
Fig. 1. Identifying epigenomic and other features associated with mutations in Arabidopsis.
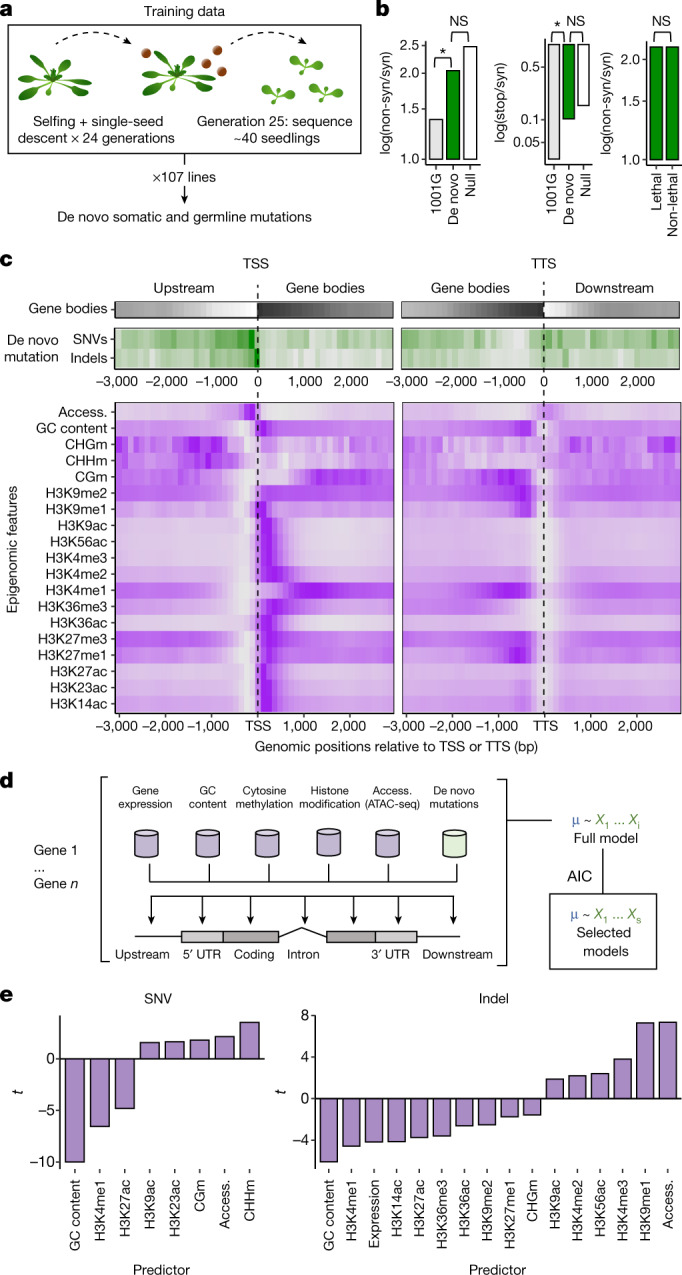
a, Experimental design for identifying germline and somatic mutations in the main dataset12. b, Relaxed purifying selection in de novo mutation calls: rates of non-synonymous (non-syn) and stop codon variants (stop) as compared with polymorphisms detected in 1,135 natural accessions from the 1001 Genomes (1001G) project35 and to a null model based on mutation spectra and nucleotide composition of coding sequences. Comparison of de novo mutations between genes predicted to have or not have lethal effects when mutated is also shown37. P values from χ2 test; *P < 0.05. NS, not significant. c, Genome-wide distributions in gene body density, observed mutation rates and candidate predictive features in relation to transcription start sites (TSS) and transcription termination sites (TTS). Darker shading represents greater density. SNV, single-nucleotide variant; CHGm, CHHm, CGm, methylation in the CHG, CHH and CG contexts, respectively. d, Modelling approach to predict mutation probability from a range of features. ATAC-seq, assay for transposase-accessible chromatin using sequencing; AIC, Akaike information criterion. e, Predictive models and t-values of predictor variables from the generalized linear model.