

Summary
Infiltration of arterial intima by foamy macrophages is a hallmark of early atherosclerotic lesions. Here, we investigated the potential role of Ser/Thr phosphatase PHLPP1 in foam cell development. PHLPP1 levels were elevated in OxLDL-exposed macrophages and high-fat diet (HFD)-fed zebrafish larvae. Using overexpression and knockdown approaches, we show that PHLPP1 promotes the accumulation of neutral lipids, and augments cellular total cholesterol and free fatty acid (FFA) levels. RNA-Seq analysis uncovered PHLPP1 role in lipid metabolism pathways. PHLPP1 interacted with and modestly increased ChREBP recruitment to Fasn promoter. PHLPP1-mediated lipid accumulation was attenuated by AMPK activation. Pharmacological inhibition or CRISPR/Cas9-mediated disruption of PHLPP1 resulted in lower lipid accumulation in the intersegmental vessels of HFD-fed zebrafish larvae along with a reduction in total cholesterol and triglyceride levels. Deficiency of phlp-2, C. elegans PHLPP1/2 ortholog, abolished lipid accumulation in high cholesterol-fed worms. We conclude that PHLPP1 exerts a significant effect on lipid buildup.
Subject areas: Biological sciences, Lipid, Molecular physiology, Molecular biology
Graphical abstract
Highlights
-
•
PHLPP1 levels are elevated in OxLDL exposed macrophages and HFD-fed zebrafish larvae
-
•
Loss of PHLPP1 reduces neutral lipid build-up in macrophages, zebrafish, and C. elegans
-
•
PHLPP1 impairs pathways linked to lipid metabolism and inflammation
-
•
PHLPP1-dependent lipid build-up involves AMPK and ChREBP pathways
Biological sciences; Lipid; Molecular physiology; Molecular biology
Introduction
Foam cells, the hallmark of atherosclerosis, are lipid-laden macrophages that build up and form plaques leading to cardiovascular diseases (CVDs), which contribute to the highest global mortality (Aviram, 1999; Barquera et al., 2015). The formation of foam cells is initiated via macrophage engulfment of modified LDL (oxidized LDL, OxLDL) on its binding to either CD36, LDLR, and scavenger receptors class A (SR-A) promoting dramatic changes in cellular lipid metabolism (Chistiakov et al., 2017). The engulfed OxLDL enters lysosomes, is metabolized to free cholesterol and subsequently might form cytotoxic crystals (Tabas, 1997). The free cholesterol is effluxed out via ATP-binding cassette (ABC) transporters such as ABCG1 and ABCA1 or further gets converted into nontoxic cholesteryl ester by Sterol O-acyltransferase I (SOAT1) (Ouimet and Marcel, 2012). Neutral cholesteryl ester hydrolase (nCEH) aids in the reconversion of cholesteryl ester to cholesterol in blood and the effluxed cholesterol is tracked into reverse cholesterol transport (Cuchel and Rader, 2006; Jeong et al., 2017). The balance between uptake, cholesteryl ester formation, and efflux decides the cell′s fate (Yuan et al., 2012).
PH domain and leucine-rich repeat protein phosphatase (PHLPP) exist as two isozymes, PHLPP1 and PHLPP2 with 50% global amino acid sequence identity. PHLPP1 further comprises two alternatively spliced variants PHLPP1α and PHLPP1β, differing in their amino terminus. Although the tumor suppressor role of PHLPP1 is actively pursued, its involvement in different facets of metabolic syndrome remains largely unexplored. Toward this, our laboratory has previously shown that PHLPP1 augments skeletal muscle insulin resistance and impairs glucose homeostasis by inducing ER stress, a key driver of metabolic syndrome (Behera et al., 2018). Furthermore, findings from different research groups including ours highlight the importance of PHLPP1 in controlling macrophage inflammatory responses (Alamuru-Yellapragada et al., 2017a; Alamuru et al., 2014; Katsenelson et al., 2019), prompting us to investigate the significance of PHLPP1 in the development of foam cells which display abnormal lipid accumulation and altered inflammatory program.
In our current study, we investigated PHLPP1 involvement in lipid accumulation using in vitro foam cell studies and an HFD-fed zebrafish larval model, and observed that PHLPP1 augments neutral lipid accumulation, cellular free fatty acid (FFA), and total cholesterol levels through the differential regulation of AMP-activated protein kinase (AMPK) and carbohydrate response element binding protein 1 (ChREBP1)-mediated lipid uptake and fatty acid synthesis pathways.
Results
In vitro foam cells show altered PHLPP1 levels
We and others have previously reported that PHLPP1 restrains pro-inflammatory responses of macrophages which are commonly dysregulated in atherosclerosis (Alamuru-Yellapragada et al., 2017a; Katsenelson et al., 2019). Thus, we investigated the possible involvement of PHLPP1 in the development of foam cells, a hallmark of atherosclerosis. For this, we examined the expression of PHLPP1 in in vitro foam cells using OxLDL-exposed macrophages as the model system. In line with our earlier observation (Alamuru-Yellapragada et al., 2017a), stimulation of RAW 264.7 cells with bacterial endotoxin for 24 h depleted PHLPP1 protein levels (Figure 1A). Furthermore, treatment of RAW 264.7 cells with OxLDL resulted in the dose-dependent elevation of PHLPP1 protein levels up to 24 h (Figure 1A). PHLPP1 transcripts were also enhanced at 24 h as revealed by qPCR analysis (Figures 1B and 1C). Post–24 h of OxLDL exposure, PHLPP1 mRNA and protein levels were reduced (Figures 1C and 1E). Elevated circulating 7-ketocholesterol, an abundant oxysterol found in atherosclerotic plaques and processed cholesterol-rich foods, is associated with heightened cardiovascular risk and death (Hayden et al., 2002; Rao et al., 2014). Similar to OxLDL treatment, 7-ketocholesterol treatment also enhanced PHLPP1 levels up to 24 h (Figure 1D), which declined 48 h posttreatment (Figure 1E). The levels of fatty acid synthase (FASN) and the lipogenic transcription factor carbohydrate responsive element binding protein (ChREBP) were also augmented in 7-ketocholesterol exposed macrophages. Consistent with the role of 7-ketocholesterol in ER stress induction that supports foam cell formation (Pedruzzi et al., 2004), ER stress markers such as CHOP, ATF6α, and TRB3 were elevated up to 6–9 h post–7-ketocholesterol treatment (Figures 1D and S1C). In accordance with our previous findings, PHLPP1 levels increased up to 3 h and then declined at both mRNA and protein levels on exposure to the ER stress inducer thapsigargin (Figures S1A and S1B) (Behera et al., 2018). Parallel experiments confirmed the induction of ER stress in thapsigargin-treated cells (Figure S1B).
Figure 1.

ChREBP-mediated PHLPP1 regulation in in vitro foam cells
(A–C) Dose and time kinetics of PHLPP1 protein levels (A) with densitometric quantification (bottom) and mRNA (B and C) levels after OxLDL treatment in RAW 264.7 cells; Statistical analysis was performed using one-way ANOVA followed by Bonferroni multiple comparisons test for (C) (∗p < 0.05 vs Control, 0 h; ap < 0.05 vs 24 h).
(D) Protein levels of PHLPP1, ChREBP, and ER stress markers upon 7-ketocholesterol treatment under different time points. Densitometric analysis is shown in Figure S1C.
(E) PHLPP1 protein levels upon 48 h OxLDL and 7-ketocholesterol treatment along with densitometric quantification (Bottom).
(F and G) PHLPP1 protein (F) and mRNA (G) levels upon ChREBP overexpression; Statistical analysis was performed using two-tailed t-test for (G) (∗p < 0.05 vs Vector).
(H) PHLPP1 and FASN protein levels in ChREBP knockdown cells treated with 7-kC or OxLDL. Densitometric analysis is shown in Figure S1D.
(I) Recruitment of ChREBP to PHLPP1 promoter. Data are a representative of three independent experiments. Numerical data are expressed as mean ± SEM. See also Figures S1 and S2.
Carbohydrate responsive element binding protein functions as a transcriptional activator of PHLPP1
ChREBP, also known as MLX-interacting protein-like (MLXIPL), is a key transcriptional activator regulating the glucose-dependent expression of triglyceride and fatty acid synthesis genes. Immunoblot analysis of 7-ketocholesterol-treated macrophages showed similar induction patterns of PHLPP1 and ChREBP proteins suggesting the possibility of ChREBP-mediated upregulation of PHLPP1 (Figure 1D). Computational analysis of the PHLPP1 promoter revealed an evolutionarily conserved ChREBP binding site between −488 and −473 bp (Figure S2). Therefore, we next determined the effect of ChREBP on PHLPP1 levels using Western blot, qPCR, and ChIP analysis. Overexpression of flag-tagged ChREBP in RAW 264.7 cells upregulated PHLPP1 protein and mRNA levels (Figures 1F and 1G). Consistently, PHLPP1 and FASN protein levels were reduced in OxLDL and 7-ketocholesterol-treated cells upon ChREBP knockdown (Figures 1H and S1D). ChIP experiments confirmed ChREBP recruitment to Phlpp1 promoter (Figure 1I).
PHLPP1 escalates lipid accumulation in in vitro foam cells
We next examined neutral lipid accumulation (cholesteryl ester and triglycerides) in RAW 264.7 cells upon PHLPP1 overexpression or knockdown using Oil Red O staining. Forced expression of PHLPP1α enhanced neutral lipid accumulation in OxLDL-treated cells as well (Figures 2A and 2B). Neutral lipid buildup in OxLDL-exposed PHLPP1-ablated cells was robustly reduced to levels observed in untreated control cells (Figures 2A and 2C). Overexpression of PHLPP1α or PHLPP1β but not PHLPP2 increased basal levels of neutral lipids (Figures 2A–2D). Additional analysis showed that ectopic PHLPP1 expression enhanced total cholesterol and FFA levels in untreated RAW 264.7 macrophages (Figures 2E and 2F).
Figure 2.

PHLPP1 promotes foam cells in vitro
(A–C) Neutral lipid staining by Oil Red O (A) along with quantification in PHLPP1 overexpressing (A and B) or knockdown RAW 264.7 cells (A and C) with parallel immunoblot analysis (B and C top panels); Images are taken at |×20×| magnification. Each image in (A) is a representative image of four different experiments with a minimum of eight different fields in each experiment. Statistical analysis was performed using one-way ANOVA followed by Bonferroni′s post hoc test (∗p < 0.05, ∗∗∗p < 0.001 vs Vector-OxLDL; @p < 0.05 vs PHLPP1-OxLDL; $p < 0.05 vs sh-Luc+OxLDL).
(D) Neutral lipid staining in cells overexpressing PHLPP isoforms; Statistical analysis was performed using one-way ANOVA followed by Dunnett′s multiple comparisons test (∗p < 0.05 vs Vector).
(E and F) Total cellular cholesterol (E) and FFA levels (F) in RAW 264.7 cells upon PHLPP1 overexpression; Statistical analysis was performed using two-tailed t-test (∗p < 0.05, ∗∗p < 0.01 vs Vector). Data are a representative of at least three independent experiments. Numerical data are expressed as mean ± SEM.
RNA-seq analysis reveals the role of PHLPP1 in lipid and cholesterol metabolism
RNA-seq analysis of differential gene expression under three different experimental conditions was performed (Figure 3A): set 1: control cells and OxLDL-treated control cells (100 μg/mL) for 24 h; set 2: control cells and PHLPP1-knockdown cells; set 3: OxLDL-treated control cells and OxLDL-treated PHLPP1-knockdown cells ((100 μg/mL) for 24 h). Volcano plots presented in Figure 3B show robust differential regulation of genes under the three conditions. With a 2-fold change and FDR < 0.05 as the cut-off, 121, 569, and 1,303 genes were significantly altered in sets 1, 2, and 3, respectively (Figure 3C). Importantly, supporting the findings in Figure 1, fatty acid biosynthesis, fatty acid elongation, fluid shear stress & atherosclerosis, and biosynthesis of unsaturated fatty acid pathways were highly enriched among the Top 25 hits along with those associated with reported roles of PHLPP1 such as apoptosis and insulin signaling among others in set 3 (Figure 3D).
Figure 3.

RNA-seq analysis of PHLPP1 depleted in vitro foam cells
(A) Representation of experimental conditions-set 1: control cells versus OxLDL-treated control cells; set 2: control cells versus PHLPP1-knockdown cells; set 3: OxLDL-treated control cells versus OxLDL-treated PHLPP1-knockdown cells.
(B) Volcano plot analysis of set 1, 2 and 3 genes.
(C) Venn diagram showing commonly affected genes in the three sets.
(D) Top 25 affected pathways in set 3 were identified using Enrichr tool by submitting the differentially altered genes from set 3 (fold change >1.5, FDR: 0.05). KEGG 2021 human pathways library was used to analyze the possible disease conditions.
(E) Heatmap analysis of selected pathways (inflammation, cholesterol biosynthesis, and lipid metabolism) of set 3. Data are a representative of three independent experiments. See also Figure S3.
Several cholesterol biosynthesis genes were downregulated in OxLDL-treated PHLPP1-knockdown cells (Figure 3E). HMG-CoA reductase, a rate-limiting enzyme in the cholesterol biosynthesis pathway and the target of statins and AMPK-mediated inhibitory phosphorylation, showed a prominent decrease. Furthermore, the expression of another rate-limiting enzyme, squalene epoxidase (SQE), and terminal enzymes in cholesterol biosynthesis (DHCR7 and DHCR24) were also reduced in PHLPP1-ablated cells. Fatty uptake and synthesis-associated genes such as CD36 (OxLDL uptake), Fasn (fatty acid synthesis), ELOVL6 (fatty acid elongation), and SCD1 (fatty acid desaturation) were reduced in PHLPP1-deficient cells (Figures 3E and S3). Supporting the role of PHLPP1 in restraining macrophage pro-inflammatory responses, PHLPP1-knockdown cells showed significant upregulation of genes including IL6, IL1-β, CD40, and CD86 on OxLDL exposure (Figure 3E). Expectedly, pathway analysis for set one also listed atherosclerosis condition as one of the top 20 hits; Furthermore, we observed that synthesis of unsaturated fatty acids and AMPK signaling, both pathways associated with lipid metabolism, are among the top 20 hits in untreated PHLPP1 knockdown cells (Figures S3A and S3B).
PHLPP1 promotes OxLDL uptake through CD36
We next examined the impact of PHLPP1 on OxLDL uptake and cholesterol efflux using commercially available kits. Forced PHLPP1 expression in RAW 264.7 cells enhanced fluorescent OxLDL uptake (Figure 4A). PHLPP1-dependent OxLDL uptake was abolished by prior treatment with myricetin, a hexahydroxyflavone that inhibits CD36 expression, suggesting that PHLPP1-mediated OxLDL uptake requires CD36. Likewise, PHLPP1 knockdown substantially blocked OxLDL uptake by RAW 264.7 cells (Figure 4B). Protein levels of CD36, a selective receptor for cholesteryl ester uptake, and other lipid biosynthesis genes SOAT1 and FASN were elevated in macrophages upon PHLPP1 overexpression. Consistently, depletion of PHLPP1 by shRNA robustly reduced expression of CD36, SOAT1, and FASN (Figure 4E). Very high PHLPP1 expression caused a reduction in protein levels of ABCG1, a lipid efflux transporter and as expected, PHLPP1 knockdown increased ABCG1 protein levels (Figure 4E). However, levels of another cholesterol efflux transporter ABCA1 were unchanged upon PHLPP1 ectopic expression. Surprisingly, neither PHLPP1 overexpression nor PHLPP1 knockdown showed any major impact on fluorescent cholesterol efflux (Figures 4C and 4D).
Figure 4.

PHLPP1 deficiency suppresses lipid metabolism gene expression
(A and B) Fluorescent images of DyLight™488 OxLDL-treated Hoechst-stained PHLPP1 overexpressing (A) or knockdown (B) RAW 264.7 cells along with quantification (bottom); Each image in (A and B) is a representative image of three different experiments with a minimum of 10 different fields in each experiment. Images are taken at |×20×| magnification; Statistical analysis in (A) was performed using one-way ANOVA followed by Bonferroni′s post hoc test (∗p < 0.05, vs Vector-Myr; $p < 0.05 vs PHLPP-Myr); Statistical analysis in (B) was performed using two-tailed t-test (∗p < 0.05, vs sh-Luc).
(C and D) BODIPY-labeled cholesterol efflux quantification in PHLPP1 overexpressing (C) or knockdown cells (D); statistical analysis was performed using one-way ANOVA followed by Bonferroni′s post hoc test (∗p < 0.05 vs Vector).
(E) Protein levels of foam cell markers in PHLPP1 overexpressing or knockdown cells along with densitometric quantification (bottom). Data are a representative of three independent experiments unless specified. Numerical data are expressed as mean ± SEM [except mean ± SD for (E)-CHOP (n = 2)].
Prolipogenic effects of PHLPP1 are mediated through carbohydrate responsive element binding protein and AMPK
Based on the transcriptomics analysis, we reasoned that PHLPP1 may target a key lipid-metabolism-associated transcription factor. We analyzed the enrichment for transcription factors based on differentially regulated genes in OxLDL-treated PHLPP1 knockdown cells and prioritized ChREBP for further experimentation (Figures 5A–5C and S4A and S4B) based on the following observations: (1) ChREBP is a key lipogenic transcriptional factor and was earlier reported to be critical for the regulation of lipid metabolism in liver, β-cells, and so forth. (Iizuka et al., 2020; da Silva Xavier et al., 2006). (2) PHLPP1 promoted the expression of ChREBP targets Fasn, ELOVL6, and so forth. (Figures 4E and S3C). (3) AMPK, a target of PHLPP1, was earlier shown to inhibit the DNA binding activity of ChREBP (Behera et al., 2018; Kawaguchi et al., 2002). (4) Analysis of phosphorylation sites of ChREBP by PhosphoSitePlus identified a putative PHLPP1 target residue pSer196 (Figure S4B), previously reported to regulate its nuclear translocation (Sakiyama et al., 2008). (5) ChREBP facilitated OxLDL/7-ketocholesterol-dependent increase in PHLPP1 levels (Figure 1H) prompting us to envisage that PHLPP1 may modulate the activity of ChREBP, as an integral feedback control loop. Overexpression of ChREBP resulted in the robust accumulation of neutral lipids which was stronger than the accumulation observed in PHLPP1 overexpressing macrophages (Figure 5D). Ectopic expression of ChREBP resulted in a subtle increase of cholesterol but a more prominent increase in FFAs (Figures 5E and 5F) along with the induction of FASN (da Silva Xavier et al., 2006) (Figure 5G). However, CD36, SOAT1, and ABCG1 protein levels remain unchanged (Figures 5G and S5A). Immunoprecipitation experiments revealed an interaction between ectopically expressed PHLPP1 and ChREBP in HEK 293T cells (Figure 5H). Pull-down of endogenously expressed ChREBP in RAW 264.7 cells also coprecipitated PHLPP1 validating the interaction between them (Figure 5I). Incubation of PHLPP1 and ChREBP proteins enriched from HEK 239T cells resulted in higher phosphate release, suggesting that ChREBP may be a direct PHLPP1 substrate (Figure 5J). Previous studies showed that the nuclear translocation and DNA binding activity of ChREBP is controlled by PKA and AMPK-mediated phosphorylation of ChREBP residues Ser196 and Ser568 residues, respectively (Sakiyama et al., 2008). Thus, to further examine the effect of PHLPP1 on ChREBP, ChIP assay was performed to monitor ChREBP recruitment to Fasn promoter. ChIP-qPCR analysis revealed an increase in ChREBP recruitment to Fasn promoter under basal conditions (Figure 5L). The enrichment of ChREBP at Fasn promoter under basal conditions was modestly enhanced in PHLPP1-overexpressing cells. Overexpression of PHLPP1 or ChREBP reduced the phosphorylation of AMPK pThr172 phosphorylation (Figures 5K and S5B). AICAR-mediated AMPK activation lowered CD36 and FASN protein levels in PHLPP1 overexpressing cells (Figures 5M and S5C). Treatment of cells with activator-3 (Act-3), which we recently described (Bung et al., 2018), to directly activate AMPK, also resulted in the reduction of CD36 and FASN protein levels despite PHLPP1 overexpression (Figures 5M and S5D). Consequently, AICAR and Act-3 led to a significant reduction in lipid accumulation in OxLDL-treated PHLPP1 overexpressing cells (Figure 5N). To our surprise, we noticed that ChREBP-dependent neutral lipid buildup in macrophages was not influenced by AMPK activation (Figure 5N).
Figure 5.

PHLPP1-dependent neutral lipid accumulation involves ChREBP and AMPK
(A) Scatterplot of enriched transcription factors in set 3: Differentially altered gene-set from set 3 (cut-off: FC > 1.5, FDR 0.05) was analyzed by “Enrichr” tool to analyze the possible transcription factors involved in the gene alterations using ChIP-X enrichment analysis (ChEA) gene set library (The brighter the color, the more significant the transcription factor).
(B and C) Plot and table indicating the –log10 p value (B) and terms odds ratio, and combined score (C) used by the Enrichr software to represent the output (scatterplot). p values are computed based on alterations in the number of genes that are known primary or secondary targets of transcription factors; Odds ratio indicates a measure of association between input and outcome; combined score: log(p) ∗z, p is p value, and z = deviation from the expected rank.
(D) Neutral lipid staining by Oil Red O in ChREBP overexpressing cells along with quantification (top); Images are taken at |×20×| magnification. Each image is a representative image of four different experiments with a minimum of eight different fields in each experiment. Statistical analysis was performed using two-tailed t-test (∗p < 0.05, vs Vector).
(E and F) Total cellular FFA (E) and cholesterol levels (F) upon ChREBP overexpression. Statistical analysis was performed using two-tailed t-test (∗p < 0.05, vs Vector).
(G) Protein levels of foam cell markers in ChREBP overexpressing cells. Densitometric analysis is shown in Figure S5A.
(H and I) Pull-down assay: HA-PHLPP1 and Flag-ChREBP interaction in HEK293T cells (H) and endogenous interaction (I) between PHLPP1 and ChREBP in RAW 264.7 cells;
(J) Malachite green assay: phosphate release upon the incubation of Flag-ChREBP with SFB-PHLPP1 in vitro. Statistical analysis was performed using two-tailed t-test (∗p < 0.05, vs Vector).
(K) AMPK Thr172 phosphorylation in ChREBP or PHLPP1 overexpressing RAW 264.7 cells. Densitometric analysis is shown in Figure S5B.
(L) Graph showing enriched ChREBP binding to FASN promoter upon PHLPP1 overexpression in ChIP-qPCR. Statistical analysis was performed using two-tailed t-test (∗p < 0.05 vs vector).
(M) Effect of AICAR and Act-3 on CD36 and FASN protein levels in PHLPP1 overexpressing cells. Densitometric analysis is shown in Figures S5C and S5D.
(N) Oil Red O quantification of neutral lipids in PHLPP1 and ChREBP over-expressed RAW 264.7 cells treated with AICAR and Act3. Images from three different experiments with a minimum of eight different fields in each experiment were used to quantify the data. Statistical analysis was performed using one-way ANOVA followed by Bonferroni′s post hoc analysis (∗p < 0.05 versus vector-untreated; δp < 0.05, δδp < 0.01 versus corresponding control-untreated; ζp < 0.05 versus corresponding sample OxLDL-treated). Data are a representative of three independent experiments. Numerical data are expressed as mean ± SEM. See also Figures S4 and S5.
Pharmacological inhibition of PHLPP1 reduced neutral lipid accumulation in high-fat diet-fed zebrafish larvae
BLAST analysis showed high global sequence identity (Similarity 73.7; Identity 62.8%) between zebrafish and human PHLPP1, indicating high conservation and the utility of zebrafish as an in vivo model for investigating PHLPP1 function. Neutral lipid staining of 13-dpf HFD-fed larvae (Figure 6A) using Nile Red revealed robust lipid accumulation in the head, trunk, tail, and intersegmental vessels (ISV) regions (Figures 6B–6D). HFD feeding of zebrafish larvae resulted in an increase in total cholesterol and FFAs in larval homogenates (Figures 6E and 6F). In addition, HFD-fed zebrafish larvae showed higher PHLPP1 protein levels (Figure 6G). We assessed the importance of PHLPP1 for lipid accumulation in HFD-fed zebrafish larvae. Exposure of HFD-fed larvae to 10 μM NSC117079, a commonly used small molecule PHLPP inhibitor (Jackson et al., 2013), resulted in a substantial reduction in neutral lipid accumulation in the head, trunk, and tail regions of zebrafish larvae (Figure 6H). Additionally, a marked reduction in the neutral lipid buildup of the ISV region of HFD-fed PHLPP1 inhibitor-treated larvae was evident (Figure 6I). Although NSC117079 treatment resulted in a modest decrease in total cholesterol levels, the triglyceride levels were strikingly reduced in inhibitor-treated larvae (Figure 6J).
Figure 6.

Pharmacological PHLPP1 inhibition attenuates lipid accumulation in HFD-fed zebrafish larvae
(A) Zebrafish larval experiment design;
(B–D) Fluorescent microscopic images of head, trunk, and tail (B) along with intersegmental blood vessels (ISVs) (C) with ImageJ quantification (D). Statistical analysis was performed using two-tailed t-test (∗p < 0.05 vs CD).
(E and F) Estimation of cholesterol (HPLC-based; (E) and FFA (F) levels in CD vs HFD-fed zebrafish larvae; each experimental data point was from 10 pooled larvae. Statistical analysis was performed using two-tailed t-test (∗p < 0.05, ∗∗∗p < 0.001 vs CD).
(G) Protein levels of indicated proteins in HFD fed zebrafish larvae.
(H and I) Fluorescent microscopic images of head, trunk, and tail (H) along with intersegmental blood vessels (ISVs; I) with ImageJ quantification (Right). Statistical analysis was performed using two-tailed t-test (∗p < 0.05 vs HFD).
(J) Kit-based estimations of whole-body triglyceride and cholesterol levels in NSC117079-treated zebrafish larvae. Each experimental data point was from 10 pooled larvae. Statistical analysis was performed using two-tailed t-test (∗p < 0.05 vs HFD). Each microscopic image in (B, C, H, and I) represents three to four experiments, each studying minimum five larvae; Magnification: Head, trunk, and tail - |×4×|; ISVs– 10X. Data are a representative of at least three independent experiments, unless specified. Numerical data are expressed as mean ± SEM.
PHLPP1 deficiency attenuates lipid accumulation in high-fat diet-fed zebrafish larvae and high cholesterol diet fed C. elegans
To probe the role of PHLPP1 in the zebrafish model of lipid accumulation using a genetic approach, a CRISPR-Cas9 strategy was employed to knockout PHLPP1 function in zebrafish (Figures S6A–S6E). Neutral lipid accumulation in head, trunk, and tail regions of HFD-fed PHLPP1-knockout larvae was lower than that in their control counterparts (Figure 7A). Furthermore, quantification of the ISV Nile red staining showed diminished lipid accumulation in knockout larvae (Figure 7B). Total cholesterol levels were also reduced in HFD-fed PHLPP1-knockout zebrafish larvae (Figure 7C). Phenotypic measurements revealed that body length was reduced in HFD-fed PHLPP1-knockout larvae but this difference was not statistically significant (Figures 7D and 7E). On the other hand, linear body width of the PHLPP1-knockout larvae was significantly lowered (Figures 7D and 7F). To test the evolutionary significance of PHLPP1 in lipid metabolism, we studied its impact on lipid accumulation in HCD-fed C. elegans. phlp-2 is the only conserved nematode gene in the PHLPP family with predicted serine/threonine phosphatase activity and is orthologous to human PHLPP1 (48% similarity with PHLPP1α) and PHLPP2 (44% similarity) (Shaye and Greenwald, 2011). Consistent with in vitro and zebrafish larval findings, inactivation of phlp-2 by RNAi (Figure S6F) resulted in a robust reduction in HCD-fed neutral lipid buildup (Figures 7G and 7H). Consistently, elevated triglyceride levels in control HCD-fed worms were normalized upon phlp-2 ablation (Figure 7I).
Figure 7.

CRISPR-Cas9-mediated PHLPP1 knockout in HFD-fed zebrafish larvae and siRNA silencing of phlp-2 in C. elegans diminishes lipid accumulation
(A and B) Fluorescent microscopic images of head, trunk, and tail (A) along with intersegmental blood vessels (ISVs; B) with quantification (A, Bottom panel and B, right panel). Each microscopic image represents three to four experiments with each experiment studying minimum of five larvae; Magnification: Head, trunk, and tail - |×4×|; Intersegmental blood vessels – 10X; Statistical analysis was performed using two-tailed t-test (∗p < 0.05 vs WT-HFD).
(C) Cholesterol levels in PHLPP1-knockout HFD-fed zebrafish larvae measured by HPLC-based method. Each experimental data point indicates 20 pooled larvae; Statistical analysis was performed using two-tailed t-test (∗p < 0.05 vs WT-HFD).
(D–F) Pictorial depiction along with linear body width (E) and whole-body length (F) measurement in PHLPP1-knockout zebrafish larvae; Statistical analysis was performed using two-tailed t-test (∗p < 0.05 vs WT-HFD).
(G and H) Microscopic images (G) and quantification (H) of Oil Red O staining in cholesterol-fed phlp-2 knockdown C. elegans; data presented are from a representative experiment of two independent trials (n = 7–12 per trial); Statistical analysis was performed using one-way ANOVA followed by Bonferroni′s post hoc analysis (∗∗∗p < 0.001 vs N2/control+EtOH; δδδp < 0.001 vs N2/control+cholesterol).
(I) TLC analysis of triglyceride levels in cholesterol-fed phlp-2 knockdown C. elegans along with ImageJ quantification (Bottom). Statistical analysis was performed using one-way ANOVA followed by Bonferroni′s post hoc analysis (∗p < 0.05 vs N2/Ctrl; $p < 0.05, $$p < 0.01 vs N2/Ctrl+chol). Data are a representative of at least three independent experiments, unless specified. Numerical data are expressed as mean ± SEM. See also Figure S6.
Discussion
Atherosclerosis is a progressive inflammatory disease of large arteries. It is clinically manifested as CVDs such as myocardial infarction, unstable angina, stroke, and sudden cardiac death which collectively constitute the leading cause of death worldwide and low to middle-income groups of countries account for more than 75% of CVD deaths. The sequence of events in the pathogenesis of atherosclerosis are: accumulation of lipoprotein particles and their aggregates in the arterial intima, migration of monocytes into intima where they differentiate into macrophages and endocytose modified or aggregated lipoproteins (oxidized LDL, acetylated LDL) to develop into foam cells. Lipid-loaded macrophages undergo apoptosis and progress to secondary necrosis. When dead cells are not effectively cleared by efferocytosis, necrotic core harboring lipid deposits and dead cells is formed. Vascular smooth muscle cells migrate into the necrotic core where they proliferate and secrete fibrous elements of the extracellular matrix forming fibrous plaques. The lesions grow outward thereby obstructing the arterial lumen. Importantly, in advanced lesions, the fibrous plaque is destabilized by matrix modifying enzymes thereby exposing prothrombogenic factors present in the necrotic lipid core to circulating platelets causing clotting and subsequent myocardial infarction or stroke. Foam cells are formed owing to the imbalance in the cholesterol handling pathways of macrophages. For example, proteins involved in lipid transport (adipophilin) and lipid storage (perilipin) are increased in human atherosclerotic lesion (Nuotio et al., 2007). Although the expression of CD36 is enhanced, the protein levels of ABCA1 were drastically reduced in human atheromas (Isoviita et al., 2010). Consistently, knockout of CD36 protected against atherosclerotic lesion development in atherogenic ApoE null mice (Febbraio et al., 2000). Double knockout mice lacking both ABCG1 (ATP binding cassette subfamily member G1) and ABCA1 (ATP binding cassette transporter subfamily A1) in macrophages showed heightened inflammatory responses and accelerated atherosclerotic lesions (Yvan-Charvet et al., 2008).
Gaining a comprehensive understanding of the foam cell development is crucial for devising newer therapeutic strategies against atherosclerotic manifestations. The role of PHLPP1 in metabolic syndrome and particularly in atherosclerosis is speculated but remains largely unexplored (Mathur et al., 2017). The importance of PHLPP1 in cardio-protection is beginning to be understood. PHLPP1 knockdown has resulted in enhanced protective effects in nontransformed cardiomyocytes that are attributed to increased Akt activity (Miyamoto et al., 2010). Several studies have emphasized the importance of Akt signaling in cardioprotective effects, potentiated via PHLPP1 knockdown/deletion. PHLPP1/2 knockdown-mediated protection against Ischemia/Reperfusion in leukemia inhibitory factor-induced cardiomyocytes and protection against hypertrophy in PHLPP1 knockout mice points to a significant role of PHLPP1/2 in cardio-protection (Miyamoto et al., 2010; Moc et al., 2015).
Following up on our and other studies (Alamuru et al., 2014; Behera et al., 2018; Katsenelson et al., 2019; Lupse et al., 2021), in the current study, we explored the importance of PHLPP1 in the development of foam cells. We observed that lipid accumulation in foam cells in vitro was substantially reduced upon the depletion of PHLPP1 in RAW 264.7 cells. Consistent with the above observation, PHLPP1-knockout zebrafish larvae displayed lower lipid accumulation in intersegmental vessels, emphasizing the crucial role of PHLPP1 in lipid overload which is likely to be evolutionarily conserved. Preliminary investigations showed no role for PHLPP2 isoform in the accumulation of neutral lipids in macrophages. However, it was recently shown that adenoviral-mediated overexpression of PHLPP2 in mice resulted in the decline of hepatic triglyceride content and plasma triglyceride levels along with the reduction of Fasn and Srebp1c gene expression (Kim et al., 2016). In addition, Huang et al. uncovered the role of PHLPP2 in vascular remodeling, a vital concept in atherosclerotic lesion formation (Huang et al., 2020). Huang et al. observed that PHLPP2 levels were elevated in the human atherosclerotic plaques and blood cells isolated from patients with coronary artery disease. Endothelial-specific deficiency of PHLPP2 reduced neointima formation in carotid artery ligated mice. Furthermore, the hepatic levels of PHLPP2, but not PHLPP1, were reduced in aging and obese mice (Kim et al., 2016). On the other hand, proteins levels of PHLPP1 were elevated in the skeletal muscle and adipose tissue of obese subjects (Andreozzi et al., 2011; Cozzone et al., 2008), skeletal muscle of high sucrose diet or HFD-fed mice (Behera et al., 2018) and OxLDL/7-ketocholesterol-treated mouse macrophages and HFD-fed zebrafish larvae in the current study. The observed differences in the functions of PHLPP1 and PHLPP2 may be owing to context-dependent, tissue/cell-type-specific roles of PHLPP isoforms. In the same lines, while we recently showed that PHLPP1 interacts with and dephosphorylates AMPK in muscle cells and soleus muscle of HFD-fed mice (Behera et al., 2018), Yan et al. reported that PHLPP2, but not PHLPP1, dephosphorylates AMPK in metabolically stressed Jurkat cells (Yan et al., 2021).
The expression of PHLPP1 was enhanced during the early stages of foam cell formation but the levels declined at later time points (Figures 1A–1E). Decreased PHLPP1 levels at later stages of OxLDL treatment may amplify the inflammatory program of foam cells. An increase of PHLPP1 protein during the initial period (until 24 h) followed by a decrease in in vitro foam cells may indicate that the protein has a role in the early establishment of foam cell formation. PHLPP1 levels may have been reduced at the later time points as the lipid accumulation in foam cells is likely saturated. Such a reduction in certain lipid metabolism genes was also noticed in advanced human atherosclerotic plaques (Sulkava et al., 2017). In a study by Sulkava et al., atherosclerotic vascular samples from 29 patients subjected to carotid endarterectomy owing to occlusive atherosclerosis were compared with control samples from the left internal thoracic artery (LITA) from 28 patients who underwent coronary artery bypass surgery. Along with changes in several genes such as spp1, ApoE, MMP1, CIDEA, and ACTA1, RNA-seq analysis showed a 50% decrease in PHLPP1 and PHLPP2 transcript levels in human atherosclerotic plaques (Sulkava et al., 2017) (Figure S1E). Lipid accumulation in foam cells demands the increased expression of several lipid-metabolizing and lipid import/export-associated genes resulting in the unfolded protein response which in turn stimulates ER stress (Hotamisligil, 2010). As observed in mouse and rat muscle cells, induction of ER stress via sarco/ER Ca2⁺ ATPase inhibition by thapsigargin led to the rhythmic expression of PHLPP1 in RAW 264.7 cells (Behera et al., 2018). Both ChREBP and PHLPP1 showed parallel induction upon 7-ketocholesterol treatment, prompting us to examine the possibility of ChREBP as an upstream regulator of PHLPP1. The recruitment of ChREBP to the Phlpp1 promoter and OxLDL/7-ketocholesterol induced increase in PHLPP1 levels in ChREBP-dependent manner offers a mechanistic basis for PHLPP1 upregulation in in vitro foam cells.
Transcriptomics analysis provided the molecular basis for reduced neutral lipid accumulation in PHLPP1-ablated foam cells. Gene expression programs driving lipid metabolism, cholesterol biosynthesis and, expectedly inflammation, were perturbed in PHLPP1-deficient macrophages. Among them, CD36 is worthy of mention. Hyperlipidemic mice showed attenuated lesion progression owing to CD36 loss. Also, CD36 binds TLR2-TLR4 commencing the most important TLR4 inflammasome pathway that fuels overall plaque progression (Chávez-Sánchez et al., 2014). PHLPP1 augments CD36 expression and is required for CD36-mediated lipid uptake. Based on the role of ER stress in CD36 regulation and our findings from our current and previous studies, we speculate that PHLPP1 may be one of the mediators of ER stress-induced CD36-dependent enhanced lipid uptake (Yao et al., 2014). On the other hand, despite a strong increase in ABCG1 protein levels upon PHLPP1 knockdown, no differential cholesterol efflux was observed upon the perturbation of PHLPP1 levels (Figures 4C and 4D). A possible explanation is that the reduction in ABCG1 function might be compromised by ABCA1-mediated efflux, because ABCA1 transcript levels and protein levels remained unchanged upon PHLPP1 knockdown (Figure 4E). Besides, the kit measures BODIPY-labeled cholesterol efflux which is primarily mediated via the ABCA1 transporter (Takata et al., 2019). Passive diffusion-mediated lipid efflux also cannot be dismissed.
Transcription factor gene set enrichment analysis using Enrichr identified ChREBP as a target of PHLPP1 (Figures 5A–5C and S4B). Sarrazy et al. have reported an interesting finding that despite its role in increasing lipid-metabolizing gene expression, ChREBP functions to prevent inflammatory responses and aids in the maintenance of macrophage redox status thereby protecting against atherosclerosis development (Sarrazy et al., 2015). The contribution of ChREBP to in vitro foam cell development is not understood. We observed that forced expression of ChREBP robustly increased neutral lipid accumulation and strongly elevated FFA levels. ChREBP being a potent glucose sensor and a vital transcription factor of various lipid-metabolizing genes is very intricately regulated by various kinases and phosphatases. AMPK-mediated phosphorylation at Ser568 in ChREBP is one such regulation that inhibits the protein′s DNA-binding capacity thereby hindering its transcriptional ability (Liangpunsakul et al., 2013). Also, phosphorylation at Ser196 of ChREBP, a potential PHLPP1 dephosphorylation site, determines its cellular localization (Sakiyama et al., 2008).
Our mechanistic analysis showed that PHLPP1 interacts with ChREBP and modestly enhances its occupancy at Fasn promoter. Furthermore, it dephosphorylated ChREBP in vitro in malachite green-based phosphate release assays, suggesting that ChREBP may be a direct substrate of PHLPP1. However, owing to the unavailability of phospho-Ser196-specific commercial antibodies, we could not definitively show that ChREBP is a genuine substrate of PHLPP1. Furthermore, consistent with our previous findings in myoblasts (Behera et al., 2018), we showed that (i) PHLPP1 inactivates AMPK in macrophages as well and (ii) the activation of AMPK prevents PHLPP1-promoted neutral lipid accumulation. Based on our current and past findings (Behera et al., 2018), we speculate that PHLPP1, ChREBP, and AMPK may be a part of an integrated signaling network that regulates lipid metabolism. We propose the following model (Figure 8): ChREBP is recruited to Phlpp1 promoter in foam cells leading to its transcriptional upregulation. PHLPP1 may subsequently control lipid metabolism through AMPK and ChREBP-mediated mechanisms. First, PHLPP1 directly inactivates AMPK and may indirectly enhance the occupancy of ChREBP on its target promoters. Second, PHLPP1 likely dephosphorylates ChREBP thereby augmenting its nuclear localization and chromatin occupancy. However, the second mechanism needs further validation. Although some of the effects of PHLPP1 maybe mediated through both ChREBP and AMPK (FASN), certain pro-lipogenic effects of PHLPP1 (CD36) are possibly achieved through ChREBP-independent but AMPK-dependent mechanisms. An increase in neutral lipid accumulation upon ChREBP overexpression despite AMPK activation by AICAR or Act-3 intrigued us (Figure 5N). We previously reported that the complex formation of ectopically expressed PHLPP1α with AMPK was disrupted upon AMPK activation in HEK 293T cells (Behera et al., 2018). Therefore, the increase in lipid accumulation upon ChREBP overexpression independent of AMPK activation status suggests the existence of various other lipid metabolism control pathways that ChREBP/PHLPP1 is a part of.
Figure 8.

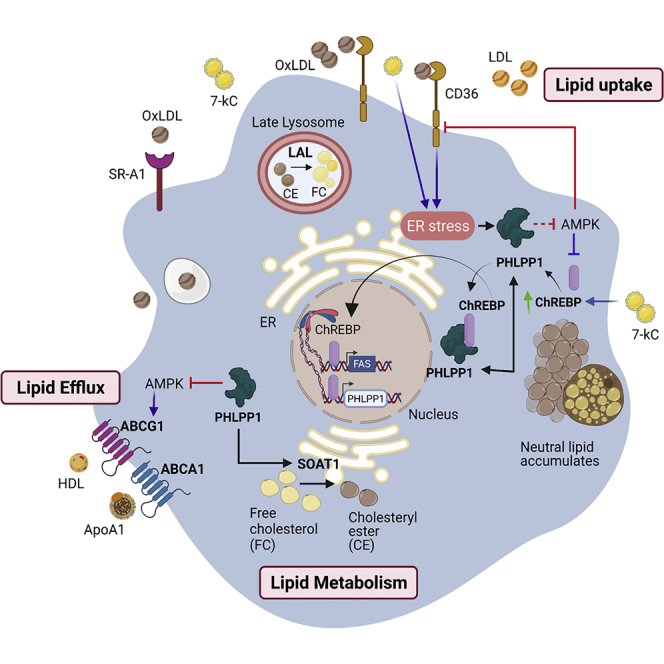
Graphical abstract of PHLPP1-mediated foam cell formation
PHLPP1 protein levels are elevated in OxLDL and ER stress-exposed macrophages and HFD-fed zebrafish larvae. PHLPP1 augmented cellular cholesterol and FFAs levels through the regulation of key lipogenic genes such as FASN, CD36, and so forth. Transcriptomic analysis revealed that loss of PHLPP1 suppresses gene expression associated with fatty acid synthesis and cholesterol metabolism. PHLPP1 promoted OxLDL-uptake in AMPK/CD36-dependent manner. Also, 7-kC treatment mediates the augmentation of ChREBP levels. Furthermore, PHLPP1 interacted with and dephosphorylated lipogenic transcription factor ChREBP and enhanced the levels of its target, FASN. Data from other studies suggest that AMPK assists in lipid efflux via upregulating ABCG1 levels. Thus, PHLPP1 mediates foam cell formation via ChREBP/AMPK axis (Green arrow indicates protein increase; purple arrow indicates data from other studies).
Taken together, our results imply that PHLPP1 promotes neutral lipid accumulation and total cholesterol levels in in vitro foam cells, HFD-diet-fed zebrafish larvae, and HCD-fed C. elegans by augmenting the expression levels of key genes involved in lipid uptake, cholesterol biosynthesis, and lipid metabolism. Collectively, our data position PHLPP1 as an important player in the pathophysiology of atherosclerosis (Figure 8).
Limitations of the study
We observed a significant reduction of neutral lipid accumulation in cultured foam cells upon PHLPP1 knockdown. Consistent with our in vitro data, knockout of PHLPP1 gene in zebrafish larvae and siRNA-mediated phlp-2 (PHLPP ortholog) gene silencing in C. elegans alleviated whole-body lipid accumulation upon HFD and HCD feeding, respectively. Despite foam cells being a hallmark of early atherosclerosis and PHLPP1 having a robust influence on foam cell formation, additional studies are required to unambiguously establish the function of PHLPP1 in the pathology of atherosclerosis. Although the functionality of PHLPP1 in the lipid accumulation of zebrafish larval blood vessels is clear, its impact on the formation of atherosclerotic plaque, if any, could not be studied in the HFD-fed zebrafish larval model. Furthermore, the current study falls short of unambiguously establishing ChREBP as a direct substrate of PHLPP1 owing to the unavailability of commercial phospho-specific antibody. In vitro phosphatase assays with purified PHLPP1 and ChREBP proteins followed by immunoblotting with pSer196 ChREBP antibody may further validate our findings. Foam cell formation is a multi-regulated process. Although our research addresses the effect of PHLPP1 on ChREBP/AMPK controlled lipid metabolism, PHLPP1-mediated regulation of other foam cell mediators is unclear at this stage. Based on our findings, we propose that PHLPP1 may play a role in the initial stages of atherosclerosis and further studies will likely address the above research gaps in the future.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal Anti-ABCA1 | Novus Biologicals | NB400-105 |
| Rabbit polyclonal Anti-ABCG1 | Novus Biologicals | NB400-132 |
| Rabbit polyclonal Anti-ChREBP | Novus Biologicals | NB400-135 |
| Rabbit polyclonal Anti-PHLPP1 | Merck Millipore | 07-1341 |
| Mouse monoclonal Anti-FLAG M2 | Sigma Aldrich | F3165 |
| Mouse monoclonal Anti-CD36 | Santa Cruz Biotechnology | sc-7309 |
| Rabbit monoclonal Anti-CD36 | Cell Signaling technologies | 14347 |
| Mouse monoclonal Anti-FASN | Santa Cruz | sc-48357 |
| Mouse monoclonal Anti-DDIT3/CHOP/GADD153 | Santa Cruz | sc-7351 |
| Rabbit polyclonal Anti-p-PERK (Thr981) | Santa Cruz | sc32577 |
| Mouse monoclonal Anti-PERK | Santa Cruz | sc-377400 |
| Mouse monoclonal Anti-ATF-6α | Santa Cruz | sc-166659 |
| Mouse monoclonal Anti-TRB3 | Santa Cruz | sc-365842 |
| Mouse monoclonal Anti-ICAM-1/CD54 | Santa Cruz | sc-8439 |
| Rabbit monoclonal Anti-GAPDH | Cell Signaling technologies | 2118 |
| Rabbit polyclonal Anti-β-Actin | Cell Signaling technologies | 4967 |
| Rabbit monoclonal Anti-p-AMPKα (Thr172) | Cell Signaling technologies | 2535 |
| Rabbit Anti-AMPKα | Cell Signaling technologies | 2532 |
| Goat Anti-Rabbit IgG (H + L)-HRP Conjugate | Bio-Rad | 170-6515 |
| Goat Anti-Mouse IgG (H + L)-HRP Conjugate | Bio-rad | 170-6516 |
| Chemicals, peptides and recombinant proteins | ||
| Polyethylenimine “Max” (PEI), linear, MW: 40,000 | Polysciences | 247G5-1 |
| Human plasma lipoprotein, low density | Sigma Aldrich | L7914 |
| 7-keto cholesterol | Cayman ChemicalCayman Chemical | Cay16339-5 |
| AICAR | Cayman Chemical | Cay10010241-50 |
| Activator-3 | generous gift from Dr. Parimal Misra; (Bung et al., 2018) | N/A |
| RNAiso Plus | Takara Bio | 9109 |
| Oil Red O | Sigma Aldrich | O0625 |
| Nile Red | Sigma Aldrich | 72485 |
| 40,6-diamidino-2-phenylindole dihydrochloride (DAPI) | Invitrogen | Cat# D1306; RRID: AB_2629482 |
| PrimeScript IV 1st strand cDNA Synthesis Mix | Takara Bio | RR037A |
| TB Green® Premix Ex Taq™ II (Tli RNase H Plus) | Takara Bio | RR820A |
| Critical commercial assays | ||
| OxLDL uptake assay kit | Cayman Chemical | 601180 |
| Cholesterol efflux assay kit | Abcam | ab196985 |
| Total cholesterol estimation kit | Erba | BLT00034; CHOL 5 × 50 |
| Free fatty acid estimation kit | Sigma Aldrich | MAK044 |
| Serum Triglyceride Assay kit | My Biosource | MBS168769 |
| Experimental models: Cell lines | ||
| RAW 264.7 | ATCC | RAW 264.7 (ATCC® TIB-71™) |
| HEK 293T | ATCC | 293T (ATCC® CRL-3216™) |
| DMEM | Lonza | BE12-604F |
| Fetal bovine serum | Himedia | RM9955 |
| Recombinant DNA | ||
| SFB-tagged human PHLPP1α | Generous gift from Dr. Maddika Subba Reddy; (Gangula and Maddika, 2013) | N/A |
| pcDNA3-HA-PHLPP1 | Addgene | Plasmid # 22404 |
| pcDNAe-HA-PHLPP2 | Addgene | Plasmid # 22403 |
| pCMV4-Flag-ChREBP | Generous gift from Dr. Howard Towle; (Stoeckman et al., 2004) | N/A |
| pLKO.1-shPHLPP1 | Sigma Aldrich | Clone ID: TRCN0000238470 |
| pSHS207 | Addgene | Plasmid # 101199 |
| pLKO.1-shChREBP | Sigma Aldrich | Clone ID: TRCN0000082206 |
| HA-PHLPP1β | Generous gift from Dr. Alexandra Newton; (Warfel et al., 2011) | N/A |
| Software and Algorithms | ||
| Fiji/ImageJ | NIH | RRID: SCR_002285 |
| GraphPad Prism 6 | GraphPad Software | RRID: SCR_002798 |
| Microsoft Excel | Microsoft | RRID: SCR_016137 |
| Biorender | Web tool for graphical illustrations | RRID: SCR_018361 |
| Enrichr | Gene list enrichment analysis tool | RRID: SCR_001575 |
| Deposited data | ||
| RNA-seq data | GEO | GSE193132 |
Resource availability
Lead contact
Requests and information regarding resources and reagents should be directed to the Lead Contact, Kishore V. L. Parsa. E-mail: kishorep@drils.org
Materials availability
This study generated sgRNAs targeting the zebrafish PHLPP1 gene. The sgRNA sequences have been reported in the STAR Methods, and any remaining aliquots are available and can be shipped. The study also generated whole body PHLPP1-knockout zebrafish, which are available for laboratories within India subject to fulfilment of Animal Ethics requirements; the knockout zebrafish cannot be shipped outside India currently because we do not yet have the required permits for animal export/import. This study did not generate any new plasmids.
Experimental model and subject details
Animal ethics guidelines were followed for all zebrafish experiments according to Institutional Animal Ethics Protocols of Dr. Reddy’s Institute of Life Sciences, Hyderabad, India (DRILS-IAEC-KP-2019-1) framed in compliance with the animal ethics laws of India and the US NIH guidelines for zebrafish experimentation. The Institutional Bio-Safety Committee (Protocol No. DRILS/IBSC/2017/30) approved general bio-safety procedures for the conduct of experiments. Zebrafish husbandry was done based on procedures described in the Zebrafish book (Westerfield, 2007). In short, wild caught zebrafish were housed and bred in-house. Maintenance of zebrafish were carried out in recirculating reverse osmosis water (with 0.2% sea salt added) at 28 ± 1°C with a 12 h light/dark cycle in a Tecniplast ActiveBlue™ standalone rack. Adults were fed three times a day with dry food or live brine shrimp. Breeding was carried out at a ratio of 3 Males: 2 females per breeding chamber and the embryos collected and washed. Embryos were maintained at 28°C in embryo medium (E3, pH ∼ 7.4). Larvae were fed twice a day beginning 5dpf using a co-culture of microalgae and rotifers. They were moved to the recirculation system when they were 3–4 weeks of age. The larvae used for experiments were of the same developmental stage in every experimental group. For generation of an atherosclerotic zebrafish larval model, wild type zebrafish or PHLPP1-knockout zebrafish larvae were obtained by crossing appropriately aged males and females. Embryos were collected and kept in E3 media for 5 days at 28°C.
For the atherosclerotic zebrafish larval model, the larvae were fed twice a day with 0.2 g dry food (Control) or 0.2 g boiled antibiotic-free egg yolk (HFD) per 100 larvae for 8 days beginning at 5 dpf.
All zebrafish larval experiments were conducted with 13–20 dpf zebrafish larvae, and gender determination is not possible at this developmental stage.
Generation of CRISPR-Cas9-mediated PHLPP1 knockout zebrafish
CRISPR single-guide RNAs (sgRNAs) that specifically target the zebrafish PHLPP1 gene were designed using Benchling software and the most recent zebrafish genome release (GRCz11):
guide1: GGGACGTATAACGTGCGGAAtaatacgactcactataGGGACGTATAACGTGCGGAAgttttagagctagaa;
guide2: GTGCATCTTCCCCGTATGGCTGGtaatacgactcactatagGTGCATCTTCCCCGTATGGCgttttagagctagaa.
Adult zebrafish were mated and embryos collected and washed; the CRISPR gRNAs along with purified Cas9 protein was microinjected into apparently viable embryos within 30 min of hatching, and the embryos incubated at 28°C. After 24h, the presence of indels in the PHLPP1 gene was assessed using heteroduplex mobility assay (HMA) in randomly selected microinjected embryos. Embryos were lysed in lysis buffer containing Tris buffer pH 7.0 and EDTA pH 8. The crude extract was used as template to amplify the CRISPR-targeted region of the PHLPP1 gene (Espino-Saldaña et al., 2020; Sorlien et al., 2018). Indels were visualized using the heteroduplex mobility assay (HMA) on 10% native PAGE. Differential amplicon migration compared to wild type control indicated indels. Following the confirmation of indel presence in at least 50% of the sampled embryos, the remaining embryos were allowed to grow for 2 months. To identify potential F0 founders with indels in PHLPP1, adult fish were anesthetized using Tricaine and the tips of their tails were clipped. The clipped tails were lysed and subject to HMA as described above. Fish showing indels were considered potential founders and were mated with wild type fish to yield potential F1 heterozygote embryos. Following HMA of a random set of embryos, PCR amplicons from representative embryos were sequenced, and the sequence data was analyzed using the Synthego ICE web-based tool yielding an indel prediction score of 40, confirming germline transmission and heterozygote production. The remaining embryos were grown into adults and tail clip HMA analysis was performed to verify and select heterozygotes. Of these, one male and one female were mated, and a random selection of the produced embryos were analyzed via mixing HMA to verify homozygote production and Mendelian inheritance. All the homozygous embryos obtained were grown into adults after HMA confirmation and representative sequencing. The PHLPP1-KO homozygous adult zebrafish were then maintained and bred for further experiments.
Generation of siRNA-mediated PHLPP1 knockdown in obese model of C. elegans
Synchronized (using sodium hypochlorite procedure) L1 stage N2 strains were grown on nematode growth medium (containing 1mM IPTG and 50 μg/mL ampicillin) seeded with HT115 bacterial strain containing either empty L4440 plasmid or phlp-2 RNAi construct. For high fat diet conditions, cholesterol was added during plates preparation at 25mM final concentration at NGM levels. All experiments were conducted with hermaphrodite worms.
Method details
Cell stimulation studies, transfection and nucleofection
For in vitro foam cell formation, RAW 264.7 cells were stimulated with OxLDL generated by CuSO4 based oxidation of human plasma lipoprotein, low density (LDL). 5 mg/mL of LDL was incubated with 10 μL of 0.01 M CuSO4 at 37°C in the dark and open to room air for 24 h followed by dialysis overnight against a 0.15 M NaCl solution. For oxidation confirmation, thiobarbituric acid-reactive substances (TBARS) were measured in randomly selected vials. For TBARS measurement, a homemade solution containing 15% thiobarbituric acid (TBA)-37.5% trichloroacetic acid (TCA) and 0.25 N HCl was made. The solution was added to blank (water), control (LDL) and test (OxLDL) samples and the samples were heated for 15 min in boiling water. The samples were then allowed to cool and centrifuged for 10 min at 1,000 g and supernatants collected. Absorbance of the supernatant was measured at a wavelength of 535 nm in a spectrophotometer. Concentrations of LDL and OxLDL were calculated using Beer’s law (Fc = FA/Fa, where Fc is the concentration, FA is the absorbency and Fa is the molar absolutivity; Fa (LDL) = 1.56 × 105). Level of oxidation was calculated using the formula FL = F1/F2 (F1- [OxLDL following 24 h of exposure to CuSO4/air]; F2- [concentration of the LDL prior to exposure to high oxidizing conditions]). Level of oxidation was calculated to be 9.7. Where indicated, cells were treated with 50–100 μg of OxLDL per mL media for 16–48 h. Macrophage cells were also stimulated with 20μM of 7-keto cholesterol for different time points as indicated. For transient HEK 293T transfection, Polyethylenimine “Max” (PEI) was used according to the manufacturer's protocol. Briefly, 3 million cells were transfected with 4μg DNA at a DNA: PEI ratio of 1:5. Transfection mix was prepared in serum-free media and the cells were incubated in serum free media for 5 h. The cells were used for further experiments 24 h post-transfection. For RAW 264.7 cells, nucleofections were performed with home-made buffer comprised of 15mM MgCl2, 5mM KCl, ,120 mM Na2HPO4/NaH2PO4 pH 7.2 supplemented with mannitol 50 mM (Alamuru-Yellapragada et al., 2017b). Briefly, 10 million cells were nucleofected with 12μg of DNA and the cells were harvested 24 h post-transfection for over-expression and 48 h post-transfection for knockdown studies. For stable PHLPP1 knockdown cell generation, RAW 264.7 cells were nucleofected with pLKO.1-shGFP or pLKO.1-shPHLPP1 vector and selection was done using 2μg of puromycin per mL media. For AICAR and Activator-3 experiments, RAW 264.7 cells were treated with 10μM of AICAR or 100nM of Activator-3 for 3 h along with OxLDL or 7-ketocholesterol stimulation.
Cell lysis and western blotting
HEK 293T cells were lysed using TENNS lysis buffer [120 mM NaCl, 1% Triton X-100, 20 mM Tris–HCl pH 7.5, 10% glycerol, 2 mM EDTA and protease inhibitor cocktail (10 μg/mL each aprotinin and leupeptin)] whereas RAW 264.7 cells were lysed in TN1 lysis buffer [50 mM Tris [pH 8.0], 10 mM EDTA, 10 mM Na2P2O7, 10 mM NaF, 1% Triton-X 100, 125 mM NaCl, 10 mM Na3VO4 and protease cocktail inhibitor]. Approximately 50μg of protein was resolved by 10% SDS-PAGE and western blot analysis was carried out using specified antibodies. The protein levels were normalized using GAPDH or β-Actin.
RNA sequencing, data analysis
RNA quality and quantity were assessed using Bioanalyzer 2100 with RNA 6000 Nano Labchips (Agilent Technologies). Samples with RNA integrity number (RIN) higher than 8.0 were used for library preparation using NEBNext Ultra RNA library Prep kit (NEB #E7770S). A total of 300 ng RNA was used for library preparation. The RNA was enzymatically fragmented and then converted to cDNA per the kit protocol (NEB #E7770S). Next, end repair and 3′ end-adenylation of the fragments was performed and bar-coded adapters were ligated to the cDNA fragments. 12 cycles of PCR were performed to produce the sequencing libraries. The PCR products were purified using AmPure XP beads. Library quality and quantity were measured using Agilent 2100 Bioanalyzer and qPCR with KAPA Library Quantification kit (KAPA Biosystems, Foster City, USA), respectively. Adapter-ligated cDNA fragment libraries were run on Illumina HiSeq. 2500 at a 150-nucleotide read length using the paired-end chemistry. The RAW reads (FASTQ) were subjected to contamination [structural RNA/low complexity sequences, adapters] removal by mapping with bowtie 2-2.2.1. The dataset after contamination removal was mapped to the Mus musculus GRCm38 using STAR. Read mapping to genes in the Mus musculus GRCm38 gene list [GTF] were counted using feature count module of sub reads package and were normalized in DESeq2-3.5 followed by differential expression analysis. Significantly up- and down-regulated genes (differentially expressed) were selected based on 2-fold change with p value < 0.05 (FDR: 0.05; Student′s t-test, unpaired). Hierarchical clustering was performed with the programs Cluster (uncentered correlation; average linkage clustering) and Treeview (Eisen et al., 1998). GO annotation was carried out using the Gorillaweb server [http://cbl-gorilla.cs.technion.ac.il]. Term enrichments of differentially regulated genes were calculated based on mouse genome database as background. GO terms with corrected p < 0.05 were considered to be significantly enriched for pathway analysis.
For ‘Enrichr’ analysis, differentially altered genes (criteria: cut-off: Fold Change >1.5 and FDR: 0.05) were submitted to ‘Enrichr’ tool. The tool used KEGG pathway 2021 human pathway to analyze the possible disease conditions in all the sets and the possible transcription factors involved in the gene alterations using ChIP-X enrichment analysis (ChEA) gene set library. The disease pathways and enriched transcription factors are calculated based on the following terms: 1) –log10 p value that are computed based on alterations in the number of genes that are known primary or secondary targets of transcription factors; 2) odds ratio that indicate measure of association between input and outcome and 3) combined score that signifies deviation from the expected rank and is calculated by log(p) ∗ z where p is p value, and z = deviation.
Immunoprecipitation and ChIP studies
In immunoprecipitation experiments, precleared 500 μg cell lysates were incubated either with Flag beads (Sigma Aldrich) or PHLPP1 antibody (4 μg) for 2 h or overnight respectively at 4°C. Post incubation, beads were washed with lysis buffer followed by boiling with Laemmli Buffer. The enriched samples were separated and probed with indicated antibodies. RAW 264.7 cells grown in 100-mm dishes in DMEM supplemented with 10% (v/v) FBS or RAW 264.7 cells over-expressing PHLPP1 post 16 h of nucleofection were obtained for ChIP experiments once they attained 80% confluency. Cells were fixed with 1% (w/v) formaldehyde in DMEM for 10 min at room temperature, followed by one wash using ice-cold PBS. The fixation reaction was stopped with the addition of glycine solution for 15 min. Cells were then scraped from flasks with 10 mL ice cold PBS and centrifuged for 5 min at 1,000 rpm. Cells were then lysed using SDS ChIP lysis buffer and the chromatin obtained was sheared by sonication at an amplitude of 45% with a 10-s on/off pulse for 2 min in ice. This optimized sonication conditions resulted in 200-1000-bp DNA fragments. Post lysis, 250 μL of the isolated chromatin was diluted three times using ChIP dilution buffer and incubated with 3 μg of ChREBP antibody or IgG antibody at 4°C overnight. ChREBP antibody and IgG rabbit polyclonal antibody was obtained from Novus biologicals and Merck respectively for ChIP studies. Region akin to ChoRE site present in -488/-473 region of the PHLPP1 promoter were amplified using the following primers
Forward: 5′CTACAACCCCCACTGTGCAA3′
Reverse: 5′GCATGACGACATTTCTGCGG3′
A product of 167-bp size was amplified at an annealing temperature of 58°C and visualized on a 1% agarose gel.
The ChoRE region present in -349/-333 region of the FASN promoter was amplified using the following primers
Forward: 5′ GCAGAGTTTCCAGTGTGACC3′
Reverse: 5′ CTGGAGCACAAGGAACGC3′
For ChIP-qPCR, samples were normalized by using equal volume. A measure of ChREBP recruitment to FASN promoter region was calculated using fold-enrichment method. Ct values were considered as ChIP signals and the fold increase in signal was calculated relative to the IgG antibody background signal.
Real-time PCR
Total RNA was isolated using Trizol and reverse transcription was performed with 2–4 μg total RNA using random hexamers and oligo dTs in a reaction volume of 20 μL using the Superscript II First Strand cDNA Synthesis System. qPCR reactions were performed using TB Green mix on QuantStudio5 (Applied Biosystems). Data were normalized to β-actin using the Ct method (ΔΔCt). Primer sequences are available upon request. The cDNAs from respective samples were diluted 1:5 for the genes PHLPP, CD36, Fasn, ChREBP, LDLR, etc., and 1:50 for reference genes (β-Actin, GAPDH). The primers were used at a 200 nM concentration in a 20 μL reaction. Relative gene expression was plotted using the control sample set as 1. The statistical significance was calculated on non-normalized and non-transformed ΔCt values.
Colorimetric and fluorescent staining studies
Oil red O staining of RAW 264.7 cells
Oil Red O (0.5% (w/v)) was dissolved in isopropanol at 56°C overnight. Pre-warmed stock was diluted at a 3:2 ratio with ultrapure water, filtered and used within an hour. RAW 264.7 cells were fixed in 10% phosphate buffered formalin for 10 min. Post rinsing the cells with 1X phosphate buffered saline (PBS) for 1 min, 60% isopropanol was added to cells for 15 s to facilitate neutral lipid staining. The cells were stained with Oil Red O for 1 min at 37°C in darkness. The cells were then de-stained with 60% isopropanol for 15 s and washed with 1X PBS thrice for 3 min each. Images were captured using a brightfield microscope (Zeiss primostar, 415500-1501-000). The images were converted to RGB stack in ImageJ software and the threshold was adjusted so that the red droplets were visible as black dots on a white background. The area of the red lipid droplets was measured using ImageJ software.
Oil red O staining of C. elegans
Day 1 adult animals were washed from the plates and centrifuged at 1,200 rpm for 30 s. After washing twice with PBS, animals were suspended in 200 μL of 1× PBS. To this an equal volume of MRWB (160 mM KCl, 40 mM NaCl, 14 mM Na2EGTA, 1 mM Spermidine HCl, 0.4 mM Spermine, 30 mM Na PIPES at pH 7.4, 0.2% BME) containing 2% paraformaldehyde was added and incubated for 15 min at room temperature. Worms were centrifuged at 12000rpm for 1 min and pellet was washed once with 1× PBS. An 60% of 5 mg/mL Oil-Red-O solution is added to the worm pellet and incubated with continuous rocking. After an overnight incubation, worms were allowed to settle and dye was removed by washing twice with 1× PBS containing 0.01% Triton X-100. Worms were mounted on glass slide and imaged with Olympus CKX53. Oil-Red-O dye intensity in stained worms was quantified using ImageJ (NIH) software and average integrated density values are plotted.
Nile red staining of zebrafish larvae
Control and HFD fed larvae were euthanized at 4°C for 30 min and fixed with 4% formaldehyde overnight and stained with Nile Red at 0.25 mg/mL concentration for 30 min. The Nile red was dissolved in 0.4% acetone. Visualization of Nile red was done using a fluorescence microscope (EVOS cell imaging) under an RFP filter. Quantification of staining was done using ImageJ software as described above. To calculate the Nile Red stain in the inter-segmental vessels, 10X trunk images were cropped using free hand tool in ImageJ and the images were processed as earlier. Data were also analyzed by a researcher blinded to the identity of samples.
In vitro phosphatase assay
HEK 293T cells were transfected with SFB-PHLPP1α and/or pCMV-Flag-ChREBP. Twenty four hours after transfection, cells were lysed in Phosphatase Lysis buffer (PLB) (50 mM Tris (pH 7.4), 1 mM DTT, 100 mM NaCl, 0.1% NP40 and 5 mM MnCl2). Proteins were enriched using FLAG beads for 3 h. The bead-bound proteins were incubated in the reaction buffer (50 mM Tris (pH 7.4), 1 mM DTT, 10 mM ATP and 5 mM MnCl2) for 45 min with gentle stirring at 30°C. Post incubation, the supernatant was collected and the released phosphate was measured using a malachite green phosphate assay kit (Cayman chemical) per the manufacturer’s protocol. The phosphate-containing supernatant was incubated with MG acidic and MG blue solution for the indicated time periods and the absorbance measured at 620 nm.
Lipid estimation assays
Lipids were isolated from cells using the Bligh and Dyer organic lipid extraction method. Approximately 10 million cells were lysed using lipid lysis buffer [20 mM Tris, 1 mM EDTA]. Post lysis, chloroform and methanol were added at a ratio of 1:2 followed by addition of chloroform and lysis buffer. The samples were incubated at 25°C for 5–10 min and centrifuged at 3000g for 5 min. The organic phase was collected and further analysed. Total Cholesterol and free fatty acids were measured using kits following manufacturer’s instructions.
For HPLC based cholesterol estimation in zebrafish larvae, lipids were extracted from larvae using Bligh and Dyer organic lipid extraction method with some modifications as specified (Miyares et al., 2014). HPLC analysis was performed using a Waters Alliance e2695 HPLC system on a SYMMETRY C18, 75∗4.6 mm 3.5 μm column. The mobile phase was: A) 10 mM NH4OAc in water B) ACN:IPA (70:30)T/%B: 0/70, 10/95, 40/95, 41/70, 45/70 at a flow rate of 1.0 mL/min. ACN:THF (60:40) was used as diluent; RT = 17.811.
Triglyceride estimation in zebrafish larvae was performed using MyBioSource kit following manufacturer’s instructions.
For Thin Layer Chromatography (TLC) based triglyceride measurements in C. elegans, about 300 Worms were washed with 2 mL of 1X PBS, taken in 2 mL microfuge tubes, centrifuged at 1,000 × g, and the supernatant was discarded. The pellet containing worms were transferred to glass tubes and resuspended in 3 mL of 2:1 chloroform: methanol solution, briefly sonicated and was shaken vigorously for 30 min, followed by vortexing for 60 s at high speed. The solution was centrifuged at 1,000 × g, the supernatant was taken and washed with 0.2X volume of water and vortexed at high speed for 60 s. The resulting solution was centrifuged at 1,000 × g for 60 s and the upper organic phase was transferred to a new glass tube. 0.2X volume of 0.034% of MgCl2 was added to the organic phase, followed by vortexing for 1 min, centrifuged and the organic phase was transferred into a new tube. The organic phase was dried under nitrogen stream, dissolved into 100 μL of chloroform and loaded on a TLC plate (HPTLC Silica gel 60 F254 AMD extra thin, Merck, HX381899), and TLC was performed using 4:1 hexane: ethyl ether as mobile phase. To visualize the triglycerides, the plates were soaked in copper staining solution (7.5% CuSO4 and 85% ortho-phosphoric acid) and charred at 180°C. The plates were scanned and densitometric analysis was performed using ImageJ software.
OxLDL uptake assay and cholesterol efflux assay
The OxLDL uptake and cholesterol efflux were quantified in PHLPP1 over-expressed and knocked-down RAW 264.7 cells using cell-based kits as per the manufacturer’s protocol.
Quantification and statistical analysis
Numerical data are expressed as mean ± SEM unless or otherwise specified. Majority of the data are expressed as non-normalized values except for densitometric protein quantifications where data are shown as mean (fold change) ± SEM. q-PCR data are expressed as fold change and their statistical significance is calculated using respective non-normalized and non-transformed ΔCt values. For comparison between two groups, two tailed t-test was used. One-way ANOVA and a subsequent Dunnett's or Bonferroni multiple comparison post-hoc tests were used to compare differences between multiple group means. p < 0.05 was considered as significant.
Acknowledgments
KVLP and KC acknowledge the financial support from Department of Biotechnology, India (BT/PR27445/MED/30/1962/2018). KVLP and KC appreciate the insightful comments from their lab members. The authors are also thankful to Dr. Maddika Subba Reddy, Dr. Alexandra Newton, and Dr. Howard Towle for their kind gifts of plasmids. KVLP, KC, and KB thank Dr. Parimal Misra for thoughtful discussions. Graphical abstract was created with BioRender.com.
Author contributions
KVLP: Overall experimentation supervision; KC, KVLP: zebrafish experiments supervision; AC: zebrafish CRISPR-Cas9 design and troubleshooting; RM, KB, PB: zebrafish experiments; MT, JK: C. elegans experiments; KB, Kc: in vitro experiments; MRK: RNA-seq analysis; RS: HPLC analysis; VAM and WBB: TLC; KVLP, KC, MB: critical insights; KB, KVLP: Writing – Original Draft; KVLP, KC, MBJ, KB: Writing – Review & Editing; KVLP, KC: Acquired funding. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Published: February 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.103766.
Contributor Information
Kiranam Chatti, Email: kiranamc@drils.org.
Kishore V.L. Parsa, Email: kishorep@drils.org.
Supplemental information
Data and code availability
-
•
RNA-sequencing datasets without raw data was deposited at Gene Expression Omnibus database (GEO) and can be accessed through the identifier GSE193132. The raw read-counts, raw data quality summary, read alignment scores, processed data identifying DE-Gs, pathway analysis are available on Mendeley data at https://doi.org/10.17632/p2h84hs6n2.1 (https://data.mendeley.com/datasets/p2h84hs6n2/1). The raw data files of sequencing could not be shared as the raw data files crashed and were corrupted in our data repository. Attempts for retrieval from backup sources were not successful.
-
•
All analyses were performed with Fiji/ImageJ as indicated in the results sections. RNA-seq analyses is explained in detail in “method details” section. No new code is generated in this study.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.
References
- Alamuru-Yellapragada N.P., Vundyala S., Behera S., Parsa K.V.L. LPS depletes PHLPP levels in macrophages through the inhibition of SP1 dependent transcriptional regulation. Biochem. Biophys. Res. Commun. 2017;486:533–538. doi: 10.1016/j.bbrc.2017.03.080. [DOI] [PubMed] [Google Scholar]
- Alamuru-Yellapragada N.P., Kapadia B., Parsa K.V.L. In-house made nucleofection buffer for efficient and cost effective transfection of RAW 264.7 macrophages. Biochem. Biophys. Res. Commun. 2017;487:247–254. doi: 10.1016/j.bbrc.2017.04.043. [DOI] [PubMed] [Google Scholar]
- Alamuru N.P., Behera S., Butchar J.P., Tridandapani S., Kaimal Suraj S., Babu P.P., Hasnain S.E., Ehtesham N.Z., Parsa K.V.L. A novel immunomodulatory function of PHLPP1: inhibition of iNOS via attenuation of STAT1 ser727 phosphorylation in mouse macrophages. J. Leukoc. Biol. 2014;95:775–783. doi: 10.1189/jlb.0713360. [DOI] [PubMed] [Google Scholar]
- Andreozzi F., Procopio C., Greco A., Mannino G.C., Miele C., Raciti G.A., Iadicicco C., Beguinot F., Pontiroli A.E., Hribal M.L. Increased levels of the Akt-specific phosphatase PH domain leucine-rich repeat protein phosphatase (PHLPP)-1 in obese participants are associated with insulin resistance. Diabetologia. 2011;54:1879–1887. doi: 10.1007/s00125-011-2116-6. [DOI] [PubMed] [Google Scholar]
- Aviram M. Macrophage foam cell formation during early atherogenesis is determined by the balance between pro-oxidants and anti-oxidants in arterial cells and blood lipoproteins. Antioxid. Redox Signal. 1999;1:585–594. doi: 10.1089/ars.1999.1.4-585. [DOI] [PubMed] [Google Scholar]
- Barquera S., Pedroza-Tobías A., Medina C., Hernández-Barrera L., Bibbins-Domingo K., Lozano R., Moran A.E. Global overview of the epidemiology of atherosclerotic cardiovascular disease. Arch. Med. Res. 2015;46:328–338. doi: 10.1016/j.arcmed.2015.06.006. [DOI] [PubMed] [Google Scholar]
- Behera S., Kapadia B., Kain V., Alamuru-Yellapragada N.P., Murunikkara V., Kumar S.T., Babu P.P., Seshadri S., Shivarudraiah P., Hiriyan J., et al. ERK1/2 activated PHLPP1 induces skeletal muscle ER stress through the inhibition of a novel substrate AMPK. Biochim. Biophys. Acta Mol. Basis Dis. 2018;1864:1702–1716. doi: 10.1016/j.bbadis.2018.02.019. [DOI] [PubMed] [Google Scholar]
- Bung N., Surepalli S., Seshadri S., Patel S., Peddasomayajula S., Kummari L.K., Kumar S.T., Babu P.P., Parsa K.V.L., Poondra R.R. 2-[2-(4-(trifluoromethyl) phenylamino) thiazol-4-yl] acetic acid (Activator-3) is a potent activator of AMPK. Sci. Rep. 2018;8:1–16. doi: 10.1038/s41598-018-27974-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez-Sánchez L., Garza-Reyes M.G., Espinosa-Luna J.E., Chávez-Rueda K., Legorreta-Haquet M.V., Blanco-Favela F. The role of TLR2, TLR4 and CD36 in macrophage activation and foam cell formation in response to oxLDL in humans. Hum. Immunol. 2014;75:322–329. doi: 10.1016/j.humimm.2014.01.012. [DOI] [PubMed] [Google Scholar]
- Chistiakov D.A., Melnichenko A.A., Myasoedova V.A., Grechko A.V., Orekhov A.N. Mechanisms of foam cell formation in atherosclerosis. J. Mol. Med. 2017;95:1153–1165. doi: 10.1007/s00109-017-1575-8. [DOI] [PubMed] [Google Scholar]
- Cozzone D., Fröjdö S., Disse E., Debard C., Laville M., Pirola L., Vidal H. Isoform-specific defects of insulin stimulation of Akt/protein kinase B (PKB) in skeletal muscle cells from type 2 diabetic patients. Diabetologia. 2008;51:512–521. doi: 10.1007/s00125-007-0913-8. [DOI] [PubMed] [Google Scholar]
- Cuchel M., Rader D.J. Macrophage reverse cholesterol transport: key to the regression of atherosclerosis? Circulation. 2006;113:2548–2555. doi: 10.1161/CIRCULATIONAHA.104.475715. [DOI] [PubMed] [Google Scholar]
- Eisen M.B., Spellman P.T., Brown P.O., Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espino-Saldaña A.E., Rodríguez-Ortiz R., Pereida-Jaramillo E., Martínez-Torres A. Modeling neuronal diseases in zebrafish in the era of CRISPR. Curr. Neuropharmacol. 2020;18:136–152. doi: 10.2174/1570159X17666191001145550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbraio M., Podrez E.A., Smith J.D., Hajjar D.P., Hazen S.L., Hoff H.F., Sharma K., Silverstein R.L. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Invest. 2000;105:1049–1056. doi: 10.1172/JCI9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangula N.R., Maddika S. WD repeat protein WDR48 in complex with deubiquitinase USP12 suppresses Akt-dependent cell survival signaling by stabilizing PH domain leucine-rich repeat protein phosphatase 1 (PHLPP1) J. Biol. Chem. 2013;288:34545–34554. doi: 10.1074/jbc.M113.503383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden J.M., Brachova L., Higgins K., Obermiller L., Sevanian A., Khandrika S., Reaven P.D. Induction of monocyte differentiation and foam cell formation in vitro by 7-ketocholesterol. J. Lipid Res. 2002;43:26–35. [PubMed] [Google Scholar]
- Hotamisligil G.S. Endoplasmic reticulum stress and atherosclerosis. Nat. Med. 2010;16:396–399. doi: 10.1038/nm0410-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Cai C., Zheng T., Wu X., Wang D., Zhang K., Xu B., Yan R., Gong H., Zhang J. Endothelial scaffolding protein ENH (enigma homolog protein) promotes PHLPP2 (pleckstrin homology domain and leucine-rich repeat protein phosphatase 2)-mediated dephosphorylation of AKT1 and eNOS (endothelial NO Synthase) promoting vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 2020;40:1705–1721. doi: 10.1161/ATVBAHA.120.314172. [DOI] [PubMed] [Google Scholar]
- Iizuka K., Takao K., Yabe D. ChREBP-mediated regulation of lipid metabolism: involvement of the gut microbiota, liver, and adipose tissue. Front. Endocrinol. 2020;11:587189. doi: 10.3389/fendo.2020.587189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isoviita P.M., Nuotio K., Saksi J., Turunen R., Ijäs P., Pitkäniemi J., Soinne L., Kaste M., Kovanen P.T., Lindsberg P.J. An imbalance between CD36 and ABCA1 protein expression favors lipid accumulation in stroke-prone ulcerated carotid plaques. Stroke. 2010;41:389–393. doi: 10.1161/STROKEAHA.109.567636. [DOI] [PubMed] [Google Scholar]
- Jackson T.C., Verrier J.D., Drabek T., Janesko-Feldman K., Gillespie D.G., Uray T., Dezfulian C., Clark R.S., Bayir H., Jackson E.K. Pharmacological inhibition of pleckstrin homology domain leucine-rich repeat protein phosphatase is neuroprotective: differential effects on astrocytes. J. Pharmacol. Exp. Ther. 2013;347:516–528. doi: 10.1124/jpet.113.206888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong S.-J., Lee M.-N., Oh G.T. The role of macrophage lipophagy in reverse cholesterol transport. Endocrinol. Metab. 2017;32:41. doi: 10.3803/EnM.2017.32.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsenelson K.C., Stender J.D., Kawashima A.T., Lordén G., Uchiyama S., Nizet V., Glass C.K., Newton A.C. PHLPP1 counter-regulates STAT1-mediated inflammatory signaling. Elife. 2019;8:e48609. doi: 10.7554/eLife.48609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi T., Osatomi K., Yamashita H., Kabashima T., Uyeda K. Mechanism for fatty acid “sparing” effect on glucose-induced transcription: regulation of carbohydrate-responsive element-binding protein by AMP-activated protein kinase. J. Biol. Chem. 2002;277:3829–3835. doi: 10.1074/jbc.M107895200. [DOI] [PubMed] [Google Scholar]
- Kim K., Qiang L., Hayden M.S., Sparling D.P., Purcell N.H., Pajvani U.B. mTORC1-independent raptor prevents hepatic steatosis by stabilizing PHLPP2. Nat. Commun. 2016;7:1–10. doi: 10.1038/ncomms10255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liangpunsakul S., Ross R.A., Crabb D.W. Activation of carbohydrate response element–binding protein by ethanol. J. Investig. Med. 2013;61:270–277. doi: 10.231/JIM.0b013e31827c2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupse B., Annamalai K., Ibrahim H., Kaur S., Geravandi S., Sarma B., Pal A., Awal S., Joshi A., Rafizadeh S. Inhibition of PHLPP1/2 phosphatases rescues pancreatic β-cells in diabetes. Cell Rep. 2021;36:109490. doi: 10.1016/j.celrep.2021.109490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur A., Pandey V.K., Kakkar P. PHLPP: a putative cellular target during insulin resistance and type 2 diabetes. J. Endocrinol. 2017;233:R185–R198. doi: 10.1530/JOE-17-0081. [DOI] [PubMed] [Google Scholar]
- Miyamoto S., Purcell N.H., Smith J.M., Gao T., Whittaker R., Huang K., Castillo R., Glembotski C.C., Sussman M.A., Newton A.C. PHLPP-1 negatively regulates Akt activity and survival in the heart. Circ. Res. 2010;107:476–484. doi: 10.1161/CIRCRESAHA.109.215020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyares R.L., de Rezende V.B., Farber S.A. Zebrafish yolk lipid processing: a tractable tool for the study of vertebrate lipid transport and metabolism. Dis. Model. Mech. 2014;7:915–927. doi: 10.1242/dmm.015800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moc C., Taylor A.E., Chesini G.P., Zambrano C.M., Barlow M.S., Zhang X., Gustafsson Å.B., Purcell N.H. Physiological activation of Akt by PHLPP1 deletion protects against pathological hypertrophy. Cardiovasc. Res. 2015;105:160–170. doi: 10.1093/cvr/cvu243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuotio K., Isoviita P.M., Saksi J., Ijäs P., Pitkäniemi J., Sonninen R., Soinne L., Saimanen E., Salonen O., Kovanen P.T. Adipophilin expression is increased in symptomatic carotid atherosclerosis: correlation with red blood cells and cholesterol crystals. Stroke. 2007;38:1791–1798. doi: 10.1161/STROKEAHA.106.478867. [DOI] [PubMed] [Google Scholar]
- Ouimet M., Marcel Y.L. Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler. Thromb. Vasc. Biol. 2012;32:575–581. doi: 10.1161/ATVBAHA.111.240705. [DOI] [PubMed] [Google Scholar]
- Pedruzzi E., Guichard C., Ollivier V., Driss F., Fay M., Prunet C., Marie J.-C., Pouzet C., Samadi M., Elbim C. NAD (P) H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol. Cell. Biol. 2004;24:10703–10717. doi: 10.1128/MCB.24.24.10703-10717.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao X., Zhong J., Maiseyeu A., Gopalakrishnan B., Villamena F.A., Chen L.-C., Harkema J.R., Sun Q., Rajagopalan S. CD36-dependent 7-ketocholesterol accumulation in macrophages mediates progression of atherosclerosis in response to chronic air pollution exposure. Circ. Res. 2014;115:770–780. doi: 10.1161/CIRCRESAHA.115.304666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakiyama H., Wynn R.M., Lee W.-R., Fukasawa M., Mizuguchi H., Gardner K.H., Repa J.J., Uyeda K. Regulation of nuclear import/export of carbohydrate response element-binding protein (ChREBP): interaction of an α-helix of ChREBP with the 14-3-3 proteins and regulation by phosphorylation. J. Biol. Chem. 2008;283:24899–24908. doi: 10.1074/jbc.M804308200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrazy V., Sore S., Viaud M., Rignol G., Westerterp M., Ceppo F., Tanti J.F., Guinamard R., Gautier E.L., Yvan-Charvet L. Maintenance of macrophage redox status by ChREBP limits inflammation and apoptosis and protects against advanced atherosclerotic lesion formation. Cell Rep. 2015;13:132–144. doi: 10.1016/j.celrep.2015.08.068. [DOI] [PubMed] [Google Scholar]
- Shaye D.D., Greenwald I. OrthoList: a compendium of C. elegans genes with human orthologs. PLoS One. 2011;6:e20085. doi: 10.1371/journal.pone.0020085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva Xavier G., Rutter G.A., Diraison F., Andreolas C., Leclerc I. ChREBP binding to fatty acid synthase and L-type pyruvate kinase genes is stimulated by glucose in pancreatic β-cells. J. Lipid Res. 2006;47:2482–2491. doi: 10.1194/jlr.M600289-JLR200. [DOI] [PubMed] [Google Scholar]
- Sorlien E.L., Witucki M.A., Ogas J. Efficient production and identification of CRISPR/Cas9-generated gene knockouts in the model system Danio rerio. J. Vis. Exp. JoVE. 2018:56969. doi: 10.3791/56969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckman A.K., Ma L., Towle H.C. Mlx is the functional heteromeric partner of the carbohydrate response element-binding protein in glucose regulation of lipogenic enzyme genes. J. Biol. Chem. 2004;279:15662–15669. doi: 10.1074/jbc.M311301200. [DOI] [PubMed] [Google Scholar]
- Sulkava M., Raitoharju E., Levula M., Seppälä I., Lyytikäinen L.-P., Mennander A., Järvinen O., Zeitlin R., Salenius J.-P., Illig T. Differentially expressed genes and canonical pathway expression in human atherosclerotic plaques–Tampere Vascular Study. Sci. Rep. 2017;7:1–10. doi: 10.1038/srep41483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I. Free cholesterol-induced cytotoxicity: a possible contributing factor to macrophage foam cell necrosis in advanced atherosclerotic lesions. Trends Cardiovasc. Med. 1997;7:256–263. doi: 10.1016/S1050-1738(97)00086-8. [DOI] [PubMed] [Google Scholar]
- Takata K., Honda S., Sidharta S.L., Duong M., Shishikura D., Kim S.W., Andrews J., Di Bartolo B.A., Psaltis P.J., Bursill C.A. Associations of ABCG1-mediated cholesterol efflux capacity with coronary artery lipid content assessed by near-infrared spectroscopy. Cardiovasc. Diagn. Ther. 2019;9:310. doi: 10.21037/cdt.2018.11.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfel N.A., Niederst M., Stevens M.W., Brennan P.M., Frame M.C., Newton A.C. Mislocalization of the E3 ligase, β-transducin repeat-containing protein 1 (β-TrCP1), in glioblastoma uncouples negative feedback between the pleckstrin homology domain leucine-rich repeat protein phosphatase 1 (PHLPP1) and Akt. J. Biol. Chem. 2011;286:19777–19788. doi: 10.1074/jbc.M111.237081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. The Zebrafish Book, 5th Edition. A guide for the laboratory use of zebrafish (Danio rerio) Eugene, University of Oregon Press; 2007. [Google Scholar]
- Yan Y., Krecke K.N., Bapat A.S., Yang T., Lopresti M.W., Mashek D.G., Kelekar A. Phosphatase PHLPP2 regulates the cellular response to metabolic stress through AMPK. Cell Death Dis. 2021;12:1–10. doi: 10.1038/s41419-021-04196-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao S., Miao C., Tian H., Sang H., Yang N., Jiao P., Han J., Zong C., Qin S. Endoplasmic reticulum stress promotes macrophage derived foam cell formation by up-regulating cluster of differentiation 36 (CD36) expression. J. Biol. Chem. 2014;289:4032–4042. doi: 10.1074/jbc.M113.524512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y., Li P., Ye J. Lipid homeostasis and the formation of macrophage-derived foam cells in atherosclerosis. Protein Cell. 2012;3:173–181. doi: 10.1007/s13238-012-2025-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvan-Charvet L., Welch C., Pagler T.A., Ranalletta M., Lamkanfi M., Han S., Ishibashi M., Li R., Wang N., Tall A.R. Increased inflammatory gene expression in ABC transporter–deficient macrophages: free cholesterol accumulation, increased signaling via Toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837–1847. doi: 10.1161/CIRCULATIONAHA.108.793869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNA-sequencing datasets without raw data was deposited at Gene Expression Omnibus database (GEO) and can be accessed through the identifier GSE193132. The raw read-counts, raw data quality summary, read alignment scores, processed data identifying DE-Gs, pathway analysis are available on Mendeley data at https://doi.org/10.17632/p2h84hs6n2.1 (https://data.mendeley.com/datasets/p2h84hs6n2/1). The raw data files of sequencing could not be shared as the raw data files crashed and were corrupted in our data repository. Attempts for retrieval from backup sources were not successful.
-
•
All analyses were performed with Fiji/ImageJ as indicated in the results sections. RNA-seq analyses is explained in detail in “method details” section. No new code is generated in this study.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon reasonable request.