

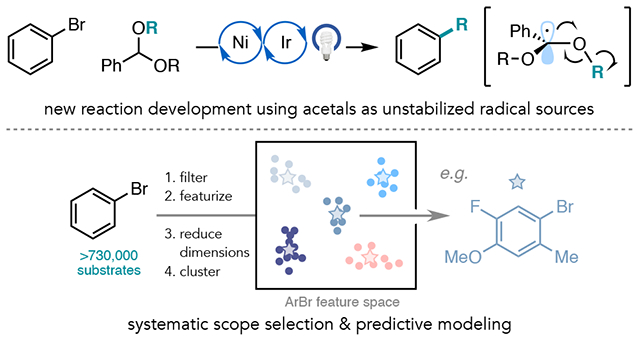
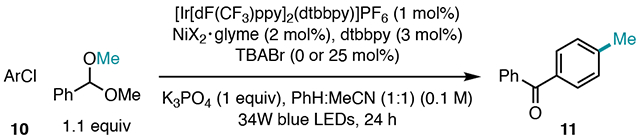
Abstract
Ni/photoredox catalysis has emerged as a powerful platform for C(sp2)–C(sp3) bond formation. While many of these methods typically employ aryl bromides as the C(sp2) coupling partner, a variety of aliphatic radical sources have been investigated. In principle, these reactions enable access to the same product scaffolds, but it can be hard to discern which method to employ because non-standardized sets of aryl bromides are used in scope evaluation. Herein we report a Ni/photoredox-catalyzed (deutero)methylation and alkylation of aryl halides where benzaldehyde di(alkyl) acetals serve as alcohol-derived radical sources. Reaction development, mechanistic studies, and late-stage derivatization of a biologically-relevant aryl chloride, fenofibrate, are presented. Then, we describe the integration of data science techniques, including DFT featurization, dimensionality reduction, and hierarchical clustering, to delineate a diverse and succinct collection of aryl bromides that is representative of the chemical space of the substrate class. By superimposing scope examples from published Ni/photoredox methods on this same chemical space, we identify areas of sparse coverage and high versus low average yields, enabling comparisons between prior art and this new method. Additionally, we demonstrate that the systematically-selected scope of aryl bromides can be used to quantify population-wide reactivity trends and reveal sources of possible functional group incompatibility with supervised machine learning.
Graphical Abstract
INTRODUCTION
Although a relatively new field, Ni/photoredox catalysis has seen widespread development as a general approach to C(sp2)–C(sp3) bond formation in chemical synthesis. In this dual catalytic strategy, a photoredox catalyst supplies substrate radicals as coupling partners to Ni and/or modulates Ni’s oxidation state in catalysis.1 Since the substrate radicals can be generated from stable, feedstock chemicals under mild conditions, a high level of functional group tolerance is often associated with these methods.2 The advances accessible with this strategy have been particularly enabling in medicinal chemistry, where introduction of C(sp3), rather than C(sp2), fragments onto arenes is a well-established strategy for preparing architecturally complex molecules with improved receptor/ligand complementarity and solubility.3
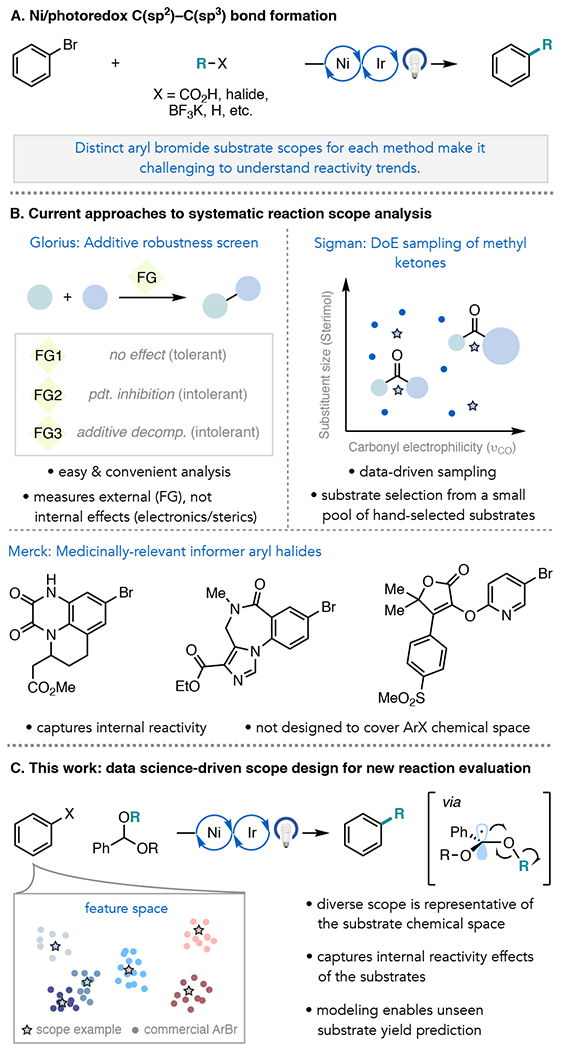
The majority of Ni/photoredox methodologies involve the coupling of aryl bromides with aliphatic radical precursors, such as carboxylic acids,4a alkyl halides,4b trifluoroborate salts,4c oxalates,4d silicon catechols,4e 1,4-dihydropyridines,4f and C(sp3)–H bonds4g(Figure 1A). In principle, these reactions afford access to the same product structures, but it is difficult to compare amongst the methods or to understand if there are general reactivity trends because each method features a different collection of aryl bromides in their substrate scope tables.5 Additionally, limitations of the methods (so-called “negative” results) are often not reported. This has made it challenging for chemists to discern how well a method will translate to a new reaction partner or to select among methods to adopt.
Figure 1.
Challenges & new approaches to surveying reactivity from scope studies.
New strategies, and a community-wide effort, are needed to address these shortcomings in substrate scope selection and reporting.6 Toward the aim of studying functional group compatibility, Glorius and co-workers have advanced a “robustness screen,” allowing for expedient examination of diverse additives for reaction inhibition or additive decomposition (Figure 1B).7 This protocol assesses an important aspect of generality, but it does not capture the impact of internal steric or electronic effects on the reactivity of a substrate. To this end, scientists at Merck have pioneered the use of informer libraries of complex, drug-like molecules for testing the medicinal applicability of cross-coupling methodologies.8 While highly enabling, these informer libraries were selected to represent the physicochemical space of marketed drugs rather than the chemical space of a substrate class. Moreover, due to the complexity of the informer molecules, they have yet to be broadly adopted by academic laboratories for reaction discovery.8b In a retrospective study, Sigman and coworkers reported a quantitative approach to substrate selection for asymmetric hydrogenation of ketones, identifying 32 ketones to span a 52-member library using Design of Experiments precepts and featurization through hand-selection of two mechanistically relevant descriptors.9 Adaptation of the workflow to accommodate the use of larger populations of substrates and higher dimensional featurization could be highly enabling and help mitigate selection bias. High-throughput experimentation (HTE) has also been used in a retrospective sense to evaluate substrate generality across synthetic methods, although the ability to gain similar information directly from reaction development manuscripts would be of immediate value to the synthetic community.10
To address these goals, and inspired by recent development in machine learning in chemistry,11 we envisioned a general and quantitative scope selection workflow informed by studies in chemoinformatics and molecular library design,12 including for informing the selection of bioactive molecules from inactive molecules,13 virtual screening of drug-like molecules based on structure or their interaction with known protein targets,14 and the generation of additive15 and ligand16 libraries for transition metal-catalyzed processes. The scope selection workflow proceeds by DFT featurization of a large virtual library of commercial substrates using our recently developed software package, auto-qchem,17 followed by dimensionality reduction with Uniform Manifold Approximation and Projection (UMAP) and hierarchical clustering to arrive at a diverse and systematically selected substrate scope.
Herein, we describe the development, application, and assessment of this workflow for aryl bromide scope evaluation in literature examples of Ni/photoredox C(sp2)–C(sp3) coupling, as well as for a new Ni/photoredox method in which we show that acetals can activate methyl, primary, and secondary alcohols as aliphatic radical precursors for cross-coupling. Alcohols are attractive cross-coupling partners owing to their abundance, low cost and toxicity, and functional group compatibility.18 However, use of alcohols as aliphatic radical precursors in Ni/photoredox catalysis has seen limited development due to their high C(sp3)–O bond strengths. Zuo and coworkers have reported C(sp3)–CH2OH activation of free alcohols in photocatalytic Ni/Ce cross-coupling.19a Alternatively, the MacMillan and Martin groups have pursued masked alcohol derivatives for Ni/photoredox cross-coupling, including oxalates4d and phthalimides.19b These methods afford access to stabilized aliphatic radicals in cross-coupling, whereas those that access methyl or primary unactivated radicals from alcohols are less developed. Very recently, Ni-catalyzed coupling reactions of free alcohols using phosphine20a–b and N-heterocyclic carbene mediators20c for C(sp3)–O bond activation have been reported.
Our lab has demonstrated that another alcohol-based coupling partner, trimethyl orthoformate, can serve as a source of high-energy methyl radicals upon β-scission of its tertiary carbon-centered radical.21 While trimethyl orthoformate is an abundant and functional group-tolerant methyl radical source, other aliphatic orthoformates are less accessible. The method also required solvent-quantity orthoformate, limiting its application as a general C(sp2)–C(sp3) cross-coupling method. In contrast, we anticipated that the facile synthesis of benzaldehyde di(alkyl) acetals from benzaldehyde and aliphatic alcohols would render them a promising source of aliphatic radicals for new reaction development.22
This article is organized as follows: first, we discuss reaction optimization and our studies aimed at elucidating the mechanism of the coupling reaction. Second, we describe a synthetic application of the method via late-stage alkylation of a biologically-relevant aryl chloride. We then discuss our approach to visualizing the chemical space of aryl bromides and superimpose the reactivity of aryl bromides reported in the Ni/photoredox literature onto this space. Fifteen aryl bromides that maximally cover this substrate space are evaluated for the new synthetic method. Finally, we assess the ability of this data science-generated aryl bromide scope to capture reactivity trends via supervised machine learning.
RESULTS AND DISCUSSION
Reaction Optimization
While developing a methylation protocol with trimethyl orthoformate, we found that acetals could also serve as sources of methyl radical, but this approach required solvent quantity of the acetal to achieve a synthetically useful yield. In these reactions, activation of the acetal was mediated by chlorine radicals generated upon photoelimination from a Ni catalyst.21 In developing a new method that employed acetals as stoichiometric coupling partners, we evaluated alternative hydrogen atom transfer (HAT) catalysts and strategies.23 DFT calculations revealed that benzaldehyde dimethyl acetal possesses a sufficiently weak tertiary C–H bond strength (bond dissociation free energy (BDFE) = 77.0 kcal/mol)24 for abstraction by a variety of HAT agents. These computations also revealed that the primary C–H bonds of benzaldehyde dimethyl acetal are significantly stronger (BDFE = 87.6 kcal/mol),24 indicating high selectivity for the weaker tertiary benzylic bond was possible.
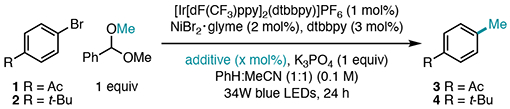
The methylation of 4’-bromoacetophenone 1 with benzaldehyde dimethyl acetal (1.5 equiv) was first investigated. Using [Ir[dF(CF3)ppy]2(dtbbpy)]PF6 (1 mol%) as a photocatalyst, NiBr2[sip]-glyme (10 mol%) and 4,4’-di-tert-butylbipyridine (dtbbpy) (15 mol%) as the cross-coupling catalyst, quinuclidine (15 mol%) as a HAT catalyst, and K3PO4 (1 equiv) in a 1:1 mixture of benzene: acetonitrile under visible light irradiation afforded 3 in 75% yield (Table 1, Entry 1). We were delighted to find that reaction performance was not affected by reducing the acetal loading to 1 equiv, in addition to lowering the catalyst and ligand loadings to 2 mol% NiBr2·glyme and 3 mol% dtbbpy. Under these conditions, methylation proceeded in 74% yield (Table 1, Entry 2). When performing control reactions, we found that omission of quinuclidine as a HAT catalyst resulted in a small boost in yield to 82% (Table 1, Entry 3), suggesting that bromide, present from the aryl bromide or Ni precatalyst, can mediate HAT with the acetal upon oxidation to bromine radical (vide infra).4b,25 This proposal is consistent with the stronger bond strength of H–Br over the tertiary C–H bond of the acetal (H–Br BDFE = 80.8 kcal/mol).24 Independent omission of Ni/dtbbpy, photocatalyst, and visible light irradiation resulted in no cross-coupling (Table 1, Entries 4-6).
Table 1.
Conditions Evaluation for Aryl Methylation
| ||||
|---|---|---|---|---|
| Entry | R | deviation | additive | % Yielda |
| 1 | Ac | 10 mol% Ni; 15 mol% dtbbpy; 1.5 equiv acetal | quinuclidine (15 mol%) | 75% |
| 2 | Ac | none | quinuclidine (15 mol%) | 74% |
|
| ||||
| 3 | Ac | none | none | 82% |
|
| ||||
| 4 | Ac | no Ni/dtbbpy | none | 0% |
| 5 | Ac | no Ir | none | 0% |
| 6 | Ac | no blue light | none | 0% |
|
| ||||
| 7 | t-Bu | none | none | 16% |
| 8 | t-Bu | 25% light intensity | TBABr (25 mol%) | 78% |
|
| ||||
| 9 | t-Bu | 0.4 mmol scale; 25% light intensity | TBABr (25 mol%) | 89% |
Reactions performed on 0.1 mmol scale with 1,3,5-trimethoxybenzene added as an external standard (GC yield).
Despite finding conditions for high-yielding methylation with an electron-deficient aryl bromide, methylation of the more electron-rich 1-bromo-4-tert-butylbenzene 2 proceeded in only 16% yield under the same conditions (Table 1, Entry 7). The mass balance for this reaction could be traced to unconverted aryl bromide and protodehalogenation of 2 to tert-butylbenzene. After a survey of HAT catalysts and bromide additives (see SI for more details), we found that employment of exogenous bromide source tetrabutylammonium bromide (TBABr) in 25 mol% furnished the methylated product in 78% yield (Table 1, Entry 8). Critical to the success of this reaction was reduction of the light intensity to 25%, which minimized protodehalogenation entirely. This modification enhances reaction yield, but also simplifies the purification process, as separation of methylated arenes from their unmethylated analogs can be challenging. A common limitation of photoredox reactions is their scalability, often due to issues of light penetration;26 however, scale-up of this reaction to 0.4 mmol provided an additional boost in yield to 89% (Table 1, Entry 9). Notably, the product derived from C–H abstraction at the primary site of benzaldehyde dimethyl acetal was never observed.
Mechanistic Investigations
Having identified conditions that enable high-yielding methylation of aryl bromides possessing varying electronic properties, we then sought to study the reaction mechanism. Our proposal for the generation of methyl radical requires stoichiometric formation of a β-scission byproduct possessing a strong C–O π bond (methyl benzoate, 7).22,27 In all reactions, this ester byproduct can be tracked in a nearly 1:1 ratio relative to the methylated product (Figure 2A). Cyclopropylmethyl radical ring opening from the corresponding acetal to afford 8 presented additional evidence for the intermediacy of organic radical species (Figure 2B).
Figure 2.
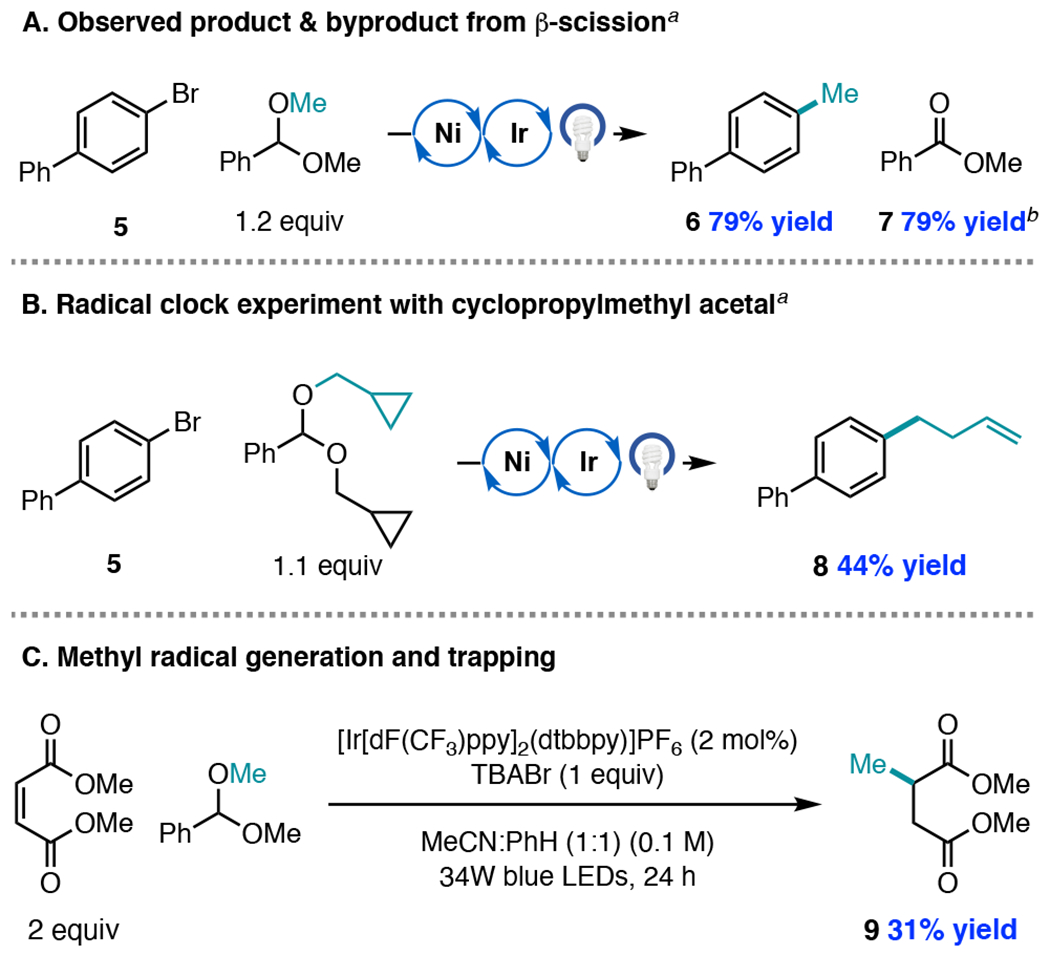
Selected mechanistic studies. a Conditions shown in Table 2 Entry 9. b Yield relative to acetal.
We next sought to understand the nature of the HAT step. According to the mechanistic proposal, bromide undergoes oxidation to bromine radical (E1/2[Br−/Br•] = +0.80 V vs SCE in DCE; E1/2[*IrIII/IrII] = +1.21 V vs SCE in MeCN)4b to mediate HAT with the tertiary C–H bond of the acetal. To evaluate the feasibility of bromine radical undergoing HAT under the coupling conditions, we performed a reaction with benzaldehyde dimethyl acetal and dimethyl maleate using 2 mol% photocatalyst and 1 equiv TBABr. This reaction produced 9, the product of methyl radical trapping by dimethyl maleate, in 31% yield, while methyl benzoate 7 was formed in 66% yield (Figure 2C). When TBABr was omitted from the reaction, neither methyl radical incorporation nor the ester byproduct was observed.
Halide identity studies were next explored to further understand the source and role of bromine radical. These studies were initiated with aryl chloride 10 using NiBr2·glyme as a precatalyst with and without the addition of catalytic TBABr (Table 2, Entries 1-2). In both cases, methylation of the aryl chloride proceeded in >80% yield. However, when NiCl2·glyme was employed as the precatalyst, thereby removing all sources of bromide from the reaction, methylation proceeded in <1% yield, likely because the photocatalyst cannot oxidize chloride to chlorine radical (Table 2, Entry 3).28–29 Reactivity was restored upon introduction of TBABr to reactions employing NiCl2·glyme, providing the methylated product in 79% yield (Table 2, Entry 4). These experiments demonstrate that catalytic bromide present from the Ni precatalyst is sufficient to produce high yields of methylated arenes.
Table 2.
Role of halide in the reaction
| |||
|---|---|---|---|
| Entry | Ni source | TBABr | % Yield |
| 1 | NiBr2·glyme | 0 mol% | 81% |
| 2 | NiBr2·glyme | 25 mol% | 85% |
| 3 | NiCl2·glyme | 0 mol% | <1% |
| 4 | NiCl2·glyme | 25 mol% | 79% |
ArCl = 4-chlorobenzophenone. Reactions performed on 0.1 mmol scale with 1,3,5-trimethoxybenzene as external standard (1H NMR yield).
An alternative pathway that is consistent with this data is direct oxidation of benzaldehyde dimethyl acetal followed by deprotonation to generate the tertiary radical of benzaldehyde dimethyl acetal. However, Stern-Volmer quenching studies revealed that the acetal does not quench the excited state of the photocatalyst; in contrast, TBABr undergoes rapid quenching. These studies also showed that TBABr undergoes more rapid quenching than (dtbbpy)Ni(o-Tol)(Br) (o-Tol = ortho-tolyl) in the reaction solvent system (1:1 mixture of PhH:MeCN), providing support for bromide oxidation over a bromine photoelimination pathway from Ni.29–30
On the basis of these studies, a catalytic cycle is shown in Figure 3. Oxidative addition of Ni(0) complex 12 into an aryl halide produces a Ni(II) aryl halide intermediate (13). Irradiation of the iridium(III) photocatalyst 16 affords the highly oxidizing, long-lived *Ir(III) triplet excited state (17), which is capable of oxidizing bromide for the production of bromine radical. Bromine then mediates selective HAT with the tertiary C–H bond of benzaldehyde dimethyl acetal, which upon β-scission affords methyl radical and methyl benzoate 7. The former species is trapped by 13, generating Ni(III)(Ar)(Me) (14) from which a facile reductive elimination forges a C(sp2)–C(sp3) bond and Ni(I) complex 15. The reduced state of the photocatalyst (18) can then reduce Ni(I) to regenerate both the Ir(III) and Ni(0) catalysts.
Figure 3.
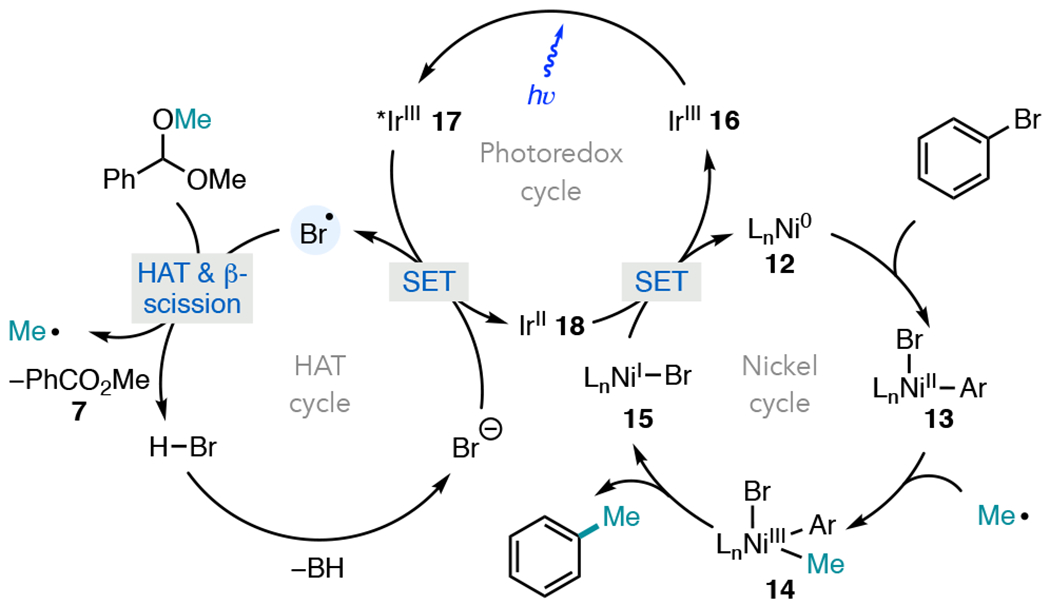
Catalytic cycle for aryl methylation from acetals.
Late-Stage Application of the Methodology
After exploration of the reaction mechanism, we examined whether acetals can serve more generally as low molecular weight radical sources. This exploration was initiated with late-stage pharmaceutical product fenofibrate, which possesses an aryl chloride. Notably, aryl chlorides represent the most inexpensive, abundant, and biologically relevant aryl halide. Thus, we aimed to demonstrate that a library of alkylated derivatives of this substrate class could be prepared on the basis of the results in Table 2 which revealed that methylation of an aryl chloride is possible in the presence of a catalytic bromide additive.31 Upon application of the reaction conditions shown in Table 2, Entry 1, methylation of fenofibrate proceeded in 75% yield (Figure 4, 19).
Figure 4.
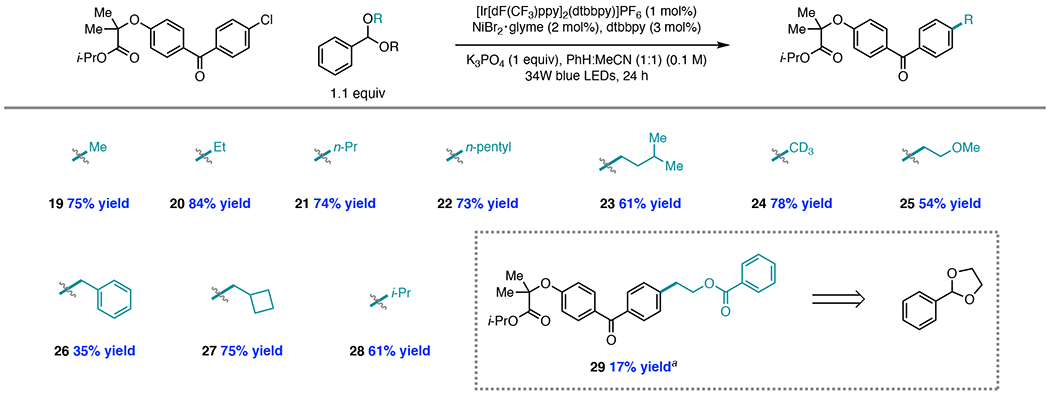
Late-stage functionalization of fenofibrate from benzaldehyde di(alkyl) acetals. Yields are isolated yields on 0.4 mmol scale. a Reaction performed with 25 mol% TBABr
While aryl methylation is an established strategy for rendering compounds with improved binding affinity, bioavailability, and metabolic stability, the ability to install a variety of aliphatic groups onto a late-stage compound would be valuable for enabling rapid access to alkylated analogs.32 Gratifyingly, employment of benzaldehyde diethyl acetal in place of the methyl analog provided ethylated product 20 in 84% yield. While benzaldehyde dimethyl and diethyl acetals are commercially available, other benzaldehyde dialkyl acetals were easily synthesized on multi-gram scale upon reaction of benzaldehyde, an aliphatic alcohol, and 5Å molecular sieves at room temperature. Installation of primary aliphatic groups, including n-propyl (21), n-pentyl (22), and iso-amyl (23) groups from the corresponding acetals proceeded in high yields. Additionally, we were excited to find that deuteromethylation of fenofibrate was possible in 78% yield (24). Limited methods exist to install deuteromethyl groups in cross-coupling, although iodomethane-d3 (CD3I),33a trideuteriomethyl p-toluenesulfonate (CD3OTs),33b and dimethyl-d6 carbonate (CO(OCD3)2)33c have previously found application as deuteromethyl sources. Acetals present a functional group-tolerant and easily synthesized alternative deuteromethyl source derived from methanol-d4.
Because alkyl halides containing β-heteroatoms are often not stable and may be prone to nucleophilic displacement of the halide, we also sought to install aliphatic groups containing heteroatoms, including the 2-methoxyethyl group to furnish 25. While benzylation of fenofibrate proceeded in 35% yield (26), installation of the cyclobutylmethyl group provided 27 in 75% yield,34 demonstrating that the diminished yield of the former product is likely a consequence of competitive abstraction at the secondary benzylic sites of the acetal or product rather than a steric effect. Indeed, iso-propylation of fenofibrate afforded 28 in 61% yield, showing that coupling to secondary aliphatic groups is possible. Finally, we found that cyclic acetal 2-phenyl-1,3-dioxolane underwent HAT at the tertiary C–H bond, followed by ring opening and trapping of the resulting radical species to produce 29 in 17% yield.
Data Science-Driven Aryl Bromide Scope Studies and Analysis of the Chemical Space Coverage
Alkylation reactions with fenofibrate demonstrated that acetals can serve as modular sources of aliphatic radicals. Since the reaction worked with aryl bromides and chlorides, but because aryl bromides are much more commonly employed in Ni/photoredox catalysis (>100 Ni/photoredox methods published with aryl bromides versus <10 with aryl chlorides), we selected aryl bromides for scope studies to evaluate the generality of this method and to best compare to prior art.21b We aimed to study how well the aryl bromide chemical space has been covered in Ni/photoredox cross-coupling, as well as to use data science to generate a maximally diverse set of commercial aryl bromides.
To initiate our studies, we turned to a Reaxys® structure search of aryl bromides, which provided >730,000 substrates (Figure 5). These aryl bromides were then filtered based on molecular weight (<400 amu), commercial availability, available spectral data, and functionality (see SI for full details), which provided us with a ~2600-member dataset to analyze aryl bromide chemical space. Because we filtered the aryl bromides on the basis of functionality known to be incompatible with Ni/photoredox catalysis (e.g. iodides were omitted due to their preferential oxidative addition to Ni over bromides; nitro groups were omitted due to their facile reduction by the photocatalyst) this aryl bromide dataset is not universally applicable, but instead designed to serve the current method. Nevertheless, the workflow and open-source code is generally applicable to other methods and substrate classes. It is also flexible, affording the ability to adapt and update as the chemical space of a substrate class changes. Indeed, we have provided the annotated code as a supporting document in order to make this approach accessible to the community.
Figure 5.
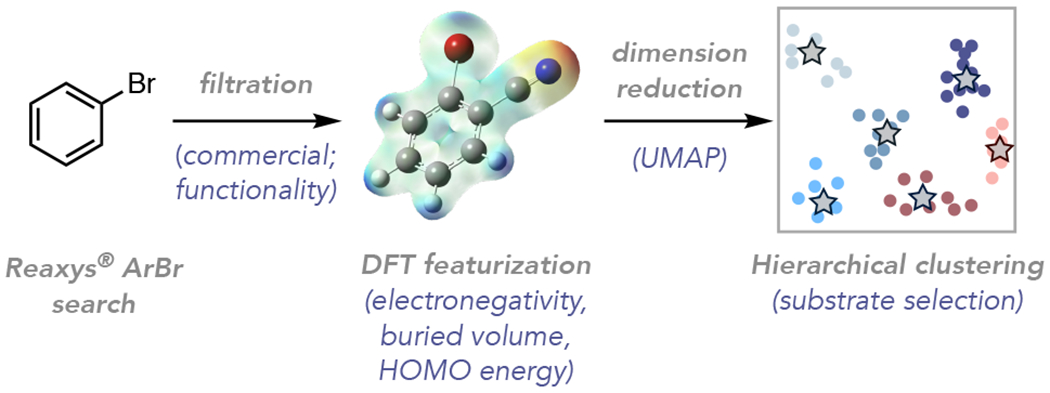
Workflow for construction of the chemical space and substrate selection via unsupervised learning.
Next, we pursued molecular featurization to quantify the similarity of the aryl bromides with the goal of visualizing the chemical space of the substrate class and systematically selecting a diverse and representative scope. Several approaches were considered, including molecular fingerprints,35 Mordred descriptors,15,36 and DFT featurization. Neither fingerprints nor Mordred features fully capture the electronic and steric properties of aryl bromides likely relevant to their activation by oxidative addition, a shared elementary step in Ni/photoredox coupling reactions with these substrates. Accordingly, we anticipated that DFT-based featurization, which captures the underlying physical organic properties (electronegativity, buried volume, etc.) would be most appropriate for capturing reactivity trends37 (see SI Section IX for a broader discussion and comparison between DFT and Mordred featurization).
We therefore performed DFT calculations and featurization using our previously published auto-qchem code17 for the set of ~2600 aryl bromides. With auto-qchem, a system that is connected to a computing cluster and a bookkeeping database, calculations of this size complete within a few days. The calculations delivered 168 features that consist of physical properties (HOMO/LUMO energies, dipole moment, etc.), as well as partial charges and NMR shifts on the bromide and six aromatic carbon atoms (C6) (see SI Table S22 for the complete list of features). Notably, the atoms of peripheral functional groups are not directly featurized; only their impact on the common core atoms (six aromatic carbons and bromine) and the whole molecule is accounted for (see Section IX part E in the SI for an alternative featurization that includes heteroaryl bromides).38
Next, we conducted a literature survey of aryl bromides employed in Ni/photoredox methodologies. Our literature search produced 116 papers (see SI for details),39 from which the yield and the frequency with which each aryl bromide appeared in substrate scope tables or supporting information documents were recorded. With the molecular featurization at hand, we used Uniform Manifold Approximation and Projection (UMAP)40 to reduce the featurization to two-dimensions for chemical space visualization (for alternative visualization of chemical space using principal components analysis (PCA) see SI Figure S11). We then overlaid all aryl bromides that appeared three or more times in the literature on the chemical space (Figure 6A). Generally, examples from the literature cover the right half of the chemical space well, while the left half is sparsely covered. By virtue of using reported yields from the literature, the average yields shown are likely to be inflated as unsuccessful substrates are seldom reported. Nevertheless, a trend is observed that the more sparsely covered chemical space corresponds to relatively lower yielding aryl bromide substrates. The five substrates covering this left region of the space all possess ortho substitution, revealing that steric hindrance represents a limitation in the overall methodology (see SI, Figure S28). Another trend that is apparent in surveying the full space is that more electron-donating aryl bromides provide lower yields than aryl bromides with electron-withdrawing substituents at the same position.41
Figure 6.
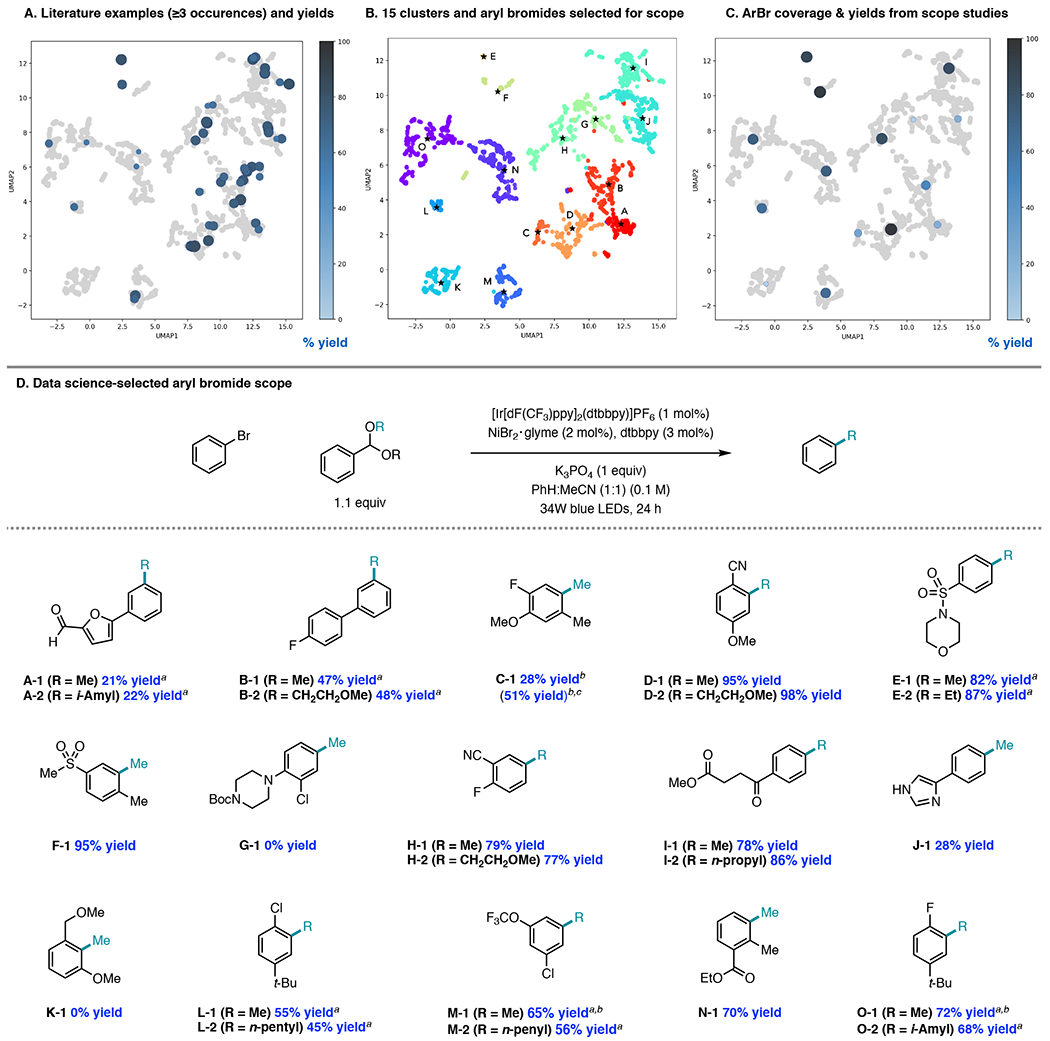
Evaluation of the literature coverage, clustering of the chemical space for scope selection, and scope studies (0.4 mmol scale). a 25 mol% TBABr added; b 1H NMR yield; c 5 mol % Ni/7.5 mol% dtbbpy; d GC yield.
To generate a diverse substrate scope for the alkylation of aryl bromides from acetals, we sought to cluster the chemical space for substrate selection. Although no universal definition of clustering exists, it is a statistical tool used to arrange large quantities of multivariate data into natural groups.42 We chose agglomerative hierarchical clustering with the Ward connectivity criterion.43 This algorithm starts with each aryl bromide as a separate cluster and then iteratively connects clusters such that the total within-cluster variance of all clusters is minimized until a desired number of clusters is reached. We optimized the clustering workflow by maximizing the average Silhouette score,44 a clustering quality index, with respect to an experimentally tractable number of clusters in the range of 3-40, and the level of dimensionality reduction with either UMAP or PCA. Silhouette score values lie in the range of −1 and 1: large values indicate that a sample matches its assigned cluster well, while at the same time does not match any other cluster. Optimization revealed that UMAP dimensionality reduction is largely superior to PCA (average silhouette score up to ~0.5, compared to ~0.3 for PCA), and a stable and broad maximum silhouette score was reached at about fifteen clusters (average silhouette score = 0.53) with UMAP. In addition, the silhouette score varies widely as a function of the dimension for PCA, while little dependence on the dimension of UMAP is observed (See SI Figure S10). Ultimately, we selected 10-dimensional UMAP and 15 clusters as our optimal parameters. From the fifteen chemically diverse clusters, we selected the center-most molecule per cluster for experimental evaluation, as indicated in Figure 6B.
To a chemist’s eye, the data science-driven scope features a diverse array of functional groups, including ester, nitrile, chloride, ether, trifluoromethoxy, sulfonyl, morpholine, imidazole, and furan functionality (Figure 6D). Substituents on the aryl bromide are present on all positions of the aryl ring through mono-, di-, and tri-substitution patterns. The frequency of multi-substitution in the data science-generated scope contrasts the literature scope, which is dominated by mono-substituted aryl bromides. In fact, Clusters C and K, both of which contain highly substituted aryl bromides, were not represented in the overlay with aryl bromides from the literature (Figure 6A). The data science scope also captures a variety of steric features: among ortho substituents, fluoride (O-1), chloride (L-1), methyl (C-1, F-1, N-1), and nitrile (D-1) groups are represented. In addition, an ortho,ortho-disubstituted aryl bromide that would produce K-1 is present in the scope.
Subjecting this collection of aryl bromides to the optimized Ni/photoredox coupling conditions with acetals demonstrated quite broad tolerance of the methodology (Figure 6D). Methylation reactions were performed for each substrate. Additional alkylation reactions were also conducted and showed that methyl and primary aliphatic acetals generally perform similarly in the cross-coupling method. The products are labeled with a letter (A-O) that corresponds to their cluster indicated in Figure 6B. With the exception of G-1 and ortho-, ortho-disubstituted K-1, all aryl bromides underwent productive alkylation to generate at least 21% yield of the desired product. The analytical methylation yields of these products and their distribution over the chemical space are shown in Figure 6C. Figures 6A and 6C present overall similar reactivity profiles, suggesting that this method compares favorably to the collection of methods published in the literature. In particular, this method appears to be more tolerant of steric hindrance than examples reported in the literature when examining reaction performance across clusters comprised of substrates possessing ortho-substitution (Clusters L, N, and O).
Juxtaposition of the reactivity and substrate space of this method with other Ni/photoredox methods offers an opportunity to identify global limitations and inspire new reaction development. Preliminarily, we have found that the yield of C-1, derived from a tri-substituted aryl bromide, could be boosted from 28% to 51% yield by increasing the cross-coupling catalyst loading to 5 mol% NiBr2·glyme and 7.5 mol% dtbbpy. However, further reaction development will likely be required to identify conditions that enable coupling of substrates in Cluster K that are generally both electronically-deactivated and sterically encumbered.
Yield Regression from the Aryl Bromide Scope Studies
The intention of using data science techniques for substrate scope generation was to maximally cover the aryl bromide chemical space using a conserved number of aryl bromides. Given the diversity of the aryl bromides, as well as the range of yields observed when applied to the cross-coupling methodology, we wondered if this small set of aryl bromides could provide predictive generalizations on the performance of unseen aryl bromides. To do so, we evaluated univariate relationships with supervised machine learning. Using a combination of Boruta45 and mRMR46 feature selection methodologies on our dataset of 15 aryl bromides from the clustering (A-1 to O-1, using 1H NMR yields from the methylation of each substrate), we found that electronegativity (χ) of the aryl bromide, defined as the average of the HOMO and LUMO energies, serves as a predictive feature.47 This finding is in line with chemical intuition, where more electron-rich substrates undergo a more challenging oxidative addition to Ni.41,48 We then pursued training and validation of the electronegativity model using a generalized additive model (GAM).49 The validation set is comprised of 37 additional aryl bromides present in our laboratory that were subjected to methylation using the conditions shown in Figure 6.
The regression model displayed in Figure 7A generalizes well to the validation data. The root-mean-squared-error (RMSE) is 21% and 19% in the clustering and validation data, respectively (R-squared = 0.57). By contrast, the high yield of the common literature substrates found in the validation set (outlined in gray in Figure 7A) amounts to a selection bias, making them less effective as a training set for building predictive models. Indeed, despite the literature set being larger (22 aryl bromides), a univariate regression model with these examples as a training set is worse at generalizing: training/validation RMSE of 14%/29% (see SI, Figure S19). Thus, this analysis demonstrates that with a small and systematically diversified scope, one can provide predictive generalizations applicable to a larger population of substrates.
Figure 7.
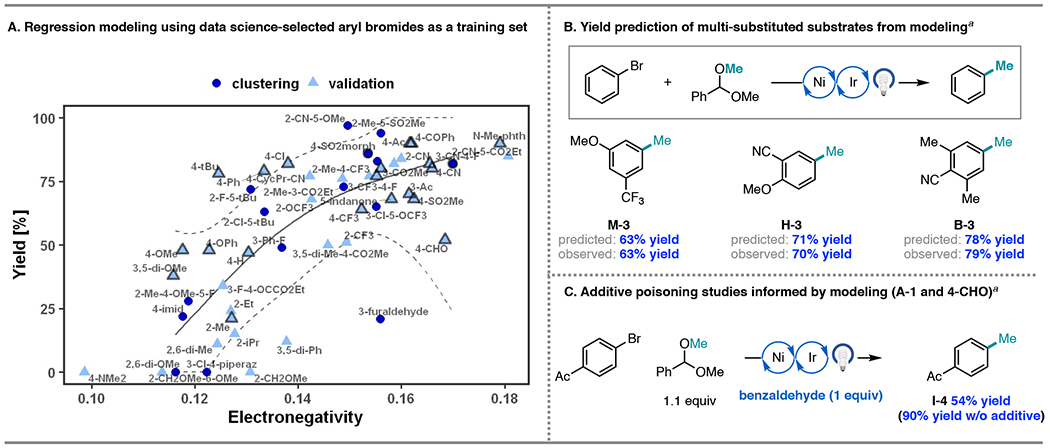
(A) Methylation yield as a function of aryl bromide electronegativity. The model and a 2 standard error band are trained only with the 15 data science-selected aryl bromides. Aryl bromides that appear in our Ni/photoredox literature search at least 3 times are outlined. (B) Utility of the model as a predictive tool for unseen substrate yield. (C) Additive poisoning studies using the conditions. a 1H NMR yield using 1,3,5-trimethoxybenzene as an external standard.
Despite the simplicity of the univariate model, we anticipated that it would have predictive value for multi-substituted substrates, where it is more challenging to intuit the overall electronic profile of an aryl bromide and its resulting reactivity. At the same time, multi-substitution dominates late-stage coupling examples in chemical synthesis but is severely under-represented in literature scope tables (85% of the literature aryl bromides plotted in Figure 6A are mono-substituted). To test the performance of the model on unseen substrates with complex substitution, we selected three aryl bromides possessing di-or tri-substitution with varying, non-intuitive electronic properties (Figure 7B). A 63% yield was predicted for M-3, which possesses meta-trifluoromethyl and meta-methoxy groups. We were pleased to find that this product was formed in 63% analytical yield under the standard reaction conditions. Two additional substrates, H-3 possessing meta-nitrile and para-methoxy functionality, as well as B-3 with meta-methyl groups and a para-nitrile, delivered methylated product within 1% of the predicted yield. These results suggest that the systematic substrate selection and modeling workflow is highly effective at revealing substituent impacts that affect relative reactivity (internal effects).
Nevertheless, a few substrates fall outside the 2-standard error band of the regression model in Figure 7A.50 For example, aldehyde-containing substrates that produced A-1 and 4-methylbenzaldehyde (I-3) provided noticeably lower yields than predicted, suggesting that a factor other than the internal reactivity of the substrate was overriding performance in the cross-coupling. 4-Bromobenzaldehyde belongs to Cluster I, one that is well covered and high-yielding in literature examples; in the methylation, two additional substrates belonging to this cluster afforded I-1 and 4’-methylacetophenone (I-4) in 86 and 90% yields, respectively. Thus, we hypothesized that the aldehyde functionality was leading to reaction poisoning by an off-cycle pathway.25a–b,51 To test this, we conducted a robustness experiment, evaluating the methylation of 4’-bromoacetophenone with 1 equiv benzaldehyde as an additive.7 While the reaction was not shut down by the additive, the yield of I-4 was reduced to 54% from 90% yield (Figure 7C). This reaction was also marked by consumption of benzaldehyde (53% conversion), indicating that the reduced yields of A-1 and I-3 could arise from an external effect of the aldehyde functionality rather than its influence on the electronegativity of the substrate.25a–b,51
More broadly, this analysis shows how substituent effects can be de-coupled using this workflow, with those that affect off-cycle steps (external effects) arising as outliers to the model, something that cannot be accomplished solely by using a fragment additive screen. Evaluation of only external effects on reactivity has been shown to result in false negatives and false positives.52 As an example within this dataset, robustness screening indicated that the preparation of imidazole-containing J-1 would fail: methylation of 4-bromobenzonitrile with 1 equiv imidazole delivered methylated product in <5% yield (compared to 80% yield without the additive). However, the data science clustering workflow showed that preparatively useful yield is possible: J-1 was produced in 28% yield. Taken together, these data suggest that neither the robustness screening nor clustering workflow on its own is enough to sample the reactivity and functional group compatibility encompassed in a reaction space. However, we anticipate that these tools can be used synergistically for expedient and comprehensive modeling of internal and external effects on reactivity.
CONCLUSION
In conclusion, we have developed a C(sp2)–C(sp3) cross-coupling reaction using acetals as sources of aliphatic coupling partners. Bromine-mediated HAT with the tertiary C–H bond of benzaldehyde di(alkyl) acetals, followed by β-scission of the resultant radical, enables the generation of carbon-centered radicals for coupling with aryl bromides and chlorides. The reaction platform works with 1:1 stoichiometry of acetal and aryl halide and installs (deutero)methyl, primary aliphatic, and secondary aliphatic groups efficiently with high functional group tolerance. By integrating DFT featurization and data science techniques, including hierarchical clustering, we were able to design a substrate scope that is representative of the diverse chemical space of commercial aryl bromides. While fifteen substrates are not enough to sample the reactivity of all regions of the space, we show that substrate selection using this workflow is better suited than traditional literature scopes for quantifying population-wide reactivity trends with supervised machine learning. Indeed, regression modeling enables population-wide yield prediction of unseen coupling partners and visually indicates possible sources of functional group incompatibility in a method. As such, we anticipate that this approach could serve as a model for standardizing scope analysis, enabling chemists to compare amongst methods, reduce the time and cost associated with scope evaluation, and afford literature data better suited to quantitative modeling of reactivity.
Supplementary Material
ACKNOWLEDGMENT
This material is based on work supported by the National Science Foundation Graduate Research Fellowship Program under Grant Number DGE-1656466 (to S.K.K.). A.M.Z. acknowledges financial support from the Schmidt DataX Fund at Princeton University made possible through a major gift from the Schmidt Futures Foundation. A.G.D. gratefully acknowledges the Princeton Innovation Fund, NIGMS (R35 GM126986) (Ni/photoredox method development), and the CCI Center for Computer Assisted Synthesis (CHE-1925607) (data science contributions) for financial support. We thank Jose Garrido Torres for helpful discussions on the data science workflow and Wendy Williams for computational support.
Funding Sources
The authors declare no competing financial interest.
Footnotes
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, spectroscopic data, aryl bromide chemical space construction and substrate selection details, information about the literature search and regression modeling (PDF).
Smiles (Smiles_list_2683.txt).
Annotated Code (Bromide_clustering.ipynb, Bromide_regression.ipynb).
The authors declare no competing financial interest.
REFERENCES
- [1].(a) Twilton J; Le C; Zhang P; Shaw MH; Evans RW; MacMillan DWC The Merger of Transition Metal and Photocatalysis. Nat. Rev. Chem 2017, 1, 0052. [Google Scholar]; (b) Milligan JA; Phelan JP; Badir SO; Molander GA Alkyl Carbon–Carbon Bond Formation by Nickel/Photoredox Cross-Coupling. Angew. Chem., Int Ed 2019, 58, 6152–6163. [Google Scholar]
- [2].Cannalire R; Pelliccia S; Sancineto L; Novellino E; Tron GC; Giustiniano M Visible light photocatalysis in the late-stage functionalization of pharmaceutically relevant compounds. Chem. Soc. Rev 2021, 50, 766–897. [DOI] [PubMed] [Google Scholar]
- [3].Lovering F; Bikker J; Humblet C Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- [4].(a) Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DWC Merging Photoredox Catalysis with Nickel Catalysis: Coupling of α-Carboxyl sp3-Carbons with Aryl Halides. Science 2014, 345, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang P; Le C; MacMillan DWC Silyl Radical Activation of Alkyl Halides in Metallaphotoredox Catalysis: A Unique Pathway for Cross-Electrophile Coupling. J. Am. Chem. Soc 2016, 138, 8084–8087. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tellis JC; Primer DN; Molander GA Single-Electron Transmetalation in Organoboron Cross-Coupling by Photoredox/Nickel Dual Catalysis. Science 2014, 345, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhang X; MacMillan DWC Alcohols as Latent Coupling Fragments for Metallaphotoredox Catalysis: sp3–sp2 Cross-Coupling of Oxalates with Aryl Halides. J. Am. Chem. Soc 2016, 138, 13862–13865. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Corcé V; Chamoreau L-M; Derat E; Goddard J-P; Ollivier C; Fensterbank L Silicates as Latent Alkyl Radical Precursors: Visible-Light Photocatalytic Oxidation of Hypervalent Bis-Catecholato Silicon Compounds. Angew. Chem., Int. Ed 2015, 54, 11414–11418. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Gutiérrez-Bonet Á; Tellis JC; Matsui JK; Vara BA; Molander GA 1,4-Dihydropyridines as Alkyl Radical Precursors: Introducing the Aldehyde Feedstock to Nickel/Photoredox Dual Catalysis. ACS Catal. 2016, 6, 8004–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Perry IB; Brewer TF; Sarver PJ; Schultz DM; DiRocco DA; MacMillan DWC Direct arylation of strong aliphatic C–H bonds. Nature 2018, 560, 70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].This is also true of the aliphatic coupling partner, but in this case there may not be the same set of available substrates. For example, certain alkyl halides might not be available as trifluoroborate salts, and vice versa.
- [6].(a) Gensch T; Glorius F The straight dope on the scope of chemical reactions. Science 2016, 352, 294–295. [DOI] [PubMed] [Google Scholar]; (b) Nguyen T The substrate scope conundrum C&EN News; 2017, 95, 16–18. [DOI] [PubMed] [Google Scholar]
- [7].(a) Collins KD; Glorius F A robustness screen for the rapid assessment of chemical reactions. Nat. Chem 2013, 5, 597–601. [DOI] [PubMed] [Google Scholar]; (b) Collins KD; Glorius F Intermolecular Reaction Screening as a Tool for Reaction Evaluation. Acc. Chem. Res 2015, 48, 619–627. [DOI] [PubMed] [Google Scholar]; (c) Gensch T; Teders M; Glorius F Approach to Comparing the Functional Group Tolerance of Reactions. J. Org. Chem 2017, 82, 9154–9159. [DOI] [PubMed] [Google Scholar]
- [8].(a) Kutchukian PS; Dropinski JF; Dykstra KD; Li B; DiRocco DA; Streckfuss EC; Campeau L-C; Cernak T; Vachal P; Davies IW; Krska SW; Dreher SD Chemistry Informer Libraries: A Chemoinformatics Enabled Approach to Evaluate and Advance Synthetic Methods. Chem. Sci 2016, 7, 2604–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dreher SD; Krska SW Chemistry Infomer Libraries: Conception, Early Experience, and Role in the Future of Chemoinformatics. Acc. Chem. Res 2021, 54, 1586–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bess EN; Bischoff AJ; Sigman MS Designer substrate library for quantitative, predictive modeling of reaction performance. Proc. Natl. Acad. Sci. U.S.A 2014, 111, 14698–14703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dombrowski AW; Gesmundo NJ; Aguiree AL; Sarris KA; Young JM; Bogdan AR; Martin MC; Gedeon S; Wang Y Expanding the Medicinal Chemist Toolbox: Comparing Seven C(sp2)–C(sp3) Cross-Coupling Methods by Library Synthesis. ACS Med. Chem. Lett 2020, 11, 597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].(a) Jorner K; Tomberg A; Bauer C; Sköld C; Norrby P-O Organic reactivity from mechanism to machine learning. Nat. Rev. Chem 2021, 5, 240–255. [DOI] [PubMed] [Google Scholar]; (b) Coley CW; Eyke NS; Jensen KF Autonomous Discovery in the Chemical Sciences Part I: Progress. Angew. Chem., Int. Ed 2020, 59, 22858–22893. [DOI] [PubMed] [Google Scholar]; (c) Strieth-Kalthoff F; Sandfort F; Segler MHS; Glorius F Machine learning the ropes: principles, applications and directions in synthetic chemistry. Chem. Soc. Rev 2020, 49, 6154–6168. [DOI] [PubMed] [Google Scholar]; (d) de Almeida AF; Moreira R; Rodrigues T Synthetic organic chemistry driven by artificial intelligence. 2019, 3, 589–604. [DOI] [PubMed] [Google Scholar]
- [12].Xu J; Hagler A Chemoinformatics and Drug Discovery. Molecules 2002, 7, 566–600. [Google Scholar]
- [13].Brown RD; Martin YC Use of Structure–Activity Data To Compare Structure-Based Clustering Methods and Descriptors for Use in Compound Selection. J. Chem. Inf. Comput. Sci 1996, 36, 572–584. [Google Scholar]
- [14].Mok NY; Brenk R; Brown N Increasing the Coverage of Medicinal Chemistry-Relevant Space in Commercial Fragments Screening. J. Chem. Inf. Model 2014, 54, 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Saito M; Kawamata Y; Meanwell M; Navratil R; Chiodi D; Carlson E; Hu P; Chen L; Udyavara S; Kingston C; Tanwar M; Tyagi S; McKillican BP; Gichinga MG; Schmidt MA; Eastgate MD; Lamberto M; He C; Tang T; Malapit CA; Sigman MS; Minteer SD; Neurock M; Baran PS N-Ammonium Ylide Mediators for Electrochemical C–H Oxidation. J. Am. Chem. Soc 2021, 143, 7859–7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].(a) Fey N Lost in chemical space? Maps to support organometallic catalysis. Chem. Cent. J 2015, 9:38. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gensch T; dos Passos Gomes G; Friederich P; Peters E; Gaudin T; Pollice R; Jorner K; Nigam A; Lindner-D’Addario ML; Sigman MS; Aspuru-Guzik A A comprehensive discovery platform for organophosphorus ligands for catalysis. ChemRxiv 2021, Preprint. 10.26434/chemrxiv.12996665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].https://github.com/PrincetonUniversity/auto-qchem (accessed November 18, 2021).
- [18].Henkel T; Brunne RM; Müller H; Reichel F Statistical Investigation into the Structural Complementarity of Natural Products and Synthetic Compounds. Angew. Chem. Int. Ed 1999, 38, 643–647. [DOI] [PubMed] [Google Scholar]
- [19].(a) Chen Y; Wang X; He X; An Q; Zuo Z Photocatalytic Dehydroxymethylative Arylation by Synergistic Cerium and Nickel Catalysis. J. Am. Chem. Soc 2021, 143, 4896–4902. [DOI] [PubMed] [Google Scholar]; (b) Cong F; Lv X-Y; Day CS; Margin R Dual Catalytic Strategy for Forging sp2–sp3 and sp3–sp3 Architectures via β-Scission of Aliphatic Alcohol Derivatives. J. Am. Chem. Soc 2020,142, 20594–20599. [DOI] [PubMed] [Google Scholar]
- [20].(a) Li Z; Sun W; Wang X; Li L; Zhang Y; Li C Electrochemically Enabled, Nickel-Catalyzed Dehydroxylative Cross-Coupling of Alcohols with Aryl Halides. J. Am. Chem. Soc 2021, 143, 3536–3543. [DOI] [PubMed] [Google Scholar]; (b) Chi BK; Widness JK; Gilbert MM; Salgueiro DC; Garcia KJ; Weix DJ Cross-Electrophile Coupling of Alcohols with Aryl and Vinyl Halides. hemRxiv 2021, Preprint. 10.33774/chemrxiv-2021-33jp3. [DOI] [PubMed] [Google Scholar]; (c) Dong Z; MacMillan DWC Metallaphotoredox-enabled deoxygenative arylation of alcohols. Nature 2021, 598, 451–456. [DOI] [PubMed] [Google Scholar]
- [21].(a) Kariofillis SK; Shields BJ; Tekle-Smith MA; Zacuto MJ; Doyle AG Nickel/Photoredox-Catalyzed Methylation of (Hetero)aryl Chlorides Using Trimethyl Orthoformate as a Methyl Radical Source. J. Am. Chem. Soc 2020, 142, 7683–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kariofillis SK; Doyle AG Synthetic and Mechanistic Implications of Chlorine Photoelimination in Nickel/Photoredox C(sp3)–H Cross-Coupling. Acc. Chem. Res 2021, 54, 988–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].(a) Kuhn LP; Wellman C Reaction of t-Butyl Peroxide with Acetals. J. Org. Chem 1957, 22, 774–776. [Google Scholar]; (b) Hartzell GE; Huyser ES Generation of Methyl Radicals by Decomposition of Bibenzyl Compounds Containing α-Methoxy Substituents. J. Org. Chem 1964, 29, 3341–3344. [Google Scholar]
- [23].Capaldo L; Ravelli D Hydrogen Atom Transfer (HAT): A Versatile Strategy for Substrate Activation in Photocatalyzed Organic Synthesis. Eur. J. Org. Chem 2017, 2056–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].All bond strength calculations performed at the uM062x/def2tzvpp//uB3LYP/CBSB7 level of theory.
- [25].Selected proposals invoking bromine radical generation in Ni/photoredox catalysis:; (a) Kawasaki T; Ishida N; Murakami M Dehydrogenative Coupling of Benzylic and Aldehydic C–H Bonds. J. Am. Chem. Soc 2020, 142, 3366–3370. [DOI] [PubMed] [Google Scholar]; (b) Kawasaki T; Ishida N; Murakami M Photoinduced Specific Acylation of Phenolic Hydroxy Groups with Aldehydes. Angew. Chem. Int. Ed 2020, 59, 18267–18271. [DOI] [PubMed] [Google Scholar]; (c) Heitz DR; Tellis JC; Molander GA Photochemical Nickel-Catalyzed C–H Arylation: Synthetic Scope and Mechanistic Investigations. J. Am. Chem. Soc 2016, 138, 12715–12718. [DOI] [PubMed] [Google Scholar]; (d) Santos MS; Corrêa AG, Paixão MW; König B C(sp3)–C(sp2) Cross-Coupling of Alkyl Bromides and Ethers Mediated by Metal and Visible Light Photoredox Catalysis. Adv. Synth. Catal 2020, 362, 2367–2372. [DOI] [PubMed] [Google Scholar]; (e) Huang L; Rueping M Direct Cross-Coupling of Allylic C(sp3)–H Bonds with Aryl- and Vinylbromides by Combined Nickel and Visible Light Catalysis. Angew. Chem., Int. Ed 2018, 57, 10333–10337. [DOI] [PubMed] [Google Scholar]; (f) Shu X; Huan L; Huang Q; Huo H Direct Enantioselective C(sp3)–H Acylation for the Synthesis of α-Amino Ketones. J. Am. Chem. Soc 2020,142, 19058–19064. [DOI] [PubMed] [Google Scholar]
- [26].Harper KC; Moschetta EG; Bordawekar SV; Wittenberger SJ A Laser Driven Flow Chemistry Platform for Scaling Photochemical Reactions with Visible Light. ACS Cent. Sci 2019, 5, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].(a) Kochi JK Chemistry of Alkoxy Radicals: Cleavage Reactions. J. Am. Chem. Soc 1962, 84, 1193–1197. [Google Scholar]; (b) Bacha JD; Kochi JK Polar and Solvent Effects in the Cleavage of t-Alkoxy Radicals. J. Org. Chem 1965, 30, 3272–3278. [Google Scholar]; (c) Walling C Some Aspects of the Chemistry of Alkoxy Radicals. Pure Appl. Chem 1967, 15, 69–80. [Google Scholar]
- [28].(a) Isse AA; Lin CY; Coote ML; Gennaro A Estimation of Standard Reduction Potentials of Halogen Atoms and Alkyl Halides. J. Phys. Chem. B 2011, 115, 678–684. [DOI] [PubMed] [Google Scholar]; (b) Shields BJ; Doyle AG Direct C(sp3)–H Cross Coupling Enabled by Catalytic Generation of Chlorine Radicals. J. Am. Chem. Soc 2016, 138, 12719–12722. [DOI] [PubMed] [Google Scholar]; (c) Rohe S; Morris AO; McCallum T; Barriault L Hydrogen Atom Transfer Reactions via Photoredox Catalyzed Chlorine Atom Generation. Angew. Chem., Int. Ed 2018, 57, 15664–15669. [DOI] [PubMed] [Google Scholar]
- [29].The reaction performed under the conditions shown in Table 2 Entry 3, but in a 1:1 mixture of benzaldehyde dimethyl acetal:benzene as solvent, enabled methylation in 28% yield. This result suggests that the photoelimination pathway can be turned on with an excess of the acetal coupling partner.
- [30].Although more rapid quenching was observed with TBABr than Ni, MeCN can displace the bromide ligand on Ni, leading to comparable rates of quenching:; Feth MP; Klein A; Bertagnolli H Investigation of the Ligand Exchange Behavior of Square-Planar Nickel(II) Complexes by X-ray Absorption Spectroscopy and X-ray Diffraction. Eur. J. Inorg. Chem 2003, 839–852. [Google Scholar]
- [31].Aryl chloride methylation has previously enabled preparation of the first selective COX-2 NSAID:; Sun S; Fu J Methyl-containing pharmaceuticals: Methylation in drug design. Bioorg. Med. Chem. Lett 2018,28, 3283–3289. [DOI] [PubMed] [Google Scholar]
- [32].(a) Barreiro EJ; Kümmerle AE; Fraga CAM The Methylation Effect in Medicinal Chemistry. Chem. Rev 2011, 111, 5215–5246. [DOI] [PubMed] [Google Scholar]; (b) Schönherr H; Cernak T Profound Methyl Effects in Drug Discovery and a Call for New C–H Methylation Reactions. Angew. Chem., Int. Ed 2013, 52, 12256–12267. [DOI] [PubMed] [Google Scholar]; (c) Aynetdinova D; Callens MC; Hicks HB; Poh CYX; Shennan BDA; Boyd AM; Lim ZH; Leitch JA; Dixon DJ Installing the “magic methyl” – C–H methylation in synthesis. Chem. Soc. Rev 2021, 50, 5517–5563. [DOI] [PubMed] [Google Scholar]
- [33].(a) Hu L; Liu X; Liao X Nickel-Catalyzed Methylation of Aryl Halides with Deuterated Methyl Iodide. Angew. Chem., Int. Ed 2016, 55, 5517–5563. [DOI] [PubMed] [Google Scholar]; (b) Komeyama K; Yamahata Y; Osaka I Nickel and Nucleophilic Cobalt-Catalyzed Trideuteriomethylation of Aryl Halides Using Trideuteriomethyl p-Toluenesulfonate. Org. Lett 2018, 20, 4375–4378. [DOI] [PubMed] [Google Scholar]; (c) Wu Z; Wei F; Wan B; Zhang Y Pd-Catalyzed ipso,meta-Dimethylation of ortho-Substituted Iodoarenes via a Base-Controlled C–H Activation Cascade with Dimethyl Carbonate as the Methyl Source. J. Am. Chem. Soc 2021, 143, 4524–4530. [DOI] [PubMed] [Google Scholar]
- [34].Minor cyclobutylmethyl ring opening was observed in this reaction.
- [35].(a) Senese CL; Duca J; Pan D; Hopfinger AJ; Tseng YJ 4D-Fingerprints, Universal QSAR and QSPR Descriptors. J. Chem. Inf. Comput. Sci 2004, 44, 1526–1539. [DOI] [PubMed] [Google Scholar]; (b) Rogers D; Hahn M; Extended-Connectivity Fingerprints. J. Chem. Inf. Model 2010, 50, 742–754. [DOI] [PubMed] [Google Scholar]; (c) Pattanaik L; Coley CW Molecular Representation: Going Long on Fingerprints. Chem 2020, 6, 1203–1207. [DOI] [PubMed] [Google Scholar]; (d) Sandfort F; Strieth-Kalthoff F; Kühnemund M; Beecks C; Glorius F A Structure-Based Platform for Predicting Chemical Reactivity. Chem 2020, 6, 1379–1390. [DOI] [PubMed] [Google Scholar]
- [36].Moriwaki H; Tian Y-S; Kawashita N; Takagi T Mordred: a molecular descriptor calculator. J. Cheminformatics 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Żurański AM; Martinez Alvarado JI; Shields BJ; Doyle AG Predicting Reaction Yields via Supervised Learning. Acc. Chem. Res 2021, 54, 1856–1865. [DOI] [PubMed] [Google Scholar]
- [38].Our first report here focuses on the generation of an aryl bromide scope using molecular and atomic (C6Br) features. However, this workflow can be applied to heteroaromatic bromides by changing the atomic features to the conserved C–Br bond (see SI for full details and a collection of 15 heteroaryl bromides generated from clustering, including N-, O-, and S-containing heterocycles, Figures S16–S17).
- [39].Literature search performed on December 10, 2020.
- [40].(a) McInnes L; Healy J; Melville J UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv Preprint Feb 9, 2018, arXiv:1802.03426 [stat.ML]. https://arxiv.org/abs/1802.03426 (accessed May 15, 2021). [Google Scholar]; (b) McInnes L; Healy J; Saul N; Groβberger L UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw 2018, 3, 861. [Google Scholar]
- [41].Foà M; Cassar L Oxidative Addition of Aryl Halides to Tris(triphenylphosphine)nickel(0). J. Chem. Soc. Dalton Trans 1975, 2572–2576. [Google Scholar]
- [42].Izenman AJ Modern Multivariate Statistical Techniques, Regression, Classification, and Manifold Learning. Springer Texts Statistics 2008. [Google Scholar]
- [43].(a) Hastie T; Tibshirani R; Friedman J Cluster Analysis. In The Elements of Statistical Learning, 2nd ed.; Springer, 2009; pp 501–527. DOI: 10.1007/b94608_2. [DOI] [Google Scholar]; (b) Kowalski BR; Bender CF Pattern Recognition. A Powerful Approach to Interpreting Chemical Data. J. Am. Chem. Soc 1972,94, 5632–5639. [DOI] [Google Scholar]
- [44].Rousseeuw PJ Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math 1987, 20, 53–65. [Google Scholar]
- [45].Kursa MB; Jankowski A; Rudnicki WR Boruta – A System for Feature Selection. Fundam. Inform 2010, 101, 271–285. [Google Scholar]
- [46].Peng H; Long F; Ding C Feature Selection Based on Mutual Information: Criteria of Max-Dependency, Max-Relevance, and Min-Redundancy. IEEE Trans. Pattern Anal. Mach. Intell 2005, 27, 1226–1238. [DOI] [PubMed] [Google Scholar]
- [47].Hastie T; Tibshirani R Generalized Additive Models. Stat Sci 1986, 1, 297–310. [DOI] [PubMed] [Google Scholar]
- [48].Zhu C; Yue H; Jia J; Rueping M Nickel-Catalyzed C-Heteroatom Cross-Coupling Reactions under Mild Conditions via Facilitated Reductive Elimination. Angew. Chem., Int. Ed 2021, 60, 17810–17831. [DOI] [PubMed] [Google Scholar]
- [49].Using cross-validation we found that multivariate models do not offer increased predictive power, indicating that a higher fidelity regression model requires more data or dedicated feature engineering.
- [50].It is interesting to note that the most positive outliers (4-tBu and 4-Ph) in the model are the substrates that we subjected to intensive reaction optimization.
- [51].Poisoning by an aldehyde additive could arise from activation by Ni, HAT, or photosensitization by a photoredox catalyst. See:; (a) Hoshimoto Y; Ohashi M; Ogoshi S Nickel-Catalyzed Selective Conversion of Two Different Aldehydes to Cross-Coupled Esters. J. Am. Chem. Soc 2011,133, 4668–4671. [DOI] [PubMed] [Google Scholar]; (b) Zhang L; Si X; Yang Y; Zimmer M; Witzel S; Sekine K; Rudolph M; Hashmi ASK The Combination of Benzaldehyde and Nickel-Catalyzed Photoredox C(sp3)–H Alkylation/Arylation. Angew. Chem., Int. Ed 2019, 58, 1823–1827. [DOI] [PubMed] [Google Scholar]
- [52].Lin S; Dikler S; Blincoe WD; Ferguson RD; Sheridan RP; Peng Z; Conway DV; Zawatzky K; Wang H; Cernak T; Davies IW; DiRocco DA; Sheng H; Welch CJ; Dreher SD Mapping the dark space of chemical reactions with extended nanomole synthesis and MALDI-TOF MS. Science 2018, 361, 1–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.