

Abstract
On March 10, 2021, the FDA granted regular approval to tivozanib for treatment of patients with relapsed or refractory (R/R) advanced renal cell carcinoma (RCC) following two or more prior systemic therapies. Approval was based on the TIVO-3 study, a randomized trial of tivozanib versus sorafenib in patients with R/R advanced RCC. In TIVO-3, patients were randomized to receive either tivozanib 1.34 mg orally once daily for 21 consecutive days of every 28-day cycle or sorafenib 400 mg orally twice daily continuously. The primary endpoint was progression-free survival (PFS) per RECIST v1.1. Tivozanib demonstrated efficacy compared with sorafenib with an improvement in PFS [HR: 0.73; 95% confidence interval (CI): 0.56–0.95; p=0.016]. The estimated median PFS was 5.6 months and 3.9 months in the tivozanib and sorafenib arms, respectively. There was no evidence of a detrimental effect on overall survival: HR 0.97 (95% CI: 0.75–1.24). The most common grade 3–4 adverse reaction on the tivozanib arm was hypertension (24%). Compared to sorafenib, tivozanib was associated with lower rates of grade 3–4 diarrhea, rash, and palmar-plantar erythrodysesthesia. Patients receiving tivozanib in TIVO-3 had lower rates of dose reduction, interruption, or permanent discontinuation than those receiving sorafenib.
Introduction
In 2021, an estimated 13,780 deaths due to kidney cancer will occur in the U.S.(1) Approximately 80-90% of kidney cancers are renal cell carcinoma (RCC), and clear cell RCC is the most common histologic subtype in adults, comprising approximately 70-80% of RCC cases.(2, 3) Kidney cancer deaths comprise approximately 2.4% of cancer deaths in the U.S. The 5-year survival of newly diagnosed metastatic kidney cancer is 13%.(4)
The standard of care for the first and second-line treatment of patients with advanced RCC has evolved over the past 2 decades. The approval of sorafenib and sunitinib in 2005 and 2006 offered patients the first kinase inhibitors (KIs) for advanced RCC. In subsequent years, additional kinase inhibitors, including pazopanib, axitinib, and cabozantinib were approved, mostly in line-agnostic settings. The U.S. Food and Drug Administration (FDA) has subsequently approved several checkpoint inhibitor (CPI)-containing combinations in frontline RCC since 2018: nivolumab + ipilimumab, pembrolizumab + axitinib, avelumab + axitinib, and nivolumab + cabozantinib. Standard second-line therapy has not been established since these approvals. Given the multiple options in the frontline setting, guidelines suggest considering any frontline option to which the patient has not been exposed, as well as KIs used as monotherapy, as reasonable choices.(5)
On March 10, 2021, the FDA approved tivozanib (Table 1), a KI for the treatment of adult patients with relapsed or refractory advanced RCC following two or more prior systemic therapies.(6) This was based on the results of the randomized clinical study TIVO-3, which demonstrated improved progression-free survival (PFS) in patients randomized to tivozanib vs. sorafenib, and a similar or more tolerable safety profile. During the review of TIVO-3, FDA also considered TIVO-1, a randomized trial in a KI-naïve advanced RCC population which randomized patients to tivozanib vs. sorafenib. The Applicant submitted a marketing application based on TIVO-1 in 2013, which FDA did not approve.
Table 1.
FDA Benefit-Risk Assessment
| Dimension | Evidence and Uncertainties | Conclusions and Reasons |
|---|---|---|
| Analysis of Condition | Advanced RCC with disease progression on two prior lines of therapy represents a setting of unmet medical need and is considered incurable. | Advanced RCC is a serious and life-threatening condition |
| Current Treatment Options | • While there is no therapy approved
specifically in the third-line setting for advanced RCC, there are
several approved TKIs which are active and which can be used (including
sorafenib). • Checkpoint inhibitor (CPI)-containing combinations are approved in the first-line setting. Nivolumab is a CPI approved in the second-line setting. Most contemporary patients have therefore been treated and have progressed or are intolerant to a CPI by the time they have reached the later lines of therapy. • Although infrequently used, other treatment options for patients with advanced RCC include mammalian target of rapamycin inhibitors and bevacizumab. |
Although treatment options exist for advanced RCC, none is curative and none is approved specifically for patients whose disease has progressed after treatment with two prior lines of therapy. Therefore, there is an unmet medical need for effective treatments for these patients. |
| Benefit | • In TIVO-3, the estimate for the HR
for PFS was 0.73 (95% CI: 0.56 to 0.95, p-value of 0.016). The median
PFS estimate was 5.6 months (95% CI: 4.8, 7.3) for tivozanib vs. 3.9
months (95% CI: 3.7 to 5.6) for sorafenib. Subgroup analyses were
supportive. • ORR was 18% (95% CI: 12%, 24%) for tivozanib vs. 8% (95% CI: 4%, 13%) for sorafenib. • The final median OS estimate was 16.4 months for tivozanib vs. 19.2 months for sorafenib, with a HR estimate of 0.97 (95% CI: 0.75 to 1.24). |
Tivozanib demonstrated a PFS advantage over
sorafenib in patients with advanced RCC whose disease has progressed
after treatment with 2 prior lines of therapy. The submitted evidence met the statutory evidentiary standard for regular approval of tivozanib for this indication. |
| Risk and Risk Management | • The safety profile of tivozanib
was consistent with the known safety profile of TKIs. • Compared to patients treated with sorafenib, patients treated with tivozanib had lower rates of dose reduction, interruption, and/or permanent discontinuation. |
The safety profile of tivozanib is
manageable and is acceptable in this setting of advanced
disease. Product labeling addresses recommendations for dose modifications. The Warnings and Precautions section addresses identified significant and potentially serious adverse reactions of tivozanib. A Risk Evaluation and Mitigation Strategy (REMS) was not required for this application. |
Herein, we discuss FDA’s benefit-risk analysis of the recent marketing application that led to approval in third-line RCC.
Chemistry, Manufacturing, and Control
Tivozanib hydrochloride capsules are supplied with two strengths: 1.34 mg of tivozanib (equivalent to 1.5 mg tivozanib hydrochloride) and 0.89 mg of tivozanib (equivalent to 1.0 mg tivozanib hydrochloride) with inactive ingredients of mannitol and magnesium stearate.(7)
Nonclinical Pharmacology and Toxicology
Tivozanib is a tyrosine KI. Tivozanib inhibits vascular endothelial growth factor receptor (VEGFR) 1, 2 and 3 and other kinases including c-kit and platelet-derived growth factor receptor (PDGFR) β at clinically relevant concentrations. In tumor xenograft models in mice and rats, tivozanib inhibited tumor growth, angiogenesis, and vascular permeability.
In 13-week repeat-dose toxicology studies in monkeys and rats, the predominant target organs/systems of toxicity included adrenal gland, gastrointestinal system (stomach, duodenum, pancreas), kidney, bone marrow, liver, brain, pituitary gland, lymphoid organs, heart, incisors and female reproductive system. In addition, growth plate hypertrophy effects on the femur and tibia were reported in young monkeys and adult rats. Tivozanib was not mutagenic or clastogenic. In a male and female fertility study in rats, tivozanib produced infertility at a clinically relevant dose. In embryo-fetal developmental studies, tivozanib administered to pregnant animals during the period of organogenesis resulted in embryo-fetal lethality and malformations at a clinically relevant dose. Based on findings in animals, tivozanib can cause fetal harm when administered to a pregnant woman and may impair fertility.
Clinical Pharmacology
The recommended dose of tivozanib is 1.34 mg taken orally, with or without food, once daily for 21 days on treatment followed by 7 days off treatment for a 28-day cycle.
The clinical pharmacology review focused on the drug-drug interaction potential for tivozanib as a victim and dose adjustments for hepatic impairment. Tivozanib is a substrate for CYP3A4, and the product label recommends patients to avoid concomitant use of strong CYP3A inducers.
The recommended dosage in patients with hepatic impairment was based on the assessment of the combined results from a dedicated hepatic impairment trial, as well as a population pharmacokinetic (PK) analysis and reclassification of organ impairment from Child-Pugh to National Cancer Institute (NCI) Organ Dysfunction Working Group (ODWG) criteria. For patients with moderate hepatic impairment, the product label recommends a tivozanib dosage of 0.89 mg taken orally once daily for 21 days on treatment followed by 7 days off treatment for a 28-day cycle. The recommended dosage of tivozanib in patients with severe hepatic impairment has not been established.
Clinical Studies
The efficacy of tivozanib in patients with RCC that progressed on 2 or more prior systemic regimens was supported by the multinational, open-label, randomized controlled clinical study TIVO-3 (NCT02627963)(8) that enrolled patients with metastatic RCC and progression after 2 or 3 prior systemic regimens, one of which included a VEGFR KI excluding sorafenib or tivozanib. Patients were required to have measurable disease per Response Evaluation Criteria in Solid Tumors (RECIST) and Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. Patients with active brain metastases, HIV infection, poorly-controlled hypertension, or history of recent thromboembolism, heart failure, severe angina, or acute coronary syndrome were excluded. Patients were randomized 1:1 to either tivozanib 1.34 mg (1.34 mg tivozanib free base is equivalent to 1.5 mg tivozanib hydrochloride) orally once daily for 21 consecutive days of 28-day cycles or sorafenib 400 mg orally twice daily continuously.
The primary efficacy endpoint was PFS per blinded independent review committee (BIRC) assessment based on RECIST v 1.1. Secondary endpoints included overall survival (OS) and objective response rate (ORR). The OS analysis plan was changed at the time of the PFS analysis in 2018: HR 1.12 (95% CI: 0.4-1.51) based on 183 OS events (70% information fraction). The Applicant revised the SAP at that time by adding a new final OS analysis with longer follow-up. The final OS analysis was conducted using a cutoff date of May 1, 2020. However, results of all secondary endpoints including OS were considered exploratory by FDA due to no multiplicity adjustment.
Efficacy Results
TIVO-3 enrolled 350 patients; 175 on the tivozanib arm and 175 on the sorafenib arm. Eight patients (2 on the tivozanib arm and 6 on the sorafenib arm) did not receive treatment after randomization. The demographic and disease characteristics of the intention-to-treat population (n=350) are presented in Table 2.
Table 2.
Demographic and Baseline Characteristics
| Tivozanib (n=175) |
Sorafenib (n=175) |
|
|---|---|---|
| Age in years, n (%) | ||
| Median (min, max) | 62 (34, 88) | 64 (30, 90) |
| Sex, n (%) | ||
| Male | 126 (72) | 128 (73) |
| Female | 49 (28) | 47 (27) |
| Race, n (%) | ||
| White | 165 (94) | 167 (95) |
| African American | 0 | 2 (1) |
| Other | 3 (2) | 2 (1) |
| Unknown | 7 (4) | 4 (2) |
| Ethnicity, n (%) | ||
| Hispanic/Latino | 10 (6) | 10 (6) |
| Not Hispanic/Latino | 156 (89) | 162 (93) |
| Unknown | 9 (5) | 3 (2) |
| Region, n (%) | ||
| North America | 31 (18) | 27 (15) |
| Europea | 144 (82) | 148 (85) |
| IMDC risk category (eCRF), n (%) | ||
| Favorable | 26 (15) | 35 (20) |
| Intermediate | 120 (69) | 107 (61) |
| Poor | 29 (17) | 33 (19) |
| Prior Therapies (eCRF), n (%) | ||
| Two VEGFR KIs | 80 (46) | 85 (49) |
| Checkpoint inhibitor + VEGFR KI | 48 (27) | 44 (25) |
| VEGFR KI + other systemic agentb | 47 (27) | 46 (26) |
| Number of metastatic sites, n (%) | ||
| 1 | 13 (7) | 25 (14) |
| 2 | 53 (30) | 45 (26) |
| 3+ | 109 (63) | 105 (60) |
| Baseline systolic BP, n (%) | ||
| ≤140 mmHg | 140 (80) | 138 (79) |
| Baseline diastolic BP, n (%) | ||
| ≤90 mmHg | 164 (94) | 158 (90) |
Includes Italy (n=62), Spain (n=55), Poland (n=46), Hungary (n=33), Czech Republic (n=30), United Kingdom (n=29), France (n=13), Germany (n=11), Denmark (n=7), Belgium (n=6)
Most patients in this category received an mTOR inhibitor.
Source: NDA 212904 Multi-Discipline Review (6).
Abbreviations: BP, blood pressure; eCRF, electronic case report form; IMDC, International Metastatic RCC Database Consortium; KI, kinase inhibitor; VEGFR, vascular endothelial growth factor receptor;
TIVO-3 met its primary endpoint. The improvement in PFS observed in the tivozanib arm was statistically significant with the comparison of sorafenib, an active control: hazard ratio (HR) for PFS was 0.73 (95% CI: 0.56 to 0.95, p-value = 0.016). The median PFS estimates were 5.6 months and 3.9 months for tivozanib and sorafenib, respectively (Table 3).
Table 3.
Progression-Free Survival and Overall Survival in TIVO-3
| Tivozanib (N=175) |
Sorafenib (N=175) |
|
|---|---|---|
| Progression-Free Survival | ||
| Number of events, n (%) | 123 (70.3%) | 123 (70.3%) |
| Progression | 103 (58.9%) | 109 (62.3%) |
| Death | 20 (11.4%) | 14 (8.0%) |
| Median (95% CI), in months | 5.59 (4.83, 7.33) | 3.88 (3.71, 5.55) |
| HR (95% CI)1 | 0.73 (0.56, 0.95) | |
| P-value2 | 0.016 | |
| Overall Survival | ||
| Number of events, n (%) | 125 (71.4) | 126 (72.0) |
| Median (95% CI), in months | 16.39 (13.44, 21.95) | 19.15 (14.92, 24.21) |
| HR (95% CI)1 | 0.97 (0.75, 1.24) | |
HR was from a Cox proportional hazards model stratified by IMDC risk categories and prior therapy collected from the Interactive Web Response System (IWRS).
P-value was from a log-rank test stratified by IMDC risk categories and prior therapy collected from IWRS.
Source: Drugs@FDA (7).
At the time of the final OS analysis, a total of 251 deaths occurred with a median follow-up of 38 months on the tivozanib arm and 40 months on the sorafenib arm. The results of the OS final analysis showed a HR 0.97 (95% CI: 0.75, 1.24) with a final median OS estimate of 16.4 months for tivozanib vs. 19.2 months for sorafenib (Table 3). ORR per BIRC was 18% in the tivozanib arm and 8% in the sorafenib arm; all partial responses. Median duration of response was not reached vs. 5.7 months in the tivozanib vs. sorafenib arms, respectively.
Safety Results
FDA’s safety review primarily focused on the 173 patients treated with at least 1 dose of tivozanib in TIVO-3. Supportive safety data was obtained from 5 other trials, for a total of 1008 patients with advanced RCC who received tivozanib monotherapy at the proposed dose and schedule.
Deaths due to adverse events occurred in 12 tivozanib-treated patients (7.5%), including 4 deaths that may have been related to tivozanib, in the FDA review team’s assessment. In the 1008-patient population, the most common fatal adverse events were cardiac failure (0.6%), arterial thromboembolic events including stroke (0.5%), and venous thromboembolism (0.3%). Two-thirds of tivozanib-treated patients in TIVO-3 experienced grade 3-4 treatment-emergent adverse events (TEAEs) compared to 72% of sorafenib-treated patients, with the most common event being hypertension (grade 3-4, 24% vs. 17%). Tivozanib was associated with lower rates of grade 3-4 diarrhea (1.7% vs. 11.2%), rash (1.2% vs. 15%), and palmar-plantar erythrodysesthesia (1.2% vs. 10%).
Patient-Reported Outcomes and Overall Tolerability
Patient-reported outcome (PRO) data was not collected in TIVO-3. However, they were collected in TIVO-1, which used the same treatment arms as TIVO-3, was conducted in a patient population who was similar to the population studied in TIVO-3 other than line of therapy, and used similar dose modification and safety monitoring guidelines. Therefore, FDA requested PRO analyses from TIVO-1 to characterize the patient experience with tivozanib vs. sorafenib. Patients randomized to sorafenib reported greater side effect bother (Figure 1). As PRO data was collected on day 1 of each cycle, following a one-week washout period for tivozanib, patients’ overall sense of treatment bother may therefore have been incompletely captured, as the PRO questionnaire asks about the last 7 days of symptoms. In contrast, sorafenib was given on a continuous treatment schedule.
Figure 1.
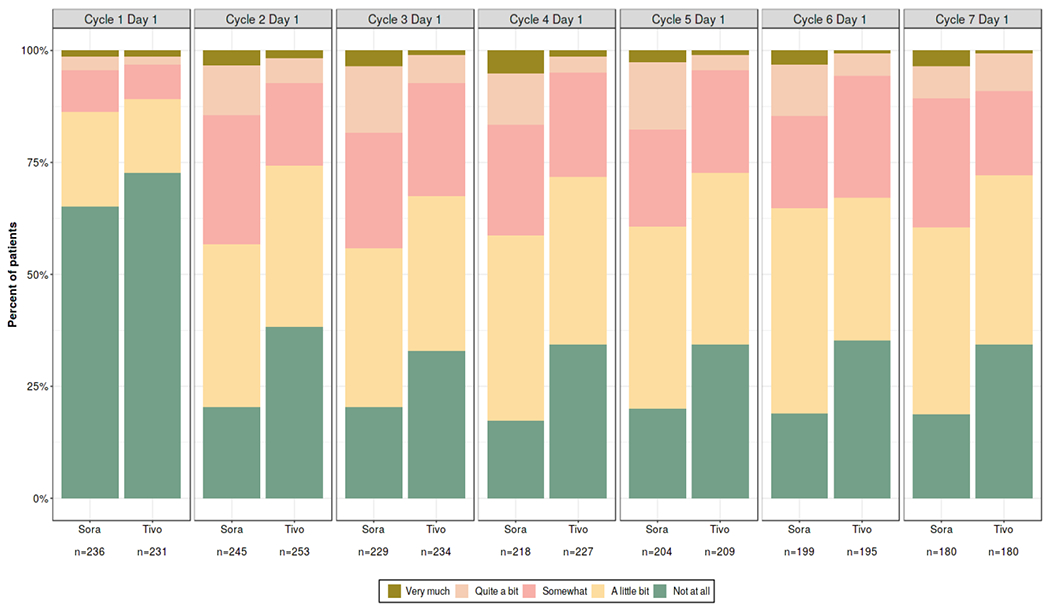
Patient-Reported Outcomes from TIVO-1: Distribution of Responses to “I Am Bothered by Side Effects of Treatment”
Notes: n = number of respondents to this question
In TIVO-3, tivozanib-treated patients experienced lower rates of dose reduction, interruption, or permanent discontinuation vs. sorafenib-treated patients. These findings, supported by the analyses of TIVO-1 PRO data, suggest possible relatively improved tolerability of tivozanib compared to sorafenib. However, FDA considers the results of the PRO analyses to be exploratory and recognizes the limitations of the TIVO-1 results, as they were from a different trial and may be impacted by timing of assessments. Therefore, no information on PROs was included in the U.S. prescribing information for tivozanib.
Regulatory Insights
During the review of this marketing application, FDA considered the results of TIVO-1 (NCT01030783), described above, originally reviewed in a marketing application in 2012. Although TIVO-1 met its primary endpoint (PFS HR 0.80 [95% CI: 0.64, 0.99], p = 0.04), a trend towards a detrimental effect on OS was observed (HR = 1.25 [95% CI: 0.95, 1.62], p = 0.11).(9, 10) An Oncologic Drugs Advisory Committee (ODAC) vote was 13 “no” votes to 1 “yes” vote in response to the question, “Has the Applicant demonstrated a favorable benefit to risk evaluation for the treatment of renal cell carcinoma in an adequate and well-controlled trial?” Members’ concerns included the decrement in OS and confounding factors in the trial design (9) including the one-way crossover design, in which patients with progression on sorafenib could receive tivozanib through an extension study (NCT01076010), but patients with progression on tivozanib were not given the opportunity to receive second-line therapy as part of a protocol. As a result, 154 (60%) patients randomized to sorafenib received subsequent VEGFR KI therapy, the majority having crossed over to tivozanib, compared to 31 (12%) patients randomized to tivozanib. While crossover may have been an important reason for the decrement in OS, other reasons for the OS results could not be ruled out. Therefore, a line-agnostic indication was not granted during review of the TIVO-3 data, as the TIVO-1 OS results were still uninterpretable.
The totality of the new data from TIVO-3 warranted an approval for a third-line indication, considering the PFS advantage, supportive ORR, acceptable safety for the intended population, and the HR for OS that did not demonstrate a decrement in patients randomized to tivozanib, despite the decrement in OS observed in TIVO-1. In contrast to the accelerated approval pathway, regular approval does not require a new drug to demonstrate improvement over all available therapies in order to provide evidence of efficacy and safety. For regular approval, a drug could demonstrate efficacy above a placebo-only control arm or be approved based on demonstration of non-inferiority to an active control therapy. In this case, a key regulatory consideration was that sorafenib is an active control.(11, 12) Although only a small PFS benefit was demonstrated by tivozanib, TIVO-3 used a superiority design, thus at minimum providing assurance that tivozanib conferred similar benefit as sorafenib.
The Applicant’s original proposed indication was for patients with relapsed or refractory RCC, which would have included patients with only one prior systemic therapy. This reflected not only the TIVO-3 population but also the extension study from TIVO-1, a single-arm study with no primary efficacy endpoint. FDA did not consider the results from the extension study to be substantial evidence of effectiveness of tivozanib for the second-line indication, and thus the indication was narrowed to patients who had received two or more prior systemic therapies.
The Applicant revised the OS analysis plan in the TIVO-3 SAP following interim results. FDA generally does not recommend non-prespecified revisions to the SAP of major endpoints based on unblinded data. However, in TIVO-3, analyses of OS were supportive evidence of efficacy and safety, and not the basis of efficacy claims. Furthermore, given the uncertainty of the OS results at the time of PFS analysis, an OS analysis based on more mature data is expected to have a more accurate overall estimate of survival for the totality of the trial. Therefore, the SAP revision appeared acceptable in this application.
A major review issue was the OS results: HR close to 1 and median estimate approximately 3 months shorter for the tivozanib arm vs. the sorafenib arm. However, the median is only one timepoint on the survival curve and the difference at one timepoint may not reflect the entirety of the Kaplan-Meier curves. The FDA review team also conducted a post-hoc exploratory analysis using the restricted mean survival time (RMST) method to estimate the treatment effect, which suggested that tivozanib does not have a detrimental effect on OS. FDA’s additional sensitivity analyses also confirmed similar HRs as the primary OS findings.
Approximately 25% of patients (between 44 and 48 patients in each arm) received a CPI prior to enrolling on the trial. While this is low relative to current usage patterns in the combination frontline CPI era, the proportion of patients who had received prior CPI in TIVO-3 is still higher than that of other recent randomized trials demonstrating clinical benefit in refractory RCC.(13–16) The FDA review team performed an exploratory analysis of efficacy of tivozanib specifically in those patients who received prior checkpoint (PD-1 or PD-L1) inhibitor therapy. This analysis showed an unstratified PFS HR 0.67 (95% CI 0.39, 1.16), median PFS 7.3 vs. 5.9 months for tivozanib and sorafenib, respectively. These results were supportive of the relevance of the TIVO-3 results in real-world patients receiving third-line treatment in the contemporary treatment setting.
Conclusion
The totality of the data as summarized in the risk–benefit assessment in Table 1 met the statutory evidentiary standard for regular approval of tivozanib for patients with RCC that has progressed on 2 or 3 prior systemic regimens. This provides the first approval in the third-line setting in the post-frontline CPI combination era.
Footnotes
This is a U.S. Government work. There are no restrictions on its use.
Disclosure of Potential Conflicts of Interest: The authors report no financial interests or relationships with the commercial sponsors of any products discussed in this report.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA: a cancer journal for clinicians. 2021;71(1):7–33. [DOI] [PubMed] [Google Scholar]
- 2.National Cancer Institute. Clear Cell Renal Cell Carcinoma. 2020. Available at https://www.cancer.gov/pediatric-adult-rare-tumor/rare-tumors/rare-kidney-tumors/clear-cell-renal-cell-carcinoma.
- 3.American Cancer Society. What Is Kidney Cancer? 2020. Available at https://www.cancer.org/cancer/kidney-cancer/about/what-is-kidney-cancer.html
- 4.National Cancer Institute. Cancer Stat Facts: Kidney and Renal Pelvis Cancer. 2020. Available at https://seer.cancer.gov/statfacts/html/kidrp.html.
- 5.NCCN: NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines), Version 3.2021. March 23, 2021. Available at https://www.nccn.org/professionals/physician_gls/pdf/kidney.pdf.
- 6.Drugs@FDA. MULTI-DISCIPLINE REVIEW. NDA 212904: tivozanib. April 6, 2021. Available at https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/212904Orig1s000MultidisciplineR.pdf.
- 7.Drugs@FDA. US Prescribing Information: tivozanib. March 10, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/212904s000lbl.pdf.
- 8.Rini BI, Pal SK, Escudier BJ, Atkins MB, Hutson TE, Porta C, et al. Tivozanib versus sorafenib in patients with advanced renal cell carcinoma (TIVO-3): a phase 3, multicentre, randomised, controlled, open-label study. The Lancet Oncology. 2020;21(1):95–104. [DOI] [PubMed] [Google Scholar]
- 9.FDA. Summary Minutes - ODAC 5.2.13. Available from: https://wayback.archive-it.org/7993/20170404153302/https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM359160.pdf.
- 10.Motzer RJ, Nosov D, Eisen T, Bondarenko I, Lesovoy V, Lipatov O, et al. Tivozanib versus sorafenib as initial targeted therapy for patients with metastatic renal cell carcinoma: results from a phase III trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31(30):3791–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, et al. Sorafenib in Advanced Clear-Cell Renal-Cell Carcinoma. New England Journal of Medicine. 2007;356(2):125–34. [DOI] [PubMed] [Google Scholar]
- 12.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Staehler M, et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27(20):3312–8. [DOI] [PubMed] [Google Scholar]
- 13.Motzer RJ, Hutson TE, Glen H, Michaelson MD, Molina A, Eisen T, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. The Lancet Oncology. 2015;16(15):1473–82. [DOI] [PubMed] [Google Scholar]
- 14.Motzer RJ, Escudier B, Tomczak P, Hutson TE, Michaelson MD, Negrier S, et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: overall survival analysis and updated results from a randomised phase 3 trial. The Lancet Oncology. 2013;14(6):552–62. [DOI] [PubMed] [Google Scholar]
- 15.Choueiri TK, Escudier B, Powles T, Tannir NM, Mainwaring PN, Rini BI, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. The Lancet Oncology. 2016;17(7):917–27. [DOI] [PubMed] [Google Scholar]
- 16.Escudier B, Sharma P, McDermott DF, George S, Hammers HJ, Srinivas S, et al. CheckMate 025 Randomized Phase 3 Study: Outcomes by Key Baseline Factors and Prior Therapy for Nivolumab Versus Everolimus in Advanced Renal Cell Carcinoma. European Urology. 2017;72(6):962–71. [DOI] [PubMed] [Google Scholar]