

Abstract
Objective
Enhanced de novo lipogenesis (DNL) in hepatocytes is a major contributor to nonalcoholic fatty liver disease (NAFLD). Fatty acid translocase (FAT/CD36) is involved in the pathogenesis of NAFLD through facilitating free fatty acids uptake. Here, we explored the effects of CD36 on DNL and elucidated the underlying mechanisms.
Methods
We generated hepatocyte-specific CD36 knockout (CD36LKO) mice to study in vivo effects of CD36 on DNL under high-fat diet (HFD). Lipid deposition and DNL were analyzed in primary hepatocytes isolated from CD36LKO mice or HepG2 cells with CD36 overexpression. RNA sequence, co-immunoprecipitation, and proximity ligation assay were carried out to determine its role in regulating DNL.
Results
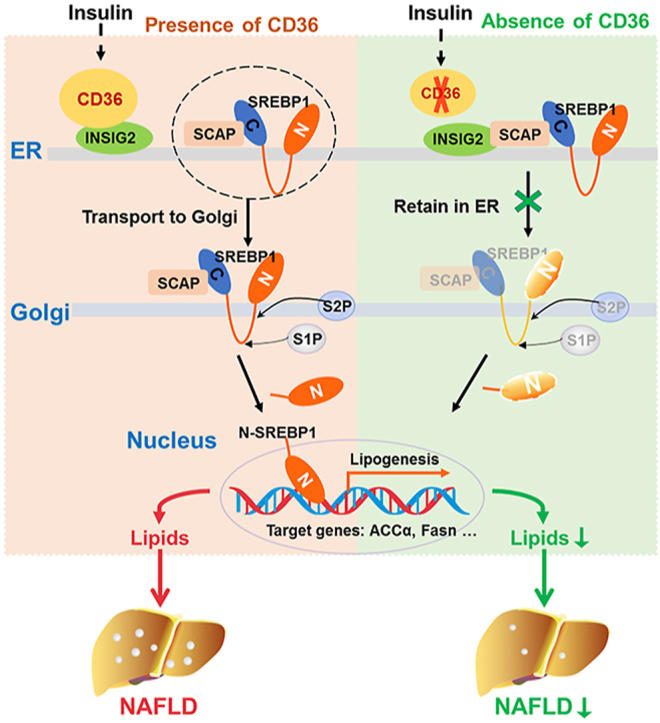
Hepatic CD36 expression was upregulated in NAFLD mice and patients, and CD36LKO mice exhibited attenuated HFD-induced hepatic steatosis and insulin resistance. We identified hepatocyte CD36 as a key regulator for DNL in the liver. Sterol regulatory element-binding protein 1 (SREBP1) and its downstream lipogenic enzymes such as FASN, ACCα, and ACLY were significantly downregulated in the liver of HFD-fed CD36LKO mice, whereas overexpression CD36 stimulated insulin-mediated DNL and lipid droplet formation in vitro. Mechanistically, CD36 was activated by insulin and formed a complex with insulin-induced gene-2 (INSIG2) that disrupts the interaction between SREBP cleavage-activating protein (SCAP) and INSIG2, thereby leading to the translocation of SREBP1 from ER to Golgi for processing. Furthermore, treatment with 25-hydroxycholesterol or betulin molecules shown to enhance SCAP–INSIG interaction, reversed the effects of CD36 on SREBP1 cleavage.
Conclusions
Our findings identify a previously unsuspected role of CD36 in the regulation of hepatic lipogenic program through mediating SREBP1 processing by INSIG2, providing additional evidence for targeting CD36 in NAFLD.
Keywords: NAFLD, CD36, Lipogenesis, SREBP1 processing, INSIG2
Graphical abstract
Highlights
-
•
CD36 plays a novel role in DNL of hepatocytes beyond its known FFA transport function.
-
•
CD36 regulates DNL via SREBP1 processing through interaction with INSIG2.
-
•
Hepatocyte-specific intervention of CD36 is a hopeful therapeutic strategy for NAFLD.
Abbreviations
- 25-HC
25-hydroxycholesterol
- ACCα
acetyl-CoA carboxylase α
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- AUC
area under the curve
- DEGs
differentially expressed genes
- DNL
de novo lipogenesis
- ER
endoplasmic reticulum
- eWAT
epididymal adipose tissue
- FASN
fatty acid synthase
- FAT /CD36
fatty acid translocase
- FFA
free fatty acids
- GTTs
Glucose tolerance tests
- H&E
hematoxylin & eosin
- HFD
high-fat diet
- INSIG
insulin-induced gene
- ITTs
insulin tolerance tests
- NAFLD
nonalcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- NCD
normal chow diet
- PCK1
phosphoenolpyruvate carboxykinase 1
- PLA
proximity ligation assay
- Q-PCR
real-time quantitative PCR
- SCAP
SREBP cleavage-activating protein
- SREBPs
sterol regulatory element-binding proteins
- TC
total cholesterol
- TG
triglycerides
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) has become the leading chronic liver disease worldwide without an effective treatment strategy [1,2]. The hallmark feature of NAFLD is the excessive accumulation of triglycerides (TG) in hepatocytes [3]. These accumulated lipids may arise via increased de novo lipogenesis (DNL), esterification of plasma free fatty acids (FFA), or dietary fatty acids intake [4]. Increasing evidence suggests that enhanced hepatic DNL is an important contributor to the development of NAFLD [[4], [5], [6], [7]]. Human isotope-labeling studies demonstrated a threefold increase of DNL contributed to hepatic TG accumulation in patients with NAFLD than those without [5]. Moreover, the expression of DNL enzymes, such as acetyl-CoA carboxylase α (ACCα) and fatty acid synthase (FASN), were upregulated in NAFLD patients [8,9]. However, the mechanisms responsible for the regulation of hepatic DNL are not fully understood.
Fatty acid translocase (FAT/CD36) is widely expressed in multiple cell types and identified as a FFA transport protein [10]. CD36 is closely associated with the development of NAFLD, which increased concomitantly with hepatic TG content [11,12]. Increased hepatic CD36 expression is significantly related to insulin resistance, hyperinsulinemia, and increased steatosis in patients with non-alcoholic steatohepatitis (NASH) [13]. In addition, soluble CD36 was emerging as a new potential biomarker for NAFLD [14]; therefore, CD36 deletion ought to prevent or reverse hepatic lipid deposition theoretically. Unexpectedly, work from our laboratory and others have found that the deletion of CD36 resulted in exacerbated hepatic steatosis in mice [[15], [16], [17]]. In humans, CD36 deficiency, which is relatively common (2–7%) in person of Asian and African descent, has been reported to exhibit insulin resistance, hyperlipidemia, and a propensity to develop NAFLD [[18], [19], [20], [21]]. These conflicting findings limit the application of targeting CD36 as a therapeutic approach for NAFLD. Our previous study found that CD36 deficiency promoted NASH by inducing macrophage infiltration [16], suggesting CD36-mediated cell–cell interaction may involve in the pathogenesis of NAFLD. In addition, it has been recently reported that the deletion of CD36 in endothelial cell unexpectedly increased liver TG content [22]. These findings indicated that the role of CD36 in NAFLD may be cell type dependent. Importantly, Wilson et al. found that hepatocyte-specific CD36 deletion was protected against HFD-induced hepatic steatosis [23]. Thus, the hepatocyte-specific intervention of CD36 may be more attractive for its further application, although the exact mechanism has not yet been elucidated. Growing evidence indicated that the role played by CD36 extends far beyond the transport of FFA, including FFA oxidation, VLDL secretion, and autophagy [17,24,25]. Given that hepatocyte DNL shows an important function on hepatic steatosis [23,26], it is necessary to clarify the regulatory mechanisms of hepatocyte-specific CD36 involved in DNL.
Sterol regulatory element-binding proteins (SREBPs) are key transcriptional factors for genes in the DNL pathway and play important roles in the pathogenesis of NAFLD [27,28]. SREBPs are synthesized as precursor proteins and retained in inactive forms in the endoplasmic reticulum (ER), where they are bound to two other proteins, insulin-induced gene (INSIG) and SREBP cleavage-activating protein (SCAP). Under the condition of sterol deprivation, SREBP/SCAP are released from INSIG and transported from ER to the Golgi complex, where SREBPs undergo a two-step proteolytic process, liberating the N-terminal, transcriptionally active (∼70Kd) form of SREBPs (N-SREBPs). The released N-SREBPs translocate to nuclear and stimulate lipogenic gene expression [29,30]. It has been reported that SREBP1 is primarily regulated at the transcriptional level by nutritional signals [31,32]. SREBP1 transcription and proteolysis could be induced by insulin and high glucose and inhibited by unsaturated FFAs, particularly polyunsaturated FFAs [31,33,34]. Moreover, it has been demonstrated that saturated FFAs such as palmitate showed an increase or no effect on the cleavage of SREBP1 [35,36], suggesting a potential but unrevealed role of lipid signals in the proteolysis of SREBP1.
In this study, by generating the hepatocyte-specific CD36 knockout (CD36LKO) mice, we elucidated the molecular mechanism of how CD36 regulates hepatic DNL. Hepatocyte CD36 deficiency ameliorated obesity, hepatic steatosis, and insulin resistance in mice were fed with an HFD. Beyond facilitating fatty acid uptake as known, CD36 played an important regulatory role in the proteolysis of SREBP1. We found that CD36 directly interacted with INSIG2 to relieve its inhibitory effect on SREBP1 processing. Our findings identify DNL as a previously unsuspected mechanism by which the fatty acid receptor CD36 regulates hepatic steatosis and suggest the lipogenic role of CD36 in hepatocyte may provide an intervention strategy for the hepatic steatosis.
2. Materials and methods
2.1. Animal model and diets
Hepatocyte-specific CD36 gene knockout (CD36LKO) mice were generated using the Cre-Loxp system. The CD36flox/flox (fl/fl) mice were generated by using a plasmid with Loxp sites flanking CD36 exons 5. After electroporation, selection, and screening, properly targeted embryonic stem cell clones were injected into blastocyst for the generation of chimeric mice. Then, fl/fl mice were crossed with Albumin-cre transgenic mice (Shanghai Model Organisms Center, China) to generate CD36LKO mice. All mouse lines were backcrossed with C57BL/6. Mice were maintained under a constant 12-light/dark cycle with unrestricted access to diet and water. All experimental procedures were approved by the animal care and use committee of Chongqing Medical University.
For induction of NAFLD, male 6- to 8-week-old mice received either a normal chow diet (NCD, D12450B, Research Diets, USA) containing 10 kcal% fat or an HFD (D12492, Research Diets, USA) containing 60 kcal% fat for 16 weeks (n = 12). For fasted (non-fed), fed, and refed experiments, the fed group was placed on an NCD, while the fasted group was fasted for 24 h and the refed group was fasted for 24 h and then refed for 12 h. The liver and epididymal adipose tissue (eWAT) were collected for further analyses.
2.2. Glucose tolerance tests (GTTs) and insulin tolerance tests (ITTs)
GTTs were performed in 12-h-fasted fl/fl and CD36LKO mice following an intraperitoneal injection of glucose (1 g/kg body weight), and ITTs were performed in 4-h-fasted fl/fl and CD36LKO mice following an intraperitoneal injection of insulin (0.7 units/kg body weight). Blood glucose levels from tail vein blood were measured at 0, 15, 30, 60, and 120 min with an ACCU-CHEK Advantage glucometer (Roche Diagnostics, USA).
2.3. Histological analysis
Liver and eWAT were fixed in 4% paraformaldehyde in PBS. Histological changes were examined by hematoxylin & eosin (H&E) stain. Fibrosis changes were examined by Sirius red stain. Lipid accumulation in the liver was analyzed by Oil red O staining on the frozen liver tissues. Micrographs were captured using an automated whole-slide scanning device (3DHISTECH, China). The quantitative analysis of Oil red O staining was performed using ImageJ.
2.4. Measurement of TG and total cholesterol (TC)
Liver tissues were homogenized and extracted in isopropanol for 48 h and then centrifuged at 3000 rpm for 10 min at 4 °C. TG and TC levels were measured by TG kit and TC kit (Biosino, China). Measurements of liver lipids were normalized to those of liver weight.
2.5. Serum biochemistry analysis
Serum TG, serum TC, serum glucose, serum FFA, serum aspartate aminotransferase (AST), and alanine aminotransferase (ALT) were determined by an automatic biochemistry analyzer as previously described [15].
2.6. Cell lines and cell treatment
Human HepG2 cell were maintained in a complete medium containing high-glucose DMEM, 10% fetal bovine serum, and 1% penicillin-streptomycin. The lentiviral (GV341) particles carrying negative control (NC) and CD36 cDNA (CD36OE) were constructed by Genechem (China). The lentiviral human full-length SREBP1 (FL-SREBP1) expression vector with an N-terminal HA tag was purchased from GeneCopoeia (China) and then packaged into lentiviral particles by Obio technology (China). The transfected cells stably expressing CD36 and/or SREBP1 were cultured for further assays.
For insulin-induced lipogenesis, cells were cultured in serum-free medium overnight before being treated without or with 100 nM insulin for 0.5, 1, 3, 6, 12, or 24 h. For inhibition of SREBP1 translocation, cells were cultured in serum-free medium overnight before being treated with DMSO, 15 μM betulin (HY-N0083, MCE, USA), or 5 μM 25-hydroxycholesterol (25-HC, HY-113134, USA) plus 100 nM insulin for 24 h. For small interfering RNA (siRNA) knockdown, HepG2 cells were transfected with siCD36 (GGCUGUGUUUGGAGGUAUUCUTT) and/or siINSIG2 (CUCACACUGGCUGCACUAUTT) using Lipofectamine RNAiMAX (13778030, Life Technologies, USA). The transfected cells were cultured in serum-free medium for 12 h and then treated with 100 nM insulin for 12 h.
2.7. Primary hepatocytes culture
Primary hepatocytes were isolated from the livers of 8- to 10-week-old fl/fl and CD36LKO mice by a modification of the two-step collagenase perfusion method, as previously described [15]. Briefly, perfusion through the hepatic portal vein commenced successively with buffer A (2.5 mM EGTA, 0.1% glucose, and 2% penicillin/streptomycin) and buffer B (5 mM CaCl2 and 0.5 mg/ml IV collagenase). The isolated mouse hepatocytes were then plated in collagen I-coated 6-well plates. For insulin treatment, hepatocytes were maintained in serum-free medium overnight and then treated with 100 nM insulin for 12 h.
2.8. BODIPY493/503 staining
HepG2 or primary hepatocytes were washed twice with PBS and fixed with 4% paraformaldehyde. BODIPY493/503 (D3922, Invitrogen, USA) was used to stain intracellular lipid droplets. DAPI was used to stain nuclei. Images were obtained using a Leica confocal microscope (Nikon, Japan). Fluorescent intensity was analyzed using ImageJ.
2.9. Co-immunoprecipitation and immunoblotting
Tissue or cell lysates were prepared with RIPA buffer. Immunoprecipitation and/or immunoblotting were performed as described previously [15]. Antibody information is listed as follows: rabbit anti-CD36 (NB400-144), mouse anti-CD36 (NB600-1423), and mouse anti-SREBP1 (NB600-582) were purchased from Novus Biological (USA); rabbit anti-SREBP1 (14088-1-AP), rabbit anti-SREBP2 (28212-1-AP), rabbit anti-Golgin97 (12640-1-AP), mouse anti-GM130 (66662-1-Ig), rabbit anti-INSIG2 (24766-1-AP), rabbit anti-lamin b1 (12987-1-AP), and mouse anti- GAPDH (60004-1-Ig) were purchased from Proteintech Group (USA); rabbit anti-ACLY (4332), rabbit anti-HA-tag (3724), rabbit anti-FLAG-tag (14793), mouse anti-FLAG-tag (8146), and rabbit anti-Calnexin (2433) were purchased from Cell Signaling Technology (USA); rabbit anti-ACCα (sc-30212), mouse anti-FASN (sc-48357), mouse anti-SCAP (sc-13553), and rabbit anti-INSIG1 (sc-25124-R) were purchased from Santa Cruz Biotechnology (USA); rabbit anti-SCD1 (E-AB-66816) was purchased from Elabscience Biotechnology (China); rabbit anti-HMGCR (bsm-52822R) and rabbit anti-β-ACTIN (bs-0061R) were purchased from Bioss Biotechnology (China).
2.10. Nuclear and cytoplasmic extraction
Nuclear and cytoplasmic extraction were prepared from HepG2 cells according to the manufacturer's instructions of NE-PER nuclear and cytoplasmic extraction kit (78833, Thermo Fisher Scientific, USA). Subsequently, proteins were used for immunoblots.
2.11. Isolation of ER and Golgi
ER and Golgi fractions were performed according to a previous report [37]. The distribution patterns of the subcellular compartment markers were determined by immunoblots (ER marker: Calnexin; Golgi apparatus marker: GM130) and pooled together as ER and Golgi fractions.
2.12. Immunofluorescence and proximity ligation assay (PLA)
HepG2 cell or primary hepatocytes were washed with PBS, fixed with 4% paraformaldehyde, permeabilized with 0.3% Triton X-100, and then blocked with 3% BSA. Cells were incubated overnight at 4 °C with primary antibodies and incubated with fluorescently labeled secondary antibodies. PLA analysis was performed according to the manufacturer's instructions of Duolink® PLA kit (Sigma, USA). Briefly, paraformaldehyde-fixed cells were permeabilized with 0.3% Triton X-100 and blocked with blocking solution. The primary rabbit and mouse antibodies were applied, and the cells were incubated with PLA probes, anti-rabbit PLUS, and anti-mouse MINUS. The incubation was followed by hybridization and ligation, and amplification was performed. Fluorescence images were obtained using a Leica confocal microscope (Nikon, Japan).
2.13. RNA isolation, real-time quantitative PCR (Q-PCR) analysis, and RNA sequence
Liver tissues or cells were lysed in RNAiso Plus (9108, TaKaRa, Japan) to extract total RNA according to the manufacturer's protocols. cDNA was synthesized by a PrimeScript® RT reagent kit (DRR037A, TaKaRa, Japan). Q-PCR was performed using the SYBR Green PCR Mix kit (TaKaRa, Japan) and the CFX connect real-time system (Bio-Rad, USA). Gene expression levels were normalized to β-actin, and relative levels were compared to control samples using the 2-ΔΔCt method.
For RNA sequence analysis, fl/fl and CD36LKO mice were fed with HFD for 16 weeks, and the liver mRNA sequence analysis was performed by the Illumina Hiseq2000 platform of Majorbio Biotech (China). The data were analyzed on the free online Majorbio I-Sanger Cloud Platform (www.i-sanger.com). RNA sequencing data were submitted to the GEO database at NCBI (GSE191059).
2.14. mRNA expression profiling from public data
Hepatic CD36 gene expression analysis was performed using the publicly available NCBI GEO dataset (www.ncbi.nlm.nih.gov/geo, GSE151158). Within the dataset, a normal control group (control, n = 21) and a group of patients diagnosed with NAFLD (NAFLD, n = 40) were selected, and CD36 mRNA expression levels were evaluated between the groups.
2.15. Quantification and statistical analysis
Differences between 2 groups were analyzed by 2-tailed Student's t-test. Comparisons among more than 3 groups were analyzed by 1-way ANOVA (GraphPad Prism 8). Statistical differences were considered significant at P-value < 0.05. Data are reported as mean ± SEM.
3. Results
3.1. The expression of CD36 in liver is correlated with feeding and insulin level
To evaluate the potential role of CD36 in NAFLD pathogenesis, we firstly analyzed hepatic CD36 levels in mice and humans with NAFLD. Liver protein and mRNA levels of CD36 were increased by more than twofold in mice that were fed with HFD for 16 weeks, which displayed simple steatosis, obesity, and insulin resistance (Figure 1A,B, Figure S1). In publicly available human liver datasets, the gene expression of CD36 is upregulated in subjects with early, non-fibrotic NAFLD (Figure 1C). These findings were consistent with our previous study showing higher CD36 expression in NASH patients [13], but indicated a role of CD36 in the early stages of NAFLD.
Figure 1.

Hepatic CD36 is regulated by nutritional signals. A–B: C57BL/6J male mice were fed with either the normal chow diet (NCD) or high-fat diet (HFD) for 16 weeks. The hepatic expression of CD36 was analyzed by immunoblotting (A) or real-time quantitative PCR (Q-PCR) (B). n = 3, ∗P < 0.05 vs. NCD group. C: The hepatic CD36 mRNA levels in the nonalcoholic fatty liver disease group (NAFLD, n = 40) and normal control group (control, n = 21) from the GEO database (GSE151158). ∗P < 0.05 vs. control group. D–E: C57BL/6J male mice were overnight-fasted and then fed again 12 h. Livers were collected and the protein level of CD36 (D) and SREBP1 signaling (E) were analyzed by immunoblotting. n = 3, ∗P < 0.05 vs. fasted group. F: C57BL/6J male mice were fasted overnight and injected with PBS or insulin (1U/kg body weight) for 4 h. Livers were collected and the protein level of CD36 was analyzed by immunoblotting. n = 3, ∗P < 0.05 vs. PBS group. G: HepG2 cells were treated with insulin (100 nM) for indicated time points. The protein levels of CD36 were analyzed immunoblotting. n = 3.
To investigate whether CD36 is a nutritional sensor, we measured the expression of CD36 in response to fasting and feeding. Hepatic CD36 expression was reduced in overnight-fasted state but was increased by feeding signals (Figure 1D). Feeding simultaneously activated hepatic SREBP1-related lipogenesis, as reported in previous studies [32], indicating a potential link between CD36 and lipogenesis (Figure 1E). Given that NAFLD nutritional status and lipogenesis are all strongly associated with insulin levels, we examined the effect of insulin on CD36 expression. In fasted mice, hepatic CD36 expression was increased by insulin injection intraperitoneally (Figure 1F). Consistent with in vivo results, the expression of CD36 was increased by insulin in a time-dependent manner in liver cells (Figure 1G). Together, these results suggest that CD36 can be stimulated by insulin and may play a role in the regulation of lipogenesis.
3.2. CD36 depletion of hepatocyte improves HFD-induced hepatic steatosis
To investigate the hepatocyte-specific role of CD36 in hepatic steatosis, we generated CD36LKO mice using the Cre/Loxp system. The fl/fl mice were crossed with Albumin-cre mice to generate CD36LKO mice (Figure S2A). The knockout efficiency was verified by immunoblot and Q-PCR (Figure 2B,C, E). We confirmed that CD36 was deleted effectively in the liver but not in other tissues (Figure S2D and E). No obvious difference in body weight, food intake, and liver weight was observed between fl/fl and CD36LKO mice that were fed with NCD (Figure S2F-H), as well as serum levels of glucose, TG, TC, ALT, and AST (Figure S2I-M). Consistently, liver histology revealed no obvious morphological changes in NCD-fed CD36LKO mice compared with fl/fl mice (Figure S2N). GTTs and ITTs also displayed no significant difference between the two genotypes (Figure S2O and P). These data demonstrate comparable metabolic phenotype in CD36LKO mice and fl/fl mice under NCD condition.
We then placed CD36LKO mice and their littermates on HFD for 16 weeks (Figure 2A). Hepatocyte CD36 deficiency attenuated HFD-induced body weight gain, without affecting liver weight and food intake (Figure 2B–D). The fasting serum TG levels in CD36LKO mice exposed to HFD were significantly lowered than those in fl/fl mice, although serum glucose and TC levels were similar (Figure 2E–G). Serum activities of ALT and AST, reflecting liver function, tend to be decreased in the HFD-fed CD36LKO mice (Figure 2H,I). Despite the GTTs being similar between genotypes, the ITTs were improved in the HFD-fed CD36LKO mice (Figure 2J,K). Histological examination of the livers from the HFD-fed CD36LKO mice showed reduced lipid deposition and no detectable hepatic fibrosis (Figure 2L,M). H&E-stained sections of eWAT from CD36LKO mice displayed decreased adipocyte size, in accordance with less body weight gain (Figure 2N). Furthermore, Oil red O staining and TG/TC quantitation revealed less lipid accumulation in the livers of CD36LKO mice than those of fl/fl mice (Figure 2O–Q). These data suggest that hepatocyte-specific knockout of CD36 prevents HFD-induced obesity, hepatic steatosis, and insulin resistance.
Figure 2.

CD36LKO mice were protected from HFD-induced hepatic steatosis. The hepatocyte-specific CD36 knockout (LKO) mice and control mice (fl/fl) were fed a HFD for 16 weeks (n = 12). A: Schematic experimental design to determine the effects of hepatocyte-specific CD36 on NAFLD. B-C: Body weight and daily food intake during the 16-week HFD condition were measured. D: Liver weight of 16-week-HFD-fed was measured. E–I: Overnight-fasted serum glucose levels (E), total triglycerides (TG) levels (F), total cholesterol (TC) levels (G), as well as ALT (H) and AST (I) levels were analyzed (n = 6). J–K: Glucose tolerance tests (GTTs, J) and insulin tolerance tests (ITTs, K) in HFD-fed mice. Quantification of the area under the curve (AUC) was shown in the right (n = 6). L–N: Representative images of hematoxylin & eosin (H&E)-stained livers (L), Sirius red-stained livers (M), and H&E-stained epididymal white adipose tissues (eWAT, N). Scale bar, 100 μm. O-P: Liver contents of TG and TC were determined (n = 6). Q: Representative images of Oil red O-stained livers. Quantification of Oil red O-stained area was shown in the right (n = 4; Scale bar, 100 μm). ∗P < 0.05 vs. fl/fl mice.
3.3. CD36 deficiency of hepatocyte attenuates the hepatic lipogenic program
To further explore the potential mechanisms for reduced lipid accumulation in CD36LKO mice, we performed RNA sequence analysis of the livers from HFD-fed fl/fl and CD36LKO mice. We identified 222 upregulated genes and 222 downregulated genes (>1 fold change and adjusted P-value<0.05). Through GO analysis, we found these differentially expressed genes (DEGs) are enriched in lipid metabolic pathways (Figure 3A). We mapped the DEGs from the top 20 enriched GO terms to the STRING database and found that the hub genes were involved in fatty acid metabolism (Srebp1, Fasn, Acly, Ppara, Pparg) and cholesterol metabolism (Ldlr, Vldlr, Apoa1, Apoa4) (Figure 3B). Volcano plot analysis showed that CD36LKO mice exhibited different gene expression patterns versus fl/fl mice with the downregulation of genes involved in fatty acid metabolism (Figure 3C). Furthermore, heatmap analysis of fatty acid metabolism-related genes showed that the expression of lipogenic genes was markedly suppressed in the CD36LKO mice (Figure 3D).
Figure 3.

Decreased hepatic lipogenesis in LKO mice. RNA sequence analysis of the livers from fl/fl and LKO mice under 16-week-HFD (n = 3). A: Top 20 biological process gene ontology (GO) terms enriched by differentially expressed genes (DEGs) in fl/fl and LKO mice. The DEGs were selected with a criterion at adjusted P-value < 0.05. B: The protein–protein interactions (PPI) network analysis of genes including in top 20 BP GO terms. The top 15 hub genes were selected and visualized with Cytoscape software. The size and color of map nodes were determined by degree score. C: Heatmap representation of genes involved in fatty acid metabolism. The gradual color ranged from pink to blue represented the mRNA expression changing from up-regulation to down-regulation. D: Volcano plot comparison of genes in fl/fl versus LKO mice, showing the distribution of significance [-log10 (P adjust value)] vs. fold change (FC) [log2 (FC)] for all genes. The red points represented up-regulated genes (log2 (FC) > 1 and P adjust value < 0.05, 222 genes) and green points represented down-regulated genes (log2 (FC) < −1 and P adjust value < 0.05, 222genes), while gray points indicated genes without significant differential expression (|log2 (FC) | < 1 or a P adjust value > 0.05). E–G: The liver mRNA levels of lipogenic genes (E), inflammatory genes (F), and fatty acid β-oxidation genes (G) determined by Q-PCR (n = 3). H: Hepatic expression of the SREBP1 and downstream lipogenic enzymes determined by immunoblotting. Quantitation of protein levels was shown in the right (n = 3). ∗P < 0.05 vs. fl/fl mice.
As shown in Figure 3E–G, the Q-PCR results agreed with those of RNA sequence analysis. The mRNA levels of lipogenic genes, including Srebp1c, Accα, Fasn, Scd1, Acly, and Pparg, were lower in the CD36LKO mice (Figure 3E). In contrast, the expression of genes that regulate fatty acid β oxidation (Cpt1 and Acox1) or inflammation (Mcp-1, Tnf-α, and Il-6) were comparable between fl/fl and CD36LKO mice (Figure 3F,G). We further confirmed these results in the livers by immunoblot analysis. Hepatic protein levels of SREBP1, ACCα, FASN, SCD1, and ACLY were all markedly decreased in the CD36LKO mice compared with fl/fl mice (Figure 3H). Besides, we found that the mRNA and protein expression of SREBP2 and its target gene HMGCR were also decreased in the CD36LKO mice (Figure S3A and B). Thus, hepatocyte CD36 deficiency presumably attenuates hepatic lipid deposition through the inhibition of lipogenesis.
3.4. CD36 mediates insulin-stimulated DNL in liver cells
The facilitation of FFA uptake by CD36 has been investigated extensively. Under in vivo conditions, it is hard to determine whether the attenuated liver steatosis in HFD-fed CD36LKO mice is due to reduced FFA uptake or decreased lipogenesis. To directly verify the contribution of CD36-mediated lipogenesis to lipid deposition, we used an in vitro model to study the effects of CD36 on lipid accumulation without exogenous FFA involvement. HepG2 cells or primary hepatocytes were serum deprived and stimulated with insulin to induce DNL (Figure 4A). As expected, insulin-induced SREBP1 activation and increased ACLY and ACCα protein levels, the rate-limiting enzymes of lipogenesis (Figure 4B). Consistently, insulin increased lipid droplet formation as evidenced by staining with neutral lipid dye BODIPY493/503 in both HepG2 cells and primary hepatocytes (Figure 4C,D). Overexpressing CD36 caused increased lipid droplet accumulation in response to insulin, while CD36 deletion prevented the lipogenic effects of insulin under serum-free condition (Figure 4E,G). At the molecular level, CD36 overexpression stimulated SREBP1 and its downstream lipogenic enzymes, including FASN, ACCα, SCD1, and ACLY, while CD36 deletion showed the opposite effects (Figure 4F,H). Besides, we found that CD36 deletion also inhibited SREBP2 and its downstream target HMGCR (Figure S3C). These data suggest that CD36 promotes insulin-mediated DNL, which contributes to lipid deposition in liver cells.
Figure 4.

CD36 is involved in insulin-stimulated DNL in vitro. A: Experimental design for insulin-stimulate DNL in vitro. B: HepG2 cells were treated with insulin (100 nM) for indicated time points. The protein levels of SREBP1 and downstream lipogenic enzymes were analyzed by immunoblotting. C–D: BODIPY staining for lipid droplets in HepG2 cells (C) or primary mouse hepatocytes (D) with or without insulin (100 nM) treatment for 12 h. Quantitative of lipid droplets was shown in the right. ∗P < 0.05 vs. control group. E: BODIPY staining for lipid droplets in CD36 overexpression (CD36OE) or NC cells with insulin (100 nM) treatment for 12 h. Quantitative of lipid droplets was shown in the right. ∗P < 0.05 vs. NC or fl/fl group. F: BODIPY staining for lipid droplets in primary hepatocytes isolated from fl/fl or LKO mice with insulin (100 nM) treatment for 12 h. Quantitative of lipid droplets was shown in the right. ∗P < 0.05 vs. fl/fl group. G–H: Protein levels of SREBP1 and downstream lipogenic enzymes in CD36OE cells (G) or primary hepatocytes lacking CD36 (H). Quantitation of protein levels was shown in the bottom. n = 3, ∗P < 0.05 vs. NC, or fl/fl group.
3.5. CD36 regulates proteolytic processing of SREBP1
SREBP1 activation requires its translocation from ER to the Golgi for subsequent cleavage and release of the active N-SREBP1 [38]. To gain further insights into the underlying mechanisms by which CD36 regulates SREBP1 activation, we accessed the subcellular distribution of SREBP1 in the liver of fl/fl and CD36LKO mice. In the separated ER fractions, the protein levels of SREBP1 were similar between fl/fl and CD36LKO mice, but in the Golgi fractions, there was a decrease of SREBP1 in CD36LKO mice compared with that in the fl/fl mice (Figure 5A). Consistent with the reduction of transport-dependent proteolysis of SREBP1 in the liver of CD36LKO mice, immunofluorescence staining observed decreased nuclear and Golgi localization of SREBP1 in primary hepatocytes from CD36LKO mice (Figure 5B,C). Conversely, CD36 overexpression increased nuclear and Golgi localization of SREBP1 (Figure 5D–F). These results clearly show that CD36 is needed for SREBP1 transport to Golgi.
Figure 5.

CD36 regulates ER-to-Golgi transport of SREBP1. A: The protein level of SREBP1 in Golgi and ER of organelle fractions from fl/fl and LKO mice was analyzed by immunoblotting. B: Nuclear location of SREBP1 (red-TRITC) was determined by immunofluorescence staining and confocal analysis in primary hepatocytes from fl/fl and LKO mice. Scale bars, 25 μm. C: Localization of SREBP1 (green-FITC) in the Golgi (red-TRITC) was determined by immunofluorescence staining and confocal analysis in primary hepatocytes from fl/fl and LKO mice. Scale bars, 25 μm. D: The protein levels of SREBP1 of cytoplasmic and nuclear fractions from NC and CD36OE cells were analyzed by immunoblotting. E: Nuclear location of SREBP1 (red-TRITC) was determined by immunofluorescence staining and confocal analysis in NC and CD36OE cells. Scale bars, 25 μm. F: The localization of SREBP1 (green-FITC) in the Golgi (red-TRITC) was determined by immunofluorescence staining and confocal analysis in NC and CD36OE cells. Scale bars, 25 μm. G–H: HepG2 cells were co-transfected with stably expressing HA-tagged full-length SREBP1 (FL-SREBP1) and FLAG-tagged-CD36. The precursor and active nuclear form of SREBP1 were visualized using HA antibody (G) and the SREBP1 downstream targets, including ACCα, FASN, SCD1, and ACLY, were analyzed by immunoblotting (H). Quantitation of protein levels was shown in the bottom. n = 4, ∗P < 0.05 vs. SREBP1OE group without CD36OE.
SREBP1 trans-activated its own gene expression, forming a positive feedback loop [39]. To directly confirm CD36 regulates SREBP1 processing, we generated an exogenous HA-tagged FL-SREBP1 expression system, which is driven by a cytomegalovirus promoter. We introduced this FL-SREBP1 into NC and CD36OE liver cells, respectively. The FL-SREBP1 was similarly overexpressed in NC and CD36OE cells, but the expression of nuclear form of SREBP1 was markedly increased in CD36OE cells (Figure 5G). At the same time, the expression of SREBP1 downstream targets, including ACCα, FASN, SCD1, ACLY, were elevated in CD36OE cells compared with those in NC cells (Figure 5H). Together, these data suggest CD36 regulates the proteolytic processing of SREBP1.
3.6. CD36 interacts with INSIG2
Given that INSIGs play a negative role in SREBP1 processing [40], changing the expression of INSIGs may modulate the proteolysis of SREBP1. Firstly, we examined whether CD36 affects the expression of INSIGs. The protein levels of INSIG1 and INSIG2 were unchanged between HFD-fed CD36LKO and fl/fl mice, although these mRNA levels showed slightly reduced in CD36LKO mice (Figure 6A,B). As SREBP1 processing was reduced in CD36LKO mice, thus, these results excluded the potential contribution of altered INSIGs expression to CD36-mediated SREBP1 activation. Then, since CD36 can be localized to ER where INSNG is anchored [41], we tested whether CD36 could bind to INSIGs to change their function. Immunoprecipitation assay demonstrated that CD36 interacted with INSIG2 (Figure 6C). Similarly, immunofluorescence showed that CD36 co-localized with INSIG2 (Figure 6D). PLA results demonstrated close proximity (<40 nm) between CD36 and INSIG2 but not INSIG1 (Figure 6E). Finally, we investigated whether the CD36–INSIG2 interaction would affect the binding of SCAP to INSIG2. PLA signals for SCAP–INSIG2 interaction were observed to be reduced in CD36OE cells compared with NC cells, suggesting that CD36 competitively inhibits the combination of SCAP to INSIG2 (Figure 6F). Consistent with decreased interaction of INSIG2 with SCAP, we observed increased Golgi-located SCAP in CD36OE cells, while CD36 deletion decreased Golgi-located SCAP (Figure 6G,H). Taken together, these data indicate that CD36–INSIG2 interaction disrupts the binding of SCAP to INSIG2.
Figure 6.

CD36 interacts with INSIG2 but not INSIG1. A–B: The hepatic protein (A) and mRNA (B) levels of INSIG1 and INSIG2 in fl/fl and LKO mice under 16-week-HFD. Quantitation of protein levels was shown in the right. n = 4, ∗P < 0.05 vs. fl/fl mice. C: HepG2 cells expressing FLAG-CD36 were subjected to immunoprecipitation with FLAG (top), INSIG1 (middle), or INSIG2 (bottom) antibodies, and the levels of the co-immunoprecipitated protein were immunoblotted with INSIG1, INSIG2, or CD36 antibodies. D: The co-localization of CD36 (red-TRITC) and INSIG2 (green-FITC) in NC and CD36OE cells was determined by confocal microscopy. Scale bars, 25 μm. E: The physical interactions of CD36 with INSIG1 (top) or INSIG2 (bottom) in CD36OE cells were detected by proximity ligation assay (PLA). Representative PLA images where the PLA signal (red) represented close proximity (<40 nm) between two proteins. Scale bars, 25 μm. F: The physical interactions of INSIG2 with SCAP in NC and CD36OE cells were detected by PLA. Scale bars, 25 μm. G–H: The localization of SCAP (green-FITC) in the Golgi (red-TRITC) was determined by immunofluorescence staining and confocal analysis in NC and CD36OE cells (G) or fl/fl and LKO mouse primary hepatocytes (H). Scale bars, 25 μm.
3.7. CD36 is involved in the regulation of INSIG2 on SREBP1 processing
To confirm the effects of CD36 on SREBP1 processing and lipogenesis are due to loss function of INSIG2, knockdown of both CD36 and INSIG2 using small interfering RNA (siCd36 or siInsig2) was performed in HepG2 cells with insulin treatment. Compared with siCon cells, knockdown of CD36 inhibited the cleavage of SREBP1 and caused a reduction of lipogenic gene expression, including SCD1, ACCα, FASN, and ACLY (Figure 7A–C). Strikingly, the effects of siCd36 on SREBP1 and lipogenesis were restored by the inhibition of INSIG2 (Figure 7A–C). These data demonstrate that INSIG2 is a key pathway by which CD36 regulates SREBP1 processing.
Figure 7.

INSIG2 is a key pathway by which CD36 regulates SREBP1 processing. A–C: After a 24 h period of transfection with siCon, siCd36, and/or siInsig2, HepG2 cells were cultured in serum-free medium for 12 h and then treated with 100 nM insulin for 12 h. The expression of SREBP1 and downstream targets were analyzed by Q-PCR (A). n = 4–6. Nuclear localization of SREBP1 was determined by immunofluorescence staining and confocal analysis (B). Scale bars, 25 μm. Quantitative of nuclear SREBP1 density was shown in the right. The expression of SREBP1 and downstream targets were analyzed by immunoblotting (C). Quantification of protein levels was shown in the bottom. n = 3, ∗P < 0.05 vs. siCon, #P < 0.05 vs. siCd36. D–E: CD36OE or NC cells were cultured in serum-free medium overnight and then treated with DMSO, 25-hydroxycholesterol (25-HC, 5 μM), or betulin (10 μM) in the presence of insulin (100 nM) for 24 h. Nuclear localization of SREBP1 was determined by immunofluorescence staining and confocal analysis (D). Quantitative of nuclear SREBP1 density was shown in the bottom. Scale bars, 25 μm. The expression of SREBP1 and downstream targets were analyzed by immunoblotting (E). n = 3. Quantification of protein levels was shown in the right. ∗P < 0.05 vs. NC, #P < 0.05 vs. NC + DMSO, †P < 0.05 vs. CD36OE + DMSO.
As the above data indicated CD36 restricted the binding of SCAP to INSIG2 (Figure 6F), we speculated CD36-inhibted SCAP–INSIG2 interaction accelerates the proteolytic processing of SREBP1. To validate this hypothesis, we incubated CD36 OE cells with 25-HC and betulin, two small molecules shown to promote SCAP–INSIGs interaction and prevent SCAP-SREBP transportation from ER to Golgi [42,43]. As expected, treatment with 25-HC or betulin blocked CD36 overexpression-induced SREBP1 cleavage and lipogenic genes expression (Figure 7D,E). These results suggest that the effects of CD36 on SREBP1 processing are dependent on the regulation of INSIG2–SCAP interaction.
4. Discussion
Exploiting the mechanisms leading to the activation of DNL may be beneficial in finding potential therapeutic strategies for NAFLD. In the current study, our data showed that hepatic CD36 is upregulated in mice and humans with early NAFLD. We revealed a stimulatory effect for hepatocyte-specific CD36 in the development of HFD-induced NAFLD. More importantly, a novel role of CD36 in regulating the SREBP1 activation and subsequent DNL in the liver were elucidated. Our study provides a molecular mechanism of how CD36 regulates SREBP1 processing via binding with INSIG2, which contributes to increased DNL in the development of NAFLD.
The significance of CD36 in the pathogenesis of NAFLD has been indicated, proposing CD36 as a potential target for treating NAFLD. Upregulated CD36 expression is a general property of steatotic livers compared with healthy livers [11]. Overexpression of CD36 has been linked to exacerbated steatosis by mechanisms involving increased hepatic FFA uptake and TG storage [12]. However, CD36 deficiency paradoxically promoted the development of NAFLD. In humans, patients with genetic CD36 deficiency have a propensity to develop fatty liver by unknown mechanisms [44]. CD36 deletion in ob/ob mice exacerbated liver steatosis by suppressing hepatic VLDL output [17]. Similarly, our previous study showed that CD36 knockout mice displayed hepatic insulin resistance and lipid deposition even fed a low-fat diet [15] and exhibited aggravated hepatic inflammation, steatosis, and fibrosis when fed an HFD [16]. The enigmatic discrepancy between CD36 deficiency protecting from and predisposing to NAFLD may be partially explained by tissue or cell specificity. Therefore, the cell type-specific intervention of CD36 may be more attractive for its further application. Our previous results have shown reduced palmitic acid-induced lipid accumulation by CD36 knockdown, while increased lipid accumulation by CD36 overexpression in liver cells [25]. Consistent with the in vitro results, in the present study, we found that hepatocyte-specific CD36 deficiency in mice markedly attenuated the development of hepatic steatosis. Our findings suggested that hepatocyte-specific intervention of CD36 could be a promising therapeutic strategy to treat NAFLD.
CD36, well known as a FFA transport protein, facilitates FFA uptake in adipocyte, cardiac, and skeletal muscle cells [45,46]. However, the contribution of CD36 to liver FFA uptake remains inconclusive. Hepatocyte-specific CD36 disruption in mice was shown to reduce hepatic uptake of BODIPY-FL C16 [23]. Compared with fatty acid analogs conjugated to fluorescent, such as BODIPY-FL C16, FFA uptake assays with radiolabeled FFAs have the additional advantage of faithfully mimicking the biochemical properties of natural FFAs. An observation in humans with CD36 deficiency showed impaired [11C] palmitate uptake by the heart but no restriction in the liver [47]. In the CD36 knockout mice, the rate of [3H] oleic acid uptake was only reduced in the heart, skeletal muscle, and adipose tissues, without changes in the liver [48,49]. These findings suggested that CD36 might not be critically required for FFA uptake in the liver. There is increasing evidence indicating that CD36 not only acts as a FFA transporter, but also involves in many other metabolic process. For example, Nassir et al. have found that CD36 deletion reduced VLDL secretion in genetic-obese mice [17]. Our previous study reported that CD36 played a negative role in regulating autophagic degradation of lipid droplets through an AMPK-dependent pathway [25]. A significant observation in the present study is the identification of CD36 as a key regulator for SREBP1-mediated DNL in the liver. Our findings were consistent with the previous studies showing decreased expression of lipogenic genes in CD36 deficient mice in response to different environmental stimuli [23,49]. The overexpression of CD36 caused an increased SREBP1 processing and lipogenesis, while CD36 deficiency resulted in the opposite effects. Thus, our study extended previous research by elucidating the role of CD36 in the regulation of SREBP1 processing.
INSIGs are ER anchor proteins and are reported to play negative feedback roles in the SREBP processing [40]. When SCAP binds to INSIGs, COPII proteins can no longer bind to SCAP, retain the SCAP/SREBP complex in the ER, and block the activation of SREBP. However, limited studies investigated the upstream regulation of INSIGs. AMPK has been shown to stabilize INSIG protein via association and phosphorylation, which results in the ablation of SREBP1 proteolytic processing and the attenuation of lipogenesis [50]. Moreover, a recent study reported that phosphoenolpyruvate carboxykinase 1(PCK1) bound to INSIGs and mediated phosphorylation of INSIGs, which disrupted the interaction between INSIGs and SCAP, thereby activating SREBP-dependent lipogenesis [51]. The above studies have shown that INSIGs could be controlled by post-translational regulation. Here, we identified a lipid mediator CD36 that is directly bound to INSIG2, and then the SCAP/SREBP1 complex dissociated from INSIG2 and transported from ER to Golgi, leading to lipogenesis. In addition, a reduction of hepatic SREBP2 and hepatic TC content was also found in the CD36 LKO mice; thus, it is conceivable that CD36 regulates cholesterol metabolism by INSIG2/SREBP2 pathway. Our findings suggest that INSIG2 and CD36 interaction regulates SREBP1 processing in the liver to maintain lipid homeostasis. Interestingly, no interaction between CD36 and INSIG1 was found. The precise mechanism by which CD36 preferentially binds to INSIG2 has not yet been resolved. INSIG1 and INSIG2 share 59% amino acid sequence homology with the differences mainly in the hydrophilic NH2- and COOH-terminal regions and are functionally similar in blocking SREBP processing. However, functional distinctions may exist between INSIG1 and INSIG2 in that they are differentially regulated by metabolic hormone and metabolites [52,53]. Several studies have found that INSIG2 genetic polymorphisms are associated with the metabolic syndrome [[53], [54], [55]], while the common variation in INSIG1 is unlikely to have a major effect on type 2 diabetes and obesity risk [56]. Further understanding of the regulatory and functional differences between INSIG1 and INSIG2 may provide a new way to explain the preferential interaction between CD36 and INSIG2.
In conclusion, our findings demonstrate that (1) a novel role of CD36 in mediating DNL, which is beyond its known function as a transporter for FFA; (2) CD36 contributes hepatic lipid homeostasis via the regulation of SREBP1 processing; (3) CD36 couples with INSIG2 to abolish the interaction between INSIG2 and the SCAP–SREBP complex. Altogether, our study suggests that CD36, by interacting with INSIG2, is involved in SREBP1 processing and lipogenic program in the liver. Thus, hepatocyte CD36-mediated DNL is a critical factor in the development of NAFLD and provides an intervention strategy for the treatment of hepatic steatosis.
Author contributions
HZ, HQ, ENZ, XQL ML, AHX, YYL, and LC performed in vitro and in vivo experiments. PY, HZ, and HQ analyzed the data, prepared figures, and contributed to the drafting of the manuscript. LZ, LW, and XZR supervised this work and edited and revised manuscript. PY and YXC initiated the project, design the experiments, and approved the final version of manuscript.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (32030054, 81873569); National Key R&D Program of China (2018YFC1312700); the Chongqing Research Program of Basic Research and Frontier Technology (cstc2020jcyj-zdxmX0007, cstc2020jcyj-msxmX0205); the Science and Technology Project of Yuzhong District of Chongqing (20190120); Kuanren Talents Program of the second affiliated hospital of Chongqing Medical University [2021]24; the 111 Project (No. D20028).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2021.101428.
Contributor Information
Ping Yang, Email: yp_apple@yeah.net.
Yaxi Chen, Email: chenyaxi@cqmu.edu.cn.
Conflict of interest
None declared.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Figure S1.

Phenotype of WT mice fed with HFD. C57BL/6 mice were fed with NCD or HFD for 16 weeks (n = 3–5). A-B: Body weight (A) and liver weight (B) were measured. C-H: Overnight-fasted serum ALT levels (C), AST levels (D), TC levels (E), TG levels (F), glucose levels (G), and insulin levels (H) were measured. I-J: Representative images of H&E-stained (I) and Sirius red stained (J) livers. Scale bar, 100 μm. K: Representative images of Oil red O-stained livers. Quantification of Oil red O-stained area was shown in the right. Scale bar, 100 μm ∗P < 0.05 vs. NCD group.
Figure S2.

Phenotype of LKO mice fed with NCD. A: Schematic diagram showing the targeting strategy for generating LKO mice. B-C: The expression of hepatic CD36 was analyzed by Q-PCR (B) and immunoblotting (C). D: The expression of CD36 in heart, kidney, and adipose tissues was analyzed by immunoblotting. E: Quantitation of protein levels in (B) and (C). n=3, ∗P < 0.05 vs. fl/fl mice. F-P: The fl/fl and LKO mice were fed with NCD for 16 weeks (n=6-12). Body weight (F), daily intake (G), and liver weight (H) were measured. Overnight-fasted serum glucose levels (I), TG levels (J), TC levels (K), as well as ALT (L) and AST levels (M) were measured. Representative images of H&E-stained livers (N). Scale bar, 100 μm. GTTs (O) and ITTs (P) in NCD-fed mice, respectively. Quantification of AUC was shown in the right.
Figure S3.

Effects of CD36 on liver SREBP2 activation. A: The mRNA levels of Srebp2 and downstream targets Hmgcr and Ldlr in HFD-fed fl/fl and LKO mice were analyzed by Q-PCR (B). n=8, ∗P < 0.05 vs. fl/fl mice. B: The hepatic expression of SREBP2 and downstream target HMGCR in HFD-fed fl/fl and LKO mice were analyzed by immunoblotting. Quantitation of protein levels was shown in the right. n=3, ∗P < 0.05 vs. fl/fl mice. C: The expression of SREBP2 and downstream target HMGCR in primary hepatocytes from fl/fl and LKO mice were analyzed by immunoblotting. Quantitation of protein levels was shown in the right. n=3, ∗P < 0.05 vs. fl/fl mice. E: The protein levels of SREBP2 of cytoplasmic and nuclear fractions from NC and CD36OE cells were analyzed by immunoblotting.
References
- 1.Loomba R., Friedman S.L., Shulman G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184(10):2537–2564. doi: 10.1016/j.cell.2021.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedman S.L., Neuschwander-Tetri B.A., Rinella M., Sanyal A.J. Mechanisms of NAFLD development and therapeutic strategies. Nature Medicine. 2018;24(7):908–922. doi: 10.1038/s41591-018-0104-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cai J.J., Zhang X.J., Li H.L. Progress and challenges in the prevention and control of nonalcoholic fatty liver disease. Medicinal Research Reviews. 2019;39(1):328–348. doi: 10.1002/med.21515. [DOI] [PubMed] [Google Scholar]
- 4.Donnelly K.L., Smith C.I., Schwarzenberg S.J., Jessurun J., Boldt M.D., Parks E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. Journal of Clinical Investigation. 2005;115(5):1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lambert J.E., Ramos-Roman M.A., Browning J.D., Parks E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146(3):726–735. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Softic S., Cohen D.E., Kahn C.R. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Digestive Diseases and Sciences. 2016;61(5):1282–1293. doi: 10.1007/s10620-016-4054-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.ter Horst K.W., Serlie M.J. Fructose consumption, lipogenesis, and non-alcoholic fatty liver disease. Nutrients. 2017;9(9) doi: 10.3390/nu9090981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kohjima M., Enjoji M., Higuchi N., Kato M., Kotoh K., Yoshimo T., et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. International Journal of Molecular Medicine. 2007;20(3):351–358. [PubMed] [Google Scholar]
- 9.Softic S., Gupta M.K., Wang G.X., Fujisaka S., O'Neill B.T., Rao T.N., et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. Journal of Clinical Investigation. 2017;127(11):4059–4074. doi: 10.1172/JCI94585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang X.C., Okamura D.M., Lu X.F., Chen Y.X., Moorhead J., Varghese Z., et al. CD36 in chronic kidney disease: novel insights and therapeutic opportunities. Nature Reviews Nephrology. 2017;13(12):769–781. doi: 10.1038/nrneph.2017.126. [DOI] [PubMed] [Google Scholar]
- 11.Miquilena-Colina M.E., Lima-Cabello E., Sanchez-Campos S., Garcia-Mediavilla M.V., Fernandez-Bermejo M., Lozano-Rodriguez T., et al. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut. 2011;60(10):1394–1402. doi: 10.1136/gut.2010.222844. [DOI] [PubMed] [Google Scholar]
- 12.Koonen D.P.Y., Jacobs R.L., Febbraio M., Young M.E., Soltys C.L.M., Ong H., et al. Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes. 2007;56(12):2863–2871. doi: 10.2337/db07-0907. [DOI] [PubMed] [Google Scholar]
- 13.Zhao L., Zhang C., Luo X.X., Wang P., Zhou W., Zhong S., et al. CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non-alcoholic steatohepatitis. Journal of Hepatology. 2018;69(3):705–717. doi: 10.1016/j.jhep.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 14.Heeboll S., Poulsen M.K., Ornstrup M.J., Kjaer T.N., Pedersen S.B., Nielsen S., et al. Circulating sCD36 levels in patients with non-alcoholic fatty liver disease and controls. International Journal of Obesity. 2017;41(2):262–267. doi: 10.1038/ijo.2016.223. [DOI] [PubMed] [Google Scholar]
- 15.Yang P., Zeng H., Tan W., Luo X.Q., Zheng E.Z., Zhao L., et al. Loss of CD36 impairs hepatic insulin signaling by enhancing the interaction of PTP1B with IR. The FASEB Journal. 2020;34(4):5658–5672. doi: 10.1096/fj.201902777RR. [DOI] [PubMed] [Google Scholar]
- 16.Zhong S., Zhao L., Wang Y., Zhang C., Liu J., Wang P., et al. Cluster of differentiation 36 deficiency aggravates macrophage infiltration and hepatic inflammation by upregulating monocyte chemotactic protein-1 expression of hepatocytes through histone deacetylase 2-dependent pathway. Antioxidants and Redox Signaling. 2017;27(4):201–214. doi: 10.1089/ars.2016.6808. [DOI] [PubMed] [Google Scholar]
- 17.Nassir F., Adewole O.L., Brunt E.M., Abumrad N.A. CD36 deletion reduces VLDL secretion, modulates liver prostaglandins, and exacerbates hepatic steatosis in ob/ob mice. Journal of Lipid Research. 2013;54(11):2988–2997. doi: 10.1194/jlr.M037812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirano K., Kuwasako T., Nakagawa-Toyama Y., Janabi M., Yamashita S., Matsuzawa Y. Pathophysiology of human genetic CD36 deficiency. Trends in Cardiovascular Medicine. 2003;13(4):136–141. doi: 10.1016/s1050-1738(03)00026-4. [DOI] [PubMed] [Google Scholar]
- 19.Curtis B.R., Ali S., Glazier A.M., Ebert D.D., Aitman T.J., Aster R.H. Isoimmunization against CD36 (glycoprotein IV): description of four cases of neonatal isoimmune thrombocytopenia and brief review of the literature. Transfusion. 2002;42(9):1173–1179. doi: 10.1046/j.1537-2995.2002.00176.x. [DOI] [PubMed] [Google Scholar]
- 20.Lee K., Godeau B., Fromont P., Plonquet A., Debili N., Bachir D., et al. CD36 deficiency is frequent and can cause platelet immunization in Africans. Transfusion. 1999;39(8):873–879. doi: 10.1046/j.1537-2995.1999.39080873.x. [DOI] [PubMed] [Google Scholar]
- 21.Yamashita S., Hirano K., Kuwasako T., Janabi M., Toyama Y., Ishigami M., et al. Physiological and pathological roles of a multi-ligand receptor CD36 in atherogenesis; insights from CD36-deficient patients. Molecular and Cellular Biochemistry. 2007;299(1–2):19–22. doi: 10.1007/s11010-005-9031-4. [DOI] [PubMed] [Google Scholar]
- 22.Son N.H., Basu D., Samovski D., Pietka T.A., Peche V.S., Willecke F., et al. Endothelial cell CD36 optimizes tissue fatty acid uptake. Journal of Clinical Investigation. 2018;128(10):4329–4342. doi: 10.1172/JCI99315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson C.G., Tran J.L., Erion D.M., Vera N.B., Febbraio M., Weiss E.J. Hepatocyte-specific disruption of CD36 attenuates fatty liver and improves insulin sensitivity in HFD-fed mice. Endocrinology. 2016;157(2):570–585. doi: 10.1210/en.2015-1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samovski D., Sun J.Y., Pietka T., Gross R.W., Eckel R.H., Su X., et al. Regulation of AMPK activaticin by CD36 links fatty acid uptake to beta-oxidation. Diabetes. 2015;64(2):353–359. doi: 10.2337/db14-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y., Yang P., Zhao L., Chen Y., Zhang X.Y., Zeng S., et al. CD36 plays a negative role in the regulation of lipophagy in hepatocytes through an AMPK-dependent pathway[S] Journal of Lipid Research. 2019;60(4):844–855. doi: 10.1194/jlr.M090969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bai J.Y., Xia M.F., Xue Y.Q., Ma F.G., Cui A.Y., Sun Y.X., et al. Thrombospondin 1 improves hepatic steatosis in diet-induced insulin-resistant mice and is associated with hepatic fat content in humans. EBioMedicine. 2020;57 doi: 10.1016/j.ebiom.2020.102849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimano H., Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nature Reviews Endocrinology. 2017;13(12):710–730. doi: 10.1038/nrendo.2017.91. [DOI] [PubMed] [Google Scholar]
- 28.Moon Y.A. The SCAP/SREBP pathway: a mediator of hepatic steatosis. Endocrinology and Metabolism. 2017;32(1):6–10. doi: 10.3803/EnM.2017.32.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adams C.M., Reitz J., De Brabander J.K., Feramisco J.D., Li L., Brown M.S., et al. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and insigs. Journal of Biological Chemistry. 2004;279(50):52772–52780. doi: 10.1074/jbc.M410302200. [DOI] [PubMed] [Google Scholar]
- 30.Nohturfft A., DeBose-Boyd R.A., Scheek S., Goldstein J.L., Brown M.S. Sterols regulate cycling of SREBP cleavage-activating protein (SCAP) between endoplasmic reticulum and Golgi. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(20):11235–11240. doi: 10.1073/pnas.96.20.11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakakuki M., Kawano H., Notsu T., Imada K., Mizuguchi K., Shimano H. A novel processing system of sterol regulatory element-binding protein-1c regulated by polyunsaturated fatty acid. Journal of Biochemistry. 2014;155(5):301–313. doi: 10.1093/jb/mvu019. [DOI] [PubMed] [Google Scholar]
- 32.Horton J.D., Bashmakov Y., Shimomura I., Shimano H. Regulation of sterol regulatory element binding proteins in livers of fasted and refed mice. Proceedings of the National Academy of Sciences of the U S A. 1998;95(11):5987–5992. doi: 10.1073/pnas.95.11.5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng C.M., Ru P., Geng F., Liu J.F., Yoo J.Y., Wu X.N., et al. Glucose-mediated N-glycosylation of SCAP is essential for SREBP-1 activation and tumor growth. Cancer Cell. 2015;28(5):569–581. doi: 10.1016/j.ccell.2015.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu J., Cho H., O'Malley S., Park J.H.Y., Clarke S.D. Dietary polyunsaturated fats regulate rat liver sterol regulatory element binding proteins-1 and-2 in three distinct stages and by different mechanisms. Journal of Nutrition. 2002;132(11):3333–3339. doi: 10.1093/jn/132.11.3333. [DOI] [PubMed] [Google Scholar]
- 35.Hannah V.C., Ou J.F., Luong A., Goldstein J.L., Brown M.S. Unsaturated fatty acids down-regulate SREBP isoforms 1a and 1c by two mechanisms in HEK-293 cells. Journal of Biological Chemistry. 2001;276(6):4365–4372. doi: 10.1074/jbc.M007273200. [DOI] [PubMed] [Google Scholar]
- 36.Lin J.D., Yang R.J., Tarr P.T., Wu P.H., Handschin C., Li S.M., et al. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1 beta cactivation of SREBP. Cell. 2005;120(2):261–273. doi: 10.1016/j.cell.2004.11.043. [DOI] [PubMed] [Google Scholar]
- 37.Su L., Zhou L.K., Chen F.J., Wang H.M., Qian H., Sheng Y.Y., et al. Cideb controls sterol-regulated ER export of SREBP/SCAP by promoting cargo loading at ER exit sites. The EMBO Journal. 2019;38(8) doi: 10.15252/embj.2018100156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown M.S., Radhakrishnan A., Goldstein J.L. Retrospective on cholesterol homeostasis: the central role of scap. Annual Review of Biochemistry. 2018;87 87:783–807. doi: 10.1146/annurev-biochem-062917-011852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amemiya-Kudo M., Shimano H., Yoshikawa T., Yahagi N., Hasty A.H., Okazaki H., et al. Promoter analysis of the mouse sterol regulatory element-binding protein-1c gene. Journal of Biological Chemistry. 2000;275(40):31078–31085. doi: 10.1074/jbc.M005353200. [DOI] [PubMed] [Google Scholar]
- 40.Goldstein J.L., DeBose-Boyd R.A., Brown M.S. Protein sensors for membrane sterols. Cell. 2006;124(1):35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 41.Wang J.C., Li Y.S. CD36 tango in cancer: signaling pathways and functions. Theranostics. 2019;9(17):4893–4908. doi: 10.7150/thno.36037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun L.P., Li L., Goldstein J.L., Brown M.S. Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. Journal of Biological Chemistry. 2005;280(28):26483–26490. doi: 10.1074/jbc.M504041200. [DOI] [PubMed] [Google Scholar]
- 43.Tang J.J., Li J.G., Qi W., Qiu W.W., Li P.S., Li B.L., et al. Inhibition of SREBP by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques. Cell Metabolism. 2011;13(1):44–56. doi: 10.1016/j.cmet.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 44.Furuhashi M., Ura N., Nakata T., Shimamoto K. Insulin sensitivity and lipid metabolism in human CD36 deficiency. Diabetes Care. 2003;26(2):471–474. doi: 10.2337/diacare.26.2.471. [DOI] [PubMed] [Google Scholar]
- 45.Su X., Abumrad N.A. Cellular fatty acid uptake: a pathway under construction. Trends in Endocrinology and Metabolism. 2009;20(2):72–77. doi: 10.1016/j.tem.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Glatz J.F.C., Luiken J.J.F.P., Bonen A. Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiological Reviews. 2010;90(1):367–417. doi: 10.1152/physrev.00003.2009. [DOI] [PubMed] [Google Scholar]
- 47.Hames K.C., Vella A., Kemp B.J., Jensen M.D. Free fatty acid uptake in humans with CD36 deficiency. Diabetes. 2014;63(11):3606–3614. doi: 10.2337/db14-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Febbraio M., Abumrad N.A., Hajjar D.P., Sharma K., Cheng W.L., Pearce S.F.A., et al. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. Journal of Biological Chemistry. 1999;274(27):19055–19062. doi: 10.1074/jbc.274.27.19055. [DOI] [PubMed] [Google Scholar]
- 49.Clugston R.D., Yuen J.J., Hu Y.Y., Abumrad N.A., Berk P.D., Goldberg I.J., et al. CD36-deficient mice are resistant to alcohol- and high-carbohydrate-induced hepatic steatosis. Journal of Lipid Research. 2014;55(2):239–246. doi: 10.1194/jlr.M041863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han Y.M., Hu Z.M., Cui A.Y., Liu Z.S., Ma F.G., Xue Y.Q., et al. Post-translational regulation of lipogenesis via AMPK-dependent phosphorylation of insulin-induced gene. Nature Communications. 2019;10 doi: 10.1038/s41467-019-08585-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu D.Q., Wang Z., Xia Y., Shao F., Xia W.Y., Wei Y.K., et al. The gluconeogenic enzyme PCK1 phosphorylates INSIG1/2 for lipogenesis. Nature. 2020;580(7804):530–+. doi: 10.1038/s41586-020-2183-2. [DOI] [PubMed] [Google Scholar]
- 52.Wang H., Zhao M.Y., Sud N., Christian P., Shen J., Song Y.Y., et al. Glucagon regulates hepatic lipid metabolism via cAMP and Insig-2 signaling: implication for the pathogenesis of hypertriglyceridemia and hepatic steatosis. Scientific Reports. 2016;6 doi: 10.1038/srep32246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dong X.Y., Tang S.Q. Insulin-induced gene A new regulator in lipid metabolism. Peptides. 2010;31(11):2145–2150. doi: 10.1016/j.peptides.2010.07.020. [DOI] [PubMed] [Google Scholar]
- 54.Le Hellard S., Theisen F.M., Haberhausen M., Raeder M.B., Ferno J., Gebhardt S., et al. Association between the insulin-induced gene 2 (INSIG2) and weight gain in a German sample of antipsychotic-treated schizophrenic patients: perturbation of SREBP-controlled lipogenesis in drug-related metabolic adverse effects? Molecular Psychiatry. 2009;14(3):308–317. doi: 10.1038/sj.mp.4002133. [DOI] [PubMed] [Google Scholar]
- 55.Zhang H.Q., Huang R., Tian S., An K., Zhu W.W., Shi J.J., et al. The CC genotype of insulin-induced gene 2 rs7566605 is a protective factor of hypercholesteremia susceptible to mild cognitive impairment, especially to the executive function of patients with type 2 diabetes mellitus. BioMed Research International. 2020;2020 doi: 10.1155/2020/4935831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Szopa M., Meirhaeghe A., Luan J., Moreno L.A., Gonzalez-Gross M., Vidal-Puig A., et al. No association between polymorphisms in the INSIG1 gene and the risk of type 2 diabetes and related traits. American Journal of Clinical Nutrition. 2010;92(1):252–257. doi: 10.3945/ajcn.2010.29422. [DOI] [PubMed] [Google Scholar]