

Abstract
SV2A, an essential transporter-like synaptic vesicle protein, is a major target for antiepileptic drugs and a receptor for clostridial neurotoxins including Botox. While SV2A is required for normal levels of evoked neurotransmitter release, the mechanism underlying this role remains unclear. Here, we introduce a new chemogenetic approach for all-optical monitoring of excitation-secretion coupling, and we demonstrate its use in characterizing the SV2A KO phenotype in cultured hippocampal neurons. This method employs the HaloTag system to target a robust small-molecule Ca2+ indicator, JF646-BAPTA, to the presynaptic compartment. The far-red fluorescence of this indicator enables multiplexing with the fluorescent glutamate sensor iGluSnFR for detection of presynaptic Ca2+ influx and glutamate release at the same axonal boutons. Evoked glutamate release probability was reduced in SV2A KO neurons without a change in presynaptic Ca2+ entry, suggesting that SV2A supports vesicle fusion by increasing the functional availability, or efficiency, of the Ca2+-regulated membrane fusion machinery.
Graphical Abstract
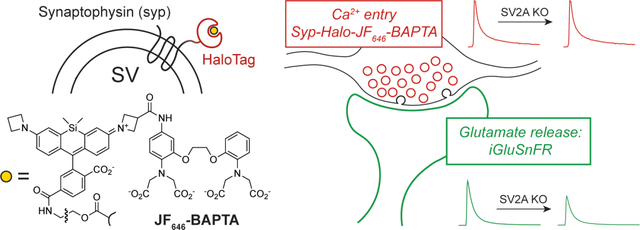
Introduction
SV2A, one of the first-cloned synaptic vesicle (SV) proteins (Bajjalieh et al., 1992; Buckley and Kelly, 1985; Feany et al., 1992), is homologous to transport proteins in the major facilitator superfamily (Jacobsson et al., 2007). Mice lacking SV2A (Sv2a KO) fail to gain weight, develop seizures, and die by ~14 days of age (Crowder et al., 1999; Janz et al., 1999). Action-potential-dependent neurotransmitter release is attenuated in Sv2a KO neurons both in culture (Chang and Sudhof, 2009; Custer et al., 2006) and in brain slice (Crowder et al., 1999), suggesting a mechanism for epileptogenesis, but how SV2A modulates the release machinery is unknown. While some evidence suggests a role for SV2A in trafficking the exocytotic Ca2+ sensor synaptotagmin-1 (syt1) to nerve terminals (Yao et al., 2010), the functional importance of this finding is unclear (Nowack et al., 2010). It is noteworthy that SV2A, along with the closely-related family members SV2B and SV2C, serve as protein receptors for clostridial neurotoxins (Dong, 2006; Yeh et al., 2010). Among these, botulinum neurotoxin type A, or Botox, is in widespread medical use for a growing variety of neurological and cosmetic indications.
Strikingly, SV2A is also a major drug target in the treatment of epilepsy (Löscher et al., 2016). The anti-epileptic drugs levetiracetam (Keppra) and brivaracetam (Briviact) bind SV2A (Klitgaard et al., 2016; Lynch et al., 2004), and it is well-established that this interaction underlies the antiepileptic action of these drugs (Kaminski et al., 2009, 2008). However, the antiepileptic mechanism of these drugs is otherwise undefined at the molecular level. While most antiepileptic drugs directly inhibit the excitatory machinery of neurons or enhance inhibitory processes (Löscher et al., 2013), several clues indicate an unusual mechanism of action for racetam antiepileptics. Their profile in rodent epilepsy models is atypical, and they have only subtle effects on neurotransmission even at high concentrations (García-Pérez et al., 2015; Löscher et al., 2016; Yang et al., 2015). The clinical presentation of levetiracetam overdose is usually benign (Kartal, 2017), suggesting an unconventional mechanism of action that relies specifically on epileptic processes.
At nerve terminals, Ca2+ influx into the cytoplasm triggers neurotransmitter release, but whether SV2A affects presynaptic Ca2+ entry has not been conclusively established. While one report has suggested role for the related protein SV2B in presynaptic Ca2+ signaling in bipolar neurons of the mouse retina (Wan et al., 2010), other reports have used indirect methods to suggest that SV2A does (Janz et al., 1999) or does not (Chang and Sudhof, 2009; Custer et al., 2006) modulate presynaptic Ca2+ signaling at small boutons in cultured mouse neurons. To clarify the relationship between SV2A and presynaptic Ca2+, we developed and applied an all-optical approach to monitor excitation-secretion coupling. These experiments revealed that, during evoked neurotransmission in cultured hippocampal neurons, SV2A supports SV fusion independently of presynaptic Ca2+ entry.
Results
Because it is unclear at what step SV2A acts in exocytosis, we sought to establish whether this SV protein modulates presynaptic Ca2+ dynamics. The poor development of Sv2a−/− mice (Crowder et al., 1999) makes brain slice studies impractical and potentially confounded by a global failure to thrive. Neuronal cultures from neonatal mouse hippocampus thus provide an ideal model to study the role of SV2A in synaptic transmission. However, the irregular morphology, long processes, and absence of tissue organization in neuronal cultures present obstacles to measuring presynaptic Ca2+ fluxes using Ca2+indicators loaded either by bath application or by whole-cell dialysis. Published approaches have involved combining bath-loaded Ca2+ indicators with presynaptic markers (Hoppa et al., 2012) or targeting genetically-encoded Ca2+ sensors to the nerve terminal (de Juan-Sanz et al., 2017), but these techniques suffer from poor rejection of non-presynaptic Ca2+ signals or limited temporal resolution of Ca2+ signals, respectively.
We thus took advantage of the HaloTag system (Los et al., 2008) along with recent advances in synthetic Ca2+ indicators (Deo et al., 2019) to target a sensitive, small-molecule Ca2+ sensor to nerve terminals using the synaptic vesicle protein synaptophysin (syp) (Fig. 1). The indicator used here, JF646-BAPTA, offers several advantages over existing Ca2+ indicators. In particular, this far-red indicator undergoes substantial brightness increases when bound to the HaloTag protein via a chloroalkane linker, enabling spatially-resolved measurements of Ca2+ depending on the subcellular targeting of the HaloTag protein (Deo et al., 2019). In neurons expressing a synaptophysin-HaloTag fusion protein (Fig. 1A), incubation with the acetoxymethyl ester of JF646-BAPTA-HaloTag ligand generated a presynaptic Ca2+ sensor with the punctate presynaptic localization of synaptophysin (Fig. 1B) along with the speed and sensitivity of small-molecule Ca2+ ligands (Fig. 1C). While JF646-BAPTA is a high-affinity Ca2+ indicator (KD ~ 150 nM) (Deo et al., 2019) and thus tracks Ca2+ fluxes slightly less faithfully than lower-affinity dyes (Sabatini and Regehr, 1998), this approach provided a substantial improvement in kinetic fidelity over a similarly targeted Syp-GCaMP6f construct (Fig. 1C). Calibration of the sensor using a dye-saturating stimulus (Fig. 1D) and the known properties of the dye (de Juan-Sanz et al., 2017; Deo et al., 2019; Maravall et al., 2000) enabled determination of absolute [Ca2+]i in individual boutons (Fig. 1E), which were readily identified in an automated manner using the resting fluorescence of the indicator (see Methods).
Fig. 1. A chemogenetic sensor for presynaptic Ca2+.

(A) Ca2+ sensor scheme. The HaloTag protein was targeted to nerve terminals by expression as a synaptophysin fusion construct. JF646-BAPTA bearing a HaloTag chloroalkane ligand (Deo et al., 2019) was added to the bath in AM ester form and allowed to undergo fluorogenic binding to Syp-HaloTag. (B) The reaction yields Syp-HaloTag-JF646-BAPTA, a Ca2+ sensor with bright resting fluorescence that matches the expected punctate distribution of synaptophysin labeling and readily reports presynaptic Ca2+ fluxes from single action potentials. Scale bar, 10 μm. Numbered ROIs correspond to traces shown in panels (D-E). (C) Comparison of exemplary single-stimulus, full field-of-view responses between Syp-HaloTag-JF646-BAPTA and Syp-GCaMP6f. The chemogenetic HaloTag-based approach demonstrates a substantial improvement in temporal fidelity. (D) A high-frequency stimulus train was used to obtain maximal fluorescence values for the sensor, which allows for calculation of [Ca]i (see Methods). The trace shown depicts the average fluorescence for the six labeled ROIs in panels (B) and (E). (E) Exemplary single-bouton, single-stimulus [Ca2+]i traces. Among the boutons shown here, the baseline [Ca2+]i was 102 ± 62 (s.d.) nM.
Co-expression of the fluorescent glutamate sensor iGluSnFR (Marvin et al., 2018), which is spectrally well-separated from JF646-BAPTA, permitted paired measurements of glutamate release from the same fields of view as used for the Ca2+ measurements. In this all-optical approach to measuring excitation-secretion coupling, cultured hippocampal neurons were transduced with lentiviruses to express both the synaptophysin-Halotag fusion protein and iGluSnFR (Fig. 2A–E). We applied this method to hippocampal neurons cultured from Sv2a WT and KO mouse pups (Fig. 2C–F, n = 3 cultures per genotype). Given the excellent presynaptic selectivity of the Ca2+-sensing construct (Fig. 1) and the need to repeatedly image multiple fields of view per coverslip, these experiments were conducted at lower resolution, and the resulting sequences were analyzed as full fields of view rather than bouton-by-bouton. All experiments were conducted on an Sv2b−/− background for consistency with previous reports (Chang and Sudhof, 2009; Custer et al., 2006), one of which found that neurons lacking SV2B do not have obvious deficits in neurotransmitter release (Custer et al., 2006). Resting [Ca2+]i was unchanged between Sv2a WT and KO (i.e., Sv2b KO and Sv2a/b DKO) neurons (Fig. 2B), consistent with the absence of a change in spontaneous synaptic current frequency in Sv2a KO neurons in slice (Crowder et al., 1999) and in culture (Chang and Sudhof, 2009; Custer et al., 2006). When single action potentials were evoked by electrical field stimulation, Syp-HaloTag-JF646-BAPTA reported peak [Ca2+]i values that increased linearly with [Ca2+]e under all conditions studied here, and Ca2+ entry was unchanged between WT and KO (Fig. 2F). By contrast, glutamate release was attenuated in the Sv2a KO at every [Ca2+]e studied, in accordance with previous reports (Chang and Sudhof, 2009; Custer et al., 2006) (Fig. 2G). For both genotypes, the relationship between iGluSnFR dF/F0 and [Ca2+]e (Fig. 2G) was well-fitted by a cooperative binding model with a Hill parameter of ~2.3 (WT: 2.2, 95% CI 1.7–2.9; KO: 2.5, 95% CI 1.9–3.0). Plotting the paired data for iGluSnFR versus peak [Ca2+]i in each field of view (Fig. 2H) yielded a similar relationship, which was noisier but converged on values closer to 4 (WT: 4.8, 95% CI 3.2–6.9; KO: 4.0, 95% CI 2.5–5.9). These results are comparable to those obtained in young cultured hippocampal neurons using flash photolysis and measurement of Ca2+ in dendrites (Burgalossi et al., 2010), which estimate this parameter to be approximately 3. As with the Hill parameter, values for [Ca2+]i at half-maximal glutamate release were nearly identical between Sv2a WT and KO neurons (WT: 337 nM, 95% CI 295–523 nM; KO: 341 nM, 95% CI 294–531 nM), indicating that the reduction in glutamate release in the Sv2a KO can be well-approximated by linearly scaling down the Ca2+-glutamate release curves observed for WT neurons (Fig. 2G–H). Our results thus demonstrate that SV2A does not change the Ca2+ dependence of glutamate release, in accordance with prior studies (Chang and Sudhof, 2009; Custer et al., 2006).
Fig. 2: SV2A supports release probability independently of presynaptic Ca2+.

(A) Representative field of view for Syp-HaloTag-JF646-BAPTA. Shown is the average fluorescence image for a single image sequence. (B) Resting [Ca2+]i at 2 mM [Ca2+]e did not differ between Sv2a WT and KO neurons (p = 0.53, Mann-Whitney test). (C) Fluorescence responses from the field of view shown in (A). Single action potentials were triggered at increasing [Ca2+]e. (D) The same field of view as in (A) was imaged in the GFP channel, showing the plasma-membrane localized iGluSnFR construct (left) and corresponding fluorescence responses from action potentials triggered at increasing [Ca2+]e (E). (F) Peak [Ca2+]i was plotted for each field of view at each [Ca2+]e examined. No differences were observed between SV2A WT and KO cultures (p = 0.97, Aikike Information Criteria). (G) iGluSnFR responses were plotted against [Ca2+]e for each field of view. Lines show the fit using a cooperative binding model with a Hill coefficient of ~2.3. (H) iGluSnFR responses were plotted against peak [Ca2+]i for each field of view. Lines show the fit of a cooperative binding model with Hill coefficient of ~4. In panels (G) and (H), no difference in the [Ca2+]e (G) and [Ca2+]i (H) at which glutamate release was half-maximal was observed for the WT and KO. The value of the Hill coefficient likewise did not differ between WT and KO. P values for panels (F), (G) and (H) are given as the probability that a single curve adequately fits the data as determined via the Aikike Information Criteria. (I) Averaged iGluSnFR signals from SV2A WT and KO neurons in response to 10-AP, 10-Hz stimulus trains. These experiments were carried out at 2 mM [Ca2+]e. (J-M) Parameters describing short-term plasticity calculated from each stimulus train response and plotted. The exocytosis deficit in SV2A KO neurons normalized over the course of the stimulus train, indicating that SV2A acts to support initial release probability. In panels (J-M), values stated indicate results of Mann-Whitney tests. For panels (A-F), n = 24 fields of view from 3 neuronal cultures per genotype; for panels (I-M), n = 24 fields of view from 2 neuronal cultures per genotype.
Application of 10-Hz stimulus trains (Fig. 2I) demonstrated that the reduced glutamate release resulted from a reduction in initial release probability (Fig. 2J–M), as supported by an increase in the paired-pulse ratio in Sv2a KO neurons (Fig. 2K) as well as a normalization of evoked glutamate release over the course of the stimulus train (Fig. 2I, L–M). Our results are in accordance with previous electrophysiological studies of glutamatergic and GABAergic transmission in Sv2a KO neurons (Chang and Sudhof, 2009; Custer et al., 2006). Together, these results strongly suggest that SV2A does not modulate Ca2+ influx or the direct action of Ca2+ ions, but instead controls the efficacy of Ca2+ effector molecules in a dynamic manner influenced by ongoing activity.
Discussion
While SV2A is well-known to support neurotransmitter release (Chang and Sudhof, 2009; Custer et al., 2006), the mechanism underlying this role has remained enigmatic. The ubiquity and physiologic importance of this putative SV transporter, which does not appear to transport canonical neurotransmitters, suggests important aspects of SV biology are yet to be defined. Because it is already a major drug target (Dong, 2006; Kaminski et al., 2008; Löscher et al., 2016; Lynch et al., 2004), defining a molecular function for SV2A at the synapse would likewise have important clinical ramifications.
A role for SV2 proteins as Ca2+ regulators has been suggested by some studies (Janz et al., 1999; Wan et al., 2010) but not others (Chang and Sudhof, 2009; Custer et al., 2006). None of these studies, however, directly assessed Ca2+ dynamics in small nerve terminals lacking SV2A. The chemogenetic Ca2+-sensing construct described here (Fig. 1) represents a useful tool for probing presynaptic Ca2+, with a favorable compromise among speed, spatial resolution, and temporal resolution versus existing techniques (Fig. 1). Future studies that capitalize on the spatial resolution of Syp-Halo-JF646-BAPTA, particularly using confocal imaging techniques, may expand the use of this tool for monitoring heterogeneity of synaptic Ca2+ transients among nerve terminals in genetically specified populations of neurons.
Integration of Syp-Halo-JF646-BAPTA with iGluSnFR enabled the monitoring of excitation-secretion with a convenient all-optical approach, which revealed that presynaptic Ca2+ entry is unaltered by the loss of SV2A (Fig. 2). The unchanged concentration dependence of glutamate release on extracellular [Ca2+] is consistent with this finding, as a reduction in Ca2+ entry per action potential would cause a shift in this curve (Fig. 2G–H) to the right if the function of the release machinery were otherwise preserved. Our results are consistent with previous studies in cultured hippocampal neurons (Chang and Sudhof, 2009; Custer et al., 2006), which likewise demonstrate an unchanged extracellular [Ca2+] dependence of glutamate (Custer et al., 2006) or GABA (Chang and Sudhof, 2009) release. Rather than change Ca2+ dynamics, SV2A appears to influence the release machinery itself. However, we interpret these data in favor of an indirect mechanism of action of SV2A rather than a direct allosteric effect on the Ca2+-secretion coupling process per se. The loss of a direct modulator is more likely to change the Ca2+ dependence of exocytosis, exemplified by the phenotype observed in neurons lacking the syt1-SNARE-binding protein complexin (Reim et al., 2001), synaptotagmin-1 (Burgalossi et al., 2010), or synaptotagmin-2 (Kochubey and Schneggenburger, 2011). In any case, these results establish unambiguously that SV2 modulates excitation-secretion coupling independently of Ca2+ entry. Our results differ from those obtained from synaptic terminals of retinal bipolar neurons from SV2B knockout mice (Wan et al., 2010), though it is difficult to draw meaningful comparisons across SV2 isoforms and synapse types.
While our results help to define the physiologic role of SV2A, a well-defined molecular function remains elusive. The normalization of synaptic deficits during sustained activity (Fig. 2I–M) suggests that ongoing action potential firing, Ca2+ entry, and vesicular recycling may help compensate for the absence of SV2A. One explanation for this could involve a role for SV2A in SV protein trafficking during periods of rest or development (Custer et al., 2006), and changes in abundance or localized trafficking of proteins such as syt1 (Kaempf et al., 2015; Yao et al., 2010; Zhang et al., 2015) may play a role in the Sv2a KO phenotype. Because SV2A co-immunoprecipitates with syt1 (Schivell et al., 1996), and because presynaptic syt1 is less abundant at synapses lacking SV2A (Yao et al., 2010), altered trafficking of syt1 provides an attractive candidate mechanism for SV2A’s role in exocytosis. However, certain point mutants suggest that presynaptic syt1 abundance does not correlate with SV2A’s function (Nowack et al., 2010). Finally, it is important to reaffirm that the amino acid sequence of SV2A strongly suggests some type of recognition and/or transport function for ions or small molecules (Madeo et al., 2014; Wibowo et al., 2014; Yao and Bajjalieh, 2008). Indeed, SV2A may carry out multiple important functions, such as both molecular transport and protein trafficking, and it is possible that only a subset of these functions directly supports synaptic transmission.
Methods
Ethical Approval
All procedures were carried out in accordance with protocols approved by the University of Wisconsin Institutional Animal Care and Use Committee (protocol M00586).
Cell culture and lentiviral transduction
P0-P2 newborn mouse pups of either sex from SV2B−/−, SV2A+/− breeders on a 129P2/OlaHsd background partially backcrossed to C57B6/J (Jackson Labs) were used. Pups were genotyped prior to dissection. Hippocampal formations from SV2A+/+ and SV2A−/− pups were obtained by microdissection and kept in Hibernate-A until the completion of dissection, at which point they were incubated with trypsin-EDTA (Corning, 0.25%) for 30 minutes at 37 °C with periodic agitation. Trypsin was then replaced with plating medium (DMEM +10% FBS) and tissue was triturated 10–20 times with a 1 ml pipette before plating at ~100,000 cells/cm2 (two pups per 12-well plate) onto poly-D-lysine coated glass coverslips. Plating medium was replaced with complete growth medium (Neurobasal-A medium supplemented with B-27 (1X, Gibco) and Glutamax (1X, Gibco) after 1 hour. Lentivirus encoding iGluSnFR A184V (Marvin et al., 2018), GAP43-jRGECO1a P2A synaptophysin-HaloTag, or synaptophysin-GCaMP6f were added on 5DIV. The bicistronic GAP43-jRGECO1a P2A synaptophysin-HaloTag construct was originally designed for the concurrent measurement of axonal [Ca2+]i transients using the axonally-targeted GAP43-jrGECO1a construct (Broussard et al., 2018). Even though the experiments shown here did not make use of the GAP43-jrGECO1a sensor, this construct was used because we observed more widespread and physiologic expression of Syp-HaloTag after the P2A sequence than with similar viral vectors encoding synaptophysin-HaloTag as the sole cistron under the synapsin promoter. Expression of GAP43-jRGECO1a did not affect Syp-JF646-BAPTA responses or the single-stimulus glutamate release phenotype of Sv2a KO neurons (Fig. 2). No differences were observed in axonal Ca2+ transients between WT and KO neurons (n = 2 litters, data not shown).
Molecular biology and lentiviral preparation
A modified iGluSnFR A184V bearing endoplasmic reticulum and Golgi export signals (Vevea and Chapman, 2020) was subcloned into the FUGW transfer plasmid under the synapsin promoter. GAP43-jRGECO1a (Broussard et al., 2018; Dana et al., 2016) P2A synaptophysin-HaloTag was generated by overlap extension PCR. DNA sequences were subcloned using the In-Fusion cloning system (Takara) into the FUGW lentiviral transfer plasmid (Addgene plasmid #14883, a gift from Dr. David Baltimore) modified to include the human synapsin promoter. Lentivirus was generated by CaPO4-mediated cotransfection of HEK293-T cells with transfer plasmid and the helper plasmids pCD/NL-BH*ΔΔΔ (Addgene plasmid #17531, a gift from Dr. Jakob Reiser) and pLTR-G (Addgene plasmid #17532, a gift from Dr. Jakob Reiser) followed by concentration of virus-bearing supernatant by ultracentrifugation as described (Kutner et al., 2009). We reiterate that the GAP43-jRGECO1a P2A Syp-Halo construct was used because we observed more even and widespread expression of Syp-HaloTag with this construct than with constructs expressing Syp-HaloTag alone. Fluorescence from GAP43-jRGECO did not interfere with glutamate or Ca2+ measurements, nor did it affect the well-described SV2 KO phenotype of reduced glutamate release (Fig. 2).
Glutamate and Ca2+ imaging
For the experiments shown in Fig. 2A–H, cultered neurons (15–18DIV) were washed twice with an artificial cerebrospinal fluid (ACSF) containing (in mM) NaCl 128, KCl 2.5, MgCl 1, CaCl2 1.2, HEPES 25, glucose 30 and incubated for 20 minutes at 37 °C in the same ACSF containing JF646-BAPTA-HTL-AM (a gift from Dr. Luke Lavis, Janelia Research Campus). JF646-BAPTA-HTL-AM was aliquoted as a 1 mM stock in anhydrous DMSO. For each experiment, an aliquot was diluted to 100 μM in anhydrous DMSO, and 1 μl of this diluted solution was added to 550 μl ACSF immediately prior to this incubation. After incubation with dye, the coverslip was washed (3 × 1 ml) and incubated for 20 more minutes at 37 °C in dye-free ACSF before being mounted in an imaging chamber with platinum electrodes (Warner Instruments). Imaging was carried out with a 40X 1.4 NA oil-immersion objective on an inverted epifluorescence microscope (IX81, Olympus) equipped with CMOS camera (Orca Flash 4.0 V2, Hamamatsu), motorized stage (Mad City Labs), and a custom illumination source containing three LEDs (470 nm, 530 nm, 625 nm) (Thorlabs). The system was controlled using Micro-Manager (Edelstein et al., 2010). Stimulus pulses (100V, 0.5 ms) were delivered with a stimulation box (SD9, Grass) controlled via a HEKA EPC 10 DAQ-amplifier and PatchMaster software, which was also used to synchronize the start of image sequence acquisition. For iGluSnFR measurements, the 470 nm LED, a standard GFP filter set (49002, Chroma) and a 10 ms exposure time was used, while JF646-BAPTA signals were acquired using the 625 nm LED, custom 3-band pass dichroic mirror (Chroma) and far-red emission filter with a 20 ms exposure time. Images were collected using binning at 2×2 (0.325 μm pixels, Fig. 1) or 4×4 (0.65 μm pixel size, Fig. 2). For each coverslip, four fields of view were selected, and each field of view was imaged sequentially for glutamate release and presynaptic Ca2+ entry, with at least 15 seconds allowed between stimuli. This was repeated for each field of view for each ACSF solution containing varying CaCl2, and the motorized stage was used to return to the same 4 fields of view for all external solutions. At the end of each experiment, each field of view was subjected to a saturating stimulus (50 AP @ 50 Hz) while imaging Syp-HaloTag-JF646-BAPTA to establish a maximum fluorescence (Fmax) for that field of view, which was used to derive [Ca2+]i values (see below). Given the expected presynaptic localization of Syp-HaloTag-JF646-BAPTA, the fluorogenic properties of the complete HaloTag-bound Ca2+ sensor (Deo et al., 2019) the dense expression of Syp-HaloTag, and the difficulty of obtaining the exact focal plane for each Ca2+ condition, image sequences were analyzed as entire fields of view. For the experiment shown in Fig. 1, iGluSnFR was not expressed, and the ACSF contained 2 mM CaCl2 during staining, washing, and imaging. For the data shown in Fig. 2 panels I-M, no Ca2+ sensing constructs were expressed, and coverslips were simply transferred from the incubator to the microscope and equilibrated at room temperature for at least 5 minutes before recording. In these experiments, the ACSF contained 2 mM CaCl2. At least 4 minutes were allowed between stimulus trains.
Data analysis
iGluSnFR image sequences were converted to ΔF/F0 traces after background subtraction using custom-written scripts in FIJI and R. Syp-Halo-JF646-BAPTA traces were background-subtracted, and [Ca2+]i was derived using the reported in vitro values for the sensor’s dynamic range and Ca2+ affinity after establishing the maximal fluorescence intensity (Fmax, i.e., the signal resulting from fully-occupied sensor) in each field of view (de Juan-Sanz et al., 2017; Maravall et al., 2000). The following equation was used:
Where Kd is the dissociation constant of the indicator, F is the average fluorescence across the field of view in each frame, Fmax is the fluorescence achieved with delivery of a saturating stimulus, Rf is the dynamic range of the indicator, and n is the Hill coefficient. For Kd, Rf, and n, we used in vitro measurements (KD = 140 nM, Rf = 5.5, n = 1) from Deo et al. (2019). The maximum fluorescence achieved with a 50-AP, 50 Hz train was defined as 95% of the true theoretical maximum value (Maravall et al., 2000). To determine the ROIs as shown in Fig. 1, a script was used in ImageJ to perform the following: average 10 frames prior stimulation, perform 5-pixel radius rolling-ball background subtraction, manually threshold, select of all above-threshold areas of the image with a minimum contiguous area of 5 pixels, and separate individual boutons with the watershed function. A time course for average pixel intensity in each ROI was obtained and the data were background-subtracted and converted to Ca2+ values using a script written in R. Data for glutamate release versus Ca2+ were plotted (Fig. 2) and fitted using a cooperative binding equation with Hill slope in Prism (GraphPad).
Statistical methods
For pairwise comparisons of datasets (Fig. 2B, Fig. 2J–M), the nonparametric Mann-Whitney test was used. For comparison of Ca2+ entry as a function of extracellular Ca2+, glutamate release as a function of extracellular Ca2+, and glutamate release as a function of Ca2+ entry (Fig. 2F–H), the Aikike Information Criteria were used to determine whether a single curve or two separate curves provided the most appropriate fit to the data.
Supplementary Material
Key points summary.
One of the most prescribed antiepileptic medications, levetiracetam, acts by binding a protein of uncertain molecular function
This transporter-like protein, SV2A, is trafficked to synaptic vesicles and acts to support neurotransmitter release, but the mechanism underlying this function has not been determined
In this study, we sought to establish whether SV2A changes Ca2+ signaling at nerve terminals, which is a key regulatory system for synaptic vesicle exocytosis
To do so, we adapted new chemogenetic tools to perform all-optical measurements of presynaptic Ca2+ and glutamate release in neurons lacking SV2A
Our measurements showed that loss of SV2A reduces glutamate release without reducing Ca2+ influx at hippocampal nerve terminals, demonstrating that SV2A increases the likelihood that Ca2+ will trigger a vesicular fusion event
Acknowledgments
We thank Dr. Luke Lavis (Janelia Research Campus, Howard Hughes Medical Institute) for the generous gift of JF646-BAPTA-HaloTag ligand, and we thank members of the Chapman lab for helpful comments and suggestions. This work was supported by grants from the National Institutes of Health (grants MH116580 to M.M.B., MH061876 to E.R.C.). E.R.C. is an Investigator of the Howard Hughes Medical Institute.
Author profile and photo
Bradberry & Chapman, JP-RP-2021–282601R1
Mazdak Moghaddam Bradberry is an M.D./Ph.D. student at the University of Wisconsin-Madison. His undergraduate studies in chemistry were completed at Northwestern University, and his Ph.D. thesis work was conducted under the supervision of Dr. Ed Chapman. He is broadly interested in the neurobiology of the synapse, from the mechanistic elements of neurotransmitter release to the biogenesis of synaptic vesicles and the vulnerability of nerve terminals to pathophysiologic processes. He plans to enter clinical training in psychiatry and continue his scientific career in the study of fundamental neurobiological processes.

Footnotes
Declaration Of Interests
The authors have no competing interests to declare.
Data Availability Statement
All primary data reported here are available upon reasonable request to either author (M.M.B or E.R.C.).
References
- Bajjalieh SM, Peterson K, Shinghal R, Scheller RH, 1992. SV2, a Brain Synaptic Vesicle Protein Homologous to Bacterial Transporters. Science, New Series 257, 1271–1273. [DOI] [PubMed] [Google Scholar]
- Broussard GJ, Liang Y, Fridman M, Unger EK, Meng G, Xiao X, Ji N, Petreanu L, Tian L, 2018. In vivo measurement of afferent activity with axon-specific calcium imaging. Nat Neurosci 21, 1272–1280. 10.1038/s41593-018-0211-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley K, Kelly RB, 1985. Identification of a transmembrane glycoprotein specific for secretory vesicles of neural and endocrine cells. The Journal of Cell Biology 100, 1284–1294. 10.1083/jcb.100.4.1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgalossi A, Jung S, Meyer G, Jockusch WJ, Jahn O, Taschenberger H, O’Connor VM, Nishiki T, Takahashi M, Brose N, Rhee J-S, 2010. SNARE Protein Recycling by αSNAP and βSNAP Supports Synaptic Vesicle Priming. Neuron 68, 473–487. 10.1016/j.neuron.2010.09.019 [DOI] [PubMed] [Google Scholar]
- Chang W-P, Sudhof TC, 2009. SV2 Renders Primed Synaptic Vesicles Competent for Ca2+-Induced Exocytosis. Journal of Neuroscience 29, 883–897. 10.1523/JNEUROSCI.4521-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowder KM, Gunther JM, Jones TA, Hale BD, Zhang HZ, Peterson MR, Scheller RH, Chavkin C, Bajjalieh SM, 1999. Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A). Proceedings of the National Academy of Sciences 96, 15268–15273. 10.1073/pnas.96.26.15268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custer Kenneth L., Austin NS, Sullivan JM, Bajjalieh SM, 2006. Synaptic Vesicle Protein 2 Enhances Release Probability at Quiescent Synapses. Journal of Neuroscience 26, 1303–1313. 10.1523/JNEUROSCI.2699-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana H, Mohar B, Sun Y, Narayan S, Gordus A, Hasseman JP, Tsegaye G, Holt GT, Hu A, Walpita D, Patel R, Macklin JJ, Bargmann CI, Ahrens MB, Schreiter ER, Jayaraman V, Looger LL, Svoboda K, Kim DS, 2016. Sensitive red protein calcium indicators for imaging neural activity. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Juan-Sanz J, Holt GT, Schreiter ER, de Juan F, Kim DS, Ryan TA, 2017. Axonal Endoplasmic Reticulum Ca2+ Content Controls Release Probability in CNS Nerve Terminals. Neuron 93, 867–881.e6. 10.1016/j.neuron.2017.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deo C, Sheu S-H, Seo J, Clapham DE, Lavis LD, 2019. Isomeric Tuning Yields Bright and Targetable Red Ca 2+ Indicators. J. Am. Chem. Soc 141, 13734–13738. 10.1021/jacs.9b06092 [DOI] [PubMed] [Google Scholar]
- Dong M, 2006. SV2 Is the Protein Receptor for Botulinum Neurotoxin A. Science 312, 592–596. 10.1126/science.1123654 [DOI] [PubMed] [Google Scholar]
- Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N, 2010. Computer Control of Microscopes Using μManager, in: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (Eds.), Current Protocols in Molecular Biology. John Wiley & Sons, Inc., Hoboken, NJ, USA, p. mb1420s92. 10.1002/0471142727.mb1420s92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feany MB, Lee S, Edwards RH, Buckley KM, 1992. The synaptic vesicle protein SV2 is a novel type of transmembrane transporter. Cell 70, 861–867. 10.1016/0092-8674(92)90319-8 [DOI] [PubMed] [Google Scholar]
- García-Pérez E, Mahfooz K, Covita J, Zandueta A, Wesseling JF, 2015. Levetiracetam accelerates the onset of supply rate depression in synaptic vesicle trafficking. Epilepsia 56, 535–545. 10.1111/epi.12930 [DOI] [PubMed] [Google Scholar]
- Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA, 2012. α2δ expression sets presynaptic calcium channel abundance and release probability. Nature 486, 122–125. 10.1038/nature11033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsson JA, Haitina T, Lindblom J, Fredriksson R, 2007. Identification of six putative human transporters with structural similarity to the drug transporter SLC22 family. Genomics 90, 595–609. 10.1016/j.ygeno.2007.03.017 [DOI] [PubMed] [Google Scholar]
- Janz R, Goda Y, Geppert M, Missler M, Südhof TC, 1999. SV2A and SV2B Function as Redundant Ca2+ Regulators in Neurotransmitter Release. Neuron 24, 1003–1016. 10.1016/S0896-6273(00)81046-6 [DOI] [PubMed] [Google Scholar]
- Kaempf N, Kochlamazashvili G, Puchkov D, Maritzen T, Bajjalieh SM, Kononenko NL, Haucke V, 2015. Overlapping functions of stonin 2 and SV2 in sorting of the calcium sensor synaptotagmin 1 to synaptic vesicles. Proc Natl Acad Sci USA 112, 7297–7302. 10.1073/pnas.1501627112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski RM, Gillard M, Leclercq K, Hanon E, Lorent G, Dassesse D, Matagne A, Klitgaard H, 2009. Proepileptic phenotype of SV2A-deficient mice is associated with reduced anticonvulsant efficacy of levetiracetam. Epilepsia 50, 1729–1740. 10.1111/j.1528-1167.2009.02089.x [DOI] [PubMed] [Google Scholar]
- Kaminski RM, Matagne A, Leclercq K, Gillard M, Michel P, Kenda B, Talaga P, Klitgaard H, 2008. SV2A protein is a broad-spectrum anticonvulsant target: Functional correlation between protein binding and seizure protection in models of both partial and generalized epilepsy. Neuropharmacology 54, 715–720. 10.1016/j.neuropharm.2007.11.021 [DOI] [PubMed] [Google Scholar]
- Kartal A, 2017. Can High-Dose Levetiracetam Be Safe? A Case Report of Prolonged Accidental High-Dose Levetiracetam Administration and Review of the Literature. Clin Neuropharm 40, 217–218. 10.1097/WNF.0000000000000229 [DOI] [PubMed] [Google Scholar]
- Klitgaard H, Matagne A, Nicolas J-M, Gillard M, Lamberty Y, De Ryck M, Kaminski RM, Leclercq K, Niespodziany I, Wolff C, Wood M, Hannestad J, Kervyn S, Kenda B, 2016. Brivaracetam: Rationale for discovery and preclinical profile of a selective SV2A ligand for epilepsy treatment. Epilepsia 57, 538–548. 10.1111/epi.13340 [DOI] [PubMed] [Google Scholar]
- Kochubey O, Schneggenburger R, 2011. Synaptotagmin Increases the Dynamic Range of Synapses by Driving Ca2+-Evoked Release and by Clamping a Near-Linear Remaining Ca2+ Sensor. Neuron 69, 736–748. 10.1016/j.neuron.2011.01.013 [DOI] [PubMed] [Google Scholar]
- Kutner RH, Zhang X-Y, Reiser J, 2009. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat Protoc 4, 495–505. 10.1038/nprot.2009.22 [DOI] [PubMed] [Google Scholar]
- Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV, 2008. HaloTag: A Novel Protein Labeling Technology for Cell Imaging and Protein Analysis. ACS Chem. Biol 3, 373–382. 10.1021/cb800025k [DOI] [PubMed] [Google Scholar]
- Löscher W, Gillard M, Sands ZA, Kaminski RM, Klitgaard H, 2016. Synaptic Vesicle Glycoprotein 2A Ligands in the Treatment of Epilepsy and Beyond. CNS Drugs 30, 1055–1077. 10.1007/s40263-016-0384-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W, Klitgaard H, Twyman RE, Schmidt D, 2013. New avenues for anti-epileptic drug discovery and development. Nat Rev Drug Discov 12, 757–776. 10.1038/nrd4126 [DOI] [PubMed] [Google Scholar]
- Lynch BA, Lambeng N, Nocka K, Kensel-Hammes P, Bajjalieh SM, Matagne A, Fuks B, 2004. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proceedings of the National Academy of Sciences 101, 9861–9866. 10.1073/pnas.0308208101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo M, Kovács AD, Pearce DA, 2014. The Human Synaptic Vesicle Protein, SV2A, Functions as a Galactose Transporter in Saccharomyces cerevisiae. J. Biol. Chem 289, 33066–33071. 10.1074/jbc.C114.584516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maravall M, Mainen ZF, Sabatini BL, Svoboda K, 2000. Estimating Intracellular Calcium Concentrations and Buffering without Wavelength Ratioing. Biophysical Journal 78, 2655–2667. 10.1016/S0006-3495(00)76809-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin JS, Scholl B, Wilson DE, Podgorski K, Kazemipour A, Müller JA, Schoch S, Quiroz FJU, Rebola N, Bao H, Little JP, Tkachuk AN, Cai E, Hantman AW, Wang SS-H, DePiero VJ, Borghuis BG, Chapman ER, Dietrich D, DiGregorio DA, Fitzpatrick D, Looger LL, 2018. Stability, affinity, and chromatic variants of the glutamate sensor iGluSnFR. Nat Methods 15, 936–939. 10.1038/s41592-018-0171-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowack A, Yao J, Custer KL, Bajjalieh SM, 2010. SV2 regulates neurotransmitter release via multiple mechanisms. American Journal of Physiology-Cell Physiology 299, C960–C967. 10.1152/ajpcell.00259.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reim K, Mansour M, Varoqueaux F, McMahon HT, Südhof TC, Brose N, Rosenmund C, 2001. Complexins Regulate a Late Step in Ca2+-Dependent Neurotransmitter Release. Cell 104, 71–81. 10.1016/S0092-8674(01)00192-1 [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG, 1998. Optical Measurement of Presynaptic Calcium Currents. Biophysical Journal 74, 1549–1563. 10.1016/S0006-3495(98)77867-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schivell AE, Batchelor RH, Bajjalieh SM, 1996. Isoform-specific, Calcium-regulated Interaction of the Synaptic Vesicle Proteins SV2 and Synaptotagmin. J. Biol. Chem 271, 27770–27775. 10.1074/jbc.271.44.27770 [DOI] [PubMed] [Google Scholar]
- Vevea JD, Chapman ER, 2020. Acute disruption of the synaptic vesicle membrane protein synaptotagmin 1 using knockoff in mouse hippocampal neurons. eLife 9, e56469. 10.7554/eLife.56469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q-F, Zhou Z-Y, Thakur P, Vila A, Sherry DM, Janz R, Heidelberger R, 2010. SV2 Acts via Presynaptic Calcium to Regulate Neurotransmitter Release. Neuron 66, 884–895. 10.1016/j.neuron.2010.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wibowo A, Peters EC, Hsieh-Wilson LC, 2014. Photoactivatable Glycopolymers for the Proteome-Wide Identification of Fucose-α(1–2)-Galactose Binding Proteins. J. Am. Chem. Soc 136, 9528–9531. 10.1021/ja502482a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Bognar J, He T, Mohammed M, Niespodziany I, Wolff C, Esguerra M, Rothman SM, Dubinsky JM, 2015. Brivaracetam augments short-term depression and slows vesicle recycling. Epilepsia 56, 1899–1909. 10.1111/epi.13223 [DOI] [PubMed] [Google Scholar]
- Yao J, Bajjalieh SM, 2008. Synaptic Vesicle Protein 2 Binds Adenine Nucleotides. J. Biol. Chem 283, 20628–20634. 10.1074/jbc.M800738200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Nowack A, Kensel-Hammes P, Gardner RG, Bajjalieh SM, 2010. Cotrafficking of SV2 and Synaptotagmin at the Synapse. Journal of Neuroscience 30, 5569–5578. 10.1523/JNEUROSCI.4781-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh FL, Dong M, Yao J, Tepp WH, Lin G, Johnson EA, Chapman ER, 2010. SV2 Mediates Entry of Tetanus Neurotoxin into Central Neurons. PLoS Pathog 6, e1001207. 10.1371/journal.ppat.1001207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Gordon SL, Fritsch MJ, Esoof N, Campbell DG, Gourlay R, Velupillai S, Macartney T, Peggie M, van Aalten DMF, Cousin MA, Alessi DR, 2015. Phosphorylation of Synaptic Vesicle Protein 2A at Thr84 by Casein Kinase 1 Family Kinases Controls the Specific Retrieval of Synaptotagmin-1. Journal of Neuroscience 35, 2492–2507. 10.1523/JNEUROSCI.4248-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All primary data reported here are available upon reasonable request to either author (M.M.B or E.R.C.).