

Summary
Here, we describe a protocol to set up a screening assay for ADP-ribosyl binding proteins including proteins that possess O-glycosidase or N-glycosidase activities. The FRET-based assay measures the interaction of any ADP-ribosyl binding protein fused to CFP with a cysteine-ADP-ribosylated GAP-tag fused to YFP. Recombinant PtxS1 and PARP2 are used to mono-ADP-ribosylate and poly-ADP-ribosylate the GAP-tag. The protocol does not require specialized compounds or substrates, making it accessible and easy to adapt in any laboratory or for other proteins of interest.
For complete details on the use and execution of this profile, please refer to Sowa et al. (2021).
Subject areas: High Throughput Screening, Signal Transduction, Molecular/Chemical Probes, Protein Biochemistry, Protein expression and purification
Graphical abstract
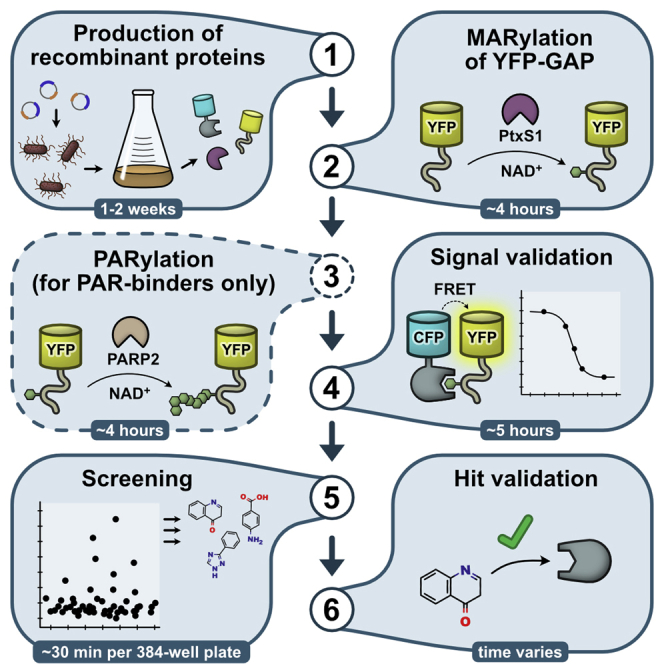
Highlights
-
•
Development of FRET-based assays for ADP-ribosyl binding proteins
-
•
An ADP-ribosylated GAP-tag is used as non-hydrolyzable binding probe
-
•
Instructions for protein production, signal validation and inhibitor screening
Here, we describe a protocol to set up a screening assay for ADP-ribosyl binding proteins including proteins that possess O-glycosidase or N-glycosidase activities. The FRET-based assay measures the interaction of any ADP-ribosyl binding protein fused to CFP with a cysteine-ADP-ribosylated GAP-tag fused to YFP. Recombinant PtxS1 and PARP2 are used to mono-ADP-ribosylate and poly-ADP-ribosylate the GAP-tag. The protocol does not require specialized compounds or substrates, making it accessible and easy to adapt in any laboratory or for other proteins of interest.
Before you begin
Three proteins are needed to facilitate the setup of the assay at the most basic level: 1) The ADP-ribosyl binder, for which the screening assay is to be designed. The protein is expressed as a fusion construct with CFP. 2) YFP with the 10-residue GAP-tag at the C-terminus, which will be ADP-ribosylated and will act as a binding probe. 3) The subunit S1 of Pertussis toxin (PtxS1) (Ashok et al., 2020). The toxin fragment is required to site-specifically mono-ADP-ribosylate a cysteine-residue located in the GAP-tag. Additionally, if an ADP-ribosyl binder will be used that only binds poly-ADP-ribosyl groups, a recombinant PARP2 construct containing the WGR and catalytic domain (residues 90–583) can be used to elongate the mono-ADP-ribosyl group on the GAP-tag to a poly-ADP-ribose chain.
Bacterial expression vectors encoding the required proteins may be prepared using standard cloning techniques or can be ordered through Addgene. All constructs should contain a hexahistidine tag for purification by immobilized metal affinity chromatography (IMAC). Removal of the hexahistidine tag from PtxS1 and PARP2 should be possible, e.g., by TEV protease. The design of the fusion constructs allows freedom in selection of the fluorescent proteins, however they should form a robust FRET pair. Common FRET pairs are for example variants of CFP/YFP or GFP/RFP (Bajar et al., 2016). The fluorescent proteins of choice should be monomeric. Fluorescent protein variants that are stable and express in high quantities are desired. In our work, we use proteins fused to mCerulean (CFP) and mCitrine (YFP). It is critical that the FRET acceptor protein (in our case YFP) contains the engineered GAP-tag (KNNLKDCGLF) at the C-terminus without additional C-terminal residues.
Recombinant protein expression
Timing: 3 days
In this step, we describe the procedure for expression of recombinant proteins as done with our expression vectors based on the pNIC28-Bsa4 plasmid backbone. These vectors encode an antibiotic resistance against kanamycin. Other vectors or expression strains may be used depending on the availability in the laboratory. Production of PARP2 is only needed if an assay for poly-ADP-ribosyl binding proteins is to be set up.
-
1.Transform chemicompetent E. coli BL21 (DE3) cells with the expression plasmids encoding YFP-GAP, PtxS1 and e.g., CFP-MacroD1.
-
a.Prepare LB-agar plates containing 50 μg/mL kanamycin.Note: The LB-agar plates may be pre-warmed prior to plating of the cells to 37°C to increase transformation efficiency.
-
b.Thaw the chemicompetent E. coli BL21 (DE3) cells on ice (25 μL).
-
c.Gently mix 1–10 ng of the respective plasmids with 25 μL of the cells in separate sterile reaction tubes.
-
d.Let the cells incubate on ice for 30 min.
-
e.Incubate the cells for 45 s in a heat-block or water bath at 42°C.
-
f.Immediately place cells on ice for 5 min.
-
g.Transfer 1 mL of sterile SOC or LB-media to the cells.
-
h.Incubate the cells in a shaker for 1 h at 37°C.
-
i.Use 20–200 μL of the suspended cells and plate them on the prepared LB-agar plates.
-
j.Incubate the agar plates for 14–18 h at 37°C.
-
a.
-
2.Express the recombinant proteins.
-
a.For each expression construct, inoculate 25 mL LB-media containing 50 μg/mL kanamycin with 3–4 average-sized colonies from the LB-agar plates.Note: Inoculation with several colonies is meant to avoid accidentally cultivating a single genetically altered colony with impaired expression levels.
-
b.Incubate the pre-cultures for 14–18 h shaking at 37°C.
-
c.Per expression construct, prepare 500 mL of sterile TB-autoinduction media supplemented with 50 μg/mL kanamycin in 5-liter Erlenmeyer flasks and inoculate with 5 mL of the respective pre-cultures. The initial OD600 of the inoculated culture is usually between 0.03 and 0.05.
-
d.Incubate the flasks in a shaker at 37°C until an OD600 of 0.6–1.0 is reached. This typically requires 3–5 h.
-
e.Set the temperature to 18°C and continue incubation for 20 h.Note: During expression, purification and handling of the fluorescent proteins, limit the exposure to bright light (e.g. direct sunlight) whenever possible to avoid photobleaching of the fluorophores. We have not observed problems with regular indoor lights.
-
f.Weigh the empty centrifugation tubes that will be used for collection of the cells.
-
g.Pellet the cells by centrifugation at 4200×g and 4°C for 15 min and discard the supernatant. Determine the weight of the cell pellets.
-
h.Resuspend the cells in 25 mL lysis buffer [50 mM HEPES pH 7.5, 0.5 M NaCl, 10%(v/v) glycerol, 15 mM imidazole, 0.5 mM TCEP] per 10 g of bacterial pellet.
 Pause Point: The resuspended cells may be stored at −20°C or below in the dark until purification of the proteins.
Pause Point: The resuspended cells may be stored at −20°C or below in the dark until purification of the proteins.
-
a.
Protein purification
-
3.Lyse the cells.
-
a.Thaw the cells if frozen in the previous step in a water bath at room temperature (20°C–25°C).
-
b.Mix the resuspended cells with Pefabloc SC and DNase I to final concentrations of 0.1 mM and 20 μg/mL, respectively. For PARP2, do not include DNase I.
-
c.Transfer the cells to a container submerged in an ice slurry. Set the sonicator to 50% intensity and lyse the cells by sonication with 10 s on/30 s off intervals (to prevent heating of the lysate) for a total of 2 min sonication time.Note: Sonication time and intensity may vary depending on the type of sonicator used.
-
d.Transfer the lysate to centrifugation tubes.
-
e.Centrifuge the lysate at 16,000×g for 30–45 min at 4°C. The time can be used to prepare the IMAC column in step 4a.
-
f.Carefully transfer the supernatant to a 50 mL syringe with a 0.45 μm PES filter attached. Filter the lysate into a clean 50 mL falcon tube placed on ice.
-
a.
-
4.Purify the protein by IMAC.Note: For PARP2, all purification steps should be done at 4°C.
-
a.Prepare a 5 mL HiTrap IMAC HP column by charging it with NiSO4 and equilibrate it with lysis buffer.
-
b.Load the cleared lysate to the column at 1 mL/min.
-
c.Wash the column with 5–10 column volumes (CV) of lysis buffer.
-
d.Wash the column with 5 CV of wash buffer [50 mM HEPES pH 7.5, 0.5 M NaCl, 10% (v/v) glycerol, 20 mM imidazole, 0.5 mM TCEP].Note: For the fluorescent proteins, washing may be extended until colored solution starts to appear.
-
e.Elute the protein using elution buffer [50 mM HEPES pH 7.5, 0.5 M NaCl, 10% (v/v) glycerol, 200 mM imidazole, 0.5 mM TCEP] in fractions of 0.5–2 mL. Keep the fractions on ice.
-
f.Analyze the fractions by SDS-PAGE (Figure 1A) and select suitable fractions based on purity. At this stage, the purity of well-expressed proteins should be >70%. The eluate may be stored overnight (up to 24 h) at 4°C until purification by size exclusion chromatography (SEC). For poorly expressed protein, see troubleshooting 1.Note: A small amount of CFP may have been proteolytically cleaved from the target protein and may be present in the elution. This impurity can usually be removed during size-exclusion chromatography. See troubleshooting 2 if extensive cleavage is observed.Note: A TEV cleavage step to remove the hexahistidine tag should be performed only for the produced PtxS1 and PARP2 proteins (see step 6). This is required for the removal of PtxS1 or PARP2 from ADP-ribosylated YFP-GAP protein by IMAC in later steps.
-
a.
-
5.Purification using Heparin (only to be done with PARP2).Note: Perform this step on the same day after purification of PARP2 by IMAC. This step removes DNA that may be bound to PARP2, which might affect the activity of the protein.
-
a.Equilibrate a 5 mL Heparin-Trap column with Heparin buffer A [50 mM HEPES pH 7.5, 400 mM NaCl, 10% (v/v) glycerol, 0.5 mM TCEP].
-
b.Pool the elution fractions of PARP2 from the IMAC purification and determine the volume.
-
c.Dilute the sample to a final NaCl concentration of 400 mM using Heparin dilution buffer [50 mM HEPES pH 7.5, 10% (v/v) glycerol, 0.5 mM TCEP].
-
d.Load the sample into the 5 mL Heparin-Trap column at 2 mL/min.
-
e.Wash the column with 5–10 CV of Heparin buffer A.
-
f.Elute the contents of the column using a gradient of Heparin buffer A to Heparin buffer B [50 mM HEPES pH 7.5, 1.5 M NaCl, 10% (v/v) glycerol, 0.5 mM TCEP] over 20 min at 2 mL/min.
-
g.Pool the fractions containing protein and proceed with the TEV cleavage.Optional: Analyze the fractions on SDS-PAGE and select based on purity.
-
a.
-
6.Perform TEV cleavage and reverse IMAC (only to be done with PtxS1 and PARP2).
-
a.Mix the protein with hexahistidine-tagged TEV protease. The final molar concentration of the TEV protease should be approximately 1:30 of the target protein concentration.Note: We prepare recombinant TEV protease (van den Berg et al., 2006), however it may also be purchased.
-
b.Dialyze the purified protein against SEC buffer overnight (up to 24 h) at 4°C using a dialysis membrane with 6–8 kDa molecular weight cutoff.Note: For PARP2, dialysis is not required as the imidazole was removed during purification at the Heparin column.
-
c.Prepare a 5 mL HiTrap IMAC HP column by charging it with NiSO4 and equilibrate it with lysis buffer.
-
d.Load the TEV reaction mixture to the column at 1 mL/min and collect the flow-through containing the target protein.
-
e.Wash with lysis buffer until no more protein appears in the flow through.
-
f.Concentrate the target protein solution to approximately 1 mL using a 15 mL Amicon centrifugal filter with 10 or 30 kDa molecular weight cutoff depending on the size of the protein used.
-
a.
-
7.Purify the protein by size-exclusion chromatography (SEC).
-
a.Equilibrate a S75 16/600 SEC column with SEC buffer [50 mM HEPES pH 7.5, 0.5 M NaCl, 10% (v/v) glycerol, 15 mM imidazole, 0.5 mM TCEP]. This can be done the day before.
-
b.Concentrate the target protein solution to approximately 1 mL using a 15 mL Amicon centrifugal filter with 10 or 30 kDa molecular weight cutoff depending on the size of the protein used.
 CRITICAL: If the protein starts to precipitate, the concentration step should be stopped. Proceed with the following steps, in which the precipitate will be removed by centrifugation. If the protein volume is too high (>1 mL) for a single size exclusion chromatography run, perform several runs.
CRITICAL: If the protein starts to precipitate, the concentration step should be stopped. Proceed with the following steps, in which the precipitate will be removed by centrifugation. If the protein volume is too high (>1 mL) for a single size exclusion chromatography run, perform several runs. -
c.Transfer the concentrated protein solution into a 1.5 mL reaction tube.
-
d.To remove potential debris and precipitation, centrifuge the solution at >16,000×g for 10–15 min at 4°C and transfer the supernatant to a new reaction tube.
-
e.Load the sample to the column and collect fractions.
-
f.Analyze the purity of the fractions by SDS-PAGE (Figure 1B) and concentrate pure fractions using a 15 mL Amicon centrifugal filter to approximately 5–20 mg/mL.
-
g.Aliquot the protein in PCR reaction tubes (5–25 μL per tube) and flash freeze the tubes in liquid nitrogen.Note: It is advised to reduce unnecessary freezing/thawing cycles with the protein aliquots used. The YFP-GAP protein may be used immediately or be kept overnight (up to 24 h) at 4°C for further modification by PtxS1 (see next steps). However, a few aliquots of YFP-GAP should be frozen without further modification. These will be used in later signal validation steps.
-
h.Store the aliquots at −70°C or below.Pause Point: The proteins can be stored frozen in aliquots until signal validation and development of the screening assay. We have not observed any issues after storing the protein for 1 year.
-
a.
Figure 1.

Representative SDS-PAGE images from the purification of CFP-MacroD1
(A) Purification of CFP-MacroD1 (MDO1) by IMAC. Samples of the crude lysate and the cleared lysate after centrifugation and filtering are included. Fractions (Fr.) of 2 mL volume were eluted and every second fraction was analyzed by SDS-PAGE. Based on this, we pooled fractions 3–13 for further purification by size exclusion chromatography.
(B) Purification of the CFP-MacroD1 IMAC elution by size exclusion chromatography. Fractions containing protein were analyzed by SDS-PAGE. The proteolytically degraded CFP impurity appeared in later fractions and could be separated from the target protein CFP-MacroD1. Fractions 17–25 were pooled, concentrated and finally stored at −70°C.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E. coli BL21 (DE3) | New England BioLabs | Cat# C2527H |
| Chemicals, peptides, and recombinant proteins | ||
| NAD+ | Sigma-Aldrich | Cat# N0632 |
| TB Auto-induction media | Formedium | Cat# AIMTB0210 |
| ADP-ribose | Sigma-Aldrich | Cat# A0752 |
| TEV protease | Merck | Cat# T4455 |
| Pefabloc SC | Merck | Cat# 11429868001 |
| DNase I | Merck | Cat# 10104159001 |
| Activating DNA | Merck | Cat# D4522 |
| Recombinant DNA | ||
| Expression vectors for CFP-fused ADP-ribosyl binders (various) | Addgene; (Sowa et al., 2021) | Various; See reference. |
| Expression vector for YFP-GAP | Addgene | 173080 |
| Expression vector for PtxS1 | Addgene | 173076 |
| Expression vector for PARP2 | Addgene | 173081 |
| Software and algorithms | ||
| Prism (ver. 8.0.2) | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| Fisherbrand 384-Well ShallowWell black polypropylene microplates | Fisher Scientific | Cat# 13595450 |
| Spark multimode microplate reader | Tecan | https://lifesciences.tecan.com/multimode-plate-reader |
| Echo 650 acoustic liquid dispenser | Beckman Coulter | https://www.beckman.com/liquid-handlers/echo-650-series |
| Mantis liquid handler | Formulatrix | https://formulatrix.com/liquid-handling-systems/mantis-liquid-handler/ |
| Branson 450 Digital Sonifier with ½-inch stepped horn. | Branson | Discontinued |
| 5 mL HiTrap IMAC HP column | Merck | Cat# GE17-0920-03 |
| 5 mL Heparin-Trap column | Merck | Cat# GE17-0407-01 |
| S75 16/600 size-exclusion chromatography column | Merck | Cat# GE28-9893-33 |
| Syringe filter PES (0.45 μm) | Sartorius | Cat# 16533 |
| Amicon Ultra-15 centrifugal filter (10 kDa) | Merck | Cat# UFC901024 |
| Amicon Ultra-15 centrifugal filter (30 kDa) | Merck | Cat# UFC903024 |
| Spectra/Por Dialysis membrane (6–8 kDa) | Fisher Scientific | Cat# 11435849 |
| Microplate foil seal | Azenta Life Sciences | Cat# 4ti-0550 |
Materials and equipment
While preparation of small-scale binding assays and validation of the signal could be easily achieved by manual pipetting, screening was done using the automated liquid dispenser Mantis (Formulatrix). Compounds were dispensed by acoustic liquid transfer using Echo 650 (Beckman Coulter).
TB autoinduction medium (including trace elements)
| Reagent | Final concentration | Amount (for 1000 mL) |
|---|---|---|
| TB autoinduction medium including trace elements (Formedium) | N/A | 55.85 g |
| Glycerol | 8 g/L | 8 g |
| Total | N/A | 1000 mL |
Autoclave according to the manufacturer’s instructions and store the sterile media at room temperature (20°C–25°C) for up to 3 months. If contamination is visible (i.e., the solution turned turbid), discard the media and prepare freshly.
Lysis buffer
| Reagent (stock concentration) | Final concentration | Amount (for 1000 mL) |
|---|---|---|
| HEPES pH 7.5 (0.5 M) | 50 mM | 100 mL |
| NaCl (5 M) | 0.5 M | 100 mL |
| Glycerol (100%) | 10%(v/v) | 100 mL |
| Imidazole pH 7.5 (2 M) | 15 mM | 7.5 mL |
| TCEP pH 7.5 (0.5 M) | 0.5 mM | 1 mL |
| Total | N/A | 1000 mL |
Store at 4°C for up to 6 months.
IMAC wash buffer
| Reagent (stock concentration) | Final concentration | Amount (for 1000 mL) |
|---|---|---|
| HEPES pH 7.5 (0.5 M) | 50 mM | 100 mL |
| NaCl (5 M) | 0.5 M | 100 mL |
| Glycerol (100%) | 10%(v/v) | 100 mL |
| Imidazole pH 7.5 (2 M) | 20 mM | 10 mL |
| TCEP pH 7.5 (0.5 M) | 0.5 mM | 1 mL |
| Total | N/A | 1000 mL |
Store at 4°C for up to 6 months.
IMAC elution buffer
| Reagent (stock concentration) | Final concentration | Amount (for 1000 mL) |
|---|---|---|
| HEPES pH 7.5 (0.5 M) | 50 mM | 100 mL |
| NaCl (5 M) | 0.5 M | 100 mL |
| Glycerol (100%) | 10%(v/v) | 100 mL |
| Imidazole pH 7.5 (2 M) | 200 mM | 100 mL |
| TCEP pH 7.5 (0.5 M) | 0.5 mM | 1 mL |
| Total | N/A | 1000 mL |
Store at 4°C for up to 6 months.
Heparin dilution buffer
| Reagent (stock concentration) | Final concentration | Amount (for 1000 mL) |
|---|---|---|
| HEPES pH 7.5 (0.5 M) | 50 mM | 100 mL |
| Glycerol (100%) | 10%(v/v) | 100 mL |
| TCEP pH 7.5 (0.5 M) | 0.5 mM | 1 mL |
| Total | N/A | 1000 mL |
Store at 4°C for up to 6 months.
Heparin buffer A
| Reagent (stock concentration) | Final concentration | Amount (for 1000 mL) |
|---|---|---|
| HEPES pH 7.5 (0.5 M) | 50 mM | 100 mL |
| NaCl (5 M) | 0.4 M | 80 mL |
| Glycerol (100%) | 10%(v/v) | 100 mL |
| TCEP pH 7.5 (0.5 M) | 0.5 mM | 1 mL |
| Total | N/A | 1000 mL |
Store at 4°C for up to 6 months.
Heparin buffer B
| Reagent (stock concentration) | Final concentration | Amount (for 1000 mL) |
|---|---|---|
| HEPES pH 7.5 (0.5 M) | 50 mM | 100 mL |
| NaCl (5 M) | 1.5 M | 300 mL |
| Glycerol (100%) | 10%(v/v) | 100 mL |
| TCEP pH 7.5 (0.5 M) | 0.5 mM | 1 mL |
| Total | N/A | 1000 mL |
Store at 4°C for up to 6 months.
SEC buffer
| Reagent (stock concentration) | Final concentration | Amount (for 1000 mL) |
|---|---|---|
| HEPES pH 7.5 (0.5 M) | 30 mM | 60 mL |
| NaCl (5 M) | 300 mM | 60 mL |
| Glycerol (100%) | 10%(v/v) | 100 mL |
| TCEP pH 7.5 (0.5 M) | 0.5 mM | 1 mL |
| Total | N/A | 1000 mL |
Store at 4°C for up to 6 months.
FRET assay buffer (mono-ADP-ribosyl binding assays)
| Reagent (stock concentration) | Final concentration | Amount (for 100 mL) |
|---|---|---|
| Bis-Tris-Propane pH 7.0 (0.5 M) | 10 mM | 2 mL |
| PEG20,000 (30%(w/v)) | 3%(w/v) | 10 mL |
| Triton X-100 (100%) | 0.01%(v/v) | 0.01 mL |
| TCEP pH 7.0 (0.5 M) | 0.5 mM | 0.1 mL |
| Total | N/A | 100 mL |
Store at 4°C for up to 3 months. Ensure that no precipitation occurred prior to use.
FRET assay buffer (poly-ADP-ribosyl binding assays)
| Reagent (stock concentration) | Final concentration | Amount (for 100 mL) |
|---|---|---|
| Tris-HCl pH 8.0 (0.5 M) | 10 mM | 2 mL |
| NaCl (5 M) | 150 mM | 3 mL |
| Tween-20 (100%) | 0.01%(v/v) | 0.01 mL |
| Total | N/A | 100 mL |
Store at 4°C for up to 6 months.
Step-by-step method details
Mono-ADP-ribosylation of YFP-GAP
In this step, the purified YFP-GAP protein will be mono-ADP-ribosylated at a specific cysteine residue at the C-terminus of the GAP-tag using PtxS1. The resulting protein will be used as the binding probe for the screening assay.
-
1.Prepare the mono-ADP-ribosylation reaction mix.
-
a.Prepare the following reagents in a 15 mL falcon tube.CRITICAL: Avoid mixing high concentrations of NAD+ and PtxS1 during preparation of the mixture. Mix all components and add PtxS1 last. Immediately gently mix by inverting the tube.
Reagent (stock concentration) Final concentration Amount Purified YFP-GAP protein (1 mM) 100 μM 500 μL Purified PtxS1 protein (500 μM) 1.5 μM 15 μL NAD+ (50 mM) 150 μM 15 μL 50 mM Sodium phosphate buffer (pH 7.5) N/A 4470 μL -
b.Let the reaction incubate at room temperature (20°C–25°C) for 1 h.Note: The incubation time can be used to prepare the IMAC column in step 2.
-
c.To ensure completeness of the reaction, add again 15 μL of PtxS1 (500 μM) and 15 μL of NAD+ (50 mM) to the reaction mixture.
-
d.Let the reaction incubate at room temperature (20°C–25°C) for 1 h.
-
a.
-
2.Remove unreacted NAD+, side-products and PtxS1 by IMAC.
-
a.Prepare a 5 mL HiTrap IMAC HP column by charging it with NiSO4 and equilibrate it with lysis buffer.
-
b.Load the reaction mixture to the column at 1 mL/min.
-
c.Wash the column with 5 column volumes (CV) of lysis buffer.
-
d.Elute the ADP-ribosylated YFP-GAP using elution buffer.
-
e.Exchange the buffer to SEC buffer using a 15 mL Amicon centrifugal filter with 10 kDa molecular weight cutoff.
-
f.Concentrate the IMAC eluate to approximately 0.5–1 mL using the same centrifugal filter.
-
g.Aliquot the protein in PCR reaction tubes (5–25 μL per tube) and flash freeze the tubes in liquid nitrogen.Note: If an assay for binders of poly-ADP-ribose is prepared, the mono-ADP-ribosylated YFP-GAP may be used immediately or be kept overnight (up to 24 h) at 4°C for further modification by PARP2 (see next steps).
-
h.Store the aliquots at −70°C or below.Optional: Analyze the modification of the protein by mass spectrometry. The addition of a single ADP-ribosyl group corresponds to an additional monoisotopic mass of 541.061 Da. If only partial modification is observed, see troubleshooting 3.Pause point: The frozen aliquots may be used at any time for validation or screening. We have not observed any issues after storing the protein for 1 year.
-
a.
Poly-ADP-ribosylation of mono-ADP-ribosylated YFP-GAP and PARP2 (required for poly-ADP-ribosyl binders only)
In this step, the mono-ADP-ribosyl group at the YFP-GAP protein from the previous step will be poly-ADP-ribosylated using PARP2. The resulting protein will be used as the binding probe for the screening assay of poly-ADP-ribosyl binders. In a similar manner, PARP2 will be auto-modified with poly-ADP-ribosyl chains, which can be used as a control during validation.
-
3.Prepare the poly-ADP-ribosylation reaction mix for YFP-GAP.
-
a.Prepare the following reagents in a 15 mL falcon tube.CRITICAL: Avoid mixing high concentrations of NAD+ and PARP2 during preparation of the mixture. Mix all components and add PARP2 last. Immediately gently mix by inverting the tube.
Reagent (stock concentration) Final concentration Amount Purified mono-ADP-ribosylated YFP-GAP protein (1 mM) 10 μM 50 μL Purified PARP2 protein (100 μM) 400 nM 20 μL NAD+ (50 mM) 1 mM 100 μL Reaction buffer: Tris pH 8.0 (50 mM), MgCl2 (5 mM) N/A 4830 μL -
b.Let the reaction incubate at room temperature (20°C–25°C) for 2 h.Note: The incubation time can be used to prepare the IMAC column in step 4.
-
a.
-
4.Remove unreacted NAD+, side-products and PARP2 by IMAC.
-
a.Prepare a 5 mL HiTrap IMAC HP column by charging it with NiSO4 and equilibrate it with lysis buffer.
-
b.Load the reaction mixture to the column at 1 mL/min.
-
c.Wash the column with 5 column volumes (CV) of lysis buffer.
-
d.Elute the poly-ADP-ribosylated YFP-GAP using elution buffer.
-
e.Exchange the buffer to SEC buffer using a 15 mL Amicon centrifugal filter with 10 kDa molecular weight cutoff.
-
f.Concentrate the IMAC eluate to approximately 0.5–1 mL using the same centrifugal filter.
-
g.Aliquot the protein in PCR reaction tubes (5–25 μL per tube) and flash freeze the tubes in liquid nitrogen.
-
h.Store the aliquots at −70°C or below.
-
a.
-
5.Prepare poly-ADP-ribosylated PARP2 to be used as a control during validation of the assay.
-
a.Prepare the following reagents in a 15 mL tube.CRITICAL: Avoid mixing high concentrations of NAD+ and PARP2 during preparation of the mixture. Mix all components and add PARP2 last. Immediately gently mix by inverting the tube.
Reagent (stock concentration) Final concentration Amount Purified PARP2 (100 μM) 10 μM 500 μL NAD+ (50 mM) 1 mM 100 μL Activating DNA (500 μM) 10 μM 100 μL Reaction buffer: Tris pH 8.0 (50 mM), MgCl2 (5 mM) N/A 4300 μL -
b.Incubate for 30 min at room temperature (20°C–25°C).
-
c.Aliquot the protein in PCR reaction tubes (5–25 μL per tube) and flash freeze the tubes in liquid nitrogen.
-
d.Store the aliquots at −70°C or below.Pause point: The frozen aliquots may be used at any time for validation or screening. We have not observed any issues after storing the protein for 1 year.
-
a.
Signal validation
In this step, a ratiometric FRET signal (rFRET) upon interaction of the CFP-fused binder and the mono-ADP-ribosylated YFP-GAP will be determined. Controls are used to show the absence or reduction of the signal.
Note: If poly-ADP-ribosyl binders are to be used, make the following changes in steps a-d: The assay buffer is [10 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.01%(v/v) Tween-20]. Substitute 5 μM mono-ADP-ribosylated YFP-GAP with 500 nM poly-ADP-ribosylated YFP-GAP. Use 250 nM CFP-fusion protein instead of 1 μM. Substitute 200 μM ADP-ribose with 2.5 μM poly-ADP-ribosylated PARP2. These changes can be used in the subsequent steps.
-
6.Validate the FRET signal by competition with ADP-ribose and unmodified protein controls.Note: Black polypropylene plates should be used for the FRET assay. The following protocol is designed for 384-well ShallowWell plates (Fisherbrand). Plates from other manufacturers may be suitable, however a higher assay volume per well may be necessary particularly if 96-well plates are used.
-
a.Prepare in assay buffer [10 mM Bis-Tris-Propane pH = 7.0, 3%(w/v) PEG20,000, 0.01% (v/v) Triton X-100, 0.5 mM TCEP] 50 μL of FRET assay mixture containing 1 μM CFP-fusion protein and 5 μM mono-ADP-ribosylated YFP-GAP.CRITICAL: The assay buffer contains PEG20,000 to promote molecular crowding thus enhancing the FRET signal. The buffer has a low ionic strength due to absence of sodium chloride. Should the proteins precipitate in the buffer, use a buffer without PEG20,000 and/or include sodium chloride in the buffer (e.g. 25–100 mM).
-
b.Prepare in assay buffer 50 μL of control mixture 1 containing 1 μM CFP-fusion protein and 5 μM unmodified YFP-GAP.
-
c.Prepare in assay buffer 50 μL control mixture 2 containing 1 μM CFP-fusion protein, 5 μM mono-ADP-ribosylated YFP-GAP and 200 μM ADP-ribose.Optional: Prepare in assay buffer 50 μL DMSO test mixture containing 1 μM CFP-fusion protein, 5 μM mono-ADP-ribosylated YFP-GAP and 1% (v/v) DMSO.
-
d.Four replicates are prepared per condition. Pipette four times 10 μL of assay buffer (blank) and each mixture prepared in the steps above into separate wells of a black 384-well plate. An example layout for this is shown in Figure 2A.
-
e.Let the plate incubate at room temperature (20°C–25°C) for 5 min.
-
f.Measure the fluorescence emission of each well used with a multimode plate reader.
-
i.Set up a protocol for two separate fluorescence intensity measurements per well.
-
ii.The first fluorescent measurement is set to an excitation wavelength of 430 nm (20 nm bandwidth) and emission wavelength of 477 nm (10 nm bandwidth).Note: In earlier reports we used 410 nm as the excitation wavelength to avoid direct excitation of YFP. However, we found that intrinsic compound fluorescence during screening is typically less apparent when an excitation wavelength of 430 nm is used, which we now recommend.
-
iii.The second fluorescent measurement is set to an excitation wavelength of 430 nm (20 nm bandwidth) and emission wavelength of 527 nm (10 nm bandwidth). All other settings such as the gain, settle time and mirror are to be kept the same as in the previous step.
-
iv.The wells containing only buffer serve as blank. The average fluorescence intensity of the blank is subtracted from the fluorescence intensity of the sample wells for both respective emission wavelengths used.
-
v.The ratiometric FRET signal of each well is calculated by dividing the fluorescence intensity at 527 nm emission wavelength by the fluorescence intensity at 477 nm emission wavelength.Note: An example for this calculation is shown in Table S1.
-
i.
-
g.Evaluate that the rFRET signal measured for the samples containing the FRET assay mixture is significantly higher compared to the signals of the control mixtures 1 and 2. In case 200 μM ADP-ribose did inhibit the signal compared to control mixture 1 significantly less, see troubleshooting 4. If prepared, the DMSO test control should not have an rFRET value significantly lower compared to the FRET assay mixture.Note: We found that a difference of 0.2–0.3 in the FRET ratio between FRET mixture and controls is a sufficient signal to set up a screening assay. If the difference in rFRET is significantly larger (e.g. 1.0–1.5), a lower concentration of the proteins may be used (e.g. 500 nM CFP-fusion and 2.5 μM mono-ADP-ribosylated YFP-GAP).Note: We describe here the minimal validation required to set up the protocol. If more extensive validation is desired, we recommend the NCBI online resource “Assay Guidance Manual” (Coussens et al., 2018) for more specific instructions on e.g. inter-day and inter-plate signal variation or spatial uniformity assessment (https://www.ncbi.nlm.nih.gov/books/NBK83783/).Pause Point: Further validation or preparation of screening assays may be continued at any later time.
-
a.
-
7.Prepare a dose-response curve with a serial dilution series of ADP-ribose. (optional)Note: This part of the validation is not required but gives additional insight into the quality of the signal. Alternatively, a decrease of rFRET signal over time by addition of TEV protease to remove CFP or phosphodiesterase I to remove the mono- or poly-ADP-ribosyl groups may be used as additional validation.
-
a.Prepare in assay buffer 250 μL of 2× FRET assay mixture containing 2 μM CFP-fusion protein and 10 μM mono-ADP-ribosylated YFP-GAP.Note: This volume is sufficient to prepare four replicates for each condition.
-
b.Prepare a 2× concentrated half-logarithmic serial dilution (1:3.16 dilution steps) starting at a concentration of 2 mM ADP-ribose. All solutions are prepared in assay buffer. Samples for each concentration should contain at least 25 μL.Note: For poly-ADP-ribosyl binding proteins, prepare a half-logarithmic serial dilution with poly-ADP-ribosylated PARP2 instead of ADP-ribose. The starting concentration should be 5 μM.
-
c.Pipette four times 5 μL for each concentration of the 2× concentrated dilution series into a 384-well assay plate (columns 3–12, Figure 2B).
-
d.Pipette four times 5 μL buffer into column 2 of the assay plate.
-
e.Pipette four times 10 μL buffer into column 1 of the assay plate.
-
f.Dispense 5 μL of the 2× FRET assay mixture into each well in columns 2–12.Note: This procedure can be done manually or with an automated low-volume liquid dispenser such as Formulatrix Mantis. When transferring the solutions manually, special care must be taken to mix the solutions without creating excessive air bubbles.Optional: Spin down the plate at 500–1000×g for 5 min to remove potential air bubbles. We recommend this step particularly if an automated dispenser has been used, as formation of air bubbles or splashing of the solution may commonly occur.
-
g.Let the plate incubate at room temperature (20°C–25°C) for 5 min.
-
h.Determine the rFRET signals as shown in step 6f.Note: Cover the plate in-between the steps with a lid or adhesive seal to prevent evaporation and accumulation of dust in the wells. Keep the plate at room temperature (20°C–25°C) throughout the measurement procedure.
-
i.Evaluate that the rFRET signal measured result in a curve with decreasing signals for increasing ADP-ribose concentrations. The curve should flatten at high ADP-ribose concentrations (>100 μM).Pause Point: Compound screening may be started at any later time.
-
a.
Figure 2.

Signal validation
An example for the signal validation of CFP-MacroD1 binding to mono-ADP-ribosylated (MAR) YFP-GAP is shown.
(A) Plate layout for comparison of rFRET signals with controls that should abolish FRET with example rFRET values determined with CFP-MacroD1.
(B) Plate layout for a dose-response curve using ADP-ribose with example dose-response curve using ADP-ribose done with CFP-MacroD1. Data are mean ± standard deviation with n = 4 replicates.
Screening
The steps to screen for compounds in a single dose format using a small chemical compound library are detailed in the following section. An example of screening 336 compounds in a 384-well plate including blanks and controls is shown with a final compound concentration of 20 μM.
-
8.Transfer the compounds dissolved in DMSO to a screening plate using Echo 650.Note: The screening plates may also be ordered and prepared externally. In this example, we assume a compound collection containing 336 compounds in Echo-compatible source plates are present.
-
a.Transfer 20 nL of the 10 mM compound stocks dissolved in DMSO to a 384-well assay plate. The compounds are dispensed to wells in columns 4–24 according to the layout in Figure 3.Optional: Add 20 nL of DMSO to the control wells. This is especially advised if the rFRET signal is affected by DMSO.
-
b.Cover the plate with a lid if used on the same day for screening. Seal the plate with an adhesive seal if the plate will be stored for a longer period.Pause Point: The plate may be stored at −20°C until use.
-
a.
-
9.Prepare the solutions required for the screening.Note: While we have not observed issues like a reduced rFRET signal after long-term storage of the protein aliquots at −70°C, we recommend to test the signal on a small scale before starting the screening. The rFRET signals should be comparable to the ones observed during the signal validation steps. If significantly lower signals are observed, the recombinant proteins would need to be prepared freshly.
-
a.Prepare 4 mL of the FRET assay mixture containing 1 μM CFP-fusion protein and 5 μM mono-ADP-ribosylated YFP-GAP.
-
b.Prepare 200 μL of the ADP-ribose control mixture containing 1 μM CFP-fusion protein, 5 μM mono-ADP-ribosylated YFP-GAP and 200 μM ADP-ribose.Note: If the results in the validation showed that 200 μM ADP-ribose did not inhibit the rFRET signal to approximately >95%, a higher concentration of ADP-ribose may be chosen as control.
-
a.
-
10.
Dispense the solutions using Formulatrix Mantis low-volume liquid dispenser (10 μL/well) according to the layout in Figure 3. Buffer is transferred to column 1, the FRET assay solution is transferred to columns 2 as well as 4–24 and the ADP-ribose control solution is transferred to column 3.
Alternatives: Any suitable low-volume liquid dispenser may be used. We recommend the use of automatic dispensers in the screening step because manual pipetting is time intensive and may lead to evaporation and introduction of air bubbles.
-
11.
Spin down the plate at 500–1000×g for 5 min to remove potential air bubbles.
-
12.
Determine the rFRET signal of each well as shown in step 6f.
-
13.
Calculate the apparent inhibition percentages for each well containing the screening compounds. As reference, the average rFRET value of the control FRET assay mixture (column 2) is set to 0% inhibition, while the average rFRET value for the ADP-ribose control mixture (column 3) is set to 100% inhibition.
Note: In the case that many compounds show an apparent inhibition significantly over 100% or below 0%, the compound library used likely contained several compounds that interfere with the signal through intrinsic fluorescence or quenching. Additional filtering steps may be used as described previously for a similar assay system (Sowa et al., 2020).
Optional: Assess the signal quality using the rFRET signals of the control FRET assay mixture and the ADP-ribose control mixture by calculating the Z′-factor (Zhang et al., 1999).
-
14.
Select compounds that show an apparent inhibition of 30% or more as hits. In our experience, this is a robust hit limit. A different cutoff percentage may be chosen depending on the desired hit rate.
Figure 3.

Screening layout
Hit validation
-
15.Prepare a dose-response curve with a serial dilution series of the hit compounds.Note: The following steps describe the validation of a single compound but can be upscaled depending on the number of compounds to be validated. Dose-response curves for 12 hit compounds can be measured per 384-well plate. In the following, duplicates are used for each condition, however a higher number of replicates may also be used if desired.
-
a.Prepare in assay buffer 90 μL of 2× FRET assay mixture containing 2 μM CFP-fusion protein and 10 μM mono-ADP-ribosylated YFP-GAP.
-
b.Prepare 25 μL of 400 μM ADP-ribose in assay buffer.
-
c.Prepare a 2× concentrated half-logarithmic serial dilution (1:3.16 dilution steps) starting at a concentration of 200 μM compound. All solutions are prepared in assay buffer. Samples for each concentration should contain at least 25 μL.Note: Compounds for the dose-response curve may be dispensed directly from compound stocks using an acoustic dispenser such as Echo 650.
-
d.Transfer four times 5 μL for each concentration of the prepared 2× concentrated compound dilution series into a 384-well assay plate (columns 3–8, Figure 4).
-
e.Transfer 5 μL of 400 μM ADP-ribose to each well in column 2.
-
f.Dispense 5 μL of the 2× FRET assay mixture into each well in rows A and B.
-
g.Fill each well to a final volume of 10 μL using assay buffer.Note: This procedure can be done manually or with an automated low-volume liquid dispenser such as Formulatrix Mantis. When transferring the solutions manually, special care has to be taken to mix the solutions without creating excessive air bubbles.
-
a.
-
16.
Spin down the plate at 500–1000×g for 5 min to remove potential air bubbles.
-
17.
Determine the rFRET signals as shown in step 6f. For each condition containing compounds, use the respective wells containing the compound in buffer as a blank (i.e., subtract the average fluorescence intensities of wells C8 and D8 from the fluorescence intensities obtained for wells A8 and B8).
-
18.
Calculate the apparent inhibition percentages as described in step 13. Dose-response curve analysis to determine the IC50 values for each compound may be performed (see “quantification and statistical analysis”).
-
19.
Compounds that show desired inhibition may now be taken for further validation and testing such as functional assays, isothermal titration calorimetry, surface plasmon resonance, differential scanning fluorimetry or protein X-ray crystallography. These methods are not part of this protocol and may be adapted for the specific target protein used.
Optional: Once a target compound was validated to bind to the desired protein, the assay described above may be established with other ADP-ribosyl binding or hydrolyzing proteins to test specificity of the inhibitor. Through the simple and homogenous setup of the assay, it is possible to test inhibition of compounds against many binders simultaneously as described previously (Sowa et al., 2021).
Figure 4.

Dose-response hit validation
Expected outcomes
Upon completion of the protocol, the user should have set up a robust FRET-based binding assay for a mono- or poly-ADP-ribosyl binding protein. Once the required proteins have been prepared and frozen in aliquots, preparation of the homogenous binding or screening assays can be facilitated with little effort. The compounds identified through screening should reduce the rFRET signal in a dose-dependent manner. Identified hit compounds can be taken for further validation using orthogonal biophysical techniques and biological assays. The selectivity of the hit compounds can also be tested using the same FRET-based assay but with different CFP-fused ADP-ribosyl binding proteins.
Quantification and statistical analysis
An example for calculation of the ratiometric FRET signals can be found in Table S1. In dose-response experiments where the decrease of the rFRET signal by addition of ADP-ribose or compound was measured, a non-linear fit was used in GraphPad Prism 8 with the model “log(inhibitor) vs. response – Variable slope (four parameters)”.
Limitations
While the concentrations of proteins used in the assay (1 μM ADP-ribosyl binder & 5 μM ADP-ribosylated YFP-GAP) are comparatively high for screening assays, the amount of expressed proteins is very large due to fusion of the well-expressing CFP and YFP variants. For many proteins, a single preparation from 1 L culture is sufficient for screening efforts comprising 10,000–100,000 compounds. However, a few of the proteins tested did not express very well (Sowa et al., 2021), hindering the applicability to high-throughput screening campaigns. It is worth noting that binding assays for these proteins could still be established and be used to test inhibition of selected compounds.
The rFRET signal generated depends highly on the binding affinity of the ADP-ribosyl binding protein to the cysteine-linked ADP-ribosyl group at the YFP-GAP protein. Though proteins may functionally bind to ADP-ribosyl groups in the cell, they may display low affinity to ADP-ribosyl groups and possibly to cysteine-linked ADP-ribosyl groups in particular. This was seen with the glycohydrolase ARH1, where no signal could be measured likely due to the low binding affinity (Sowa et al., 2021). The family-member ARH3 on the other hand possesses higher affinity to ADP-ribose and a good rFRET signal was measured in the assay for this protein.
Troubleshooting
Problem 1
Low or no expression of the CFP-fusion protein in E. coli or the protein is expressed insolubly in inclusion bodies. (Before you begin, step 4f)
Potential solution
Should the target protein express well without the CFP, it indicates a possible folding issue introduced due to the presence of the CFP protein. A different linker between ADP-ribosyl binding protein and CFP may be considered (Chen et al., 2013). CFP could be fused to the C-terminus instead of the N-terminus. Different CFP-variants may be utilized.
Should no information about the expression of the target protein without CFP be available or it is known that the target protein shows also poor expression in absence of CFP, general strategies for the improvement of protein expression may be considered (Correa and Oppezzo, 2015; Rosano and Ceccarelli, 2014).
Problem 2
A large amount of the CFP without the target protein is present in the lysate and after purification by IMAC. (Before you begin, step 4f)
Potential solution
Proteolytic cleavage may have occurred during expression or purification of the protein. We observed this for several constructs to some extent, however we could in most cases remove the cleaved CFP sufficiently during size exclusion chromatography. If the cleavage occurred excessively and significantly impacts the yield of the final fusion protein preparation, other solutions may be considered. Lowering the temperature (to e.g., 10°C) during expression may help. Use protease inhibitors in the cell lysis step. During purification, ensure that the lysate and protein is kept on ice whenever possible. It may help to perform the whole purification procedure at +4°C.
Problem 3
Analysis by mass spectrometry shows only partial mono-ADP-ribosylation of YFP-GAP. (Step-by-step method details, step 2)
Potential solution
A complete ADP-ribosylation of YFP-GAP yields the best-possible signal for the assay. It is still possible to measure a good rFRET signal with almost complete ADP-ribosylation (e.g., 80% or 90%).
An incomplete modification has occasionally happened to us and has been partially linked to upscaling of the PtxS1 reaction mixture. The reaction can be repeated after initial purification using the partially modified YFP-GAP to yield complete modification.
Problem 4
During signal validation of the CFP-fusion protein with mono-ADP-ribosylated YFP-GAP, the control containing 200 μM ADP-ribose did not show a signal as low as the control containing non-modified YFP-GAP. (Step-by-step method details, step 6g)
Potential solution
The CFP-fusion may display higher affinity to the protein-linked ADP-ribosyl group than to ADP-ribose. Use more ADP-ribose as control (e.g., 1 mM) to ensure complete or almost complete reduction of the rFRET signal. This higher ADP-ribose concentration should be used instead of 200 μM ADP-ribose in the control during the screening and validation steps.
Problem 5
A very potent inhibitor was identified, however it is not possible to determine IC50 values below half of the concentration of the CFP-fused ADP-ribosyl binder. For example, the lowest IC50 value possible to be determined with 1 μM CFP-MacroD1 would be 0.5 μM. (Step-by-step method details, step 18)
Potential solution
Higher concentrations of the CFP-fused binders are recommended when performing screening to achieve a good signal separation which should result in fewer false positive hits. However, the concentration of the CFP-fused ADP-ribosyl binder can be reduced in IC50 experiments as the signal should be robust enough, particularly if replicates are measured. E.g. using 100 nM CFP-MacroD1 and 500 nM ADP-ribosylated YFP-GAP, IC50 values as low as 50 nM can be determined. For binding experiments of such potent inhibitors, we however recommend the use of biophysical methods such as ITC, SPR or BLI.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Lari Lehtiö (lari.lehtio@oulu.fi).
Materials availability
Expression constructs generated in this study are available through Addgene.
Acknowledgments
This work was funded by the Jane and Aatos Erkko Foundation and by the Sigrid Jusélius Foundation. The use of the facilities of the Biocenter Oulu Structural Biology core facility, member of Biocentre Finland, Instruct-ERIC Centre Finland and FINStruct, as well as of Proteomics and Protein Analysis and Sequencing core facilities are gratefully acknowledged.
Author contributions
S.T.S. wrote the initial draft. L.L. conceived the study. S.T.S., A.G.P., S.W., H.I.A., and M.M.M. optimized the experimental conditions for steps described in the protocol. All authors contributed to the final version of the manuscript.
Declaration of interests
The authors declare the following competing financial interests: S.T.S., A.G.P., and L.L. are inventors listed in a patent application related to the described methods and these authors declare no additional interests. The remaining authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2022.101147.
Contributor Information
Sven T. Sowa, Email: sven.sowa@oulu.fi.
Lari Lehtiö, Email: lari.lehtio@oulu.fi.
Supplemental information
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ashok Y., Miettinen M., de Oliveira D.K.H., Tamirat M.Z., Näreoja K., Tiwari A., Hottiger M.O., Johnson M.S., Lehtiö L., Pulliainen A.T. Discovery of compounds inhibiting the ADP-ribosyltransferase activity of pertussis toxin. ACS Infect Dis. 2020;6:588–602. doi: 10.1021/acsinfecdis.9b00412. [DOI] [PubMed] [Google Scholar]
- Bajar B., Wang E., Zhang S., Lin M., Chu J. A guide to fluorescent protein FRET pairs. Sensors. 2016;16:1488. doi: 10.3390/s16091488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg S., Löfdahl P.-Å., Härd T., Berglund H. Improved solubility of TEV protease by directed evolution. J. Biotechnol. 2006;121:291–298. doi: 10.1016/j.jbiotec.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Chen X., Zaro J.L., Shen W.-C. Fusion protein linkers: property, design and functionality. Adv. Drug Deliv. Rev. 2013;65:1357–1369. doi: 10.1016/j.addr.2012.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa A., Oppezzo P. In: Insoluble Proteins. García-Fruitós E., editor. Springer New York; 2015. Overcoming the solubility problem in E. coli: available approaches for recombinant protein production; pp. 27–44. [DOI] [PubMed] [Google Scholar]
- Coussens N.P., Sittampalam G.S., Guha R., Brimacombe K., Grossman A., Chung T.D.Y., Weidner J.R., Riss T., Trask O.J., Auld D., et al. Assay guidance manual: quantitative biology and pharmacology in preclinical drug discovery: assay guidance manual. Clin. Translational Sci. 2018;11:461–470. doi: 10.1111/cts.12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosano G.L., Ceccarelli E.A. Recombinant protein expression in Escherichia coli: advances and challenges. Front. Microbiol. 2014;5:172. doi: 10.3389/fmicb.2014.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa S.T., Vela-Rodríguez C., Galera-Prat A., Cázares-Olivera M., Prunskaite-Hyyryläinen R., Ignatev A., Lehtiö L. A FRET-based high-throughput screening platform for the discovery of chemical probes targeting the scaffolding functions of human tankyrases. Sci. Rep. 2020;10:12357. doi: 10.1038/s41598-020-69229-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa S.T., Galera-Prat A., Wazir S., Alanen H.I., Maksimainen M.M., Lehtiö L. A molecular toolbox for ADP-ribosyl binding proteins. Cell Rep. Methods. 2021 doi: 10.1101/2021.05.31.445082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.-H., Chung T.D.Y., Oldenburg K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Information required to reanalyze the data reported in this paper is available from the lead contact upon request.