

Abstract
Hypertension is a major cause of chronic kidney disease. However, the pathogenesis of hypertensive kidney disease is not fully understood. Recently, we have shown that CXCL16/phosphoinositide-3 kinase γ (PI3Kγ) plays an important role in the development of renal inflammation and fibrosis in angiotensin II (AngII) induced hypertensive nephropathy. In the present study, we examined the role of phosphatase and tensin homolog (PTEN), a major regulator of PI3K signaling, in the pathogenesis of renal inflammation and fibrosis in an experimental model of hypertension induced by AngII. We generated myeloid PTEN conditional knockout mice by crossing PTENflox/flox mice with LysM-driven Cre mice. Littermate LysM-Cre−/−PTENflox/flox mice were used as a control. Both myeloid PTEN knockout mice and their littermate control mice exhibited similar blood pressure at baseline. AngII treatment resulted in an increase in blood pressure that was comparable between myeloid PTEN knockout mice and littermate control mice. Compared with littermate control mice, myeloid PTEN knockout mice developed more severe kidney dysfunction, proteinuria, and fibrosis following AngII treatment. Further, myeloid PTEN deficiency exacerbated total collagen deposition and extracellular matrix protein production and enhanced myeloid fibroblast accumulation and myofibroblast formation in the kidney following AngII treatment. Finally, myeloid PTEN deficiency markedly augmented infiltration of F4/80+ macrophages and CD3+ T cells into the kidneys of AngII-treated mice. Taken together, these results indicate that PTEN plays a crucial role in the pathogenesis of renal inflammation and fibrosis through regulation of infiltration of myeloid fibroblasts, macrophages, and T lymphocytes into the kidney.
Introduction
Chronic kidney disease (CKD) is a worldwide public health problem which affects about 10% of the population in the world (Jha et al., 2013). Hypertensive kidney disease can progress to end-stage kidney disease (ESKD) (Ruiz-Ortega et al., 2006) and there is no effective therapy to prevent the progression of CKD to ESKD (Liu, 2011). Therefore, a better understanding of the molecular mechanisms of hypertensive kidney disease are essential for developing novel therapeutic strategies to prevent this serious kidney disease.
Angiotensin II (AngII) is a central mediator of hypertensive kidney disease through induction of circulating cell infiltration and upregulation of proinflammatory and profibrotic factors (Mennuni et al., 2014). We have previously shown that chemokine CXCL16 and its receptor CXCR6 recruit macrophages, T cells, and myeloid fibroblasts into the kidney resulting in kidney injury and fibrosis in AngII-induced hypertension (Xia, Entman, & Wang, 2013; Xia, Jin, Yan, Entman, & Wang, 2014). Recently, we further demonstrate that CXCL16 binding to its receptor CXCR6 activates phosphoinositide-3 kinase γ (PI3Kγ) in AngII-induced hypertension (An, Wen, Hu, Mitch, & Wang, 2020).
Phosphatase and tensin homolog (PTEN) was originally discovered as a tumor suppressor (Li et al., 1997). Subsequent studies have shown that PTEN primarily functions as a lipid phosphatase to regulate embryonic development, cell migration and apoptosis (Song, Salmena, & Pandolfi, 2012). A major target of PTEN is phosphatidylinositol 3,4,5-trisphosphate. For example, PTEN is involved in regulating phosphatidylinositol 3 kinase (PI3K)/Akt signaling (Leslie & Downes, 2002). We have reported that pharmacological inhibition of PTEN aggravates ischemia reperfusion-induced acute kidney injury (Zhou, Jia, Hu, & Wang, 2017). However, the role of PTEN in AngII-induced hypertensive kidney disease is not known.
In this study, we investigated the role of myeloid PTEN in the pathogenesis of kidney injury and fibrosis in AngII-induced hypertension using myeloid PTEN knockout mice. Our results demonstrated that myeloid PTEN deficiency exacerbated kidney dysfunction and proteinuria, augmented myeloid fibroblast accumulation and macrophage and T cell infiltration, thereby aggravating AngII-induced kidney injury and fibrosis.
Materials and Methods
Experimental Animals
The animal experiments were performed according to the national and international animal care and ethical guidelines and were approved by the institutional animal care and use committee of the University of Connecticut Health Center. LysM-Cre mice in which the Cre recombinase expression is driven by the lysozyme M promoter via gene targeting into the endogenous lysozyme M locus and PTENf/f mice on a C57BL/6 background were purchased from the Jackson Laboratory. After 2 generations of mating, LysM-Cre+/−PTENflox/flox mice were created. Littermate LysM-Cre−/−PTENflox/flox mice were used as a control. Male mice at 10 weeks of age were infused with AngII at 1.5 μg/kg/min (Sigma, St. Louise, MO) continuously by subcutaneously implanted osmotic minipumps (Durect, Cupertino, CA) for 28 days (n=6 for LysM-Cre+/−PTENflox/flox group and n=6 for LysM-Cre−/−PTENflox/flox group). Control groups of mice were implanted with sterile saline minipumps (n=6 for LysM-Cre+/−PTENflox/flox group and n=6 for LysM-Cre−/−PTENflox/flox group). To accelerate kidney injury, the AngII-treated mice were supplied with 1% saline drinking water ad libitum as reported previously (Xia et al., 2013; Xia et al., 2014). Urine and blood samples were collected at the end of experiments.
Blood Pressure
Systolic blood pressure (SBP) was recorded in conscious mice for each experimental group using a tail cuff system (Visitech Systems, Cary, NC) as described previously (Ma, Jin, He, & Wang, 2016; Xu et al., 2011).
Renal Function
Serum creatinine was measured using a commercially available creatinine assay kit (BioAssay Systems, Hayward, CA) (Jin, Chen, Hu, Chan, & Wang, 2013; Liang, Zhang, He, & Wang, 2016; Zhou et al., 2020). Urine samples were collected using metabolic cages for 12 hours. Urine albumin and creatinine were measured using commercially available kits (Exocell, Philadelphia, PA) following the manufacturer’s instruction (Liang, Ma, et al., 2016; Wu, An, Jin, Hu, & Wang, 2020). Albuminuria was expressed as milligrams of urinary albumin per gram of urinary creatinine.
Renal Histology
Mice were perfused with phosphate buffered saline (PBS) to remove blood from the kidneys. Kidney tissues were fixed in 10% buffered formalin for 24-hour then kidney tissues were transferred into 70% ethanol after washing with PBS. The tissues were stored at 4°C until embedding into paraffin. The paraffin-embedded kidney tissues were sectioned at 5-μm thickness. Kidney sections were deparaffinized in Xylene and dehydrated in decreasing concentrations of ethanol (2X 100%, 90%, 70%). Periodic acid Schiff (PAS) staining was performed with a kit (395B-1KT, Sigma-Aldrich) following the manufacturer’s instructions. For Sirius red staining, kidney sections were incubated in 0.1% direct red 80 (Sigma-Aldrich, 365548) solution for 1 hour at room temperature, and then the sections were dehydrated sections in 95% and 100% ethanol sequentially. The pathological damages in the kidney were scored as previously described (Liang, Ma, et al., 2016). Stained kidney sections were scanned using a microscope equipped with a digital camera (Nikon Instruments Inc., Melville, NY) and analyzed in a blinded manner using the NIS-Elements Br 3.0 software as described (Xia et al., 2013).
Immunohistochemistry
Paraffin-embedded kidney sections were used for immunohistochemical staining with antibodies against F4/80 (1:150, Bio-Rad, MCA497G) and CD3 (1:800, Abcam, ab134096) (An et al., 2020). Antigen retrieval was performed with antigen unmasking solution (Vector Laboratories, Burlingame, CA) for CD3. For F4/80 staining, kidney sections were treated with proteinase K (Sigma, P2308) (20 ug/ml in TE buffer, pH8.0) for 6 minutes at room temperature. Endogenous peroxidase activity was quenched with 3% hydrogen peroxide (H325–500, Fisher Scientific). Vector stain Elite ABC kits (Vector Laboratories) were used for the staining according to manufacturer’s instructions. The reaction was visualized by incubation with diaminobenzidine solution (ImmPACT DAB, Vector Laboratories). Tissue sections were then counterstained with hematoxylin (Hematoxylin Solution, Harris modified, Sigma-Aldrich). The images were taken with a microscope equipped with a digital camera (Nikon Instruments Inc., Melville, NY) and analyzed in a blinded fashion using the NIS-Elements Br 3.0 software as described (Xia et al., 2013).
Immunofluorescence
Immunofluorescence staining was performed on paraffin-embedded kidney sections. Sections were incubated with BlockAid™ blocking solution (Invitrogen, B10710) to block nonspecific binding. After blocking, the sections were stained with anti-fibronectin antibody (1:400, Sigma-Aldrich, F3648) followed by Alexa Fluor 488-conjugated donkey anti-rabbit antibody (1:400, Invitrogen, A21206), anti-Type I collagen antibody (1:400, SouthernBiotech, 1310–01) followed by Alexa Fluor 488-conjugated donkey anti-Goat antibody (1:400, Invitrogen, A11055), or anti-α-SMA antibody (1:400, Santa Cruz, SC-32251) followed by Alexa Fluor 488-conjugated donkey anti-mouse antibody (Invitrogen, A21202) (An et al., 2020). Double immunofluorescence staining was performed using frozen kidney sections. Kidney tissues were embedded in OCT compound, snap-frozen on liquid nitrogen, sectioned at 10 μm thickness, and mounted on glass slides. The frozen sections were fixed with ice-cold acetone then incubated with BlockAid™ blocking solution (Invitrogen, B10710) to block nonspecific binding. The sections were then incubated with anti-CD45 (1:200, BD Biosciences, 550539) and anti-PDGFR-β (1:100, Santa Cruz Biotechnology, SC-432) followed by Alexa Fluor 594-conjugated donkey anti-rabbit antibody (1:400, Invitrogen, A21209) and Alexa Fluor 488-conjugated donkey anti-rabbit antibody (1:400, Invitrogen, A21206), respectively. The sections were stained with appropriate fluorescence-conjugated secondary antibodies sequentially, and then mounted with mounting medium containing DAPI (VECTOR Laboratories, H1200). Images were obtained using Nikon digital camera (Nikon Instruments Inc., Melville, NY) and analyzed in a blinded manner using the NIS-Elements Br 3.0 software (Yang et al., 2012).
Western Blot Analysis
Total proteins were extracted using RIPA buffer containing a cocktail of protease inhibitors (Thermo Fisher Scientific, Rockford, IL) and protein concentration was quantified with Bio-Rad protein assay (Bio-Rad, 5000006). Equal amounts of protein were separated on SDS-PAGE gels in Bio-Rad Mini-PROTEAN TGX Stain-Free Gels (Bio-Rad). Proteins were transferred onto nitrocellulose membranes using the Trans-Blot Turbo System (Bio-Rad). FN, COLI, α-SMA and GAPDH were detected by primary and horseradish peroxidase (HRP)-tagged secondary antibodies. Membranes were incubated overnight at 4 °C with anti-fibronectin antibody (1:1000, Sigma-Aldrich, F3648), anti-Type I collagen antibody (1:1000, Southern Biotech, 1310–01), anti-α-SMA antibody (1:500, Santa Cruz, SC-32251), and anti-GAPDH (1:10000, EMD Millipore, MAB374). The membranes were incubated with secondary anti-rabbit (1:10000, Invitrogen, A16035), anti-goat (1:10000, Invitrogen, A16017), or anti-mouse (1:10000, Invitrogen, A16005) for 1 hour at room temperature. The target proteins were detected with Odyssey IR scanner (LI-COR Bioscience, Lincoln, NE) and analyzed using the Image-J imaging analysis software (NIH, Bethesda, MD) as previously described (Wang et al., 2018).
Statistical Analysis
All data were expressed as mean ± SEM. Comparisons between the multiple groups were performed using one-way ANOVA followed by the Bonferroni procedure for comparison of means. P < 0.05 was considered statistically significant.
Results
Blood Pressure
At baseline, there was no significant difference in systolic blood pressure among the four experimental groups. AngII treatment increased blood pressure in both myeloid PTEN knockout and control mice, which was comparable between the two AngII treatment groups (Figure 1A). These data suggest that PTEN is not involved in AngII-induced hypertension.
Figure 1. Effect of Myeloid PTEN deficiency on blood pressure, kidney function and proteinuria.
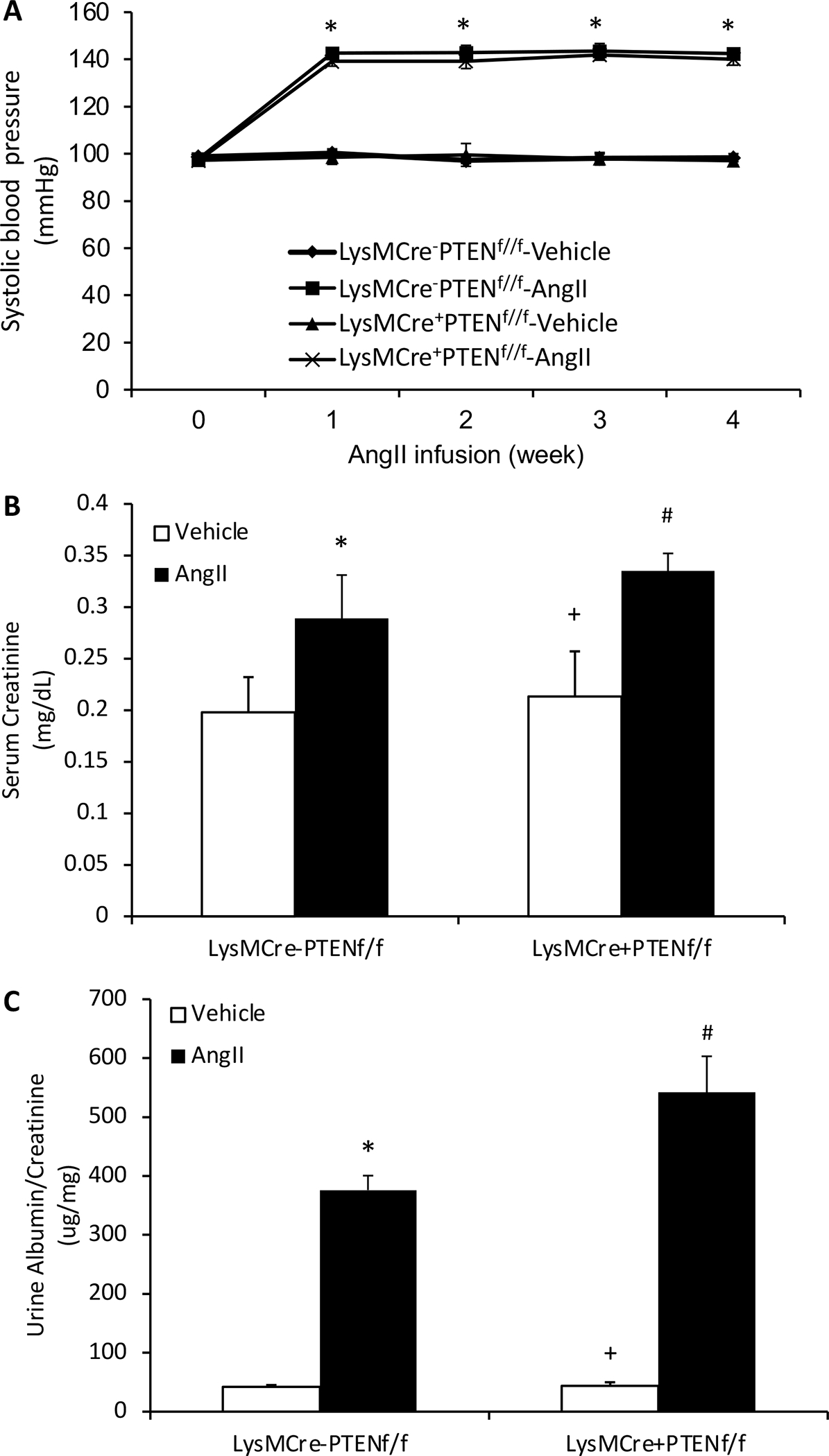
A, Myeloid PTEN deficiency has no effect on AngII-induced elevation of blood pressure. *P<0.01 between AngII-treated groups and vehicle-treated control groups. n=6 per group. B, Myeloid PTEN deficiency significantly worsens AngII-induced renal dysfunction as measured by blood creatinine. *P<0.01 vs LysMCre−PTENf//f-Vehicle; +P<0.01 vs LysMCre+PTENf//f-AngII; and #P<0.01 vs LysMCre−PTENf//f-AngII. n=6 per group. C, Myeloid PTEN deficiency significantly augments AngII-induced albuminuria. *P<0.01 vs LysMCre−PTENf//f-Vehicle; +P<0.01 vs LysMCre+PTENf//f-AngII; and #P<0.01 vs LysMCre−PTENf//f-AngII. n=6 per group.
Kidney Function and Albuminuria
After 4 weeks of AngII treatment, control mice developed significant kidney dysfunction as reflected by an increase in serum creatinine, which was worse in myeloid PTEN knockout mice (Figure 1B). Furthermore, myeloid PTEN knockout mice produced more albuminuria than control mice following AngII treatment (Figure 1C). These results indicate myeloid PTEN deficiency exacerbates AngII-induced kidney dysfunction and proteinuria.
Myeloid PTEN Deficiency Aggravates Kidney Injury and Fibrosis
To examine the effect of myeloid PTEN deficiency on kidney injury, kidney sections were stained with PAS. AngII treatment resulted in significant kidney injury in control mice, which was substantially aggravated in myeloid PTEN knockout mice (Figure 2A and B). Next, Sirius red staining was performed to assess the effect of myeloid PTEN deficiency on AngII induced total collagen deposition in the kidney. Total collagen deposition was significantly increased in the kidney of AngII-treated control mice (Figure 2C and D). In contrast, total collagen deposition was further increased in the kidney of AngII treated myeloid PTEN knockout mice (Figure 2C and D). These results suggest that PTEN functions as a negative factor in the development of AngII-induced kidney injury and fibrosis.
Figure 2. Myeloid PTEN deficiency aggravates kidney injury and fibrosis.
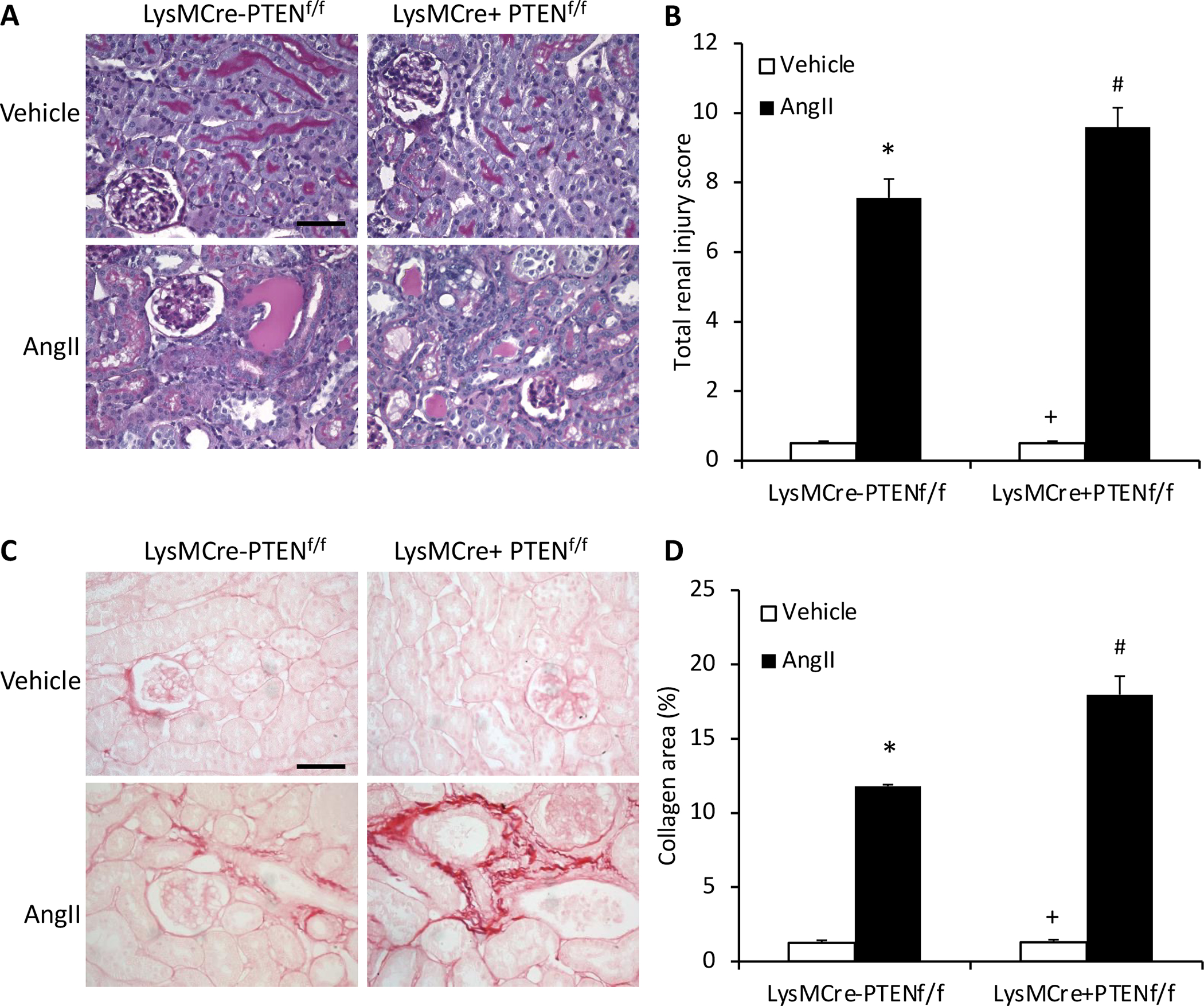
A, Representative photomicrographs of PAS-stained kidney sections showing AngII-induced kidney damage in LysMCre−PTENf//f and LysMCre+PTENf//f mice. Scale bar, 20 μm. B, Quantitative assessment of renal injury in LysMCre−PTENf//f and LysMCre+PTENf//f mice. *P<0.01 vs LysMCre−PTENf//f-Vehicle, + P<0.01 vs LysMCre+PTENf//f-AngII, # P<0.01 vs LysMCre−PTENf//f-AngII. n=6 per group. C, Representative photomicrographs of kidney sections stained with sirius red for assessment of total collagen deposition in the kidney 4 weeks after Ang II or vehicle treatment. Scale bar, 20 μm. D, Quantitative analysis of interstitial collagen content in the kidney of LysMCre−PTENf//f and LysMCre+PTENf//f mice. *P<0.01 vs LysMCre−PTENf//f-Vehicle; +P<0.01 vs LysMCre+PTENf//f-AngII; and #P<0.01 vs LysMCre+PTENf//f-AngII. n=6 per group.
Myeloid PTEN Deficiency Exacerbates AngII induced ECM Protein Production
We next assessed the effect of myeloid PTEN deficiency on the expression of ECM proteins in the kidney in response to AngII-induced hypertension. Immunofluorescence staining and Western blot analysis were performed to detect and quantify protein expression levels of two major ECM proteins, fibronectin and collagen I. The results showed that myeloid PTEN deficiency enhanced the protein expression of fibronectin and collagen I in the kidney after 4 weeks AngII treatment (Figure 3). These data suggest that PTEN deficiency promotes ECM protein expression in the kidney following AngII treatment.
Figure 3. Myeloid PTEN deficiency increases fibronectin and collagen I expression.
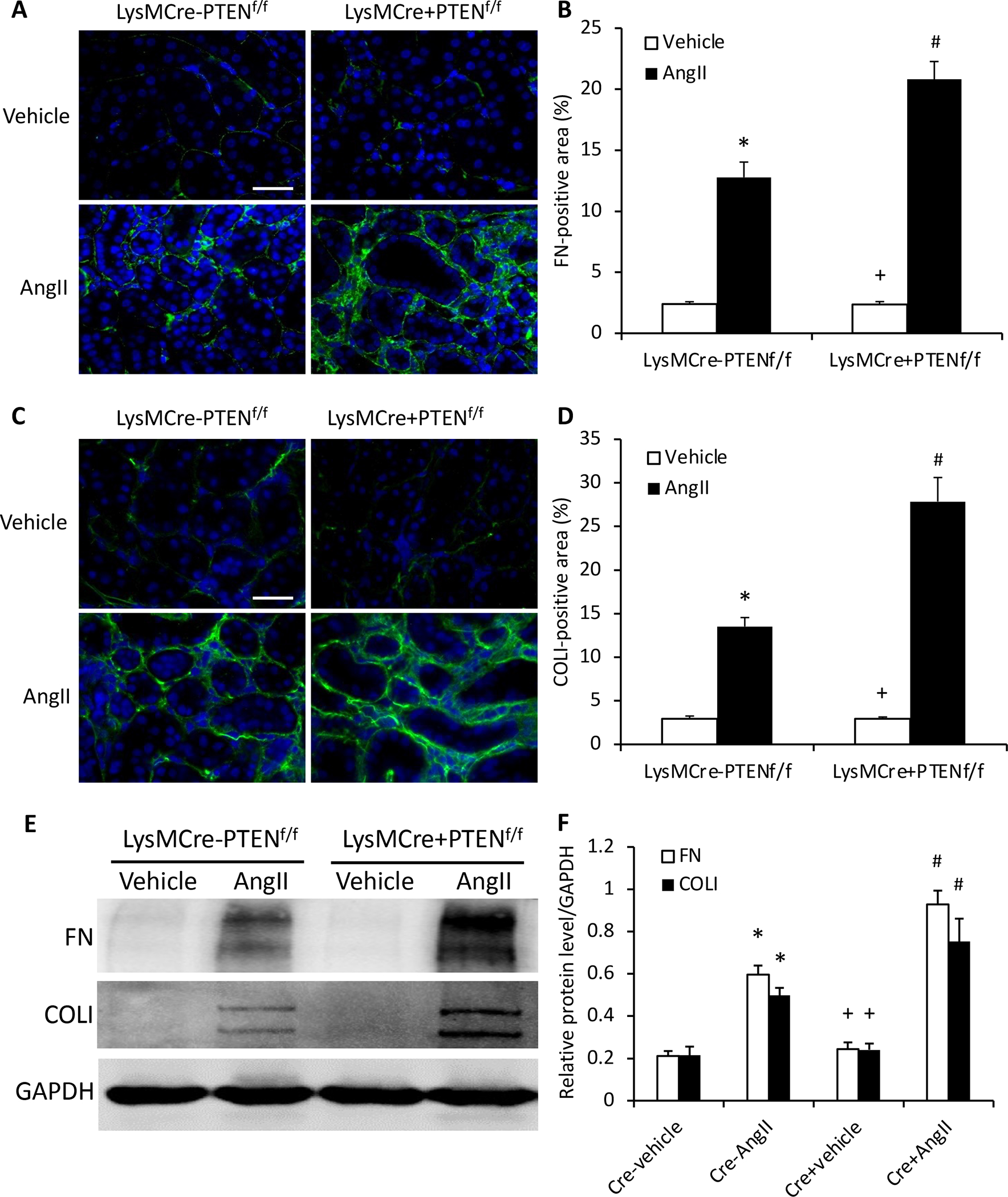
A, Representative photomicrographs of immunofluorescence staining for fibronectin (FN; green) in the kidney. Scale bar, 20 μm. B, Quantitative analysis of fibronectin (FN; green)-positive area in the kidney. *P<0.01 vs LysMCre−PTENf//f-Vehicle; +P<0.01 vs LysMCre+PTENf//f-AngII; and #P<0.01 vs LysMCre−PTENf//f-AngII. n=6 per group. C, Representative photomicrographs of immunofluorescence staining for collagen I (Col I; green) in the kidney. Scale bar, 20 μm. D, Quantitative analysis of Col I positive area in the kidney. *P<0.01 vs PTENf//f-Vehicle; +P<0.01 vs LysMCre+PTENf//f-AngII; and #P<0.01 vs LysMCre−PTENf//f-Ang II. n=6 per group. E, Representative Western blots of protein expression of FN and Col I in the kidney. F, Quantitative analysis of FN and Col I protein expression levels in the kidney. *P<0.01 vs LysMCre−PTENf//f-Vehicle; +P<0.05 vs LysMCre+PTENf//f-AngII; and #P<0.01 vs LysMCre−PTENf//f-AngII. n=6 per group.
Myeloid PTEN Deficiency Enhances Myeloid Fibroblast Accumulation
Myeloid fibroblasts are a major source of activated fibroblasts that contribute to pathogenesis of renal fibrosis (Mack & Yanagita, 2015; Yan, Zhang, Jia, & Wang, 2016). We then evaluated the effect of myeloid PTEN deficiency on myeloid fibroblast accumulation in the kidney in response to AngII-induced hypertension. Kidney sections were stained for CD45, a hematopoietic cell marker, and platelet-derived growth factor receptor β (PDGFR-β), a mesenchymal marker. The accumulation of CD45 and PDGFR-β dual positive cells were significantly augmented in AngII treated myeloid PTEN knockout mice compared with control mice (Figure 4A and B). These results suggest that PTEN is involved in recruiting myeloid fibroblasts into the kidney in response to AngII induced hypertension.
Figure 4. Myeloid PTEN deficiency induces bone marrow-derived fibroblast accumulation and myofibroblast formation in the kidney.
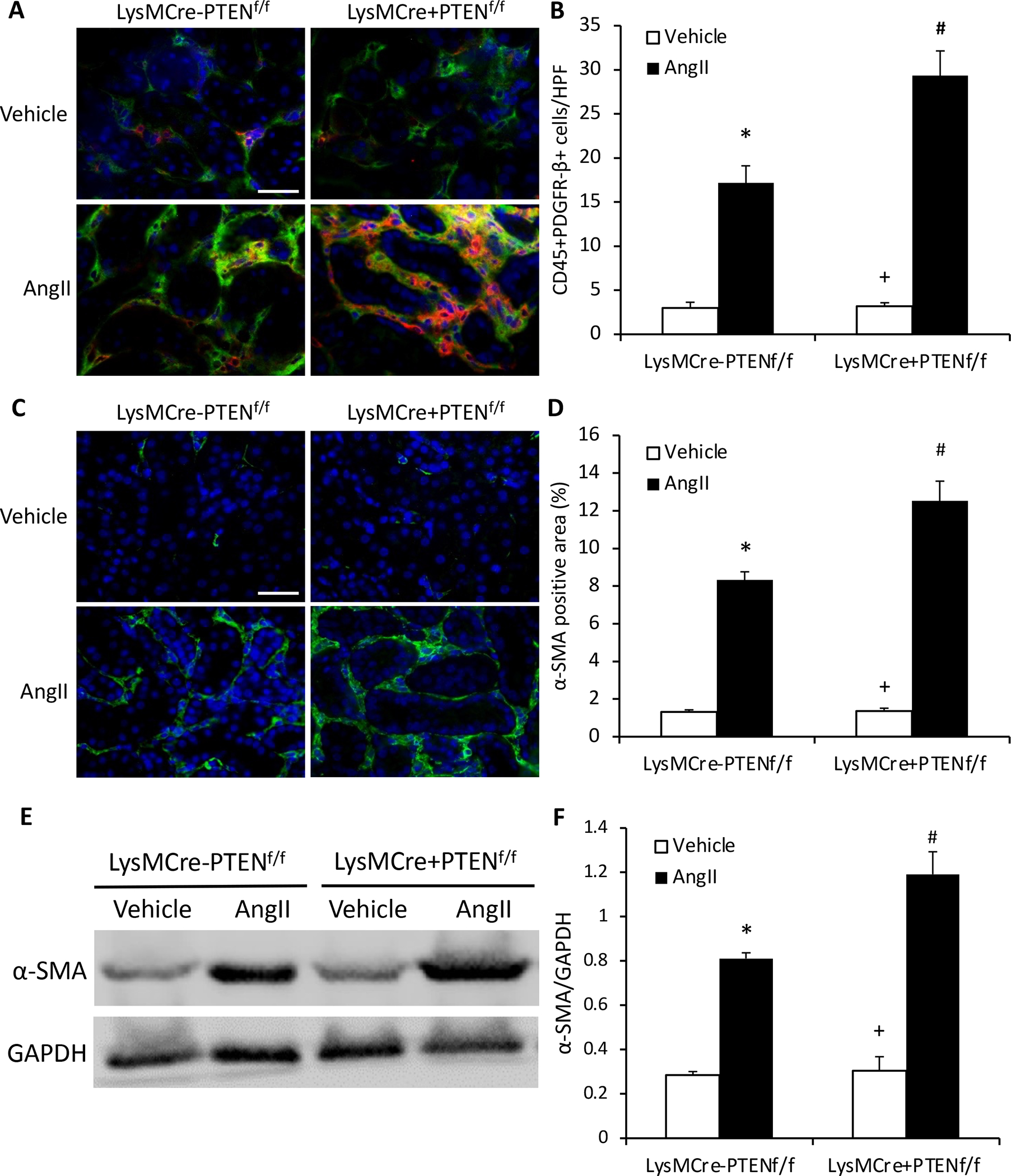
A, Representative photomicrographs of kidney sections stained for CD45 (red), PDGFR-β (green), and nuclei (DAPI; blue). Scale bar, 20 μm. B, Quantitative analysis of CD45+ and PDGFR-β+ fibroblasts in kidney. *P<0.01 vs LysMCre−PTENf//f-Vehicle; +P<0.01 vs LysMCre+PTENf//f-AngII; and #P<0.01 vs LysMCre−PTENf//f-AngII. n=6 per group. C, Representative photomicrographs of kidney sections stained for α-SMA (green) and nuclei (DAPI; blue). Scale bar, 20 μm. D, Quantitative analysis of α-SMA protein expression in kidney. *P<0.01 vs LysMCre−PTENf//f-Vehicle; +P<0.01 vs LysMCre+PTENf//f-AngII; and #P<0.05 vs LysMCre−PTENf//f-AngII. n=6 per group. E, Representative Western blots of α-SMA protein expression in the kidney. F, Quantitative analysis of α-SMA protein expression in the kidney. *P<0.01 vs LysMCre−PTENf//f-Vehicle; +P<0.01 vs LysMCre+PTENf//f-AngII; and #P<0.05 vs LysMCre−PTENf//f-AngII. n=6 per group.
To examine whether myeloid PTEN deficiency influences myofibroblast population in the kidney in response to AngII treatment, kidney sections were stained for α-smooth muscle actin (α-SMA), a marker of myofibroblasts. The results showed that myeloid PTEN deficiency markedly enhanced the number of α-SMA-positive myofibroblasts (Figure 4C and D). Consistent with these findings, Western blot analysis demonstrated that myeloid PTEN deficiency considerably augmented α-SMA protein expression in the kidney after AngII treatment (Figure 4E and F). These results indicate that myeloid PTEN deficiency enriches the population of myofibroblasts in the kidney after 4 weeks AngII treatment.
Myeloid PTEN Deficiency Augments Macrophage and T Cells Infiltration
Macrophages and T lymphocytes play a vital role in AngII target organ damage (Luft, Dechend, & Muller, 2012; McMaster, Kirabo, Madhur, & Harrison, 2015). We determined the effect of myeloid PTEN deficiency on inflammatory cell infiltration into the kidney in response to AngII induced hypertension. Kidney sections were stained for F4/80, a macrophage marker, and CD3, a T lymphocyte marker. Macrophages and T lymphocytes infiltrated into the kidney of control mice after 4 weeks AngII treatment. Myeloid PTEN deficiency considerably augmented the infiltration of macrophages and T lymphocytes into the kidney (Figure 5). These results suggest that PTEN plays a crucial role in regulating inflammatory cell infiltration into the kidney in AngII-induced hypertension.
Figure 5. Myeloid PTEN deficiency enhances macrophage and T cell infiltration into the kidney.
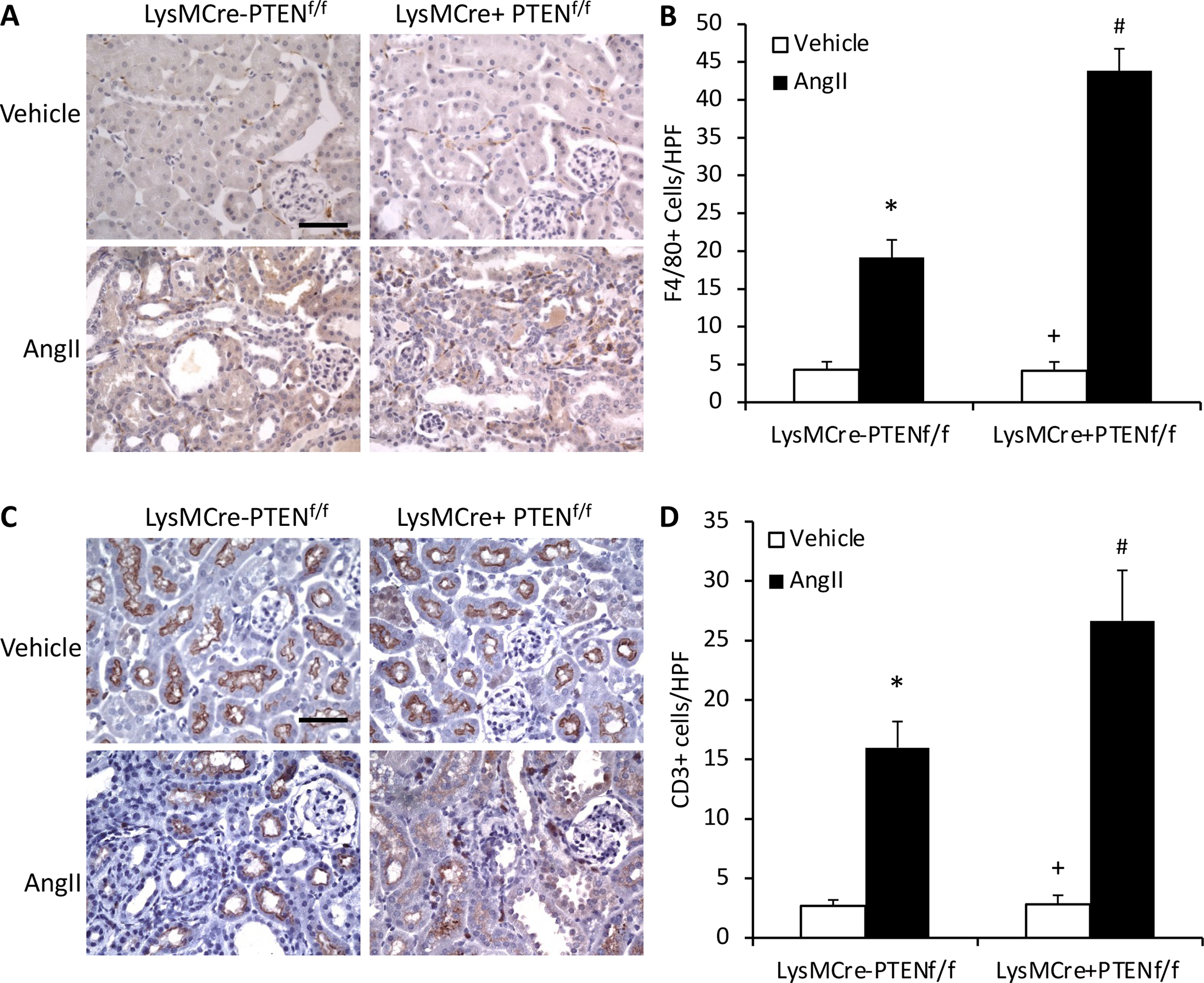
A, Representative photomicrographs of kidney sections stained for F4/80 (brown) and counterstained with hematoxylin (blue). Scale bar, 20 μm. B, Quantitative analysis of F4/80+ macrophages in the kidney. *P<0.01 vs LysMCre−PTENf//f-Vehicle; +P<0.01 vs LysMCre+PTENf//f-AngII; and #P<0.05 vs LysMCre−PTENf//f-AngII. n=6 per group. C, Representative photomicrographs of kidney sections stained for CD3 (brown) and counterstained with hematoxylin (blue). Scale bar, 20 μm. D, Quantitative analysis of CD3+ T cells in the kidney. *P<0.01 vs LysMCre−PTENf//f-Vehicle; +P<0.01 vs LysMCre+PTENf//f-AngII; and #P<0.05 vs LysMCre−PTENf//f-AngII. n=6 per group. HPF indicates high-power field.
Discussion
Chronic kidney disease is a global public health concern. Hypertension, as a major cause of chronic kidney disease plays an important role in the pathogenesis of CKD (Ruiz-Ortega et al., 2006). PTEN is originally identified as a tumor suppressor (Li et al., 1997; Simpson & Parsons, 2001). Subsequent studies have shown that PTEN plays a vital role in regulating cell growth and survival through phosphatidylinositol 3 kinase (PI3K)/AKT signaling pathway (Leslie & Downes, 2002; Song et al., 2012; Yamada & Araki, 2001). Recently, we have demonstrated that PI3 Kinase γ deficiency decreased kidney injury and fibrosis in AngII-induced hypertension (An et al., 2020). PTEN is involved in kidney development (J. K. Chen et al., 2015). Loss of PTEN promotes the development of diabetic nephropathy (Lin et al., 2015) and pharmacological inhibition of PTEN exacerbates acute kidney injury (Zhou et al., 2017). However, the role of PTEN in AngII-induced hypertensive kidney disease remains to be elucidated. In this study, we have showed that myeloid PTEN deficiency increases renal dysfunction and aggravates kidney damage in mice after 4 weeks of AngII treatment. These results demonstrate that PTEN deficiency aggravates kidney dysfunction and injury in AngII-induced hypertension.
Previous studies indicate that bone marrow-derived fibroblasts play an important role in development of renal fibrosis (Mack & Yanagita, 2015; Yan et al., 2016). We have shown that CXCL16/CXCR6/PI3 kinase signaling pathway plays an important role in recruiting bone marrow-derived fibroblasts into the kidney in AngII-induced hypertension (An et al., 2020; Xia et al., 2013; Xia et al., 2014). In the present study, our results have showed that myeloid PTEN deficiency increases kidney fibrosis development, myeloid fibroblast accumulation, and myofibroblast transformation in the kidney of AngII-induced hypertensive kidney disease. These results have demonstrated that the PTEN significantly affects the recruitment of bone marrow-derived fibroblasts into the kidney following AngII administration, contributing to the activate fibroblast population.
Inflammatory cells play an important role in the development of hypertensive kidney disease (Luft et al., 2012). AngII is a major factor of CKD which plays critical role in the regulation of inflammatory response in hypertensive end-organ damage (Mezzano, Ruiz-Ortega, & Egido, 2001; Ruiz-Ortega et al., 2006). In the present study, we have demonstrated that inflammatory cell infiltration into the kidney is significantly augmented in myeloid PTEN knockout mice in AngII-induced hypertensive kidney disease. These results suggest that PTEN is critically involved in recruiting inflammatory cells into the kidney in response to AngII-induced hypertension.
Glycogen synthase kinase 3β (GSK-3β) is a serine/threonine-protein kinase that functions as an integration point for multiple cellular pathways including PTEN/PI3K/AKT signaling, which are involved in glycogen biosynthesis, inflammation, mitochondrial dysfunction, and apoptosis (Beurel, Grieco, & Jope, 2015; Doble & Woodgett, 2003). Recent studies have shown that GSK-3β plays an important role in the development of renal fibrosis (B. Chen et al., 2021; Lu et al., 2019). Since PTEN regulates GSK-3β activities via PI3K, it will be interesting to examine if PTEN/GSK-3β signaling pathway is involved in AngII-induced renal inflammation and fibrosis in the future study.
In summary, our results demonstrate that PTEN plays an important role in the development of hypertensive kidney disease. Myeloid PTEN deficiency enhances recruitment of macrophages, T lymphocytes, and myeloid fibroblasts into the kidney exacerbating kidney injury and fibrosis in AngII-induced hypertension. These results suggest that activation of PTEN signaling may represent a novel therapeutic strategy for hypertensive kidney disease.
Acknowledgements
This work was supported by grants from the NIH (R01DK95835), the U.S. Department of Veterans Affairs (I01BX02650), and Dialysis Clinic Inc (2019-03) to YW. The contents of the article do not represent the views of the U.S. Department of Veterans Affairs or the United States government.
Footnotes
Conflict of Interest Statement
The authors declare no conflicts of interest.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Reference
- An C, Wen J, Hu Z, Mitch WE, & Wang Y (2020). Phosphoinositide 3-kinase gamma deficiency attenuates kidney injury and fibrosis in angiotensin II-induced hypertension. Nephrol Dial Transplant, 35(9), 1491–1500. doi: 10.1093/ndt/gfaa062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurel E, Grieco SF, & Jope RS (2015). Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther, 148, 114–131. doi: 10.1016/j.pharmthera.2014.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Wang P, Liang X, Jiang C, Ge Y, Dworkin LD, & Gong R (2021). Permissive effect of GSK3beta on profibrogenic plasticity of renal tubular cells in progressive chronic kidney disease. Cell Death Dis, 12(5), 432. doi: 10.1038/s41419-021-03709-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JK, Nagai K, Chen J, Plieth D, Hino M, Xu J, … Harris RC (2015). Phosphatidylinositol 3-kinase signaling determines kidney size. J Clin Invest, 125(6), 2429–2444. doi: 10.1172/JCI78945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble BW, & Woodgett JR (2003). GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci, 116(Pt 7), 1175–1186. doi: 10.1242/jcs.00384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, … Yang CW (2013). Chronic kidney disease: global dimension and perspectives. Lancet, 382(9888), 260–272. doi: 10.1016/S0140-6736(13)60687-X [DOI] [PubMed] [Google Scholar]
- Jin X, Chen J, Hu Z, Chan L, & Wang Y (2013). Genetic deficiency of adiponectin protects against acute kidney injury. Kidney Int, 83(4), 604–614. doi: 10.1038/ki.2012.408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie NR, & Downes CP (2002). PTEN: The down side of PI 3-kinase signalling. Cell Signal, 14(4), 285–295. [DOI] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, … Parsons R (1997). PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science, 275(5308), 1943–1947. doi: 10.1126/science.275.5308.1943 [DOI] [PubMed] [Google Scholar]
- Liang H, Ma Z, Peng H, He L, Hu Z, & Wang Y (2016). CXCL16 Deficiency Attenuates Renal Injury and Fibrosis in Salt-Sensitive Hypertension. Sci Rep, 6, 28715. doi: 10.1038/srep28715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Zhang Z, He L, & Wang Y (2016). CXCL16 regulates cisplatin-induced acute kidney injury. Oncotarget, 7(22), 31652–31662. doi: 10.18632/oncotarget.9386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JS, Shi Y, Peng H, Shen X, Thomas S, Wang Y, … Xu J (2015). Loss of PTEN promotes podocyte cytoskeletal rearrangement, aggravating diabetic nephropathy. J Pathol, 236(1), 30–40. doi: 10.1002/path.4508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y (2011). Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol, 7(12), 684–696. doi: 10.1038/nrneph.2011.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Wang P, Qiao Y, Jiang C, Ge Y, Flickinger B, … Gong R (2019). GSK3beta-mediated Keap1-independent regulation of Nrf2 antioxidant response: A molecular rheostat of acute kidney injury to chronic kidney disease transition. Redox Biol, 26, 101275. doi: 10.1016/j.redox.2019.101275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luft FC, Dechend R, & Muller DN (2012). Immune mechanisms in angiotensin II-induced target-organ damage. Ann Med, 44 Suppl 1, S49–54. doi: 10.3109/07853890.2011.653396 [DOI] [PubMed] [Google Scholar]
- Ma Z, Jin X, He L, & Wang Y (2016). CXCL16 regulates renal injury and fibrosis in experimental renal artery stenosis. Am J Physiol Heart Circ Physiol, 311(3), H815–821. doi: 10.1152/ajpheart.00948.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack M, & Yanagita M (2015). Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int, 87(2), 297–307. doi: 10.1038/ki.2014.287 [DOI] [PubMed] [Google Scholar]
- McMaster WG, Kirabo A, Madhur MS, & Harrison DG (2015). Inflammation, immunity, and hypertensive end-organ damage. Circ Res, 116(6), 1022–1033. doi: 10.1161/CIRCRESAHA.116.303697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennuni S, Rubattu S, Pierelli G, Tocci G, Fofi C, & Volpe M (2014). Hypertension and kidneys: unraveling complex molecular mechanisms underlying hypertensive renal damage. J Hum Hypertens, 28(2), 74–79. doi: 10.1038/jhh.2013.55 [DOI] [PubMed] [Google Scholar]
- Mezzano SA, Ruiz-Ortega M, & Egido J (2001). Angiotensin II and renal fibrosis. Hypertension, 38(3 Pt 2), 635–638. [DOI] [PubMed] [Google Scholar]
- Ruiz-Ortega M, Ruperez M, Esteban V, Rodriguez-Vita J, Sanchez-Lopez E, Carvajal G, & Egido J (2006). Angiotensin II: a key factor in the inflammatory and fibrotic response in kidney diseases. Nephrol Dial Transplant, 21(1), 16–20. doi:gfi265 [pii] 10.1093/ndt/gfi265 [DOI] [PubMed] [Google Scholar]
- Simpson L, & Parsons R (2001). PTEN: life as a tumor suppressor. Exp Cell Res, 264(1), 29–41. doi: 10.1006/excr.2000.5130 [DOI] [PubMed] [Google Scholar]
- Song MS, Salmena L, & Pandolfi PP (2012). The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol, 13(5), 283–296. doi: 10.1038/nrm3330 [DOI] [PubMed] [Google Scholar]
- Wang Y, Jia L, Hu Z, Entman ML, Mitch WE, & Wang Y (2018). AMP-activated protein kinase/myocardin-related transcription factor-A signaling regulates fibroblast activation and renal fibrosis. Kidney Int, 93(1), 81–94. doi: 10.1016/j.kint.2017.04.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, An C, Jin X, Hu Z, & Wang Y (2020). Disruption of CXCR6 Ameliorates Kidney Inflammation and Fibrosis in Deoxycorticosterone Acetate/Salt Hypertension. Sci Rep, 10(1), 133. doi: 10.1038/s41598-019-56933-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Entman ML, & Wang Y (2013). Critical Role of CXCL16 in Hypertensive Kidney Injury and Fibrosis. Hypertension, 62(6), 1129–1137. doi: 10.1161/HYPERTENSIONAHA.113.01837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Jin X, Yan J, Entman ML, & Wang Y (2014). CXCR6 Plays a Critical Role in Angiotensin II-Induced Renal Injury and Fibrosis. Arterioscler Thromb Vasc Biol, 34(7), 1422–1428. doi: 10.1161/ATVBAHA.113.303172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lin SC, Chen J, Miao Y, Taffet GE, Entman ML, & Wang Y (2011). CCR2 mediates the uptake of bone marrow-derived fibroblast precursors in angiotensin II-induced cardiac fibrosis. Am J Physiol Heart Circ Physiol, 301(2), H538–547. doi: 10.1152/ajpheart.01114.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KM, & Araki M (2001). Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis. J Cell Sci, 114(Pt 13), 2375–2382. [DOI] [PubMed] [Google Scholar]
- Yan J, Zhang Z, Jia L, & Wang Y (2016). Role of Bone Marrow-Derived Fibroblasts in Renal Fibrosis. Front Physiol, 7, 61. doi: 10.3389/fphys.2016.00061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Chen J, Yan J, Zhang L, Chen G, He L, & Wang Y (2012). Effect of interleukin 6 deficiency on renal interstitial fibrosis. PloS one, 7(12), e52415. doi: 10.1371/journal.pone.0052415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, An C, Jin X, Hu Z, Safirstein RL, & Wang Y (2020). TAK1 deficiency attenuates cisplatin-induced acute kidney injury. Am J Physiol Renal Physiol, 318(1), F209–F215. doi: 10.1152/ajprenal.00516.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Jia L, Hu Z, & Wang Y (2017). Pharmacological Inhibition of PTEN Aggravates Acute Kidney Injury. Sci Rep, 7(1), 9503. doi: 10.1038/s41598-017-10336-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.