

Abstract
KRAS mutation is prevalent in around 30% of all cancers and is an undruggable molecular target. Of seven mutations at codon 12 and 13, only one, the G12C mutant is finally proven to be druggable, as evidenced by the recent USFDA approval of sotorasib. Investigation of other small molecules targeting G12C and G12D are undergoing clinical trials. Understanding the fine structural details is a prerequisite to design specific inhibitors which also requires in depth molecular exploration. We used bioinformatics as a tool to analyze the KRAS protein’s GTP (guanosine triphosphate) binding dynamics when mutated. KRAS undergoes significant conformational changes, affecting GTP binding conformation within the active site pocket of KRAS due to high torsional strains, hydrophobicity, and altered Switch I and II regions. GTP molecule for wildtype had a low torsional strain of 10.71, and is the only molecule, in comparison to KRAS mutant bound GTP, to have a glycine at position 10 interacting with its nitrogenous base. All mutant KRAS proteins lacked the interaction of glycine with the nitrogenous base. The binding affinity of wildtype (WT) KRAS for the gamma-phosphate was lower in scoring compared to the mutated KRAS protein in multiple analyses. This study provides an insight to the GTP-KRAS protein binding detailsthat is important to define parameters required to be explored to design the appropriate inhibitor for each different type of mutant KRAS protein.
Keywords: KRAS, Codon 12 and 13, 3D-structure, kinetics, simulation
Graphical Abstract

Software
PyMOL
PyMOL is a program used for 3D protein molecular visualization. The PyMOL Molecular Graphics System, Version 2.0 Schrodinger, LLC [1].
Discovery Studio
Discovery Studio is a software for simulations and mutations of protein systems. (2012) Discovery Studio Visualizer Software, Version 4.0 [2].
Maestro
Maestro is a powerful graphics software that can perform various modifications and assessments of proteins and calculations such as simulations or binding affinity.
Schrödinger Release 2021–1: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2021. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2021.
Schrödinger Release 2021–1: Epik, Schrödinger, LLC, New York, NY, 2021.
Jorgensen, W.L.; Tirado-Rives, J., “The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin,” J. Am. Chem. Soc., 1988, 110(6), 1657–1666[3]
Schrödinger Release 2021–1: FEP+, Schrödinger, LLC, New York, NY, 2021.
Schrödinger Release 2021–1: Glide, Schrödinger, LLC, New York, NY, 2021.
Schrödinger Release 2021–1: Maestro, Schrödinger, LLC, New York, NY, 2021.
Schrödinger Release 2021–1: Prime, Schrödinger, LLC, New York, NY, 2021.[4]
Schrödinger Release 2021–1: Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY, 2021; Impact, Schrödinger, LLC, New York, NY; Prime, Schrödinger, LLC, New York, NY, 2021.
AutoDock Vina and MGL Tools
Version AutoDock 4.2. AutoDock Vina and MGL Tools are software used to visualize and perform molecular docking between protein and substrate interactions. Program uses a grid to identify a region of binding, and can calculate the RMSD(root-mean-square deviation) scoring for the affinity of binding and favorable interactions [5].
Introduction
KRAS (Kirsten rat sarcoma viral oncogene homolog) mutations are the most frequent and prevalent driver oncogenes in various cancers, occurring in about 30% of all cancer patients [6, 7]. The GTPase KRAS is a signal-transducing protein, switching between its inactive [GDP (guanosine diphosphate)-bound] and active states (GTP-bound) [8]. KRAS is frequently mutated in human cancers in the pancreas, lungs, and colon. 99.2% of the mutations are found at the 12th, 13th and 61st residue position [9]. The most prevalent mutation is at the 12th residue, responsible for 77% KRAS mutations, in which a glycine substitution is observed. This mutation, G12X, is associated with most oncogenic properties due to deficiency of intrinsic GTPase activity. Active WT KRAS plays a role as a GTPase through hydrolysis of the gamma phosphate in GTP, bringing it to an inactive state (GDP) [10, 11]. Mutant KRAS however, impairs GTPase activity, preventing hydrolysis of the gamma phosphate. Unable to switch to a GDP-bound state, mutant KRAS continues in its active state, leading to continuous downstream signaling associated with oncogenic cellular growth [12].
About 30% of all human cancers harbor a point mutation in the KRAS gene, particularly in adenocarcinomas of the pancreas, lung, and colon [13]. The most common substitutions are for glycine at the 12th and 13th positions, including G12D (33.7%), G12V (32.7%), G12C (14%), and G13D (12.5%). Oncogenic KRAS mutants are seen at positions 12 and 13 in cancer patients. G12 is the location of the protein’s active site, consisting of a p-loop (residues 10–17) along with two switch regions (SI, residues 25–40 and SII, residues 60–74). Mutation at position G12X leads to bulkier side chains in the active site, which interferes with steric activity in GTP hydrolysis.
We used bioinformatics to help understand why mutations in G12X or G13D prevent GTP hydrolysis. Our analysis targets WT KRAS and mutates the 12th and 13th positions computationally to simulate the structural changes and scoring functions ofeach mutant KRAS protein as well as its binding characteristics to GTP. We used a multi-step procedure to obtain and modify an existing crystal structure of KRAS from the RCSB database, mutate the residue of interest, dock the mutated and wildtype protein to GTP, perform a molecular dynamics simulation and then finally perform a structural assessment to observe any important local or dynamic changes. We compared each mutated simulation against wildtype KRAS to observe and compare any structural changes at a molecular level that might be affecting the hydrolysis of the gamma phosphate of GTP and how the KRAS protein binding pocket might be affected.
Materials and Methods
Protocol
KRAS 4OBE Crystal Structure Modification and Preparation
In PyMOL, the PDB entry ‘4OBE’ was used as the structure of reference [14]. This crystal structure contains chains A and B, along with the ligand magnesium and GDP (guanosine 5’-diphosphate) substrate. Chain B was removed using PyMOL. Ligands and substrates were removed from the crystal structure. This crystal structure contained 169 amino acid residues of chain A.
Introducing Mutations
Eight copies of the PDB entry, 4OBE, were created, one for each of the seven prominent mutations, and one representing the wildtype. One mutation was introduced in each mutant protein using BIOVIA Discovery Studio. The mutations were implemented in position 12 or 13. Position 12 mutated from a glycine to either: Aspartic acid, cysteine, valine, alanine, arginine, and serine. Position 13 mutated from glycine to aspartic acid. KRAS wildtype was left with no mutations, and therefore did not need to be implemented in BIOVIA Discovery Studio.
Protein-substrate Docking
Each protein was entered into AutoDock Vina. Water molecules were removed, and hydrogens were added to each polar region. Kollman charges were added, which gave the template values for each amino acid. The protein was saved as a new file extension (.pdbqt) for docking preparation. Coordinates were generated in AutoDock Vina to determine the site of binding and were saved as a text file. Coordinates were used as the standard for GTP. GTP substrates were submitted into the program and were saved as an extension (.pdbqt) file for docking preparation. These GTP molecules were selected as the primary substrates for binding. Once docking was completed, output files were generated, creating substrate conformation pdb structures. Each KRAS protein (mutant and wildtype) was docked with GTP molecule using AutoDock Vina.
Protein Preparation
Using Maestro’s protein preparation [15], KRAS mutants and wildtype went through protein preparation for molecular dynamics simulation. Bond orders were assigned using the CCD database. Original hydrogens were removed, and new hydrogens were added. Using Epik, het states were generated with a pH of 7 +/− 1 using Prime. Any missing side chains were filled.
Energy Minimization
Using Schrodinger software, we performed energy minimization using Prime. Force field OPLS3e was used. OPLS are a series of equations that are calculated when any jobs are being run for molecular dynamic simulation or minimization to calculate any bonding angles. This is important in determining the position of any atoms within the protein or substrate because this determines the arrangements of these atoms and how we can identify how these atoms interact with each other. The solvation model was a vacuum space. All the residues were selected for each respective KRAS protein. Minimization ensures the lowest energy state for each mutant and wildtype KRAS protein.
System Builder
Each GTP-bound protein went through Desmond’s system builder, creating a solvation model. An orthorhombic shaped solvation model was created around each respective GTP-bound KRAS protein (10×10×10 angstroms). Since the phosphate groups were deprotonated, ions were automatically selected to be included. However, ions were selected to not be included in this procedure. Within the orthorhombic space, the protein volume was minimized. Force field OPLS3e was implemented [3].
Minimization
Each substrate-bound protein system went through another minimization through Desmond. Simulation time was 100 picoseconds. This minimization ensured the lowest possible energy state of the protein.
Molecular Dynamics Simulation
Each GTP-bound protein system was uploaded individually in Desmond’s Molecular Dynamics Simulation [16]. Simulation time was 50 nanoseconds Temperature was at 300 Kelvin and a pressure of 1.01325, relaxation of the system model was selected before the simulation began. An interaction analysis was selected to generate data for protein-substrate contacts.
Results
Substrate MM-GBSA Torsional Strain
Torsional strain is a close approach of atoms or groups and a repulsive force of electrons. The lower the value for torsional strain, the less strain there is on an atom, therefore being more favorable. Wildtype KRAS protein had a torsional strain of 10.71, which is more favorable in comparison to KRAS mutant proteins. G12D has the highest torsional strain of 18.27 (Table 1). A low torsional strain value indicated flexible bond rotation, therefore wildtype KRAS had less constriction, while KRAS mutant variants molecules’ rotation were more constrained.
Table 1.
MM-GBSA Scoring of GTP molecule. MM-GBSA scoring was used to determine the torsional strain of each KRAS protein bound GTP molecule. WT steric strain value of 10.71, which was more favorable in comparison to the mutants. KRAS variants are listed with each of their respective torsional angle values.
| KRAS Protein Variant | GTP MM-GBSA Torsional Strain |
|---|---|
| Wildtype | 10.71 |
| G12C | 14.96 |
| G12D | 18.27 |
| G12V | 15.41 |
| G13D | 15.16 |
| G12A | 13.22 |
| G12S | 15.67 |
| G12R | 15.87 |
GTP molecule-Atom Contact with Gamma-Phosphate
Ligand (substrate) fluctuations on the Ligand RMSF graph for GTP bound KRAS wildtype had four distinct peaks: 2.31, 2.54, 2.52, and 2.68 angstroms for the phosphorous and three outer oxygen atoms. While GTP bound G12D had four distinct peaks: 1.35, 1.40, 1.73, and 1.23 angstroms on the phosphorus and three outer oxygen atoms, respectively. According to the atom index (Figure 1), this implies that those four distinct atoms had closest contact with KRAS protein in KRAS G12D, while in KRAS wildtype, the gamma phosphate had the furthest contact. KRAS mutants’ gamma phosphate’s distances are all significantly below the KRAS wildtype distance (Figure 1).
Figure 1.
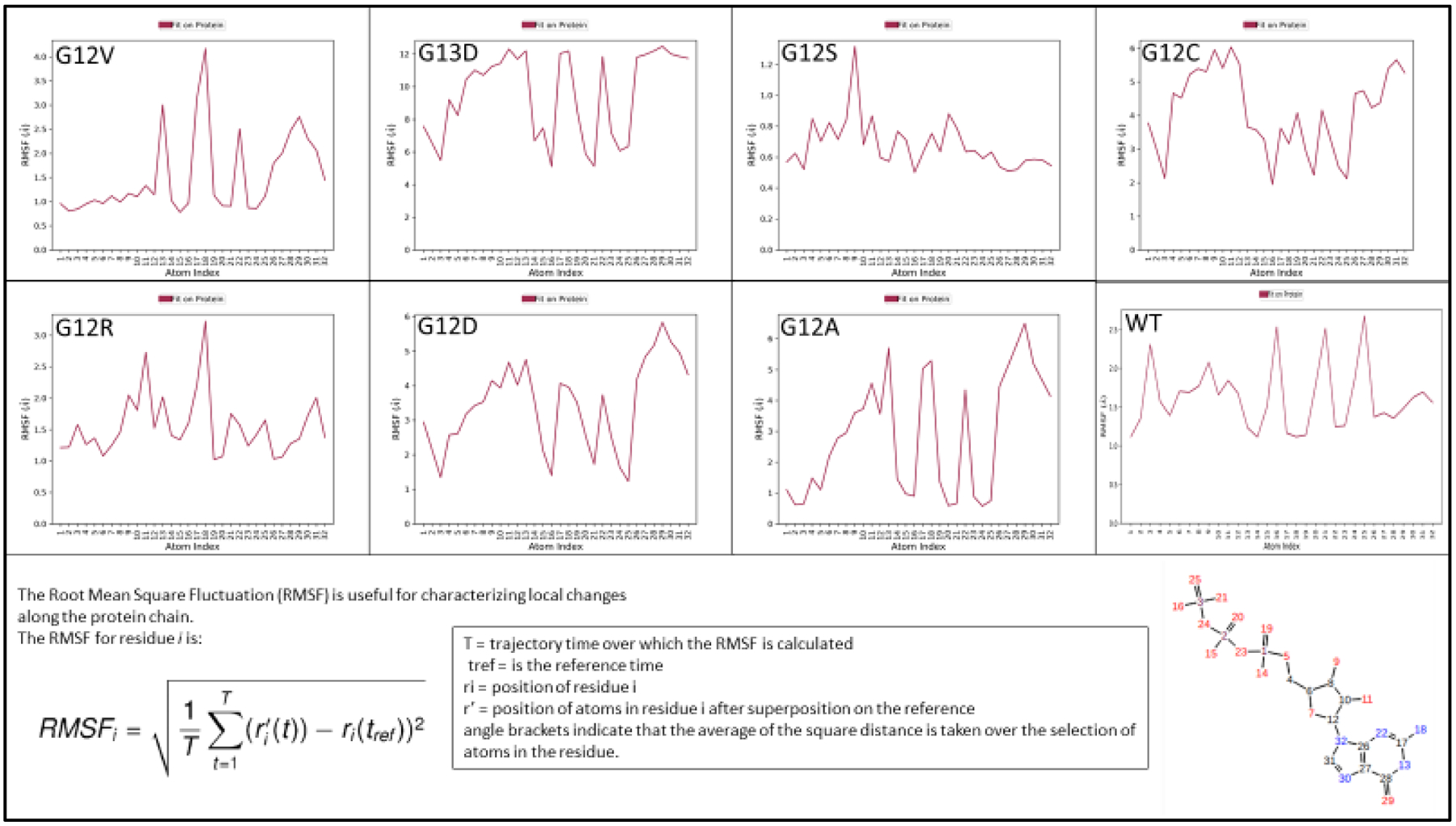
RMSF Contact. X-axis represents the atom numbers indicated on the GTP ligand on the right-hand side, the Y-axis represents the RMSF values in angstroms to determine the closeness of the GTP molecule. G12D, G13D, G12C RMSF graphs are all flipped in comparison to the WT and G12V, four significant peaks of contact are on the gamma-phosphate.
RMSD Fluctuation and Stability of KRAS Wildtype and Mutant Proteins
The Root Mean Square Deviation (RMSD) [17] graphs show the contact fluctuations and stability of the alpha carbon backbone. For KRAS wildtype, there was a brief contact at around 30 nanoseconds into the simulation, but slowly disconnected with the KRAS protein carbon backbone (Figure 2). While the KRAS wildtype shows a steady decline in contact, the KRAS G12D shows prolonged contact, indicating stronger binding stability (Figure 2). The closest similarity in contact fluctuation with KRAS wildtype was mutant G12C. The G12V and G12A mutant protein demonstrated significant stability, overlapping with the carbon backbone for the majority of the simulation time. Both G13D and G12R RMSD graphs begin the simulation with a large gap between substrate and protein alpha carbon backbone, as opposed to the other KRAS proteins, where both begin relatively close to each other. As the simulation continued, alpha carbon back of the mutant KRAS proteins for G13D and G12R came very close into contact and showed stable fluctuations.
Figure 2.
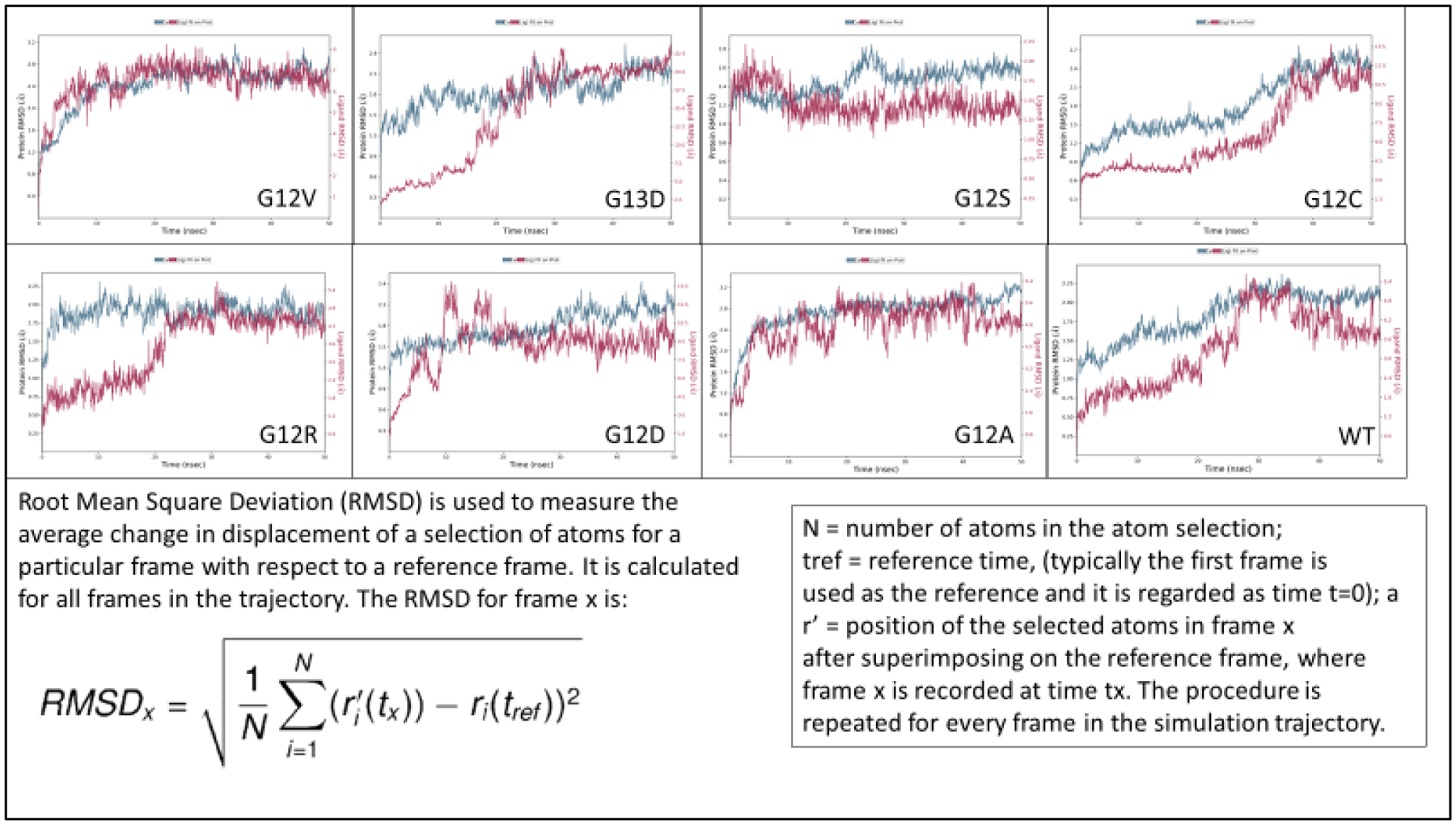
Root Mean Square Deviation (RMSD) Versus Time (nanoseconds). This graph determines the contact and fluctuation of the substrate (purple) and protein (blue). WT and G12C display similar trends of RMSD values. G12V and G12A show significant stabilization.
Gamma-Phosphate Bond Angle Measurements Varied Between KRAS Variants
Using Schrodinger’s measurement tool, bond angles were measured on the following atom angles: The oxygen linking the phosphodiester bond between the gamma phosphate and beta phosphate (1), the phosphodiester bond link between the alpha and beta phosphate (2), the oxygen linking to the beta carbon from the ribose sugar (3), the beta carbon between the oxygen atom of the alpha phosphate group and the central carbon from the ribose sugar (4), the nitrogen atom linked to the ribose sugar (5), and the central carbon forming the ribose sugar and linked to the beta carbon (6) (Table 2). On the oxygen, linking the phosphodiester bond between the beta and gamma phosphate backbone, the wildtype has an angle of 143.3 degrees. KRAS G12S had the lowest angle of 131.7 degrees.
Table 2.
Alterations in molecular angles of GTP bound to KRAS wildtype and mutants. Using Schrodinger, angles of specific atoms were measured to identify any angle conformations that could be affecting GTP hydrolysis. WT on the gamma-phosphate phosphodiester bond (1) has an angle of 143.3 degrees, while the mutants have much lower bond angles.
| Protein | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| WT | 143.3 | 142.8 | 118.2 | 111.2 | 126.2 | 109.4 |
| G13D | 139.9 | 144.2 | 122.5 | 113.5 | 128.8 | 113 |
| G12R | 138 | 141.8 | 117.1 | 108.5 | 126.2 | 112.5 |
| G12D | 132.7 | 135.6 | 121.1 | 116.4 | 129.4 | 116 |
| G12C | 141.9 | 138.1 | 121.9 | 110.3 | 124.9 | 116.2 |
| G12V | 141.7 | 139.4 | 113.4 | 110.7 | 121 | 110.5 |
| G12A | 137 | 138.6 | 118.5 | 106.2 | 130.9 | 112.6 |
| G12S | 131.7 | 141.2 | 118.9 | 115.1 | 123.5 | 119.9 |
Tetrahedral Structure Angle Distortion in KRAS Mutants
In Table 2, the angle between the ribose sugar and beta carbon (6), KRAS wildtype protein measures an angle of 109.4 degrees, which is a typical tetrahedral angle structure, near the literature value of 109.5 degrees for tetrahedral geometry. However. the values for KRAS mutant proteins surpass this value, the highest being 116.2 and 116 degrees for KRAS G12C and G12D, respectively.
Hydrogen Bonding and Hydrophobic Interactions
Noncovalent bonding was more abundant in KRAS mutations than in wildtype on the gamma-phosphate of the GTP molecule. There are noticeable structural differences of the cartoon and surface configuration of each of the mutant KRAS proteins and wildtype. Hydrophobicity was prevalent in the wildtype KRAS, while the mutants had little to no hydrophobic interactions with the GTP. Scoring on hydrogen and hydrophobic interactions are reported with Figures 3 and 4a. Interaction (IF) are scored based on the likelihood of amino acid position. Elaborated here on binding preferences emphasizing hydrophobicity within the pocket. GTP molecules are all charged in mutants. Amino acid-substrate contact was at a cut off at 80% in Figure 4a. Glycine (at position 10) binding at 90% affinity to the nitrogenous base was only observed in KRAS wildtype and defined the WT protein substrate binding conformation. The placement of the GTP molecule in figure 4b shows accessibility of the wildtype (purple) to be cleaved inside the pocket; no amino acid had contact with the nitrogenous base except for the wildtype protein, let alone having a glycine at position 10.
Figure 3.
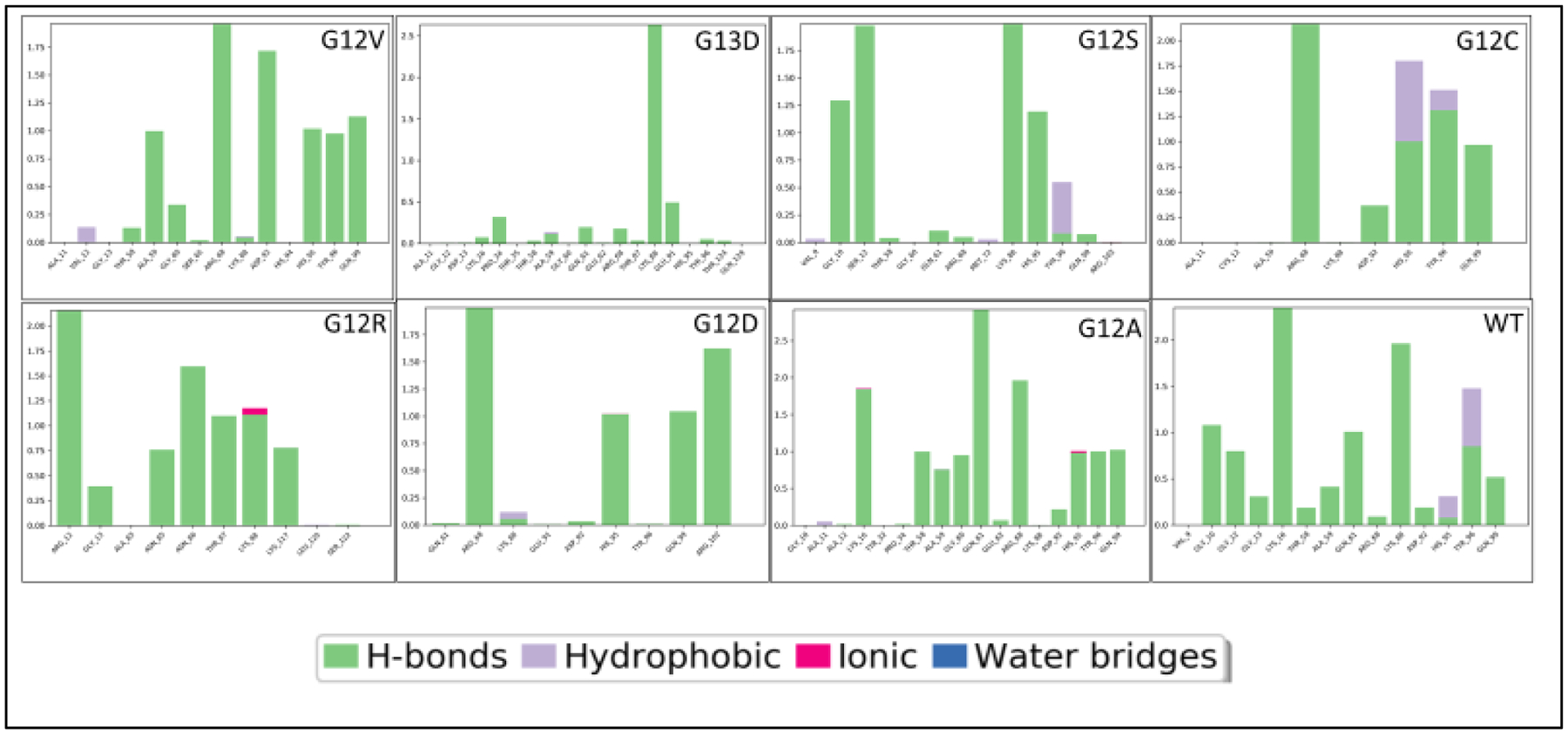
Bar graph of hydrophobic interactions and hydrogen bonding. Hydrophobic interactions have purple bars. Hydrogen bonding are green bars. WT and G12C display hydrophobicity (purple). Mutants display no hydrophobic interactions, however, have many hydrogen bonding.
Figure 4a.
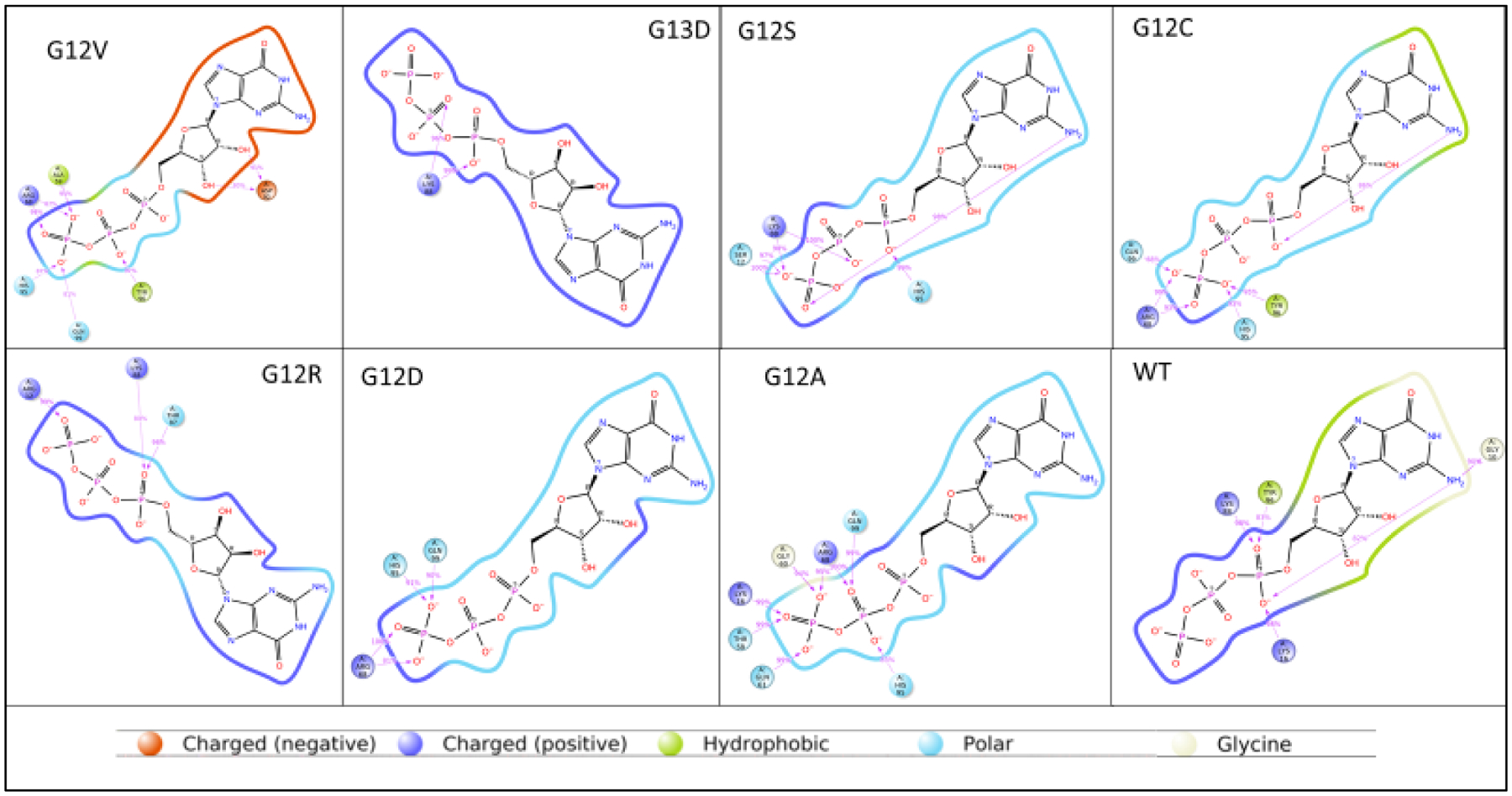
GTP and amino acid contact. 2D structure of substrate, with amino acids from each respective KRAS protein that come into contact during simulation. Contact percentage was set at 80% for amino acids that come in contact only 80% of the time during simulation. WT and G12C display hydrophobic interactions (shaded green). Mutants’ amino acid contacts with gamma-phosphate of the GTP molecule.
Figure 4b.
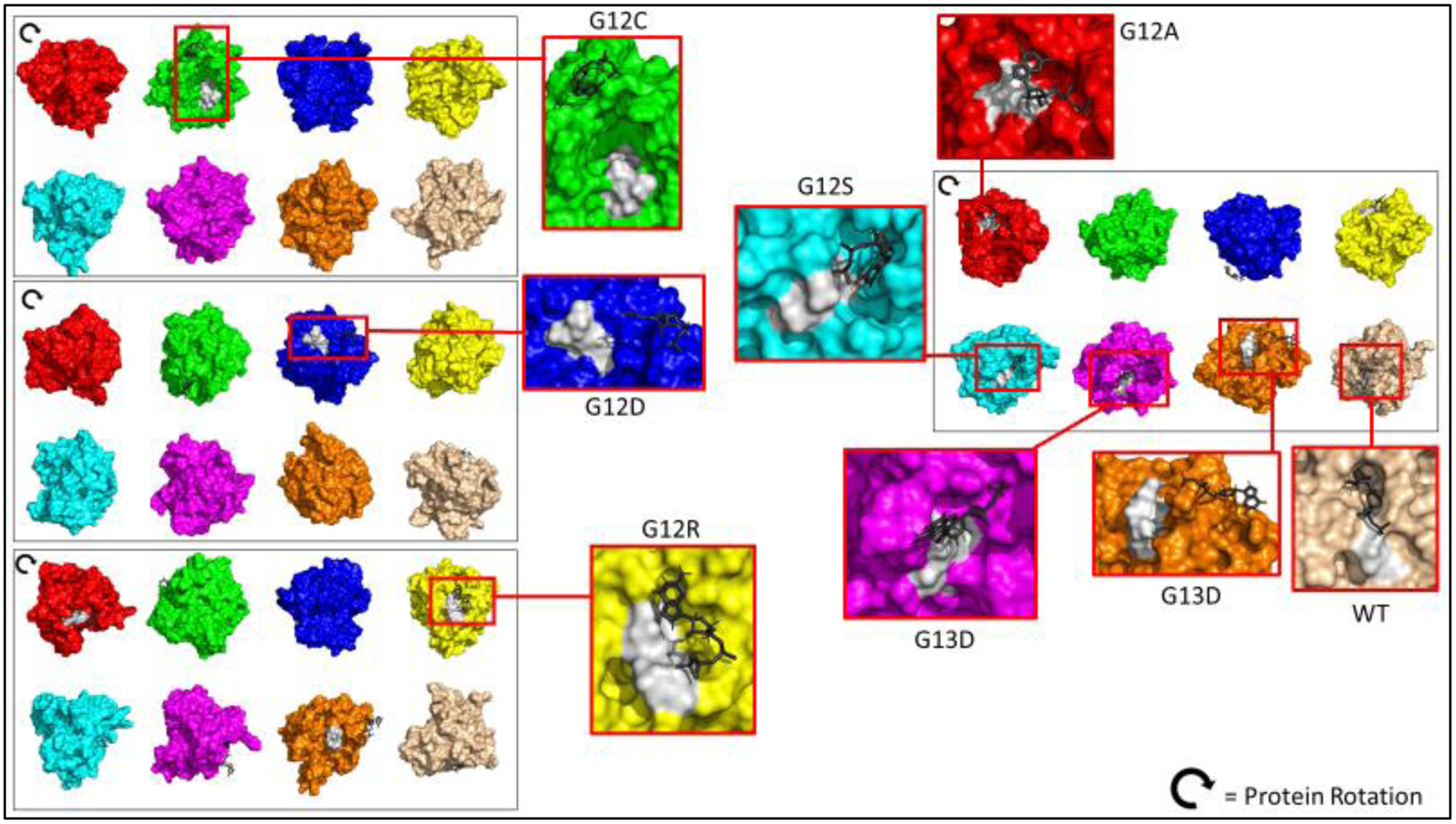
Surface KRAS in silico Protein Mutants and Wildtype 3D structure. PyMOL 3D representation of each KRAS protein bound to their respective substrate (shaded black) in its conformation. Shaded white represents the p-loop region. WT has GTP molecule sitting inside pocket of KRAS protein, while mutants are not in proper conformation.
Protein 3D Structure Conformation Changes
KRAS mutant protein has apparent conformation changes visually according to figures 4b and 4c. In both figures, the molecular conformations are viewed at the same angles using PyMOL visualization features. The binding site of GTP molecule on the wildtype (purple) varies in positioning from the rest of the KRAS mutants. To quantify this, table 3 represents a table of angles from the p-loop position (residue 12), switch I region (residue 34), and the switch II region (residue 63). KRAS wildtype GTP-bound had 134.4 degrees at residue 12, which was significantly higher than the rest of the KRAS mutant proteins. KRAS wildtype GTP-bound in comparison had a much higher score as well. Residues in the switch I and switch II showed apparent fluctuations as well between KRAS wildtype and mutant proteins in GTP-bound conditions [18]. In figures 4a, 4b, and 4c, we were able to visually inspect the changes of the structures. In figure 4d, the GTP molecule for the wildtype conformation has the gamma phosphate, in respect to its XYZ positioning, facing towards us.
Figure 4c.
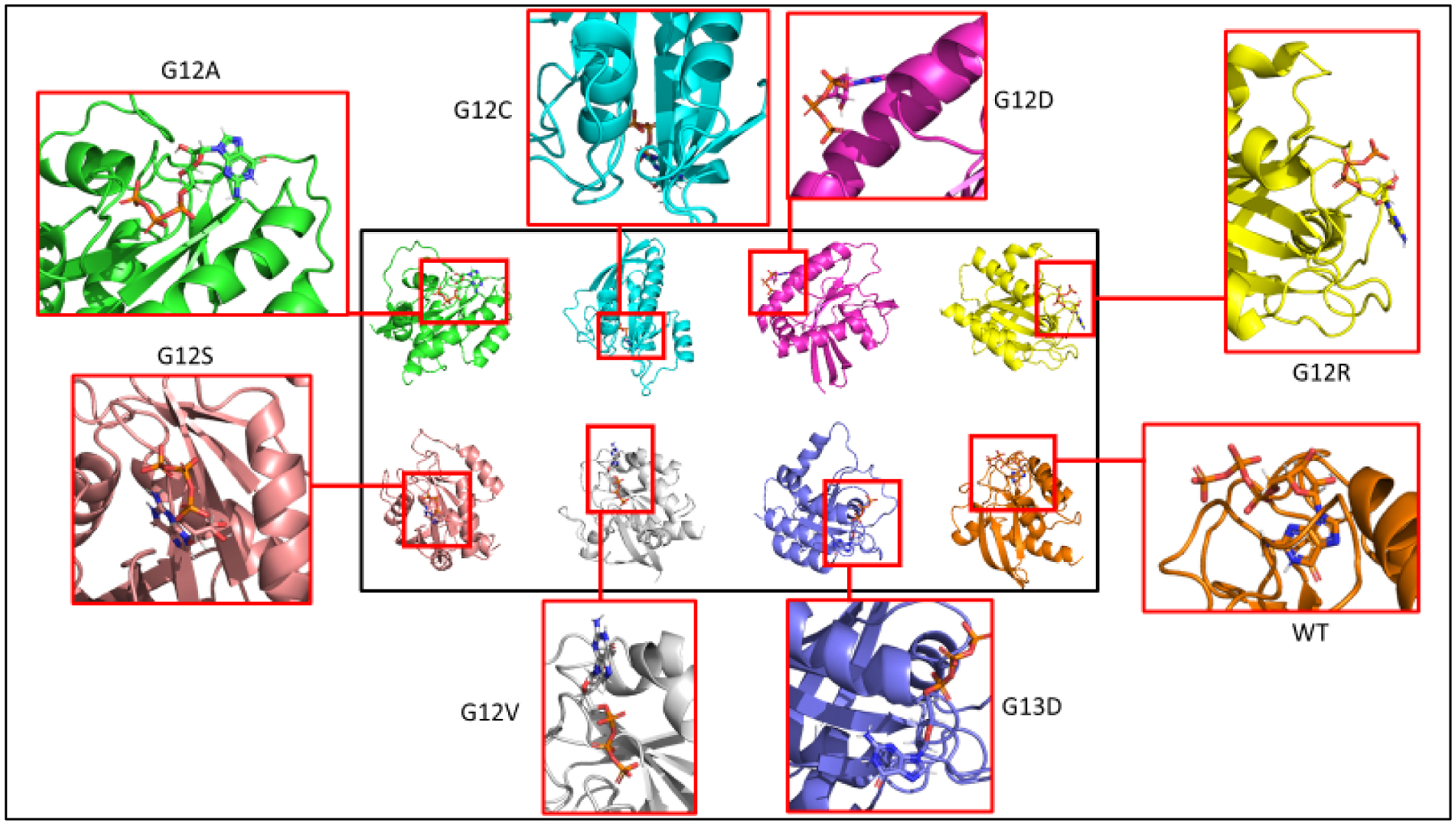
Surface KRAS in silico Protein Mutants and Wildtype 3D structure. PyMOL 3D representation of each KRAS protein bound to their respective substrate in its conformation. Structural dynamic changes in protein conformation.
Table 3.
KRAS Mutant and Wildtype Protein Angle Conformation Changes. Angles were measured in Schrodinger at position 12 to identify and determine angle differences between wildtype and mutant KRAS proteins. WT at position 12, has an angle of 134.4 degrees, while mutants displayed significant lower angle values below 134 degrees for GTP binding.
| KRAS Variant | Residue 12 | Residue 34 | Residue 63 |
|---|---|---|---|
| WT | 134.4 | 95.2 | 97.9 |
| G12C | 115.7 | 117.8 | 96.5 |
| G12D | 112.9 | 93.3 | 85.9 |
| G13D | 124.9 | 90.6 | 89.5 |
| G12V | 106.9 | 94.8 | 109.1 |
| G12A | 117.8 | 90.8 | 101.4 |
| G12S | 121.8 | 97.4 | 101 |
| G12R | 108 | 91.3 | 82.5 |
Figure 4d.
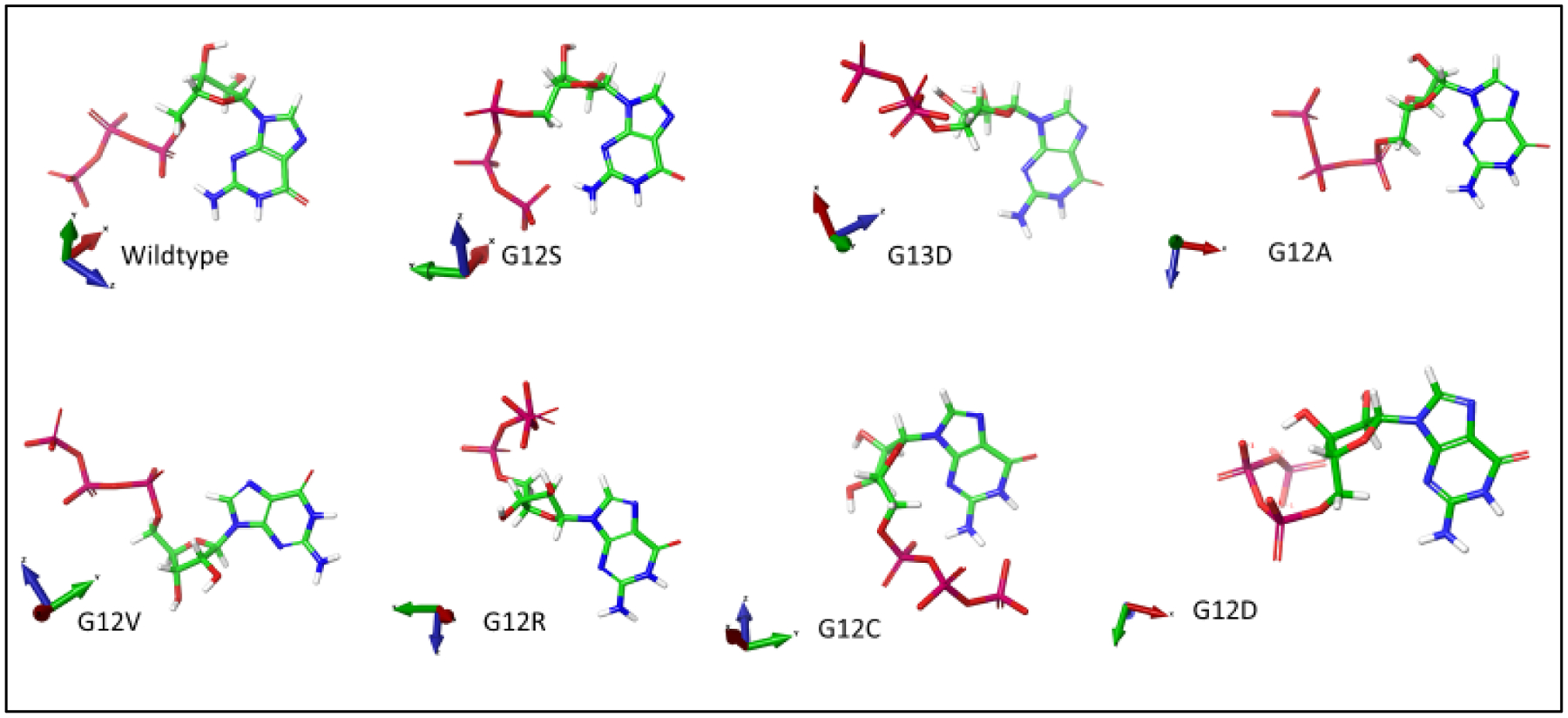
GTP Conformation Respective to its Protein Binding. XYZ coordinates are displayed on the left of each respective GTP molecule. GTP conformational changes when bound to KRAS protein. GTP molecule structures were taken post docking of their respective protein binding, and their conformational structures were compared with correlated XYZ coordinates, and using the pyrimidine rings as a point of reference.
GTP MM-GBSA Scoring for Substrate Configuration
Using Prime Energy MM-GBSA, an approach to calculating free energy of substrate and macromolecule interactions, we determined atom scores on the gamma-phosphate. Refer to table 4 for atom positioning. KRAS wildtype had lower scoring values in comparison to KRAS mutants G12C, G12D, G12V, and G12A (Table 4).
Table 4.
Prime Energy Using MM-GBSA Scoring. Prime energy visualizer was used to determine the affinity binding of the gamma phosphate of the GTP molecule. WT had less favorable affinity binding values than G12D, G12C, G12V, and G12A.
| GTP | 3 (P) | 16 (O) | 21 (O) | 24 (O) | 25 (O) |
|---|---|---|---|---|---|
| Wildtype | −5.24 | −7.21 | −8.71 | −7.42 | −7.24 |
| G12D | −38.35 | −29.45 | −37.19 | −12.96 | −15.71 |
| G12C | −34.79 | −5.67 | −27.77 | −30.41 | −17.25 |
| G12V | −37.30 | −34.78 | −28.86 | −32.78 | −26.82 |
| G13D | −5.16 | −6.99 | −5.84 | −8.32 | −7.61 |
| G12A | −25.44 | −34.44 | −24.14 | −29.18 | −42.17 |
| G12S | −4.15 | −6.19 | −5.78 | −7.16 | −5.26 |
| G12R | −5.29 | −6.08 | −7.26 | −8.61 | −7.19 |
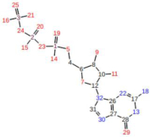
Switch I and II Regions Conformational Changes
KRAS WT had an angle of 99.7 degrees in the switch I region, and 60.6 degrees in the switch II region (Table 5). KRAS mutant G13D had the highest degree difference of 127.7 degrees, and most of the mutants had a larger degree difference than the WT, apart from mutant G12A, having a lower degree difference of 89.4 degrees (switch I region). In the switch II region, mutant G12D had the lowest degree angle of 31.4 degrees.
Table 5.
Angles of Switch I and II regions of KRAS variants. Measured in Maestro, selecting 3 points on the amino acid sequence. Switch I region, 30, 34, and 38 amino acid sequences were selected to measure angles. Switch II region 59, 64, and 67 amino acid sequences were selected to measure angles.
| KRAS Variant Proteins | Switch I Angles (30 – 38 aa) | Switch II Angles (59–67 aa) |
|---|---|---|
| WT | 99.7° | 60.6° |
| G13D | 127.7° | 100.1° |
| G12R | 105.7° | 68.7° |
| G12D | 104.8° | 31.4° |
| G12C | 119.4° | 80.1° |
| G12V | 116.6° | 97.4° |
| G12A | 89.4° | 57.8° |
| G12S | 109.5° | 60.3° |
Binding Similarities Between G12C and Wildtype KRAS
KRAS WT and G12C share prominent similarities. Both, GTP molecule of WT and G12C begin the contact time around the same RMSF values, between 0.5 and 0.6. Furthermore, in the simulation both GTP/GDP molecules from G12C and wildtype contact the alpha carbon backbone of the protein around 30 – 40 nanoseconds (Figure 2). The G12C mutant and wildtype protein-substrate contacts have hydrophobic interactions at histidine 95 and tyrosine 96 (Figure 3). It is also observed that G12C and wildtype KRAS have a hydrophobic interaction in the close vicinity of the nitrogenous base (shaded green) (Figure 4d). The binding affinity for AMG 510 for G12C is 10 kcal/mol, and only the wildtype has the highest binding affinity, in comparison to the rest of the KRAS mutant variants, −8.6 kcal/mol. These values remain consistent between in-silico docking and crystallized mutant structure docking from the RCSB. Such similarities have provided the possibility of creating an appropriate small molecule inhibitor that would successfully inhibit the under driven GTPase activity in G12C mutants.
Discussion
KRAS has been one of the most well studied oncogene that has been identified as an undruggable molecular targets in cancer. Our data explains the structural dynamics of how each mutant and wildtype KRAS protein is unique and why each protein requires a specific inhibitor. Utilizing up to date programs and bioinformatic tools like Schrodinger maestro, we were successfully able to identify intricate and intrinsic molecular properties within the wild type and 7 KRAS mutant proteins. Our goal was to identify the potential effectors with GTP hydrolysis that prevent its normal GTPase activity.
It is apparent that molecular forces at a much smaller scale are to be dealt with for drug treatment of KRAS mutant proteins. Due to a single amino acid mutation, the protein conformation and GTP affinity in KRAS mutant proteins varied and altered significantly. Binding affinity on the gamma-phosphate of the GTP molecule therefore would differ in its binding and structure conformation on the KRAS protein. Most GTP molecules did not bind directly into the pocket of the KRAS mutant proteins. Figures 4b and 4c show different conformations for each molecule but to be noted that the substrate sitting outside of the pocket for each mutant KRAS protein, whereas the wildtype KRAS protein, the GTP molecule is sitting inside the pocket with the gamma-phosphate sticking out from the protein. This pocket is where the catalytic activity for GTP hydrolysis occurs, and thus plays a significant role in determining the possibility of hydrolysis. Quantifying these changes, we took the angle of residue 12 of the alpha carbon (table 4), and successfully determined changes in the angle of the 12th amino acid residue. These angle changes impacted in the binding of the GTP and altered the total protein conformation and energy affinity for binding position of the GTP molecules as compared to the KRAS wildtype binding conformation. This configuration, visually and numerically, shows that KRAS-GTP contact with mutants are not within the pocket, and therefore its normal catalytic activity was ineffective. It is quite clear there are KRAS conformational changes based on our data, which can be further aid in discovering small drug targeting therapies. Another research study in addition makes claims to rotational dynamics [12] of the KRAS mutant proteins, and our data exhibits these changes visually and numerically.
Fluctuation of the GTP molecule bound to KRAS wildtype in figure 1 had higher hits with RMSF contact, while GTP bound KRAS mutants had significantly lower values, prominently for atom at positions: 3, 16, 21, and 25 (Figure 1). These four atom position contacts reflect having a closer proximity to KRAS protein, and all four are from the gamma-phosphate, which shows some stability in its fluctuation by the gamma-phosphate for KRAS mutants G12C, G12D, and G13D. While in the wildtype KRAS protein, the GTP molecule had high angstrom values for the gamma-phosphate. Clearly there is more affinity and stability on the gamma-phosphate for the GTP molecule on all the 7 mutants of KRAS as compared to wildtype.
G12V was the only mutated protein to have an angle of 113.4 degrees, while the rest of the mutated proteins are approximately the same angle value or higher by the alpha phosphate. According to a research study for cervical cancer [19], this study concurs with the results provided in this study (Table 3, Figures 1 & 2). G12V displayed stability in RMSD simulation out of the rest of the mutant proteins.
The oxygen connecting the phosphodiester bond measured 143.3 degrees in the wildtype, while the mutants measured a reduced bond angle value. As well as the carbon on the ribose sugar, connected to the carbon, attached to the alpha phosphate group showed the same trend. These molecular conformation changes potentially contributed to the affinity to the mutant KRAS protein, not allowing its normal binding and catalytic activity for GTP hydrolysis.
Six of the mutants’ KRAS in-silico proteins displayed a positive trend or close contact of the alpha carbon backbone to the GTP molecule. Over the duration of the 50 nanoseconds, wildtype shows a brief contact of less than 5 nanoseconds, with a declining slope, representing minimal contact. Figure 3 displays mutant KRAS proteins having stable and closer contact with GTP molecule than the wildtype.
Non-covalent bonding is an important factor in biological systems for protein and substrate interactions, Figure 3 shows hydrophobic interactions for KRAS G12C and KRAS wildtype at histidine 95 and tyrosine 96. However, all other mutants did not display any hydrophobicity in histidine 96. G12S does display hydrophobic interactions in tyrosine 96. The hydrogen bonding for all KRAS mutant proteins was abundant and could potentially explain the affinity between protein and GTP from not detaching. This is visually represented in figure 4b, KRAS wildtype and G12C have hydrophobic regions shaded in lime green. G12C and WT have been studied with inhibitor AMG 510 [20], and our results concur with the possibility of why AMG 510 binding is effective with G12C, as opposed to the other KRAS mutants, based on figures 2, 3, 4a, and table 6. G12C and WT have the highest scoring binding affinity for inhibitor AMG 510 (Table 6). AMG 510 matches our in-silico model, which proves to be an alternative method to studying protein structure dynamics for future research. Furthermore, recent USFDA approval of sotorasib (Lumacras™, Amgen Inc., Thousand Oaks, CA), selectively for G12C mutant enforces our proposition that each mutant is unique requiring specific inhibitors. MRTX849 and MRTX1133 are drugs being studied and developed for G12C and G12D inhibition, respectively. Thus, a comprehensive structural scoring is essential to move forward with drug development. G12A and G12S both serve as potential candidates for inhibitor BI 2852, which is under review for G12D. G12D was reported having low values for inhibitor 2852, however is still under review for final approval.
Table 6.
Binding affinity of inhibitors AMG 510 and BI 2852 docked with In-silico generated mutants or crystallized structures of mutant KRAS. Dockings were performed with either of the following proteins: KRAS in-silico generated mutants or crystallized structures from the RCSB (in vivo) with the following inhibitors: AMG510 or BI 2852. AutoDock Vina was used to generate these binding affinities. Between In-silico and Mutant RCSB affinity binding values, the data remains consistent between a computational and experimental approach.
| Inhibitor AMG 510 Affinity (kcal/mol) | Inhibitor 2852 Affinity (kcal/mol) | |||
|---|---|---|---|---|
| KRAS Variants | In-Silico | Mutant RCSB Structure | In-Silico | Mutant RCSB Structure |
| WT | −8.6 | −8.6 | −9.9 | −9.9 |
| G12C | −10 | −10 | −9.4 | −8.0 |
| G12D | −7.8 | −7.8 | −6.7 | −7.2 |
| G13D | −8.3 | −8.0 | −8.5 | −7.4 |
| G12V | −7.2 | −6.8 | −8.1 | −6.3 |
| G12A | −8.0 | −6.9 | −8.0 | −8.4 |
| G12S | −7.3 | −7.2 | −8.3 | −8.4 |
| G12R | −6.4 | −7.0 | −7.1 | −7.1 |
Additional information to note is the gamma-phosphate and amino acid residue contacts. Most proteins contacted the gamma-phosphate, while in KRAS wildtype the alpha phosphate has contact with amino acids. These amino acids are above the 80% contact percentage, scaled in Schrodinger platform, and therefore these amino acids from KRAS protein made contact over 80% of the time with the GTP molecule. Glycine binding to the nitrogenous base is unique only to the wildtype KRAS protein and no other amino acid contacted the nitrogenous base above 80% contact cutoff value. GTP molecule for KRAS wildtype is positioned in a specific way, that allows the gamma-phosphate to be accessible and cleaved. KRAS wildtype has a steric strain as low as 10.7, while the rest of the mutants have higher torsional strains, which is unfavorable for the mutants, compared to the wildtype. Therefore, there are hydrophobic interactions in KRAS wildtype, while charged molecules are abundant in mutant KRAS proteins (Figure 4a). These charged residues, and torsional strains affect the binding within the hydrophobic pocket of the p-loop. Due to GTP-KRAS mutant bound complexes having high torsional strains, this potentially affects pocket binding at the active site of the KRAS protein, therefore affecting its normal catalytic activity. This study can help further research to design a small molecule GTPase inhibitor. We propose that small molecules that would reduce the charged interactions of the p-loop pocket would be the most suitable inhibitor. Each mutant has its own characteristic p-loop profile creating the necessity of designing different inhibitors specific for each individual KRAS mutant.
This study has provided a molecular view of the GTP-protein interactions and the ability to visualize the effects of the mutations of KRAS, and how the conformation of the overall proteins affects binding and hydrolysis of the GTP molecule. Herein we have explored the structural characteristics of wildtype and seven KRAS mutant proteins and evaluated their binding affinity, RMSF, RMSD, hydrogen bonding and hydrophobic interactions between amino acid residues and within GTP-KRAS complexes. In the following study specific small molecules will be selected and simulated to individual mutant proteins to determine which one would restore the substrate binding conformation closest to the wildtype KRAS-GTP binding conformation. This study has provided an important insight that is essential for designing mutant specific small GTPase inhibitors to help treatment of KRAS mutated cancers.
Figure 4e.
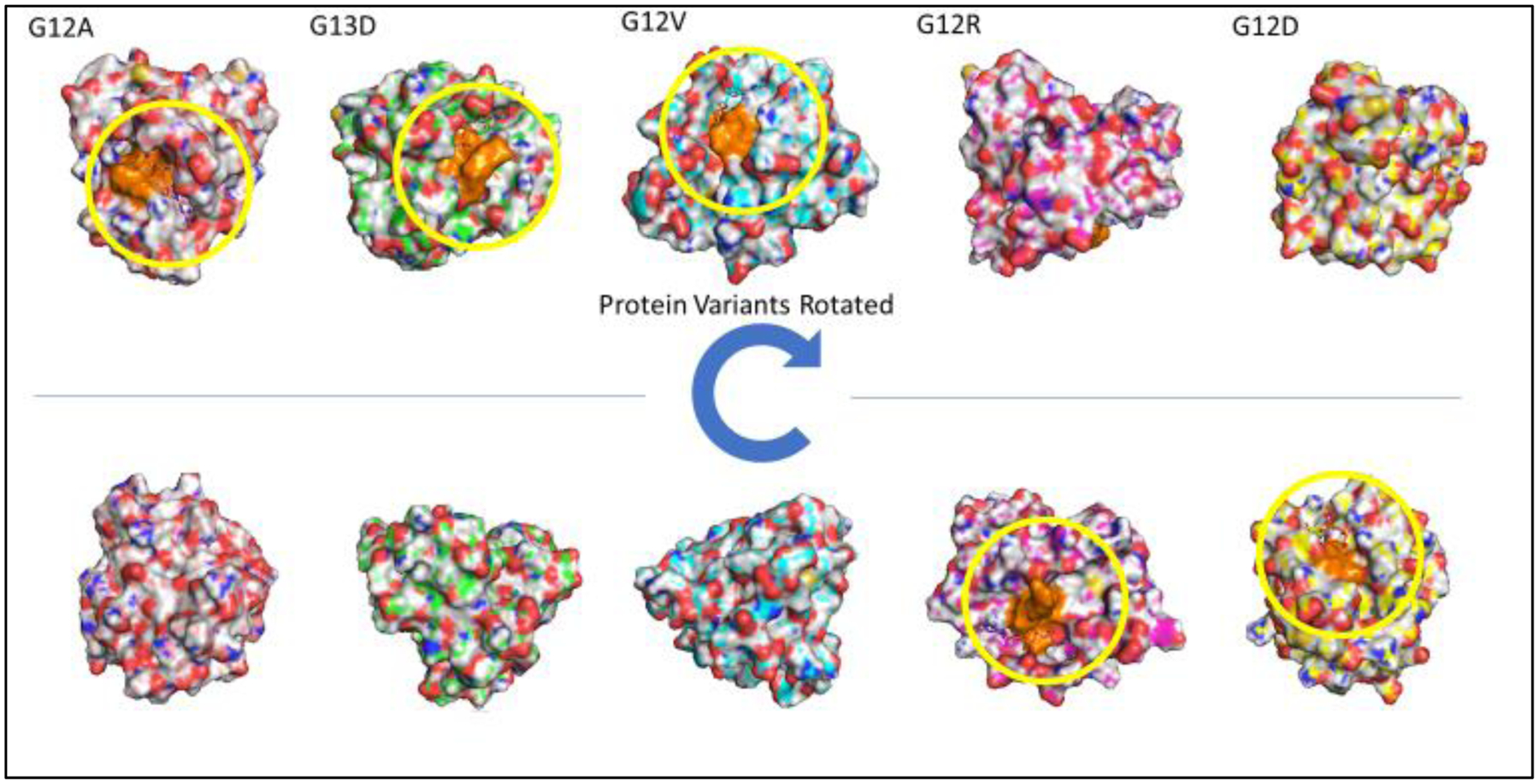
in vivo KRAS mutant proteins post molecular dynamics substrate-protein binding. Shaded in orange is the amino acid residue sequence 10 through 17. GTP is not binding clearly in the pocket of any of the mutant variants. The configuration of each of the KRAS mutants differ in structural conformation and its binding site.
Structural conformational changes in KRAS from GTP not binding in hydrophobic pocket
Switch I and II regions vary in angles, respective to its mutant counterpart, potentially affecting GAP binding
Gamma-phosphate has high hydrogen binding in mutants versus wildtype KRAS
Inhibitors approved for G12D KRAS may be an alternative for G12A and G12S
Acknowledgments:
We would like to thank Selma Botman, the Yeshiva University Provost and Vice President for Academic Affairs, for graciously providing the start-up funding to Radhashree Maitra for this research.
Funding:
This research was funded by a start-up fund #2A4108, as well as an Alexander Fund 252869-252929 provided by the Yeshiva University Office of the Provost to Radhashree Maitra.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: The authors declare that there is no conflict of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References:
- 1.Janson G and Paiardini A, PyMod 3: a complete suite for structural bioinformatics in PyMOL. Bioinformatics, 2020. [DOI] [PubMed] [Google Scholar]
- 2.Kalathiya U, Padariya M, and Baginski M, Structural, functional, and stability change predictions in human telomerase upon specific point mutations. Sci Rep, 2019. 9(1): p. 8707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harder E, et al. , OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J Chem Theory Comput, 2016. 12(1): p. 281–96. [DOI] [PubMed] [Google Scholar]
- 4.Yadav M and Khandelwal S, Homology modeling and molecular dynamics dimulation study of beta carbonic anhydrase of Ascaris lumbricoides. Bioinformation, 2019. 15(8): p. 572–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trott O and Olson AJ, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem, 2010. 31(2): p. 455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beganovic S, Clinical significance of the KRAS mutation. Bosn J Basic Med Sci, 2009. 9 Suppl 1: p. 17–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox AD, et al. , Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov, 2014. 13(11): p. 828–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dinu D, et al. , Prognostic significance of KRAS gene mutations in colorectal cancer--preliminary study. J Med Life, 2014. 7(4): p. 581–7. [PMC free article] [PubMed] [Google Scholar]
- 9.Haigis KM, KRAS Alleles: The Devil Is in the Detail. Trends Cancer, 2017. 3(10): p. 686–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu P, Wang Y, and Li X, Targeting the untargetable KRAS in cancer therapy. Acta Pharm Sin B, 2019. 9(5): p. 871–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simanshu DK, Nissley DV, and McCormick F, RAS Proteins and Their Regulators in Human Disease. Cell, 2017. 170(1): p. 17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pantsar T, The current understanding of KRAS protein structure and dynamics. Comput Struct Biotechnol J, 2020. 18: p. 189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karachaliou N, et al. , KRAS mutations in lung cancer. Clin Lung Cancer, 2013. 14(3): p. 205–14. [DOI] [PubMed] [Google Scholar]
- 14.Madej T, et al. , MMDB and VAST+: tracking structural similarities between macromolecular complexes. Nucleic Acids Res, 2014. 42(Database issue): p. D297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sastry GM, et al. , Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des, 2013. 27(3): p. 221–34. [DOI] [PubMed] [Google Scholar]
- 16.Ivanova L, et al. , Molecular Dynamics Simulations of the Interactions between Glial Cell Line-Derived Neurotrophic Factor Family Receptor GFRalpha1 and Small-Molecule Ligands. ACS Omega, 2018. 3(9): p. 11407–11414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kufareva I and Abagyan R, Methods of protein structure comparison. Methods Mol Biol, 2012. 857: p. 231–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen J, et al. , Mutation-Induced Impacts on the Switch Transformations of the GDP- and GTP-Bound K-Ras: Insights from Multiple Replica Gaussian Accelerated Molecular Dynamics and Free Energy Analysis. J Chem Inf Model, 2021. 61(4): p. 1954–1969. [DOI] [PubMed] [Google Scholar]
- 19.Chen XP, et al. , G12V KRAS mutations in cervical cancer under virtual microscope of molecular dynamics simulations. Eur J Gynaecol Oncol, 2016. 37(1): p. 69–74. [PubMed] [Google Scholar]
- 20.Canon J, et al. , The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature, 2019. 575(7781): p. 217–223. [DOI] [PubMed] [Google Scholar]