wide variety of tissues, including the heart, skeletal muscle, and brain [14]. It has been shown to play a pivotal role in maintaining the survival of cardiomyocytes in ischemic injury and inflammation [15,16]. Our group has shown that the overexpression of Hsp22 protects against diabetes-induced endothelial injury by inhibiting inflammation and oxidative stress [17]. However, the function of Hsp22 in LPS-induced myocardial injury remains unestablished. It remains unclear whether Hsp22 can protect the heart from LPS damage, and the potential protective mechanisms need investigation.
The treatment of cardiac damage caused by sepsis is not ideal, and Hsp22 displays beneficial biological functions, such as suppressing inflammation and oxidative stress. Therefore, we aimed to investigate the effect of Hsp22 overexpression on sepsis-induced myocardial dysfunction in detail. In this study, we evaluated the potential role of Hsp22 against LPS-induced cardiac dysfunction using an LPS-induced mouse model. By verifying the cardiac function and the level of inflammation, oxidative stress, and apoptosis, our results provide a potent new approach for the therapeutic intervention of SIMD.
Materials and methods
Experimental animals and treatment
All procedures conformed to the NIH Guide for Care and Use of Laboratory Animals and were approved by the Animal Ethics Committee of the Second Affiliated Hospital of Nanchang University. Male C57BL/6 mice (7–9 weeks old; 24–26 g in weight; n = 24) were sourced from the Slake Jingda Laboratory Animal Technology Company (Changsha, Hunan, China). The animals were housed in an environmentally controlled room (temperature 22 ± 1°C; relative humidity 55 ± 5%; 12 h dark-light cycle).
Following one week of adaptive feeding, the categorization of animals was done into three groups (n = 8 each): (a) Control group: tail injection with normal saline and intraperitoneal injection with normal saline (10 mg/kg body weight); (b) LPS group: tail injection with normal saline and intraperitoneal LPS injection (10 mg/kg body weight) [18]; (c) LPS + Hsp22 group: tail injection with 100 µg of Hsp22 and intraperitoneal LPS injection (10 mg/kg body weight). The protocol utilized for mouse treatment was based on an earlier study involving plasmid DNA in phosphate-buffered saline (100 μg in 2.0 mL) administered employing a hydrodynamics-based gene transfection approach using rapid tail vein injection [19].
Antibodies and reagents
A pcDNA3.1-Hsp22 plasmid (pHsp22) (Shanghai Genechem Co., Ltd., Shanghai, China) was constructed and used for mouse treatment. LPS (catalog no. L2630) derived from Escherichia coli 0111:B4 was purchased from Sigma-Aldrich Co (USA). ELISA kits for the measurement of IL-1β (MM-0040 M2), IL-6 (MM-1011M1) and TNF-α (MM-0132M1) were obtained from MEIMIAN, Jiangsu Biological Industrial Co., Ltd., China. Primary antibodies used in this study were anti-Hsp22 (Abcam, ab151552), anti-TNF-α (Abcam, ab183218), anti-NLRP3 (CST, #1510), anti-Bax (Abcam, ab32503), and anti-Bcl2 (Abcam, ab32124). The superoxide dismutase (SOD) assay kit (Cat#A001-3-2) and malondialdehyde (MDA) assay kit (Cat#A003-1-2) were procured from the Nanjing Jiancheng Bioengineering Institute, Nanjing, China.
Echocardiography
1.5% isoflurane was employed to anesthetize the animals following which echocardiography was done employing a Vevo770® imaging system (VisualSonics, Toronto, Canada) with a 30 MHz transducer-phased-array transducer to scrutinize their cardiac function. The acquisition of two-dimensional echocardiographic views of the mild-ventricular short axis and parasternal long axes along with the left ventricular end-diastolic diameter (LVEDD) and the left ventricular end-systolic diameter (LVESD) ensued. Lastly, computation of the rate of ejection fraction (EF) and fractional shortening (FS) was done employing standard formulae.
Measurement of CK-MB and LDH levels
Probing of myocardial damage entailed the quantitation of the levels of CK-MB and LDH in the blood serum. This entailed blood extraction and serum collection via 3000 centrifugation followed by enzyme detection employing a biochemical analyzer (Chemray240, Rayto, China).
Histological analysis
Following the isolation of cardiac specimens per group, rapid fixation was done A portion in 4% neutral buffered formalin solution for 24 h followed by paraffin embedding. Post-dewaxing, rehydration, hematoxylin and eosin (HE) staining, and dehydration, an inverted microscope was employed for imaging.
ELISA
The blood serum levels of (IL-1β, IL-6, TNF-α) were measured using ELISA kits (MEIMIAN, Jiangsu Biological Industrial Co., Ltd., China) adhering to the protocols outlined by the manufacturer.
Western blot analysis
Cardiac protein extraction entailed the use of a specific protein extraction kit (Beyotime, China) following pre-treatment. Following the extraction of protein samples (50 µg) employing the RIPA lysis buffer, their resolution was done on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as per the standard protocol. This was followed by the transfer of samples to a polyvinylidene fluoride (PVDF) membrane (Millipore, USA) for Western blotting. Incubation in block solution (nonfat milk) for 60 min at 37°C was done subsequent to which an overnight incubation at 4°C with the primary antibodies ensued. The HRP-conjugated secondary antibodies were added for 60 min at 37°C for target probing. This assay done in triplicate was scrutinized employing Image Lab 4.0.1 software.
TUNEL assay
Apoptotic cells were detected using the TUNEL assay (Roche, Cat#11,684,817,910) according to the manufacturer’s protocol. Quantification of TUNEL-positive cells was performed using Image J software.
DHE assay
Reactive oxygen species (ROS) in the cardiac samples were measured using DHE. Following incubation of frozen heart sections with DHE for 15 min at 37°C, the cells were visualized using fluorescence microscopy (Nikon, Japan).
Determination of oxidative stress parameters
The quantitation of the SOD activity and the MDA content in myocardial tissues entailed the use of respective kits mentioned above as per the prescribed protocols outlined by the manufacturer.
Statistical analysis
All experiments in triplicate were expressed as the mean ± SD. GraphPad Prism software 8.0 was employed for subsequent analyses. Inter-group comparisons were performed using unpaired t-tests with significance at P < 0.05.
Results
Several assays were employed to investigate the protective mechanisms of Hsp22 against LPS-induced cardiac dysfunction. The results were demonstrative of the evident amelioration of this damage by Hsp22 pre-treatment. The plausible inhibition of inflammation, oxidative stress and apoptosis by Hsp22 also emerged to pave the avenue for Hsp22 as a potential therapeutic target in SIMD.
LPS upregulates Hsp22 expression in heart tissue
The animal treatment protocols are summarized in Figure 1(a). The baseline body weights of the three groups of mice were comparable, with no significant differences observed after 12 days of the aforementioned interventions Figure 1(b). Hsp22 expression was conspicuously higher in the LPS group subsequent to LPS-stimulation as opposed to controls while they LPS+Hsp22 group showed higher Hsp22 expression than that in the LPS group Figure 1(c).
Figure 1.

LPS stimulation upregulates Hsp22 expression in C57BL/6 mice. (a) Flow diagram of animal experiment. (b) Body weight (BW) assessment once every three days. (c) The protein band of Hsp22 and its expression. *P < 0.05
Hsp22 pre-treatment improves the cardiac function in mice with LPS-induced cardiac injury
The resulting images from HE staining are shown in Figure 2(a). The control group showed normal myocardial cells with the myocardial fibers arranged in order. The LPS-treated group showed myocardial tissue injury and inflammation, as evidenced by the increased tissue edema and neutrophil infiltration. The LPS + Hsp22 treated group showed a considerable improvement in these pathological signs. The LPS group had significantly higher CK-MB and LDH levels in the blood serum as than that in the control group, whereas these levels were lower in the LPS + Hsp22 treated group than in the control group Figure 2(b,c). Echocardiography revealed a significant decrease in the left ventricular EF and FS in the LPS-treated group compared to that of the controls post-LPS administration, which was restored by Hsp22 pre-treatment Figure 2(d-f).
Figure 2.
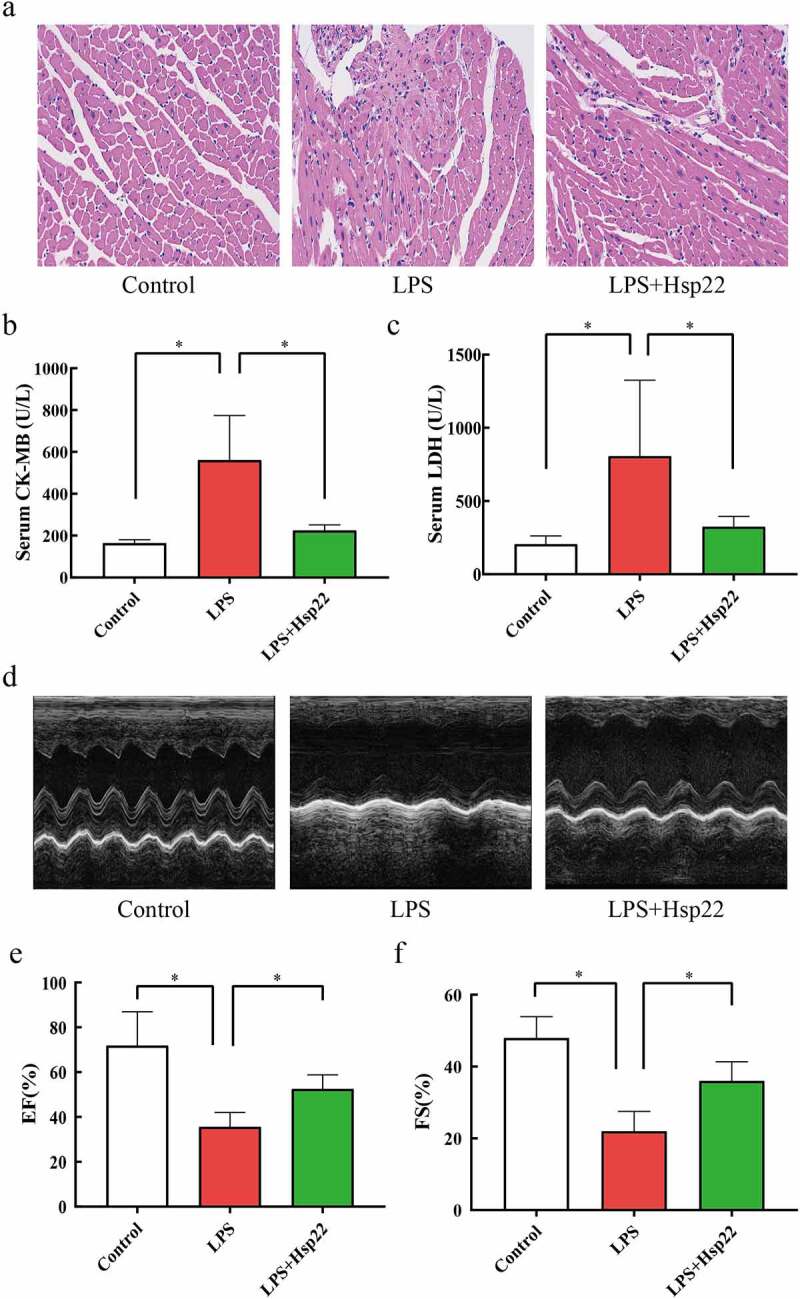
Hsp22 pre-treatment alleviates cardiac dysfunction induced by LPS. (a) Histological analysis of myocardial tissue via HE staining (×200). (b-c) CK-MB and LDH levels in the serum. (d) Echocardiographs. (e-f) Echocardiography parameters (EF% and FS%). *P < 0.05
Hsp22 pre-treatment decreases LPS-induced cardiac inflammation
The impact of Hsp22 on LPS-induced inflammation was elucidated by quantifying the cytokines in the serum and heart. IL-1β, IL-6, TNF-α, and NLRP3 levels were conspicuously more in the plasma and cardiac tissue of the LPS group than in the controls while these levels were evidently lower in the LPS+Hsp22 group (Figure 3).
Figure 3.
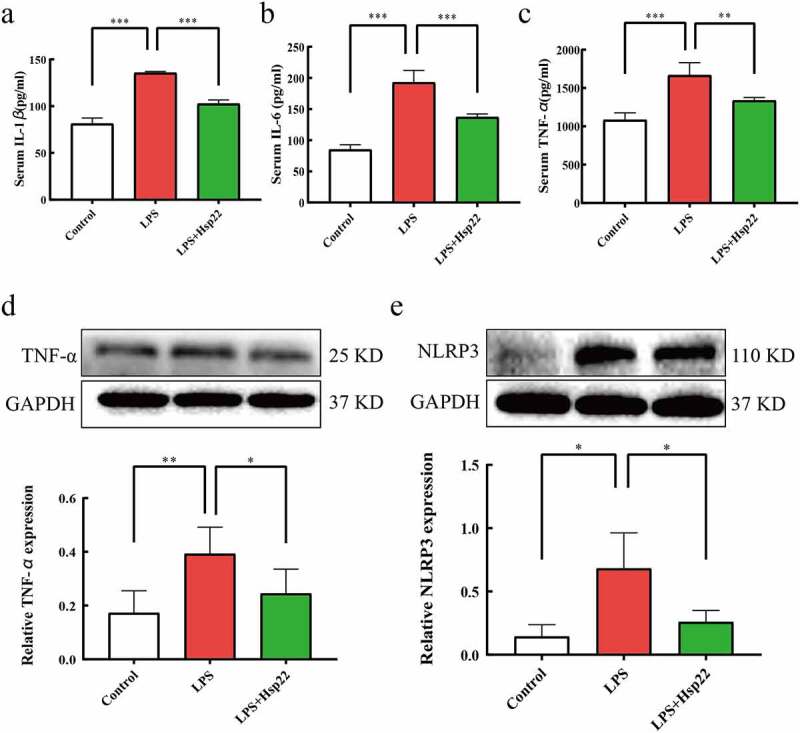
Hsp22 pre-treatment alleviates cardiac inflammation induced by LPS. (a-c) The serum levels of IL-1β (a), IL-6 (b), and TNF-α (c). (d) The protein band of TNF-α and its expression. (e) The protein band of NLRP3 and its expression. *P < 0.05, **P < 0.01 and ***P < 0.001
Hsp22 pre-treatment inhibits cardiac oxidative stress in LPS-treated mice
In this study, DHE staining was performed to examine whether Hsp22 affects the levels of oxidative stress induced by LPS. The results were demonstrative of evidently higher total superoxide levels in the LPS group than in the control group with effective lowering of the superoxide levels by the Hsp22 pre-treatment in LPS-treated mice Figure 4(a,b). Moreover, assaying of the redox activities by scoring SOD activities and MDA levels revealed a negative association for SOD and a positive association for MDA with the LPS administration that were both prevented by Hsp22 treatment, as depicted in Figure 4(c,d).
Figure 4.
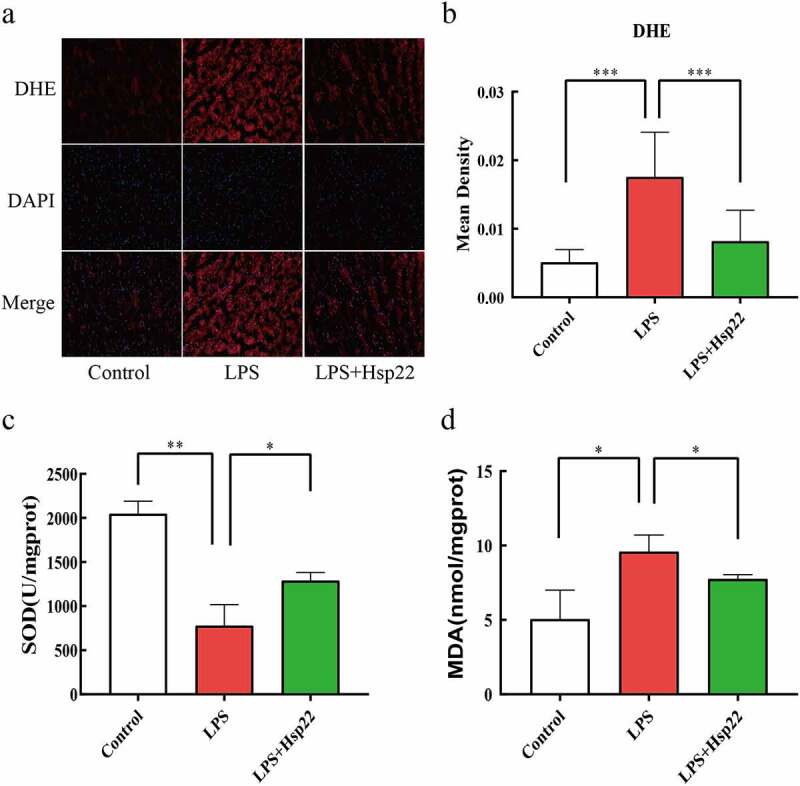
Hsp22 pre-treatment alleviates cardiac oxidative stress induced by LPS. (a-b) Representative images of DHE-stained heart sections (×200) (a); ROS expression levels (b).(c-d) SOD activity (c) and MDA levels (d) in mice heart tissues. *P < 0.05, **P < 0.01 and ***P < 0.001
Hsp22 pre-treatment inhibits cardiac apoptosis in LPS-treated mice
To determine the effects of Hsp22 treatment on LPS-induced septic myocardial apoptosis, we performed TUNEL staining to assess apoptotic cell death. LPS-treated mice showed a higher number of apoptotic myocardial cells vs. the control group, which was reversed by Hsp22 pre-treatment Figure 5(a,b). The LPS-treated group also showed upregulated Bax (pro-apoptotic) levels and downregulated Bcl2 levels, as compared to that in controls. Furthermore, Bax protein expression levels were attenuated, whereas that of Bcl2 were elevated by Hsp22 pre-treatment Figure 5(c,d).
Figure 5.
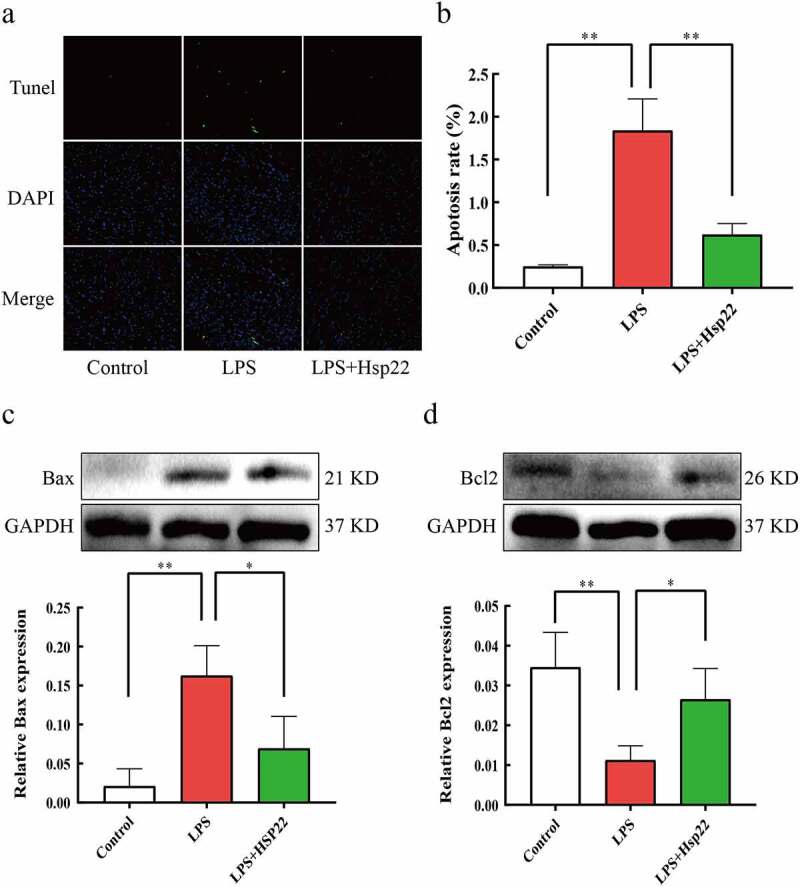
Hsp22 pre-treatment alleviates cardiac apoptosis induced by LPS. (a-b) Representative images of Tunel-stained heart sections (×200) (a); The number of apoptotic myocardial cells (b). (c-d) The protein band of Bax and its expression (c); The protein band of Bcl2 and its expression (d). *P < 0.05 and **P < 0.01
Discussion
Inflammation is a process by which the immune system responds to pathogens, harmful stimuli, irritants, and cell damage [20,21]. Sepsis is a severe inflammatory response often documented in patients with severe trauma, infection, and shock and is associated with high mortality [22]. SIMD is a central cause of mortality in septic shock patients [23]. This warrants detailed probing of the mechanisms of sepsis-induced myocardial injury to identify potential therapeutic targets facilitating improved disease management.
Previous studies have shown that lower myocardial contractility and heart rate were induced by LPS [24,25]. In line with this, Cohen et al. established a mouse septic shock model by intravenously injecting LPS (10 mg/kg body weight) resulting in reduced myocardial contractility and damage [26]. The findings of the present work are on the same lines reported by Chagnon et al. who documented a lowering of the left ventricular ejection fraction upon an intraperitoneal injection of 10 mg/kg of LPS in Wistar rats [27].
The anti-inflammatory function of Hsp22 has not been elucidated. Lan et al. have documented that the doxorubicin-induced cardiotoxicity stemming from Hsp22 pre-treatment by regulating cardiac inflammation regulation [28]. Furthermore, a reduction in ischemia/reperfusion-induced infarct size by short-term gene delivery of Hsp22 [29] paved the way for further evaluation of Hsp22 as a clinical therapeutic. These findings are hence suggestive of the plausible cardioprotective effects of Hsp22 in sepsis.
The role of NLRP3 inflammatory bodies in SIMD and in the progression of acute inflammation has been elaborated in earlier research [30,31]. This study also documented the activation of pro-inflammatory cytokines, including IL-1β, IL-6, TNF-α, and NLRP3, by LPS. This study is the first to document the potential attenuation of LPS-induced cardiac pro-inflammatory responses by Hsp22 pre-treatment through the lowering of pro-inflammatory cytokines in serum and cardiac tissue.
The stimulation of apoptosis in cardiomyocytes by LPS-induced inflammation has also been elucidated [32]. Moreover, Wu et al. have shown the dynamic balance between pro-apoptotic and anti-apoptotic processes in LPS-induced apoptosis of cardiac cells [33]. This study showed that LPS stimulation resulted in a significant increase in the apoptosis levels, with a decrease in Bcl2 expression. These observations were reversed significantly by Hsp22 pre-treatment by increasing Bcl2 levels and lowering Bax levels/apoptotic cell count. This demonstrated a reduction in the LPS-induced apoptosis of cardiac cells by Hsp22 pre-treatment in the murine system.
Oxidative stress is defined as an imbalance between boosted ROS levels and dampened antioxidant mechanisms in several physiological and pathological processes [34]. The link between LPS and oxidative stress has emerged in assays employing rodents. For example, Gorąca et al. [35] observed that rats treated with LPS derived from Escherichia coli developed oxidative stress. This work documented the lowering of LPS induced oxidative stress by Hsp22 by measuring SOD activity, MDA levels, and DHE staining.
Sakai et al. showed that Hsp22 could directly interact with mammalian target of rapamycin (mTOR) and found the binding site of the two in the immunoprecipitation experiment [36]. Knockout of HSP22 could increase the level of phosphorylated mTOR, which in turn regulates TNF-α-induced IL-6 synthesis in osteoblasts through p70 S6K. Moreover, previous studies have shown that Hsp22 directly binds to adenosine monophosphate-activated protein kinase (AMPK) and promotes its nuclear translocation and phosphorylation at Thr172, thereby reducing the level of ROS [37–39], apoptosis, and inflammation [40]. In addition, AMPK participate in the pathological processes such as inflammation, apoptosis, and oxidative stress by inhibiting the phosphorylation of mTOR [41]. In summary, we hypothesized that overexpression of Hsp22 could inhibit the expression of p-mTOR by activating the p-AMPK pathway, further reducing inflammation, oxidative stress, and apoptosis, thereby reducing sepsis-induced myocardial damage. Therefore, the Hsp22-AMPK/mTOR signaling pathway might be a potential therapeutic target.
There are several limitations to this study. First, current studies encompass only in vivo assays that warrant in vitro probing to corroborate these results. Second, the molecular mechanisms underlying Hsp22 function in LPS-induced cardiotoxicity warrant further exhaustive exploration. Three, biomarkers of myocardial injury, that is, cardiac troponins (cTnI) and brain natriuretic peptide (BNP), need to be measured for validation. Future research should increase the number of animals to eliminate individual differences and strengthen the results statistically.
Conclusion
This work demonstrates the protective effect of Hsp22 pre-treatment against LPS-induced cardiac dysfunction by inhibiting inflammation, oxidative stress, and apoptosis. This paves the way for the potential use of Hsp22 as a drug target for the treatment of SIMD, and large-scale, multi-center research for further corroboration is warranted.
Funding Statement
This work was supported by the National Natural Science Foundation of China (81760049, 82160070, 82100308), National Natural Science Foundation excellent youth cultivation project (20202ZDB01017), Jiangxi Science and Technology Innovation Platform Project (20165BCD41005), Natural Science Foundation of Jiangxi Province of China (20192BAB205033, 20202BABL216035), Science and technology planning project of Jiangxi Provincial Department of Education (GJJ200101);National Natural Science Foundation of China; Jiangxi Science and Technology Innovation Platform Project; Natural Science Foundation of Jiangxi Province of China;
Highlights
Hsp22 protects against LPS-induced cardiac dysfunction via inflammation, oxidative stress, and apoptosis inhibition.
Hsp22 has therapeutic value in treating sepsis-induced myocardial dysfunction.
Echocardiography was used to evaluate cardiac structure and function.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Author statement
Yun Yu: Animal experiment, Manuscript Preparation. Liang Liu, Ling-juan Zhu and Jing-an Rao: Tissue Collection, Statistical Analysis. Long-long Hu, Jun-pei Li, Ling-ling Yu: Animal experiment. Hui-hui Bao and Xiao-shu Cheng: Study Design and Manuscript revision.
Ethical approval
The study was was conducted according to the NIH guidelines for animal care and was approved by the Animal Ethics Committee of the Second Affiliated Hospital of Nanchang University.
References
- [1].Prasad LK, O’Mary H, Cui Z, et al. Nanomedicine delivers promising treatments for rheumatoid arthritis. Nanomedicine (Lond). 2015;10(13):2063–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].DiDonato JA, Mercurio F, Karin M, et al. NF-κB and the link between inflammation and cancer. Immunol Rev. 2012;246(1):379–400. [DOI] [PubMed] [Google Scholar]
- [3].Gotts JE, Matthay MA.. Sepsis: pathophysiology and clinical management. BMJ. 2016;353:i1585. [DOI] [PubMed] [Google Scholar]
- [4].Bosmann M, Ward PA.. The inflammatory response in sepsis. Trends Immunol. 2013;34(3):129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Frencken JF, Donker DW, Spitoni C, et al. Myocardial injury in patients with sepsis and its association with long-term outcome. Circ Cardiovasc Qual Outcomes. 2018;11(2):e004040. [DOI] [PubMed] [Google Scholar]
- [6].Ge C, Liu J, Dong S. miRNA-214 protects sepsis-induced myocardial injury. Shock. 2018;50(1):112–118. [DOI] [PubMed] [Google Scholar]
- [7].Lewis DH, Chan DL, Pinheiro D, et al. The immunopathology of sepsis: pathogen recognition, systemic inflammation, the compensatory anti-inflammatory response, and regulatory T cells. J Vet Intern Med. 2012;26(3):457–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li N, Zhou H, Wu H, et al. STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol. 2019;24:101215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Knuefermann P, Nemoto S, Misra A, et al. CD14-deficient mice are protected against lipopolysaccharide-induced cardiac inflammation and left ventricular dysfunction. Circulation. 2002;106(20):2608–2615. [DOI] [PubMed] [Google Scholar]
- [10].Kawata A, Murakami Y, Suzuki S, et al. Anti-inflammatory activity of β-carotene, lycopene and Tri-n-butylborane, a scavenger of reactive oxygen species. In Vivo. 2018;32(2):255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bose S, Song MY, Nam JK, et al. In vitro and in vivo protective effects of fermented preparations of dietary herbs against lipopolysaccharide insult. Food Chem. 2012;134(2):758–765. [DOI] [PubMed] [Google Scholar]
- [12].Tan J, Sun T, Shen J, et al. FAM46C inhibits lipopolysaccharides-induced myocardial dysfunction via downregulating cellular adhesion molecules and inhibiting apoptosis. Life Sci. 2019;229:1–12. [DOI] [PubMed] [Google Scholar]
- [13].Dubińska-Magiera M, Niedbalska-Tarnowska J, Migocka-Patrzałek M, et al. Characterization of Hspb8 in Zebrafish. Cells. 2020;9(6):1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kappé G, Verschuure P, Philipsen RL, et al. Characterization of two novel human small heat shock proteins: protein kinase-related HspB8 and testis-specific HspB9. Biochim Biophys Acta. 2001;1520(1):1–6. [DOI] [PubMed] [Google Scholar]
- [15].Lizano P, Rashed E, Kang H, et al. The valosin-containing protein promotes cardiac survival through the inducible isoform of nitric oxide synthase. Cardiovasc Res. 2013;99(4):685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hedhli N, Wang L, Wang Q, et al. Proteasome activation during cardiac hypertrophy by the chaperone H11 Kinase/Hsp22. Cardiovasc Res. 2008;77(3):497–505. [DOI] [PubMed] [Google Scholar]
- [17].Yu L, Liang Q, Zhang W, et al. HSP22 suppresses diabetes-induced endothelial injury by inhibiting mitochondrial reactive oxygen species formation. Redox Biol. 2019;21:101095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhai J, Guo Y. Paeoniflorin attenuates cardiac dysfunction in endotoxemic mice via the inhibition of nuclear factor-κB. Biomed Pharmacother. 2016;80:200–206. [DOI] [PubMed] [Google Scholar]
- [19].Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6(7):1258–1266. [DOI] [PubMed] [Google Scholar]
- [20].Ferrero-Miliani L, Nielsen OH, Andersen PS, et al. Chronic inflammation: importance of NOD2 and NALP3 in interleukin-1beta generation. Clin Exp Immunol. 2007;147(2):227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang JM, An J. Cytokines, inflammation, and pain. Int Anesthesiol Clin. 2007;45(2):27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Klingenberg C, Kornelisse RF, Buonocore G, et al. Culture-negative early-onset neonatal sepsis - at the crossroad between efficient sepsis care and antimicrobial stewardship. Front Pediatr. 2018;6:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cao Y, Wang Y, Xiao L, et al. Endothelial-derived exosomes induced by lipopolysaccharide alleviate rat cardiomyocytes injury and apoptosis. Am J Transl Res. 2021;13(3):1432–1444. [PMC free article] [PubMed] [Google Scholar]
- [24].Iqbal M, Cohen RI, Marzouk K, et al. Time course of nitric oxide, peroxynitrite, and antioxidants in the endotoxemic heart. Crit Care Med. 2002;30(6):1291–1296. [DOI] [PubMed] [Google Scholar]
- [25].Han T, Zhou Y, Li D. Relationship between hepatocellular carcinoma and depression via online database analysis. Bioengineered. 2021;12(1):8496–8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cohen RI, Wilson D, Liu SF. Nitric oxide modifies the sarcoplasmic reticular calcium release channel in endotoxemia by both guanosine-3ʹ,5ʹ (cyclic) phosphate-dependent and independent pathways. Crit Care Med. 2006;34(1):173–181. [DOI] [PubMed] [Google Scholar]
- [27].Chagnon F, Bentourkia M, Lecomte R, et al. Endotoxin-induced heart dysfunction in rats: assessment of myocardial perfusion and permeability and the role of fluid resuscitation. Crit Care Med. 2006;34(1):127–133. [DOI] [PubMed] [Google Scholar]
- [28].Lan Y, Wang Y, Huang K, et al. Heat shock protein 22 attenuates doxorubicin-induced cardiotoxicity via regulating inflammation and apoptosis. Front Pharmacol. 2020;11:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wu W, Lai L, Xie M, et al. Insights of heat shock protein 22 in the cardiac protection against ischemic oxidative stress. Redox Biol. 2020;34:101555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yang L, Zhang H, Chen P. Sulfur dioxide attenuates sepsis-induced cardiac dysfunction via inhibition of NLRP3 inflammasome activation in rats. Nitric Oxide. 2018;81:11–20. [DOI] [PubMed] [Google Scholar]
- [31].Wang L, Zhao H, Xu H, et al. Targeting the TXNIP-NLRP3 interaction with PSSM1443 to suppress inflammation in sepsis-induced myocardial dysfunction. J Cell Physiol. 2021;236(6):4625–4639. [DOI] [PubMed] [Google Scholar]
- [32].Wang Y, Wang Y, Yang D, et al. β1-adrenoceptor stimulation promotes LPS-induced cardiomyocyte apoptosis through activating PKA and enhancing CaMKII and IκBα phosphorylation. Crit Care. 2015;19(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wu D, Shi L, Li P, et al. Intermedin1-53 protects cardiac fibroblasts by inhibiting NLRP3 inflammasome activation during sepsis. Inflammation. 2018;41(2):505–514. [DOI] [PubMed] [Google Scholar]
- [34].Gu X, Ma Z, Fang J, et al. Obesity enhances antioxidant capacity and reduces cytokine levels of the spleen in mice to resist splenic injury challenged by escherichia coli. J Immunol Res. 2020;2020:5948256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gorąca A, Huk-Kolega H, Kleniewska P, et al. Effects of lipoic acid on spleen oxidative stress after LPS administration. Pharmacol Rep. 2013;65(1):179–186. [DOI] [PubMed] [Google Scholar]
- [36].Sakai G, Tokuda H, Yamamoto N, et al. Association of HSP22 with mTOR in osteoblasts: regulation of TNF-α-stimulated IL-6 synthesis. FEBS Lett. 2018;592(7):1202–1210. [DOI] [PubMed] [Google Scholar]
- [37].Depre C, Wang L, Sui X, et al. H11 kinase prevents myocardial infarction by preemptive preconditioning of the heart. Circ Res. 2006;98(2):280–288. [DOI] [PubMed] [Google Scholar]
- [38].Fan H, Ding R, Liu W, et al. Heat shock protein 22 modulates NRF1/TFAM-dependent mitochondrial biogenesis and DRP1-sparked mitochondrial apoptosis through AMPK-PGC1α signaling pathway to alleviate the early brain injury of subarachnoid hemorrhage in rats. Redox Biol. 2021;40:101856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ren Y, Shen HM. Critical role of AMPK in redox regulation under glucose starvation. Redox Biol. 2019;25:101154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jiang S, Li T, Ji T, et al. AMPK: potential therapeutic target for ischemic stroke. Theranostics. 2018;8(16):4535–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liu X, Xu Y, Cheng S, et al. Geniposide combined with notoginsenoside R1 attenuates inflammation and apoptosis in atherosclerosis via the AMPK/mTOR/Nrf2 signaling pathway. Front Pharmacol. 2021;12:687394. [DOI] [PMC free article] [PubMed] [Google Scholar]