

Abstract
Multi-enzyme assemblies composed of metabolic enzymes catalyzing sequential reactions are being increasingly studied. Here, we report the discovery of a 1.6 megadalton multi-enzyme complex from Bacillus subtilis composed of two enzymes catalyzing opposite (“counter-enzymes”) rather than sequential reactions: glutamate synthase (GltAB), and glutamate dehydrogenase (GudB), that make and break glutamate, respectively. In vivo and in vitro studies show that the primary role of complex formation is to inhibit the activity of GudB. Using cryo-electron microscopy, we elucidated the structure of the complex and the molecular basis of inhibition of GudB by GltAB. The complex exhibits unusual oscillatory progress curves and is necessary for both planktonic growth, in glutamate-limiting conditions, and for biofilm growth, in glutamate-rich media. The regulation of a key metabolic enzyme by complexing with its counter-enzyme may thus enable cell growth under fluctuating glutamate concentrations.
Keywords: Enzyme regulation, counter-enzymes, glutamate, metabolism, multi-enzyme complex, Bacillus subtilis, biofilm, cryo-electron microscopy, oscillations, enzyme kinetics
Introduction
Metabolites are key cellular resources and their homeostasis is primarily achieved by regulating enzyme activities at various levels: transcription, translation, and post-translation. While regulation of transcription and translation prevents the waste of resources and generally enables near-complete silencing of enzyme activity, post-translational enzyme regulation - including allosteric regulation via small molecules, covalent modification and degradation - enables rapid changes in metabolite levels. Regulatory modes at these various levels are crucial for the fitness of the organism1–3. Besides these modes of regulation, evidence is accumulating of transient enzyme complexes, or metabolons (typically composed of enzymes catalyzing sequential reactions)4 that increase the metabolic efficiency by compartmentalization of reaction intermediates5,6. In this study, we describe a distinct mode of regulation where two central metabolic enzymes that catalyze opposite reactions (“counter-enzymes”) form a transient complex under metabolite-limiting conditions.
Our study arose from the search for the regulatory mechanism of glutamate dehydrogenase, GudB, from B. subtilis. Glutamate dehydrogenases reside at the crossroads of carbon and nitrogen metabolism by catalyzing the interconversion of glutamate to α-ketoglutarate (AKG) and ammonia. Glutamate is a major nitrogen reservoir and can provide up to 85% of the cellular nitrogen requirement in bacteria7. Moreover, glutamate has multiple other roles, including: intracellular pH buffering, maintenance of osmolarity, and acting as a counter ion to potassium8,9. Glutamate (Glu) is also one of the most abundant metabolites with cellular concentrations up to 150 mM10–12. In contrast, AKG is maintained at low concentrations, in the range of a few millimolars, and has rapid turnover13. In most Bacilli species, the Glu/AKG balance is maintained by the activity of catabolic, NAD+-dependent, dehydrogenases that degrade glutamate, with GudB being the key player in B. subtilis14. The other enzyme that directly affects the Glu/AKG balance is glutamate synthase (GltAB), a heterodimeric enzyme composed of two subunits, GltA and GltB, which is involved in glutamate synthesis starting from glutamine and AKG (Figure 1a).. Notably, B. subtilis is known to rapidly adapt to loss/inactivation of genes involved in glutamate metabolism by suppressor mutations, suggesting the importance of glutamate homeostasis in this bacterium15.It should be noted that, GudB is cryptified and inactive in the domesticated strain, B. subtilis 16816, however, the wild-ype strain NCIB 3610 used in this study, encodes a functional copy of GudB.
Figure 1. GudB interacts with GltAB in glutamate poor growth conditions.
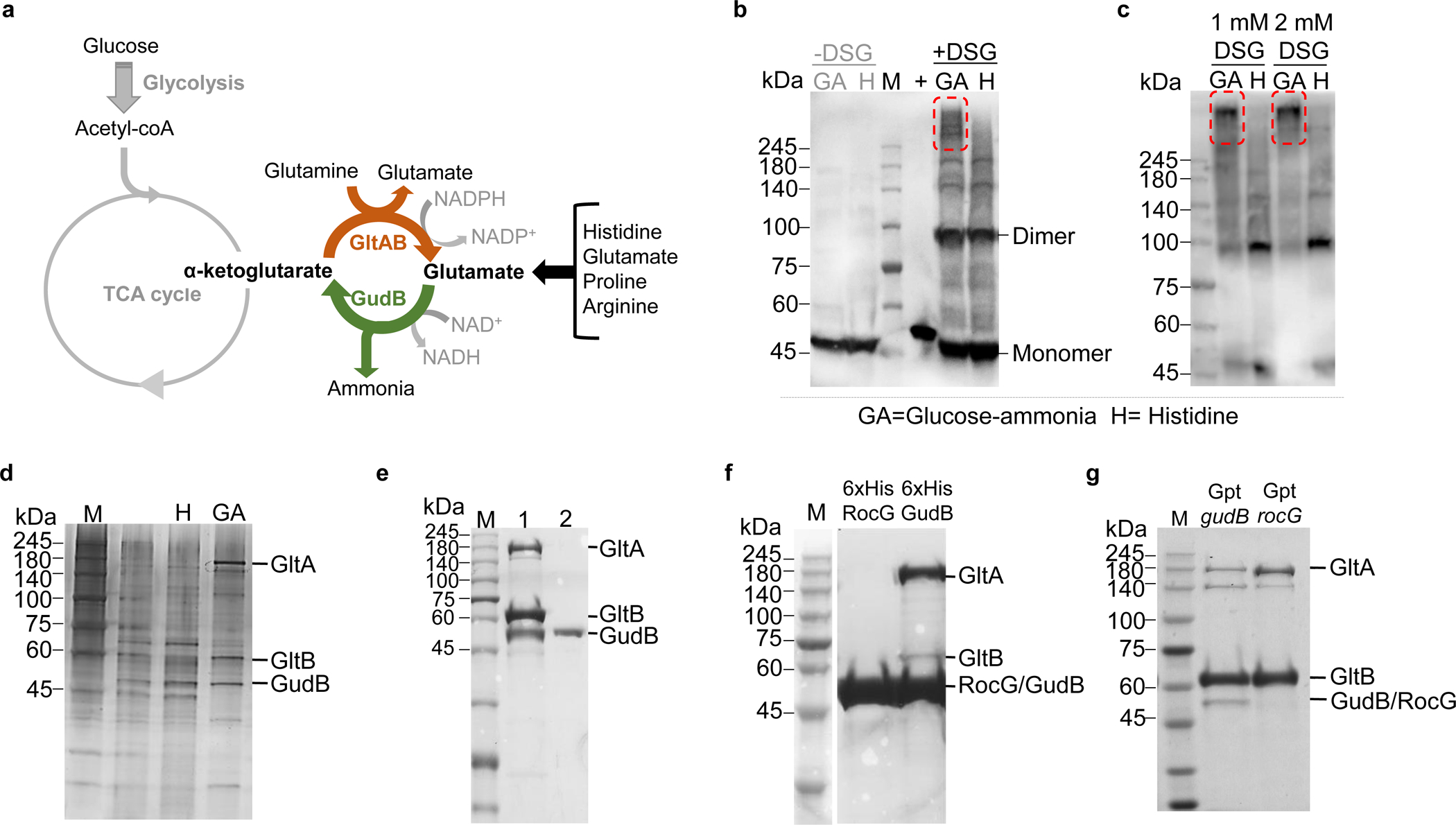
a) Key reactions involved in glutamate metabolism in Bacillus subtilis. Amino acids like proline, arginine, and histidine when provided as the sole C/N source are catabolized via glutamate. In contrast, growth on glucose as C source demands glutamate synthesis (via AKG). b) Western blot using anti-GudB antibodies indicating similar expression levels of GudB in B. subtilis cells grown in glucose-ammonia (GA) and Histidine (H) (-DSG). Upon treating with a chemical crosslinker (DSG, 0.5 mM), high molecular weight species that include GudB can be seen in cells grown on glucose-ammonia (GA, highlighted in red frame) but not on histidine (H). Recombinant GudB served as a positive control (+). c) The high molecular weight species of GudB are clearly seen in Western analysis of lysates from cells grown on glucose-ammonia (as in C) yet treated with higher concentration of DSG (1mM/2mM). d) Immunoprecipitation of GudB indicated co-elution of GltA and GltB in glucose-ammonia but not in histidine. The eluates from the pulldown was subjected to SDS-PAGE and stained with silver nitrate. e) SDS-PAGE showing co-elution of GudB and GltA upon purification of Strep-GltB from B. subtilis cells grown in glucose-ammonia (Lane 1; Lane 2 shows purified recombinant Strep tagged-GudB). f) Co-purification of GltA and GltB upon pulldown of His-tagged GudB but RocG. g) GudB but not RocG co-eluted upon pulldown of Strep-GltB from strains expressing either GudB or RocG from the constitutive gudB promoter (Gpt). Images b-g correspond to one replicate and are representative at least three independent experiments.
While not much is known about the post-translational regulation of glutamate synthases (like GltA/B), glutamate dehydrogenases (like GudB) are among the most intricately regulated enzymes – they are renowned, if not infamous, for their complex allosteric regulatory features17. The mammalian glutamate dehydrogenases are regulated by nucleotides18, while some bacterial enzymes are regulated by amino acids such as leucine19. The B. subtilis GudB is constitutively expressed14,16, and at high levels, and hence regulation is critical. However, nucleotides, amino acids, and other key metabolites we have tested in vitro have all failed to regulate GudB’s activity14.
Here we investigated the regulatory mode of GudB in B. subtilis. We found that under conditions where glutamate needs to be synthesized, GudB binds to its counter enzyme, GltAB. Growth profiling of relevant strains and steady state kinetic measurement suggested that GudB is silenced upon binding GltAB. The regulatory interplay of the enzymes in the complex results in a unique oscillatory progress curve. Native mass spectrometry, and high-resolution cryo-EM structures, revealed the molecular basis of GudB- inhibition in this unusually large (1.6 MDa) complex. Besides being crucial for planktonic growth in glutamate-limiting condition, we also show that GudB-GltAB complex formation is needed for optimal biofilm development even in a glutamate-rich medium. This counter-enzyme assembly may thus mediate the previously observed spatiotemporal glutamate catabolism in biofilms20.
Results
GudB interacts with GltAB in glutamate-poor growth conditions
B. subtilis grows in both glucose-ammonia (glutamate-poor) and in histidine/glutamate (glutamate-rich) containing medium. Besides histidine and glutamate, growth on proline, or arginine results in glutamate-rich conditions, as these amino acids are catabolized via glutamate as intermediate (Fig. 1a). Oddly, the protein levels of GudB are the same in cells grown in glucose-ammonia and in histidine (Fig. 1b, −DSG). We probed the oligomeric state of GudB in both conditions using in-vivo crosslinking of cells using dissucinimidyl glutarate (DSG) followed by western blotting using anti-GudB antibodies (Fig. 1b, +DSG). In glucose-ammonia we observed bands with molecular weight greater than hexameric GudB, the expected oligomeric state of the enzyme21, while in histidine the bands indicated dissociated monomer/dimer species. This discrepancy is even greater in high crosslinker concentration (Fig. 1c). The high molecular weight species is specific to glutamate-poor media like glucose-ammonia and was not seen in any of the glutamate-rich medium (glutamate, histidine, arginine, or glycerol-glutamate) we tested (Extended Data Fig.1a).
Immunoprecipitation of GudB from cells grown in glucose-ammonia showed two additional proteins along with GudB), with molecular weight around ~160 kDa and ~60 kDa. This interaction was specific to cells grown in glucose-ammonia but not in histidine (Fig. 1d). Shotgun proteomics of the eluates indicated that these proteins are GltA (168.7 kDa) and GltB (54.8 kDa). Both proteins were significantly enriched in eluates obtained from glucose-ammonia grown cells compared to histidine grown ones (≤167 fold with crosslinker, and ≤ 21-fold without; Supplementary Table 1, and Supplementary Datasets 1 and 2). Expression and pulldown of TwinStrep-tagged GltB (GltB-TS) from cells grown on glucose-ammonia also indicated the formation of a ternary GudB-GltAB complex (Fig. 1e). This association was surprising as GltAB, a heterodimeric glutamate synthase, is involved in glutamate synthesis, the opposing function to GudB. Unlike GudB, GltAB expression is highly regulated22,23. Indeed, GltAB is exclusively expressed in glutamate-poor conditions (Extended Data Fig. 1b). Thus, it is likely that in glutamate-rich conditions the GudB-GltAB complex is absent simply because GltAB is not present.
The interaction with GltAB is paralog specific
GudB is not the only catabolic glutamate dehydrogenase in B. subtilis. Its closely related paralog, RocG, shares 75 % sequence identity and an effectively identical hexameric structure21. Moreover, GudB and RocG share similar kinetic properties with respect to glutamate degradation14. RocG is a part of the Roc operon involved in arginine degradation and its transcription is tightly regulated. Accordingly, the levels of RocG in glucose-ammonia is up to 30-fold lower compared to GudB. Both in vitro (Fig. 1f) and in vivo (Fig. 1g) pulldown experiments indicated that RocG does not interact with GltAB. Notably, this explains the severe growth defects of the strain constitutively expressing RocG (instead of GudB) in glucose-ammonia14. Given the lack of interaction of RocG with GltAB, strains consitutively expressing RocG can be used as a control to assess the regulation of GudB by GltAB.
GltAB negatively regulates GudB activity in vivo
Having established that GudB forms a complex with its counter-enzyme, GltAB, we next aimed to decipher the role of GudB-GltAB complex formation in vivo. We profiled the growth of individual knockout strains, ΔgudB, ΔgltA, and ΔgltB in media where either glutamate synthesis or catabolism should dominate. In general, we observed that when GltAB is essential, GudB is dispensable (Fig. 2a) and vice-versa (Fig. 2b,c; note that removal of either of GltAB’s subunits leads to complete loss of the synthase activity). Specifically, the catabolic GudB is essential when glutamate (Fig. 2b), or other amino acids such as histidine (Fig. 2c) that are catabolized via glutamate, are present as the sole carbon source. Conversely, the anabolic GltAB is essential when glutamate needs to be synthesized de novo, as indicated by the inability of either ΔgltA or ΔgltB to grow on glucose-ammonia (Fig. 2b). Finally, neither GudB nor GltA/B are required when glucose-ammonia is supplemented with glutamate (Fig. 2d).
Figure 2: The phenotypic effects of GltAB and GudB and their interaction.
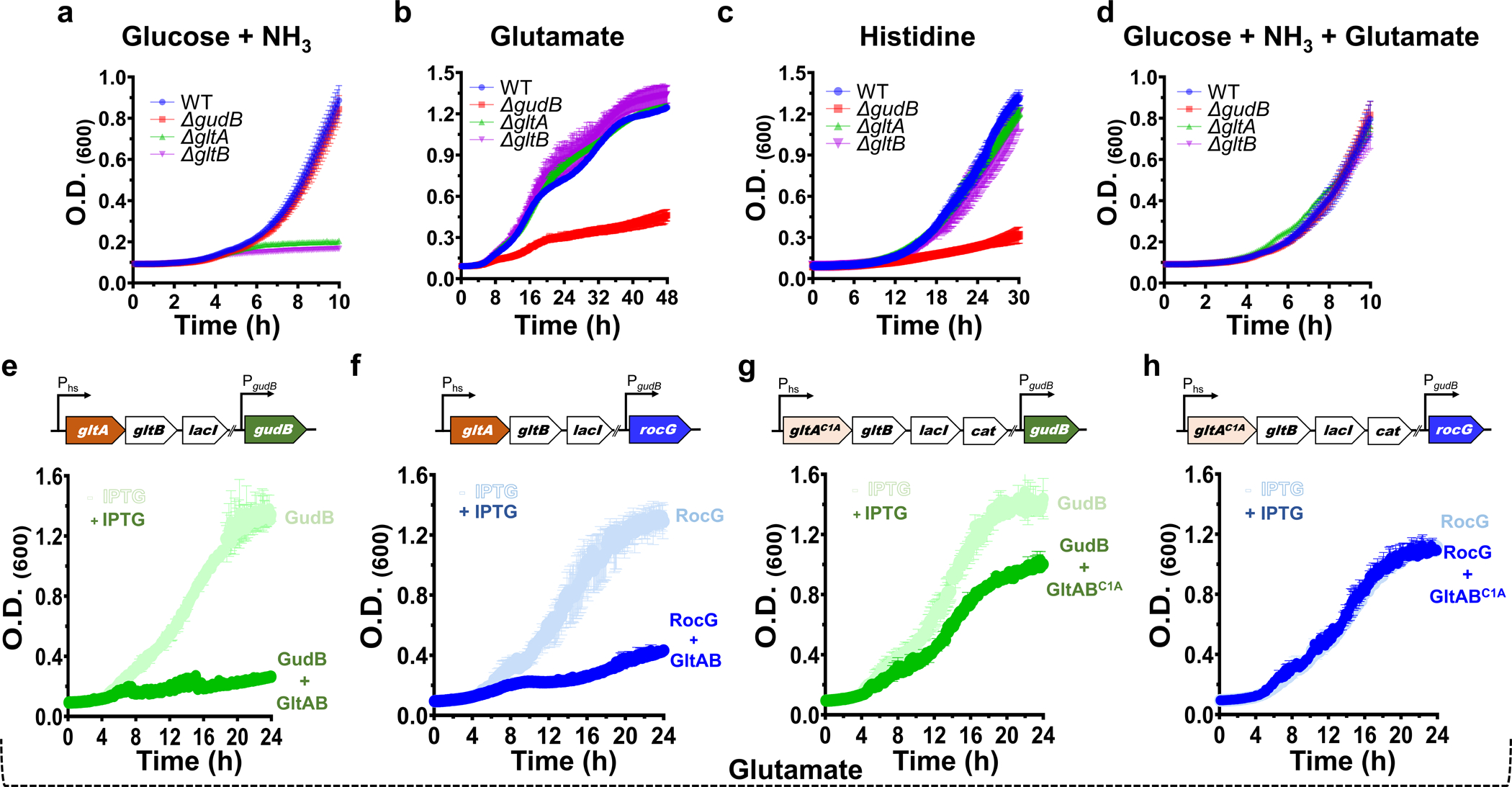
(a-d) Growth profiling of the denoted knockout strains indicate that GltAB’s glutamate dehydrogenase activity is essential for growth in glucose-ammonia (panel a) while GudB’s glutamate dehydrogenase activity is essential in glutamate (panel b) and histidine (panel c). Under conditions where both glucose and glutamate are available neither of these two activities is essential (panel d). n=3 are three independent measurements. Data is presented as mean of all measurements and error bars represent SD. (e-h) Growth inhibition in glutamate medium upon expression of wild-type GltAB, and its inactive mutant (GltAC1A), from an IPTG inducible hyper-spank promoter. The scheme above each panel shows the genotype of the corresponding strain. Expression of functional GltA causes growth suppression in strains expressing GudB (panel e) or RocG (panel f) likely due to futile cycling of making and breaking glutamate. Expression of GltABC1A causes growth suppression in a strain expressing GudB (panel G) but not in a strain co-expressing RocG (panel h). These effects are in agreement with GltA interacting with and inhibiting GudB but not RocG n=2 are two independent measurements. Data is presented as mean of all measurements and error bars represent SD..
Given the orthogonal functions, we surmised that GudB-GltAB complex formation could be inhibiting GudB’s activity to avoid the futile cycle of glutamate synthesis and breakdown. If this is indeed the case, then the complex formation should be deleterious under conditions where glutamate is the sole carbon source and GudB’s activity is pivotal. We observed that expression of GltAB in the background of a strain constitutively expressing GudB causes severe growth defect (Fig. 2e). However, similar phenotype was seen when GltAB is expressed in the strain constitutively expressing RocG (Fig. 2f), suggesting that growth inhibition in both the cases is mostly the result of futile cycling of making and breaking glutamate that will consume large amounts of NADPH and ATP due to glutamine hydrolysis. To disentangle the role of futile cycling from GltAB-GudB complex association, we expressed a catalytically inactive mutant of GltABC1A (Extended Data Fig. 1c) alongside GudB, (Fig. 2g). Here, the growth inhibition likely results from the physical interaction bewteen GudB’s and GltAB, as validated by pulldowns of the GudB-GltABC1A complex under the same growth condition (Extended Data Fig. 1d). Further, no growth inhibition was observed when the inactive GltABC1A was co-expressed with RocG (Fig. 2h; and accordingly, GltABC1A was pulled-down on its own, Extended Data Fig. 1d). Inducible expression of GltAB was verified by the IPTG-dependent growth of the strains in glucose-ammonia (Extended Data Fig. 2). Taken together, these results support a key regulatory role for the GudB-GltAB complex, in silencing GudB under glutamate-poor conditions.
GltAB binding alters the kinetic properties of GudB
To understand the effects of complex formation on the activity of the component enzymes, the complex, and the individual enzymes, were purified from B. subtilis (Fig. 3a) and used for steady-state kinetic measurements. The complex exhibited either glutamate synthase or dehydrogenase activity depending on which substrates were added (Fig. 3b), and the steady state kinetic parameters were accordingly derived from initial velocity measurements (Supplementary Table 2; Extended Data Fig. 3 a–f. The activity of GltAB is essentially the same on its own and in complex with GudB (a moderate increase in catalytic efficiency was observed when in complex with GudB). However, GudB showed altered behaviour: the KM value for glutamate is ~19 times higher in the complex (7.3 mM for free GudB, Fig. 3c, versus 138 mM for the GltAB bound form, Fig. 3d), supporting the notion that GltAB binding suppresses GudB’s activity. While the above measurements allowed us to derive kinetic parameters, the assay conditions do not reflect the cellular scenario where the substrates of both enzymes are present. Indeed, addition of GltAB’s substrates – AKG and NADPH, completely inactivates GudB at glutamate concentrations up to 50 mM (Fig. 3e). The silencing effect requires both substrates as neither AKG nor NADPH exhibited a substantial effect on their own. Using mass photometry, we observed that AKG and NADPH promote the assembly of GltA and GltB (Extended Data Fig. 3g). Moreover, we did identify AKG among other metabolites (Supplementary Table 3) extracted from the GudB-GltAB complex purified directly from B. subtilis. Overall, these results validate the hypothesis that GltAB’s binding allosterically silences GudB’s activity.
Figure 3. Enzymatic kinetics of the GltAB-GudB complex.
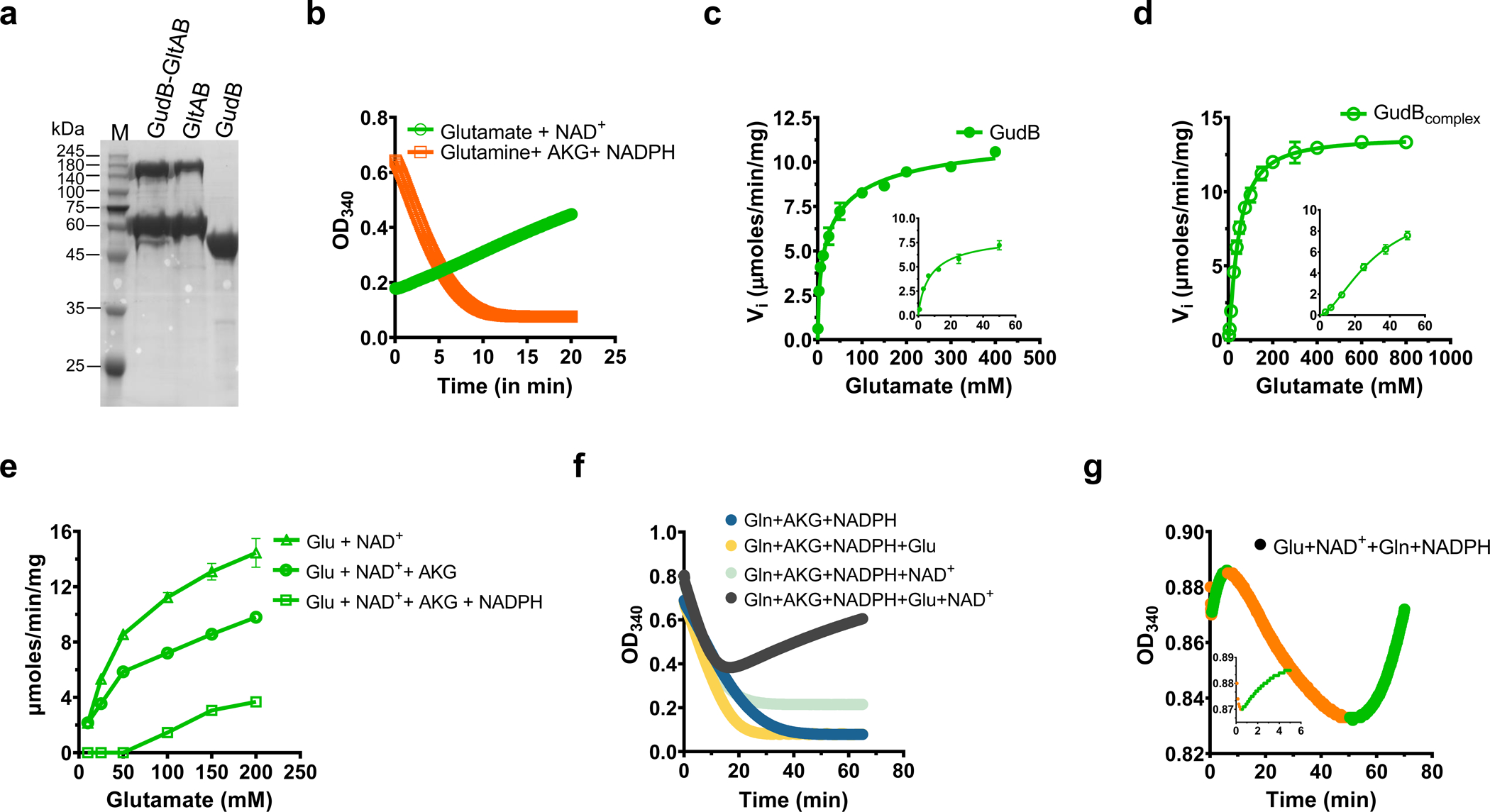
a) SDS-PAGE analysis of the GltAB-GudB complex, and its individual components, GudB, and GltAB, used in these assays. The complex and stand-alone GltAB were purified by the pulldown of Strep-GltB from respective B. subtilis strains (see methods). GudB was purified by recombinant expression in E.coli. b) The GudB-GltAB complex exhibits either glutamate dehydrogenase activity (upon addition of glutamate and NAD+; green line, indicating a drop in absorbance at 340 nm due to NAD+ reduction) or synthetase activity (upon addition of AKG, glutamine and NADPH; orange line indicating an increase in absorbance due to NADPH oxidation). c-d) Initial velocity of the dehydrogenase reaction as a function of glutamate concentration. On its own, GudB displayed a KM value of 7.3 mM for glutamate and negative cooperativity (c, Hill coefficient =0.57, Supplementary Table 2). In complex with GltAB, the KM rises to 138 mM with positive cooperativity (d, H=1.3). The insets in c and d show the initial velocities at low concentrations of glutamate. n=2 are two independent experiements. Data is presented as mean and error bars indicate standard deviation. e) Initial velocity of the dehydrogenase reaction in the GltAB-GudB complex, as is (Glu+NAD+), and in the presence of GltAB’s substrates (AKG, NADPH). n=2 are two independent experiements. Data is presented as mean and error bars indicate standard deviation. f) Reaction progress curves of the GudB-GltAB complex with various substrate combinations: substrates of GltAB only (blue), substrates of GltAB plus glutamate (yellow), substrates of GltAB plus NAD+ (light blue), all substrates for both enzymes (black). g) Multiphasic progress curve displayed by the GudB-GltAB complex in the presence of substrates of both enzymes except AKG. The phases where GltAB predominates are shown in orange and those where GudB is dominant are shown in green. The inset shows the initial few minutes of the reaction where there is a rapid drop in absorbance followed by a gradual increase. Panels a,b,f and g correspond to one experiment and are representative of atleast 4 independent experiements.
GltAB’s regulatory role was further manifested when its third substrate, glutamine, was added, unexpectedly resulting in multiphasic progress curves. We monitor GltAB’s activity through a decrease in absorbance at 340 nm (due to the reduction in NADPH levels) and GudB’s activity through an increase in absorbance due to increase in NADH formation (Fig. 3b). In principle, if both enzymes are active, a steady state would be established with a monotonic progress curve, with positive or negative slope values depending on the relative catalytic efficiency of the two enzymes24. Contrary to this expectation, in the presence of saturating concentration of substrates of both enzymes, the GudB-GltAB complex displayed a biphasic progress curve (Fig. 3f). The slope of the first phase was identical to the one observed when GltAB activity was tested separately, suggesting that in this initial phase GudB is completely inactive due to the presence of AKG and NADPH (as shown in Fig. 3e). With the drop in NADPH concentration, GudB becomes active, and this results in the onset of second phase of increasing absorbance. Indeed, when the reaction is initiated with all substrates except AKG, multiple oscillation cycles could be observed until most of the NADPH is consumed by GltAB, resulting in a final phase where GudB activity dominates (Fig. 3g and Extended Data Fig. 3 h–l). Moreover, the oscillation frequency depends non-linearly on the concentration of the complex (Extended Data Fig. 3m–o). These types of oscillations in enzyme reactions are unusual even when the assay mix contains two enzymes with opposing functions.
Structure of the GudB-GltAB complex
Native mass spectrometry (native-MS) of the individual proteins provided preliminary information about the stoichiometry of the individual enzymes. As observed previously14,21, GudB forms a hexamer (Fig. 4a). However, unlike its closest homologue with a known structure, Azospirillum GltAB, which is predominantly a hetero-dodecamer25,26, B. subtilis GltAB is a hetero-dimer (Fig. 4a). GltAB’s oligomeric state therefore resembles that of ferredoxin-dependent glutamate synthases found in photosynthetic organisms like plants or cyanobacteria27,28. The spectrum of the GudB-GltAB complex (Fig. 4b) indicated a species with a mass of ~1.6 MDa in the high m/z region, corresponding to six GudB protomers and six GltAB heterodimers (GudB6-GltA6B6).
Figure 4. The stoichiometry, oligomeric state and atomic structure of the GltAB-GudB complex.
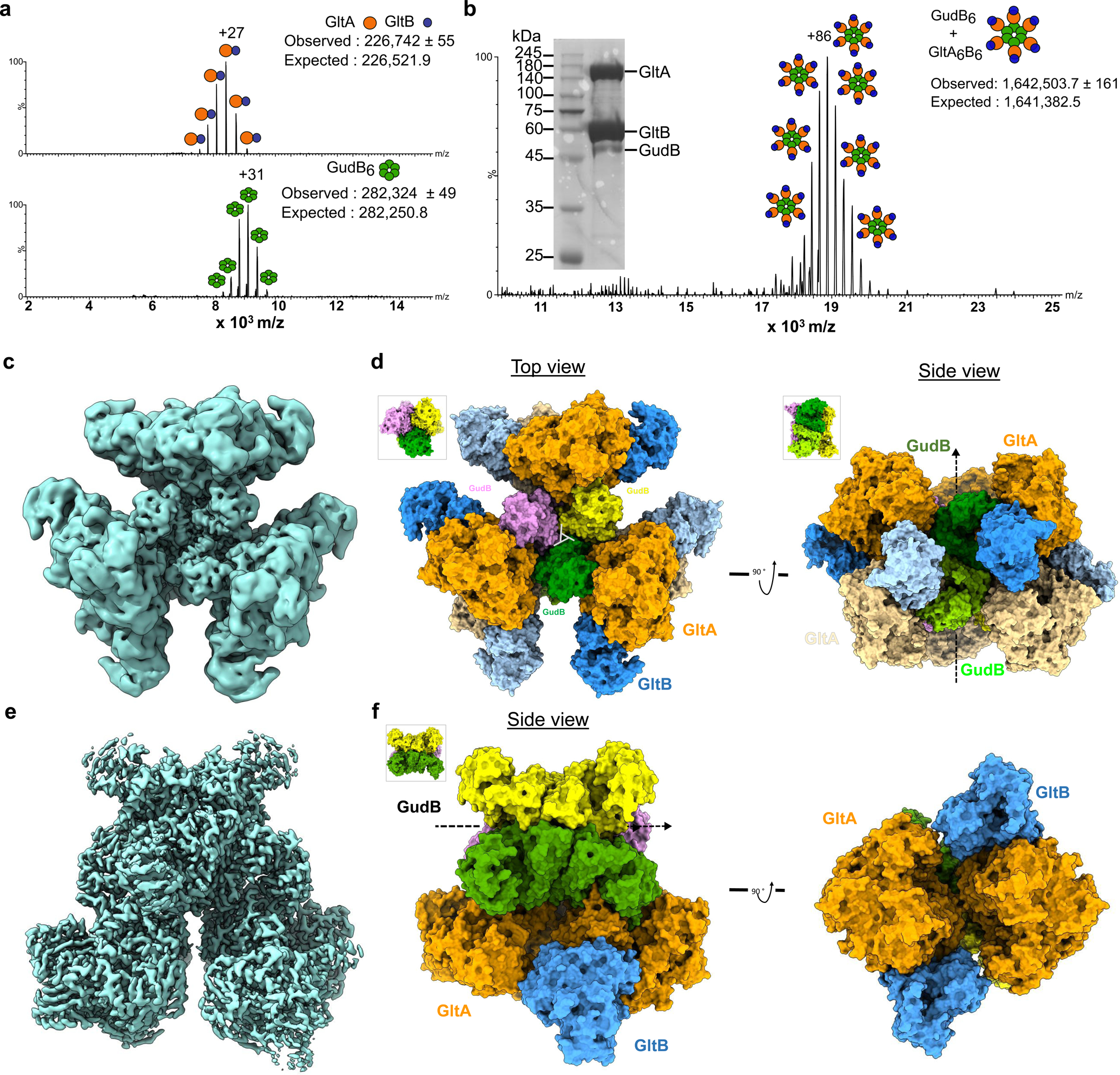
a) Native-MS of standalone GltAB (top) and GudB (bottom). The oligomeric state as inferred from the mass is shown schematically besides each charge state: GltAB’s observed mass corresponds to a heterodimer, while GudB’s mass indicated the expected hexamer. b) Native-MS of the GudB-GltAB complex. The observed mass corresponds to 6 copies of a GltAB heterodimer bound to the GudB hexamer. The low m/z region contained unbound GltAB. a). The inset shows the SDS-PAGE of the protein sample used for native-MS and cryo-EM. The image is from one batch of purification and is representative of at least 4 independent purifications. c) Density map of the GudB6-GltAB6 complex generated after applying D3 symmetry. d) Model of GudB6-GltA6B6 complex. The three dimers of the GudB hexamer are shown in three different colours (pink, yellow and green). GltA and GltB are shown in orange and blue, respectively. GltA and GltB subunits below the plane of the paper is shown in light orange and light blue, respectively. The views (top and side) are with respect to the orientation of GudB as shown in the insets. The dihedral (D3) rotation axis of GudB is depicted as a triangle in the left panel and as a dashed arrow in the right panel. e) The density map of GudB6-GltA2B2. f) On the left is shown the model of GudB6-GltA2B2 (side view) and the right panel shows the model rotated 90° along the dihedral axis of GudB. From the model on the right it is clear that there are minimal interactions between the two GltA copies.
This GudB-GltAB complex is unusually large – almost half the size of the ribosome – and we set out to determine the structure of the complex using CryoEM. We were able to obtain the structure of the fully assembled complex at an overall resolution of 3.9 Å with D3 symmetry (Fig. 4c, Extended Data Fig. 4 and Supplmentary Data Table 5). The structure unambiguously revealed that the complex comprises a GudB hexamer and six GltAB heterodimers (Fig. 4d). Each GltAB heterodimer makes contact with two protomers of GudB, with the interaction occurring solely through GltA. There are also no interactions between the GltAB hetero-dimers in the complex. Although the resolution of the map permitted assignment of the different chains, to reveal the mechanistic basis of GudB silencing by GltA, a map with higher resolution was obtained.
To increase the particle density, excess recombinant GudB was added to B. subtilis cell lysate before purifying the GudB-GltAB complex (inset in Extended Data Fig. 5a). Preliminary cryo-EM single particle analysis resulted in four 3D classes representing varied number (2–4) of GltAB hetero-dimers bound to the GudB hexamer (Extended Data Fig. 5b) in accordance with native-MS showing a range of GudB-GltAB complexes (Extended Data Fig. 5a). The four resulting maps resemble the native complex (GudB6-GltA6B6) but with varying occupancy of the GltAB heterodimers. The most abundant class corresponded to a GudB hexamer with two GltAB heterodimers (GudB6-GltA2B2). A high resolution map was prepared by collapsing these classes into a single class and refining with the preliminary GudB6-GltA2B2 map as the initial reference. This refined to high resolution (2.4 Å) with C2 symmetry (Fig. 4e,f, Extended Data Fig. 6 and Supplmentary Data Table 5). The higher resolution model allowed us to determine the precise contacts used to stabilize GudB in an inactive state (Fig. 4f). There appear to be no substantial architectural differences on GltAB with the components of the structure of the hexameric GltAB from Azospirillum brasiliense (PDB ID: 6s6x)26, but the dimerization of GltA which is a likely prerequisite for hexamer formation is not observed. Among other contacts, the GltA homo-dimer interface includes 13 putative hydrogen bonds. Examination of the corresponding residues in B. subtilis GltA suggested that 10 out of these 13 hydrogen bonds cannot be formed, which is in agreement with B. subtilis GltA not forming homo-dimers. The higher resolution did permit unambiguous assignment of two 4Fe-4S clusters in GltB, and a 3Fe-4S clusters and FMN in GltA. These co-factors play a key role is shuttling electrons from the GltB-bound NADPH to 2-iminoglurate that is formed in the synthase domain of GltA (Extended Data Fig. 7a), thus allowing the reduction of the latter to yield glutamate.
The structural basis of GudB’s inhibition by GltAB
GudB interacts with GltAB extensively through the GltA subunit (Fig. 5a) with few contacts between adjacent GltAB heterodimers (Fig. 4f, right panel). GltA is a multi-domain protein that consists of an Ntn-amidotransferase domain, a central domain, a synthase domain, and the GXGXG domain (Fig. 5b). The interactions with GudB occur primarily with GltA’s central domain that docks onto the active-site cleft of GudB, which separates GudB’s cofactor (NAD+) and substrate (glutamate) binding domains (Fig. 5a). The relative movement of these two domains results in “open” and “closed” conformations that are a key part of the catalytic cycle of glutamate dehydrogenases29. Cleft closure brings NAD+ close to glutamate thereby enabling the hydride transfer, as represented by the substrate bound structure of glutamate dehydrogenase from Aspergillus niger (PDB: 5XVX)30. Based on the distance between residues R280 and K122 as a proxy of the degree of closeness, GltA-bound GudB is in open state (d= 29.1 Å; Extended Data Fig. 7b).
Figure 5. The structural basis for inhibition of GudB by GltA binding.
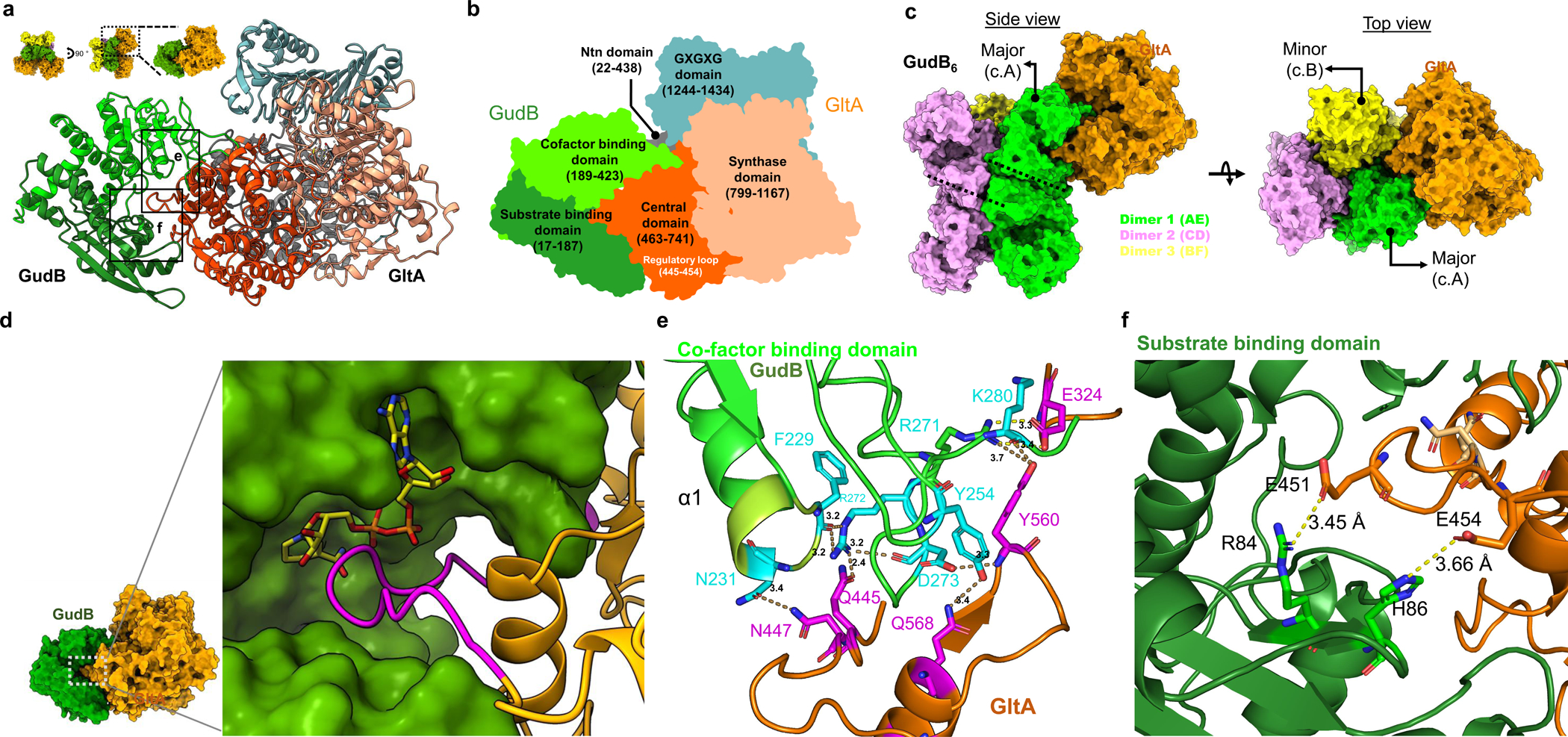
a) Zoom-in model of the GudB-GltA interaction. The colors represent the different domains of GltA and GudB as detailed in the next panel. The inset shows the perspective of the model with respect to the model shown in Fig. 4f. The dashed box (G) corresponds to the region containing residues from GudB and GltA forming a hydrogen bonding network. This is shown in detail in panel G. b) GltA consists of Ntn amidotransferase (Ntn) domain (grey), a central domain (orange), a synthase domain (sand) and a GXGXG domain (metal blue). The central domain of GltA (orange) interacts with residues from both the substrate (green) and co-factor binding domain of GudB (light green). c) GudB’s hexamer comprises a trimer of dimer; GltA (in orange) interacts with two protomers of GudB (major and minor) from two different dimers (green and yellow, respectively). The front view shows the major interacting GudB protamer, and the top view the minor one. The coloring pattern of the three dimers and the corresponding chain ID’s are also shown. The dimer interface in two of the dimers (1 and 2) is shown as dashed lines. d) Zoom-in on GudB active site cleft (surface display, in green) and the interacting loop of GltA (magenta; the remaining structure of GltA is in orange). NAD+ (in yellow sticks) is modelled into the active site cleft of GudB based on superposition of GltA-bound GudB to 1V9L (open state). The steric overlap of GltA’s loop with NAD+ would be even higher in the closed state of the enzyme. e) Zoom-in on the boxed region shown in panel A, depicting the hydrogen bonding network between residues of GltA (magenta) and the co-factor binding domain of GudB (cyan). Notably, GudB’s N231, which is located at the tip of the phosphate binding loop (and binds the NAD+’s phosphate groups) is bound to GltA’s N447, and thus directly interferes with NAD+ binding. f) The interaction of GltA’s E451 and E454 with residues of the substrate binding domain of GudB (R84 and H86).
Another key observation is that each GltA subunit interacts with two adjacent GudB subunits (Fig. 5c), which provides a molecular explanation for how GltA binding stabilizes the GudB hexamer (Extended Data Fig. 7c). The primary interacting subunit (“major”) of GudB, with an intersubunit contact area of GudB:GltA of 1931:1817 (Å2), has its active site blocked as described above. The neighboring subunit (“minor”) interacts with a much smaller surface area (360 Å2), outside the active site (Fig. 5c, top view). However, the paralogue specificity of GltA for GudB is likely explained by the lack of conservation in RocG for residues mediating this secondary “minor” interaction (underlined in Extended Data Fig. 8a, Supplementary Data Table 4) in addition to steric clashes by RocG specific residues like M277 instead of T277 in GudB The key “major” interactions between GudB and GltA involve a long loop, “the GudB regulatory loop” that connects the Ntn- and central-domains of GltA (residues 445–455). This loop extends across the active site cleft of GudB. Residues from the loop interact with both the cofactor- and substrate-binding domains of GudB (Fig. 5d–f). Foremost, a network of hydrogen bonds is observed between GltA residues and the cofactor binding domain of GudB (Fig. 5e), which in turn stabilizes multiple loops in GudB. Notably, clear density for these loops is seen in the complex but not in the apo-GudB crystal structure21 or in the GudB subunits in the complex that are not bound to GltA. The ordering of the cofactor binding domain of GudB by binding to GltA is also reflected in the lower average B-factor of GltA-bound GudB compared to unbound (Extended Data Fig. 8b). The most notable contact is between the tip of α1 and residue N231 of GudB’s NAD+ binding domain, which is typically used by Rossman folds to bind the di-phosphate group of NAD+. Consequently, GltA binding precludes NAD+ binding to GudB. Moreover, as shown in Fig. 5e, GltA’s regulatory loop occupies a specific position in the active site cleft of GudB that is otherwise occupied by the co-factor in the closed (and catalytically active) conformation of the enzyme. Thus, stabilization of the open conformer, and blocking NAD+ binding by steric hindrance are both likely key to GudB inhibition by GltAB. While mass spectrometry studies identified several ligands retained by the complex (Supplementary Table 3), the cryo-EM experiments did not reveal any density consistent with AKG or NADPH in the active site. AKG and NADPH dependence on complete inhibition of GudB could be due to long distance allosteric effect of the binding of these ligands. The relative location of AKG binding site in GltA and NADPH binding site in GltB, with respect to the regulatory loop that interacts with GudB active site cleft is shown in Extended Data Fig. 8c.
GudB-GltAB complex formation is key for the biofilm morphology
Bacillus forms biofilms in its natural environment, and glutamate metabolism plays a key role in their development31–33. Accordingly, in the laboratory, B. subtilis biofilms are usually grown in glutamate rich media containing glycerol as the carbon source (MSGG). However, unlike planktonic growth, biofilm growth on solid agar presents spatiotemporal contraints – cells in the interior depend on the peripheral cells for glutamine, while the peripheral cells depend on the interior cells for ammonia20. Thus, the regulation of the GudB-GltAB complex could be central to this division of labour.
We initially examined the morphology of biofilms that are devoid of either GudB, GltA, or GltB. Unlike for planktonic growth (Fig. 2a–d), for wild-type like biofilm growth and morphology, all three proteins seem to be important (Fig. 6a). ΔgudB produced rapidly growing biofilms (Extended Data Fig. 9a) with large “channels” running from the periphery to the interior. The ΔgltA and ΔgltB strains also showed biofilm morphologies that differ from wild-type – while ΔgltB strain had wrinkles restricted to interior, ΔgltA biofilm did not have any wrinkles. Since the enzymatic function, glutamate synthesis, is lost in the absence of either GltA or GltB, the different phenotypes are in agreement with the additional role for GltA in regulating GudB.
Figure 6. GudB-GltAB interaction is important for biofilm formation.

a) Representative images of biofilms of wild-type and mutant B. subtilis strains on glutamate-glycerol (MSGG) agar medium, monitored over a period of ~7 days. As indicated by their knockouts, both Glt subunits (GltA and B), and GudB, are essential for wild-type like biofilm morphology b) Comparision of biofilm formation by wild-type and mutants expressing GltA under an IPTG-induced promoter, at varying IPTG concentrations (0–100 μM). Wild-type (i-iii); IPTG-induced GltA plus GudB (iv-vi), or RocG (vii-ix); IPTG-induced inactive GltA mutant, GltABC1A plus GudB (x-xii), or with RocG (xiii-xv). Wild-type GltAB and the GltABC1A were expressed from an IPTG-inducible hyperspank promoter, while GudB and RocG were under gudB’s promoter. Wild-type’s biofilms are not affected by the presence of IPTG (i-iii). The strain expressing GltAB from an IPTG-induced promoter forms altered biofilms with no IPTG (iv), wild-type-like biofilms with low concentration of IPTG (v), while overexpression resulted in biofilms similar to those of the ΔgudB strain (vi). Co-expression of RocG and GltAB resulted in biofilms with no wrinkles (vii-ix). Expression of the inactive GltABC1A with GudB restores the morphology of the biofilm’s interior, in an IPTG-dependent manner (x-xii); in contrast, co-expression of GltABC1A with RocG does not (xiii-xv). The scale bars in all images correspond to 4 mm. Magnified sections are shown in the last column of panel b. c-e) A summary of the biofilm disruption phenotypes and a proposed model that accounts for them (see text). The biofilm images correspond to the following images from b: iii (c), xii (d) and xv (e).
Next we examined the role of GudB-GltAB complex formation in biofilm formation. GltAB expression even at low levels (10 μM IPTG) was sufficient to restore biofilm formation to wild-type levels (Fig. 6b (v); wild-type biofilms were not affected by addition of IPTG; Fig. 6b (i-iii)). However, overexpression of GltAB (100 μM IPTG), resulted in ΔgudB like biofilm morphology (Fig. 6b (vi), and Extended Data Fig. 9b for enlarged images) consistent with silencing of GudB by GltAB. Intriguingly, expression of the inactive GltABC1A restored wild-type-like growth, but only in the interior of the biofilm resembling biofilms formed by ΔgltB strain (Fig. 6b (x-xii); Fig. 6c and Extended Data Fig. 9c for enlarged images), suggesting that silencing of GudB by GltAB is crucial for the interior of the biofilm. In contrast, if the GudB silencing occurs in the peripheral cells, the biofilm exhibits no wrinkles in the periphery (compared to wild-type; Fig. 6c). This result is also in agreement with GltAB’s expression being the highest in the interior of the wild-type biofilm34. Indeed, when the above-described wild-type- and inactive- gltAB genes were expressed at the background of RocG, the effects differed completely from those seen at the GudB background (Fig. 6b (vii-ix, xiii-xv)). In the presence of RocG, expression of GltABC1A had no effect and the biofilms completely lacked wrinkles (Fig. 6c). This result is consistent with RocG having the same level of enzymatic activity as GudB, yet not being suppressed by GltAB binding. Overall, these results show besides enzymatically functional GudB and GltAB, regulation of GudB by GudB-GltAB counter-enzyme complex formation is critical to developing natural biofilm features.
Discussion
Our search for the regulatory mechanism of GudB, a core catabolic glutamate dehydrogenase which is constitutively expressed, yielded an unexpected answer. GudB, which is involved in glutamate catabolism, forms a transient complex with its counter-enzyme GltAB, which catalyses glutamate synthesis. The main purpose of this association seems to be silencing of GudB. The high resolution structure of the partially assembled complex clearly showed that a regulatory loop in GltA occludes the active site cleft of GudB and prevents cofactor binding. In addition, the structure revealed the mechanisms behind paralogue specificity.
GudB-GltAB complex presents a good case study for allosteric control of protomers in an oligomeric enzyme. GudB is active only as a hexamer and dimer/monomeric species are inactive14,21. The fact that physical association of GltA is needed for the inactivation of GudB protomer suggests that 6 GltA molecules are needed for the complete silencing of GudB (hexamer) and that in the partial complex, with sub-stoichiometry GltA, the unbound GudB protomers could still be active. It should be noted that GudB by itself exhibits strong negative cooperativity (H= 0.57), and GltA-bound GudB exhibits weak yet reproducible positive cooperativity (H= 1.3) (Figure 3c,d and Supplementary Table 2). This suggests that GltA binding does effect the inter-subunit allosteric communication. Future studies focusing on substrate/product/cofactor bound structure of GudB and GudB-GltAB complex should shed light on the exact mechanism behind the strong negative cooperativity of GudB and modular control of GudB protomers by GltA binding.
A notable feature of the GudB-GltAB complex is the oscillatory progress curve. Oscillatory chemical reactions35 and two-enzyme system has been described previously36–40. However, oscillations are not expected in a counter-enzyme pair. The rates of substrate binding, product release, and catalytic turnover, are typically in the sub-second range, and a steady-state would therefore be established with a constant ‘net’ rate24. Mechanistically speaking, oscillations typically involve a negative feedback loop with a delay24,41–43. The delay is key for “over-shooting” of one reaction before the next one takes over. While the GltA binding to GudB is the negative feedback here, the delay may be produced, for example, by any AKG dependent- slow event like proline cis-trans isomerization in the regulatory loop, global conformational change, domain motion or or by cycles of dis-assembly and slow re-assembly of the complex.The latter would result in GudB over-producing AKG due to a delay in GltAB’s binding. That the oscillation frequency depends on the complex’s concentration, and in a non-linear way (Extended Data Fig. 3m–o) hints at such a mechanism. The GudB-GltAB oscillatory kinetics and the regulatory mode may also have some intriguing physiological implications in biofilms. Future work may therefore enable a deeper understanding of the GltAB-GudB complex, and also of the potential correlation between GudB’s regulation and biofilm oscillations.
It is shown earlier that GudB /RocG can directly bind GltC (the transcriptional regulator of the GltAB operon) and repress GltAB expression47. This moonlighting function of GudB is undesirable in glutamate poor condition. It is therefore tempting to speculate that GudB-GltAB counter-enzyme complex formation is a fail-safe mechanism whereby, besides inhibiting the catalytic activity of GudB, it also sequesters GudB and prevent its interaction with GltC. The only other counter-enzyme complex reported previously is arginase-ornithine transcarbamylase (OTCase) complex where it was shown that the catabolic arginase inhibits the anabolic OTCase45,46. This is opposite of what we see in GudB-GltAB complex where the catabolic enzyme (GudB) is inhibited by the anabolic GltAB. Most biochemical transformations are in principle reversible. However, in vivo, many of these are effectively unidirectional due to thermodynamic and kinetic constraints. In addition, many enzymes evolved towards unidirectional catalysis, including oxidoreductases that utilize NAD+ to drive the reaction in the oxidative direction (e.g. GudB) or NADPH to direct reduction (e.g. GltAB). Consequently, cells contain multiple counter-enzymes that catalyze opposite transformations (note that the reactions themselves usually differ, as is the case with GltAB and GudB, but the primary substrate/product are the same as shown in Fig. 1a). It yields to reason that counter-enzymes would be individually regulated, as their simultaneous operation could result in futile cycles (zero net outcome and waste of energy). However, regulation via counter-enzyme pairs offers unique properties that cannot be obtained by independent regulation of the individual enzymes, such as ultrasensitivity, robustness, and temporal signaling pulses24. The opposing enzymes can be separate proteins, or fused on the same polypeptide chain (bifunctional enzymes)24,47. The GudB-GltAB example unraveled here suggests that counter-enzyme pairs could be hotspots for regulatory features and warrants a systematic investigation of different counter-enzyme pairs in core metabolism.
Methods
DNA manipulation:
All constructs used for genome modification were generated using Gibson assembly. The starting fragments were generated using Q5® High-Fidelity DNA Polymerase (New England Biolabs) and NEBuilder® HiFi DNA Assembly Master Mix (New England Biolabs) was used for assembly. The linear DNA fragments for B. subtilis transformation were generated by digestion of the plasmids (using a single restriction enzyme) or by PCR using the Gibson assembly reaction as template. The fragments had recombination homology arms ranging anywhere from 1.5 to 4 kb. All the strains generated and used in the study are listed in Supplementary Table 6 and all the oligonucleotides used for the study are listed in Supplementary Table 7. Genetic modification of B. subtilis strains were performed based on natural competence: The parental strains were streaked on an LB-agar plate and grown overnight at 37 °C. A singly colony was then used to inoculate 1 ml of modified MC medium48 containing 0.5% glucose, 1.4% K2HPO4, 0.6 % KH2PO4, 30 nM sodium citrate tribasic dehydrate, 0.2 % casein hydrolysate, 84 mM ammonium iron (III) citrate, and 3 μM MgSO4. Addition of tryptophan (0.005%) and histidine (0.005%), improved the transformation efficiency. The culture was incubated at 37 °C in a roller drum shaker for 3.5 hrs. Linear DNA/ genomic DNA (100 ng −1 μg) was added to 300 μl of this culture and growth was continued for another 3 hrs. The positive transformants were selected either on an antibiotic, or in nutrient selection media (for wild-type GltAB constructs). All transformants were verified by diagnostic PCR’s followed by DNA sequencing.
Media and growth conditions:
B. subtilis strains were grown in LB, or in MS medium (5 mM potassium phosphate, 100 mM MOPS pH 7.1, 2 mM MgCl2, 700 μM CaCl2, 50 μM MnCl2, 50 μM FeCl3, 1 μM ZnCl2, 2 μM thiamine; adapted from49. The medium was supplemented with appropriate carbon/nitrogen source (glutamate, histidine, arginine, proline) or with carbon source (glucose, glycerol) plus ammonium sulfate, all at a final concentration of 5 g/L. Media were supplemented with antibiotics when required: kanamycin (10 μg/ml), tetracycline (5 μg/ml), spectinomycin (100 μg/ml) or a mix of lincomycin (25 μg/ml) plus erythromycin (2 μg/ml). Growth profiling of B. subtilis strains in liquid MS medium was performed in a 96 well plates and absorbance at OD 600 was measured using a multi-well plate reader (Eon, Biotek Gen5). Typically, 3 μl of culture with an OD 600 of 0.8–1.2 was used as a starter to inoculate a well containing 200 μl fresh medium. Replicates were performed parallely in the same plate.
For biofilm growth, the MS media (supplemented with appropriate C/N source) was solidified using 1.5 % agar (w/v). The plates were then dried for 6–8 hrs in a laminar hood. For biofilm growth, cells stocks were streaked on to an LB agar plate and incubated overnight at 37 °C. From the confluent part of the plate, cells were swiped using a sterile loop to inoculate a 5 ml LB medium, then grown at 37 °C shaker for 2–3 hrs (OD.600 of 0.8–1.2). Following this the cells were washed twice with 1 X PBS. The cultures were normalized based on OD 600 and a 3 μl aliquot of a 0.6–1.0 OD600 culture was placed at the center of the plate for biofilm growth, and the plates were incubated at 30 °C. Photographs of the biofilms were acquired using Stereo Discovery V20 microscope with Objective Plan Apo S 0.5× FWD 134 mm or Apo S 1.0× FWD 60 mm (Zeiss) attached to an Axiocam camera. Data were captured and analyzed using ZENpro AxioVision suite software (Zeiss, Oberkochenm, Germany).
Immunoprecipitation and mass spectrometry:
Anti-GudB antibodies were generated in rabbits using recombinant GudB purified from E. coli as antigen. The IgG fraction from these polyclonal sera was purified using a Protein A column. The purified antibodies were covalently linked to CNBr beads50 and this resin was used for the pulldown of GudB from B. subtilis cell lysates. Briefly, wild-type B. subtilis (NCIB 3610) was grown in 500 ml MS medium supplemented with either glucose-ammonia or histidine. Cells were harvested by centrifugation (6100 rcf) and resuspended in 30 ml of lysis buffer (100 mM Tris. HCl, 150 mM NaCl, EDTA-free protease cocktail inhibitor (1:200, Abcam), 2 mM DTT). The cells were lysed by passing cell suspensions 3–4 times through a french press at a pressure of 15 psi. The debri was removed by centrifugation (11,648 rcf) and the resulting supernatant was mixed with the anti-GudB antibodies resin and incubated for 1 hr. After rinsing the beads, the bound proteins were eluted using a low-pH glycine buffer (100 mM, pH 2.8). Eluates were neutralized by collecting them directly into 2 M Tris buffer pH 8.5, pooled, and lyophilized. The eluted proteins were resolved on SDS-PAGE and stained with silver nitrate using established protocols51.
For MS-based identification of proteins, the dried eluates were resuspended in 5% SDS in 50 mM Tris pH 7.4 and subjected to tryptic digestion using an S-trap52. The resulting peptides were analyzed using nanoflow liquid chromatography (Acquity M-class) coupled to high resolution, high mass accuracy mass spectrometry (Q Exactive HF). Each sample was analyzed on the instrument separately, in a random order, discovery mode. Raw data were processed with MaxQuant v1.6.0.16, and searched with the Andromeda search engine against the B. subtilis proteome database appended with common lab protein contaminants, with the following modifications: Carbamidomethylation of cysteine as a fixed modification and oxidation of methionine as a variable one. The LFQ (Label-Free Quantification) intensities were calculated and used for further calculations using Perseus v1.6.0.7. Decoy hits were filtered out, as well as proteins that were identified on the basis of a modified peptide only.
Protein expression and purification:
Recombinant GudB and RocG were purified as described14. Briefly, the plasmids pET28_Strep_GudB and pET28_Strep_RocG encoding B. subtilis GudB and RocG respectively, were used for expression in the E. coli strain BL21 star/DE3 (pGRO7). Typically, the cells were grown in 1 L of terrific broth supplemented with kanamycin (50 μg/ml) and chloramphenicol (34 μg/ml) to an OD600 of 0.6–0.8 and protein expression was induced by the addition of 100 μM IPTG. Post-induction, the cultures were grown overnight at 20 °C with shaking (200 rpm). Cells were harvested by centrifugation (6100 rcf), resuspended in lysis buffer (100 mM Tris. HCl, 150 mM sodium chloride, protease cocktail inhibitor) and lysed using french press at 15 psi. The clarified cell lysate was applied on to a Strep-Tactin column, unbound proteins were removed by washing with 5 CV of lysis buffer, and the target protein was eluted using 10 mM desthiobiotin in lysis buffer. The eluted protein was then dialyzed overnight in storage buffer (50 mM HEPES pH 7.9); aliquots were flash frozen in liquid nitrogen and stored at −80 °C freezer for later use.
The GudB-GltAB complex was purified directly from B. subtilis strain in which GltB is expressed as a Twin-strep tagged protein (GltB-TS). For the purification of GltAB alone without GudB, the ΔgudB version of the strain expressing GltB-TS was used. A higher yield of the complex was obtained upon using strains containing gltAB genes under IPTG inducible-hyper spank promoter. GltAB contains Fe-S clusters that are essential for its activity; to prevent cluster oxidation, all buffers used for the purification were degassed first by applying vacuum and then purging the solution with nitrogen for ~40 min. additionally, the buffer was supplemented with 5 mM DTT to maintain reducing conditions. For purification, the relevant B. subtilis strain was grown in 1–2 L MS medium containing glucose-ammonia to an OD600 of 1.5–2. The cells were harvested by centrifugation, resuspended in lysis buffer (100 mM Tris. HCl, 150 mM sodium chloride, protease cocktail inhibitor, 2 mM DTT) and lysed using a French press. The clarified cell lysate was either directly applied on to a Strep-Tactin column or in case of GudB enriched sample, the lysate was incubated with lysate from the E. coli overexpressing 6X His-tagged GudB. The column was washed with 5 CV of lysis buffer and the bound proteins were eluted using lysis buffer containing 10 mM of desthiobiotin. Elution aliquots were immediately flash frozen and stored in −80 °C. Quantification of the eluates were performed using Bradford assay (Bio-Rad). The concentration of individual proteins in the complex was determined by densitometric analysis on ImageJ253.
Paralogue specific binding of GltAB was tested by pulldown of GltB-TS from a strain expressing either RocG or GudB. Cell lysates from the respective strains were applied onto a Strep-Tactin column, unbound proteins washed, and the bound proteins eluted using 10 mM desthiobiotin in lysis buffer. The proteins in the eluate were separated on an SDS-PAGE and Coomassie stained. For the reciprocal pulldown, purified recombinant 6XHis RocG, or GudB, were added to B. subtilis cell lysates containing GltAB-TS. Bound proteins were was pulled down as above. Interactions were also tested in vitro, by adding either recombinant 6X His tagged GudB/RocG to B. subtilis cell lysates containing GltAB-TS. The lysate was applied on to a Ni-NTA column and the column was washed with 10 CV of wash buffer. Proteins bound to the column were eluted using 500 mM imidazole in lysis buffer. The eluates were resolved on SDS-PAGE and the gel was visualized using Coomassie staining.
Crosslinking and western blotting:
Disuccinimidyl glutarate was synthesized using established protocols. Briefly, glutaric acid (2.0 g, 1.0 eq) and N-hydroxy succinimide (3.8 g, 2.2 eq) were added to an oven dried round bottom flask containing 30 ml of dimethylformamide (DMF) and kept on ice. Then, EDC.HCl (6.4 g, 2.2 eq) was added to the flask one ice, and the reaction was subsequently stirred at room temperature overnight. The reaction mixture was then poured onto an ice-cold solution of 1 N HCl (~10 volumes) and stirred for few minutes. The resultant solution was filtered and the precipitate was rinsed twice with ice-cold isopropanol. The obtained solids were dried under vacuum and further lyophilized to remove traces of water. The lyophilized powder was stored at −20 °C. For crosslinking, wild-type B. subtilis strain NCIB 3610 was grown in 10 ml of MS-medium supplemented with appropriate C/N source to an OD.600 of 1–1.5. Cells were harvested by centrifugation and washed with 1 X PBS. The cells were resuspended in 500 μl of 1 X PBS. DSG was freshly dissolving in anhydrous DMSO to 250 mM, and added to the cell suspension to a final concentration of 0.5 – 2 mM. The cells were incubated in a tumbler, at room temperature, for 30 min unless otherwise specified. The crosslinking reaction was stopped by the addition of 2 M Tris pH 8.5 (to a final concentration of 150 mM) and incubated for another 15 min. The cells were harvested by centrifugation and the cell pellets stored at −20 °C.
For western blotting, the pellets of DSG treated/untreated cells were resuspended in 1 X SDS-PAGE loading dye and heated at 95 °C for 15 min. The cell debris was removed by centrifugation and the supernatant was analyzed on 8 or 12 % SDS-PAGE gel. The loading was normalized based on OD600 of the culture before crosslinking. The resolved proteins were transferred onto a PVDF membrane and blocked with 5 % skim milk solution in 1 X PBS at room temperature for 1 hr. Anti-GudB antibody (Protein A purified) was added (1: 50,000) to the blocking solution and the membrane was incubated for 1 hr. Following this, the membrane was subjected to 3 wash cycles, 5 min each, in washing buffer containing 0.01 % Tween (in 1 X PBS). Following the washes, HRP-conjugated anti-rabbit IgG (Jackson ImmunoResearch, 111-035-144), was added (1: 50000) and incubated with shaking for 1 hr. The blot was washed again and the protein bands were visualized using the ECL system and images acquired in Amersham imager 680, General electric company.
Enzyme kinetics:
Glutamate dehydrogenase and glutamate synthase activities were monitored by a change in absorbance at 340 nm. While the dehydrogenase reaction involves NAD+ reduction and hence increase in absorbance as the reaction proceeds, the synthetase reaction involves NADPH oxidation and decrease in absorbance. All kinetic assays were performed in multiwell plates suitable for measuring in UV range (Microplate UV/VIS 96F, Eppendorf). Reactions were performed in 50 mM HEPES buffer pH 7.9, at total volume of 200 μl, and were initiated by enzyme addition (5 μg of recombinant GudB and 2.5 μg of GudB-GltAB complex). Steady-state kinetic parameters for the enzymes were derived from initial velocity measurements where one substrate was titrated keeping all other substrates saturating. For GudB, the saturating concentration for glutamate was 200 mM, and for NAD+, 4 mM. For GltAB, the saturating concentrations of glutamine was 100 mM, AKG, 2 mM, and NADPH 200 μM. In addition to these substrates, all reactions involving GltAB included 5 mM MgSO4 and 5 mM DTT. The data were fitted to Michaelis-Menten or to an allosteric sigmoidal model in GraphPad Prism (9.1.0).
Native mass spectrometry:
All native-MS measurements were preformed using Q-Exactive UHMR instrument (Thermo Fisher Scientific, Bremen, Germany). Prior to the analysis, samples were buffer exchanged into 150 mM of ammonium acetate pH 8 using Bio-Spin P6 columns (BioRad) and the final protein concentrations were adjust to 1–5 μM. The following parameters were used for MS1 experiment: capillary voltage 1.1 k, desolation voltage −100 V, source fragmentation 0 V, HCD energy 50 V, trapping pressure 7, corresponding to HV pressure of 2.04 × 10−4 mbar. All spectra were recorded at resolution of 6250 at 800 m/z and analyzed using Xcalibur and, Masslynx 4.2 (Waters) data analysis software. Spectra are shown without any smoothing, and the instrument were externally mass-calibrated using a 2 mg/mL cesium iodide solution.
Cryo-EM sample preparation:
Both GudB6-GltA2B2 and GudB6-GltA6B6 protein samples were prepared at a protein concentration of 0.5 mg/ml and applied to glow discharged R 2/2, 300 mesh, grids with an additional thin layer (~2 nm) of continuous carbon (Quantifoil). The GudB6-GltA2B2 complex solution was applied to the grid in a single 2.5 μl drop, followed by 30 sec incubation before blotting. The GudB6-GltA6B6 sample was applied in two consecutive drops of 6 and 3 μl in order to increase complex concentration on the grid. The first drop was blotted manually, followed by application of the second drop and automated blotting in the plunger. Both samples were plunge frozen in liquid ethane cooled by liquid nitrogen using a Vitrobot plunger (Thermo Fisher Scientific). The plunger was set to 3 sec blotting time and 100% humidity.
Cryo-EM data collection:
Data collection and statistics are summarized in Supplementary Table 5 and Extended Data Figures 4 and 6. Cryo-EM data were collected on a Titan Krios G3i transmission electron microscope (Thermo Fisher Scientific) operated at 300 kV. Movies were recorded on a K3 direct detector (Gatan) installed behind a BioQuantum energy filter (Gatan), using a slit of 15 eV. Movies were recorded in counting mode at a nominal magnification of 105,000x, corresponding to a physical pixel size of 0.82 Å. The GudB6-GltA6B6 data set was collected at a dose rate of 21.4 e−/pixel/sec and total exposure time of 1.5 sec, resulting in an accumulated dose of 47.7 e−/Å2. The GudB6-GltA2B2 data set was collected at a dose rate of 20.5 e−/pixel/sec and total exposure time of 2 sec, resulting in an accumulated dose of 61 e−/Å2. Each movie was split into 60 frames, and the nominal defocus range was −0.8 to −2 μm. Each movie was split into 45 frames, and the nominal defocus range was −0.7 to −1.8 μm. The microscope was optically aligned for fringe-free illumination54, enabling to reduce beam diameter to 0.7 μm on the sample. Imaging was done using an automated low dose procedure implemented in SerialEM55, in which image shift was used to collect multiple images within a single hole. The beam tilt was adjusted to achieve coma-free alignment when applying image shift.
Cryo-EM image processing and model building:
The GudB6-GltAB2 Coulomb potential density map was reconstructed in cisTEM (1.0.0-beta) using dose-weighted micrographs. These were prepared by binning the image stacks by a factor of 2, correcting for beam-induced motion, and dose-weighting with MotionCor2 (1.3.0). CTF estimation was performed using CTFFIND4 as part of the cisTEM suite. Micrographs with crystalline ice and poor CTF fits were discarded. Particles were picked in cisTEM using a featureless blob as a template, then were extracted and classified in 2D. All non-ice classes were carried forward to a two-class 3D auto refinement, using an initial reference produced by a cryoSPARC ab initio and the default starting resolution of 20 Å. The auto refinement yielded one “noise” class containing poorly aligned particles and one class containing the GudB6-GltAB2 complex. Particles from this class were carried forward in a single-class 3D auto refinement, followed by manual refinement with C2 symmetry and per-particle CTF correction. To prevent over-refinement, a final high-resolution limit of 3.14 Å was used. The final cisTEM map was auto-sharpened in PHENIX (1.19).
Image processing of the GudB6-GltA6B6 data set was performed using CryoSPARC (3.1.0)56. The processing scheme is outlined in Extended Data Fig. 4. Movies were subjected to patch motion correction, followed by patch CTF estimation. Micrographs with crystalline ice or poor CTF fits were discarded. Particles were initially picked manually from a subset of micrographs. Extracted particles were iteratively classified in 2D and their class averages used as templates for automated particle picking from all selected micrographs. The latter procedure was repeated twice. Particles from well-resolved 2D classes were used for ab initio 3D reconstruction and classification, yielding four “noise” 3D classes and one class containing the GudB6-GltA6B6 complex. Particles from this class were carried forward in a 3D non-uniform refinement with imposed D3 symmetry, followed by an additional round of 3D classification to further exclude “noise” particles. The data set was then subjected to local motion correction, per-particle defocus estimation and non-uniform refinement with D3 symmetry. In order to account for movements of asymmetric units within the complex that break the D3 symmetry, we used the ”symmetry expansion” job in CryoSPARC, in which asymmetric units are treated as single particles and rotated in 2D to one of the symmetry-related positions. The newly generated data set contains the asymmetric units of all the complexes and is therefore six times larger. Symmetry expansion was followed by 3D local refinement of the newly generated data set with a binary mask imposed on a single GudB1-GltA1B1 unit and with C1 symmetry. The final map was sharpened in CryoSPARC with a B-factor of −127 before atomic model building.
The initial model for GudB was extracted from PDB 3K8Z, but modified to re-introduce the Q71 residue which appears to be missing in GudB’s published structure. Initial models for GltA and GltB were prepared by homology modeling (SWISS-MODEL) using PDB 1OFD, chain A, as the template for GltA and PDB 6S6T, chain G, as the template for GltB. These models were aligned with the Coulomb potential density map for GudB6-GltA2B2 using rigid body docking (UCSF Chimera 1.14) then refined by performing iterative rounds of manipulation in ISOLDE (ChimeraX 1.1.1, ISOLDE 1.1.0), coot (0.9.3), and phenix.real_space_refine (1.19). The final round of refinement was with phenix.real_space_refine (1.19). From the GudB6-GltA2B2 model, one set of monomers was extracted and refined into the C1 map for the GudB6-GltA6B6 dataset described in the previous paragraph. Refinement similarly involved a round of ISOLDE (ChimeraX 1.1.1, ISOLDE 1.1.0), coot (0.9.3), and phenix.real_space_refine (1.19). To produce the biological assembly, the GudB1-GltA1B1 model was expanded into the full D3 map by applying non-crystallographic symmetry and rigid body docking. The models were inspected in coot (0.9.4.1), ChimeraX (1.1.1) and pyMol (2.4.1). 3D visualization was performed using UCSF Chimera (1.14).
Mass photometry:
The mass photometry data was acquired using a OneMP mass photometer (Refeyn Ltd, Oxford, UK). The procedure used was based on previous published protocols57. Briefly, washed and dried gaskets (with chambers for holding the protein solution) was assembled on a clean coverslip. 16 μl of 50 mM HEPES buffer was loaded in one of the chamber and this was used to find focus. To this 4 μl of protein solution (calibrant/test protein) was added and the movie recorded for a period of 120 sec. A 50 nM solution of urease was used for mass calibration. GltAB alone was used at a final concentration of 50 nM or mixed with AKG and NADPH at a final concetration of 5 mM and 200 μM, respectively. The movies were processed and analyzed using DiscoverMP.
Metabolite extraction and analysis:
Lyophilized protein pellets were mixed with 100 μL of Methanol : DDW, 1:1 (v/v). Samples were vortexed for 10 min and sonicated for 15 min. Then the samples were centrifuged twice (at maximum speed) to remove possible precipitants and were injected into LC-MS system. The metabolite analysis was performed using Acquity I class UPLC System combined with a mass spectrometer (Thermo Exactive Plus Orbitrap), which was operated in a negative ionization mode. The LC separation was performed using the SeQuant Zic pHilic (150 mm×2.1 mm) with the SeQuant guard column (20 mm×2.1 mm) (Merck). The Mobile phase B: acetonitrile and Mobile phase A: 20 mM ammonium carbonate with 0.1% ammonia hydroxide in water: acetonitrile (80:20, v/v). The flow rate was maintained at 200 μl/min, and the gradient was: 0–2 min 75% of B, 17 min 12.5% of B, 17.1 min 25% of B, 19 min 25% of B, 19.1 min 75% of B, 23 min 75% of B. The data processing was done using Thermo Scientific Xcalibur v4.1.31.9 software. Compounds were identified by accurate mass, retention time, isotope pattern, fragments and verified using in-house mass spectra library.
Extended Data
Extended Data Fig. 1. GudB interacts with GltAB.
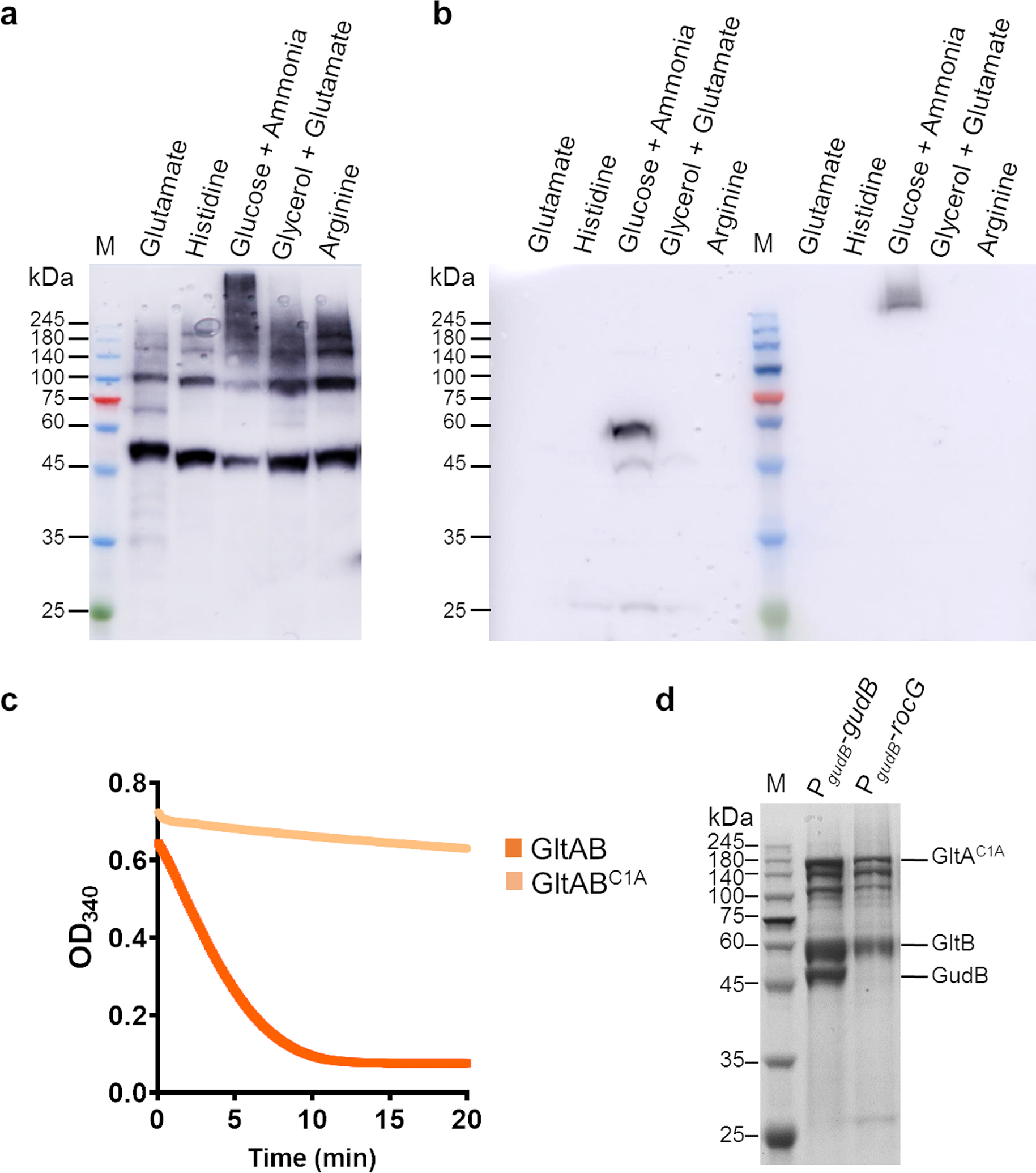
(a) Western blot analysis using an anti-GudB antibody on lysates prepared from DSG treated B. subtilis cells grown on different C/N source. The high molecular weight species of GudB are seen only in cells grown in glucose-ammonia.
(b) Western blot analysis using a Strep-Tactin-HRP antibody on the same samples used in A. Here the antibody was used on lysates prepared from both DSG-treated (lanes right to the molecular weight marker) and untreated cells (lanes left to the marker). As, can been seen, GltB is expressed only in cells grown on glucose-ammonia, and under this condition, it is also part of a high-MW complex (in the DSG treated sample) (Figure 1b).
(c) Enzyme kinetics of wild-type GltAB (orange) and its mutant GltABC1A (light orange), show that the mutant is completely inactive. A minor decrease in absorbance with mutant GltA is due to non-enzymatic oxidation of NADPH. The reaction mixture consisted of 2 mM AKG, 5 mM glutamine, 200 μM NADPH, 5 mM DTT, 5 mM MgSO4. The reaction was initiated with 2.5 μg of the wild-type or mutant enzyme.
(d) SDS-PAGE of the eluate from Strep-Tactin column to which lysate from B. subtilis strain expressing GltABC1A in the background of constitutively expressed GudB or RocG is applied. Co-elution of GudB along with the inactive GltA (GltABC1A) upon pulldown of Strep-GltB (PgudB-gudB lane), while GltABC1A elutes on its own when expressed with RocG (PgudB-rocG lane). Images in a-d are obtained from a single experiment and are a representative of at least 2 independent experiments.
Extended Data Fig. 2. Phenotyping of Phs-gltAB strain.
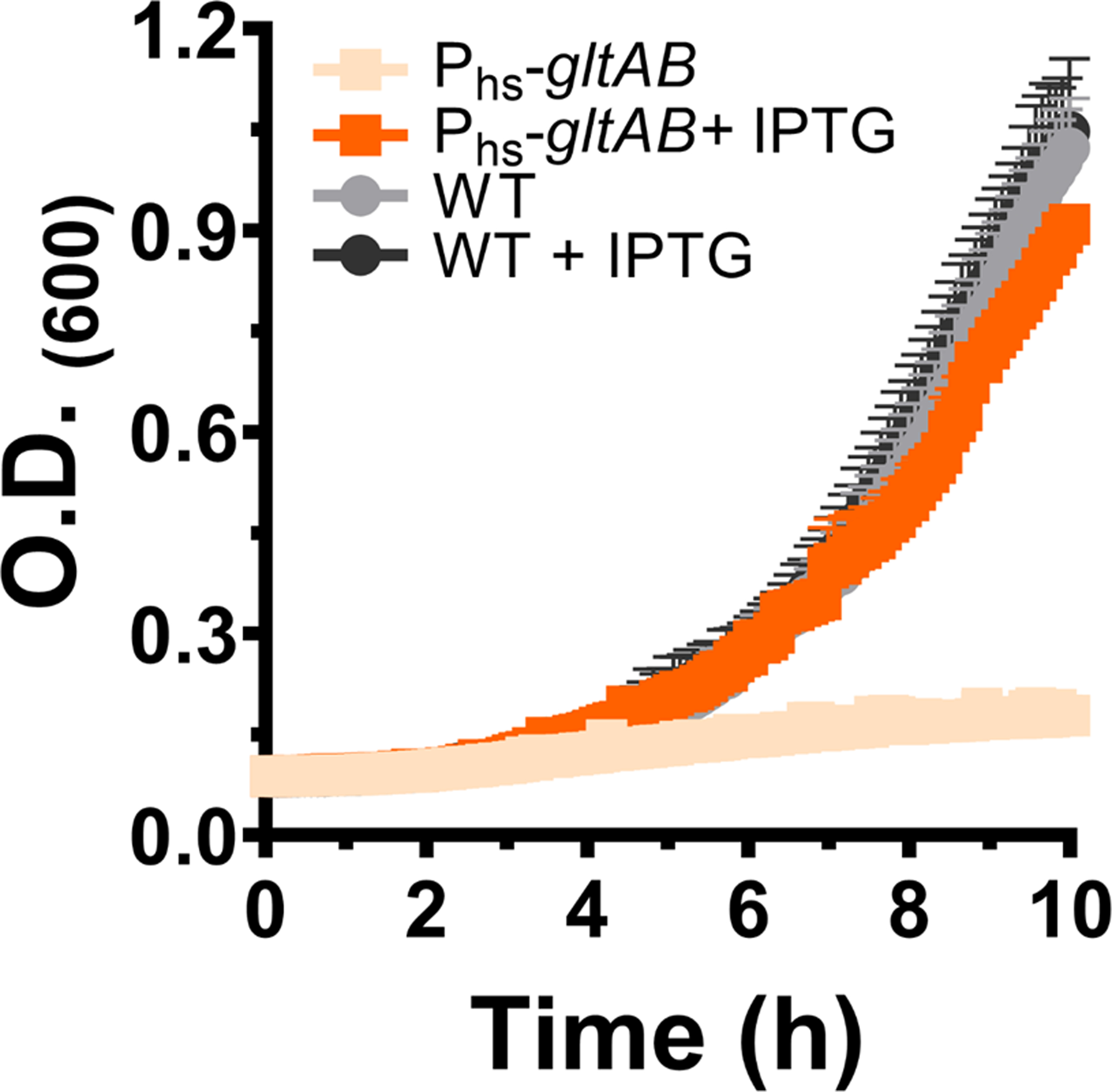
The strain expresses GltAB from the IPTG- inducible hyperspank (Phs) promoter. Without IPTG, the strain cannot grow in minimal medium containing glucose-ammonia as the C/N source (light orange); however, addition of 500 μM IPTG restores growth to almost wild-type levels (orange). Addition of IPTG does not have any effect on the growth of the parental wild-type strain. n=3 are three independent measurements. Data is presented as mean of all measurements and error bars represent SD.
Extended Data Fig. 3. Steady-state kinetics of GltAB, and GudB-GltAB complex.
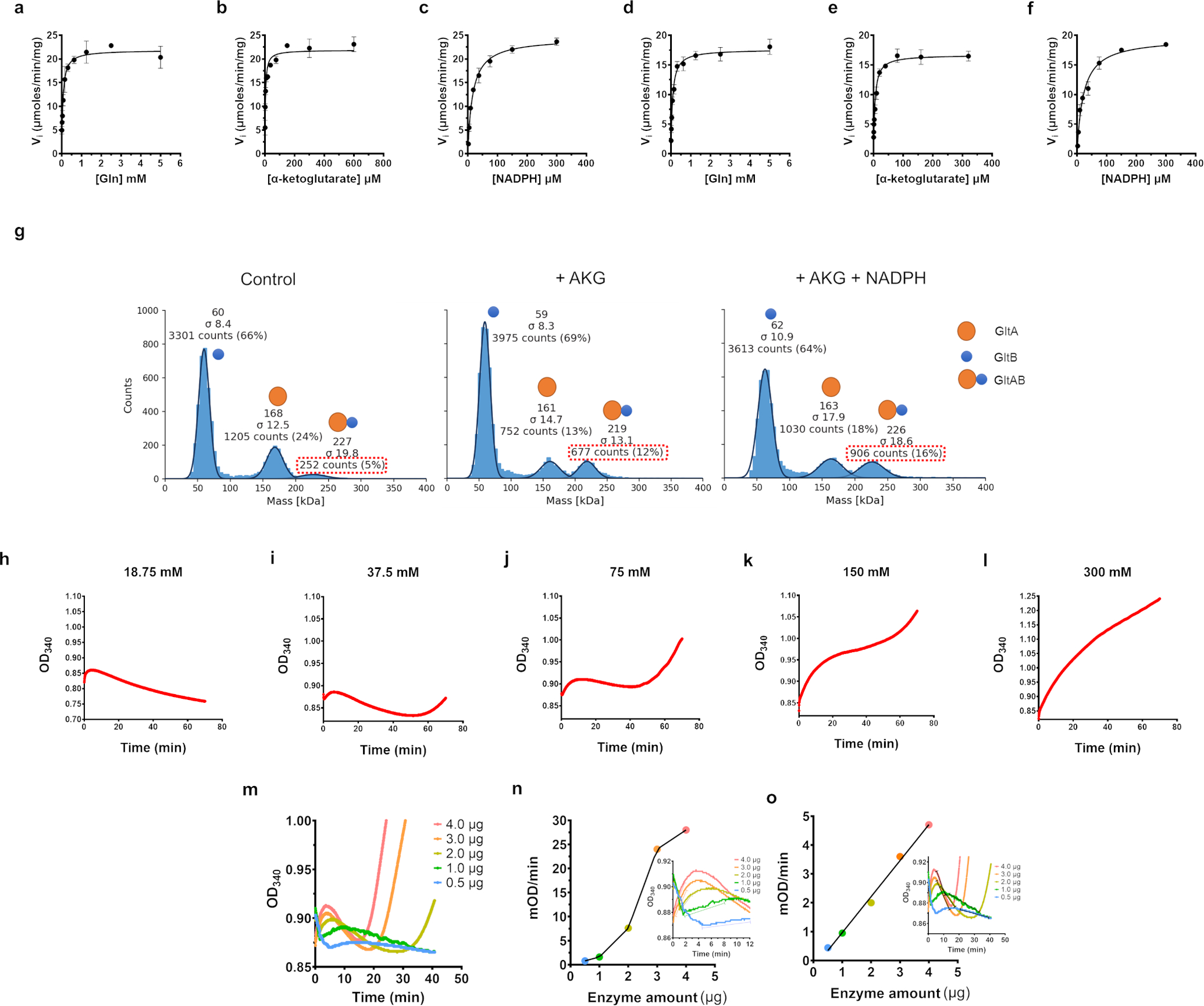
(a-f) Substrate titration plots for the glutamate synthase activity of GltAB by itself (A-C) and in the GudB bound form (D-F). One of the substrates was titrated while keeping the rest saturating: glutamine (5 mM), AKG (α-ketoglutarate, 2 mM), NADPH (200 μM). n=2 are two independent experiments. Data is presented as mean and error bars indicate SD.
(g) AKG and NADPH promote the assembly of GltA and GltB. Only 5 % of the counts corresponded to GltAB when no ligands were added (left panel). Addition of AKG increase the GltAB count to 12 % (Middle panel) and the presence of NADPH in addition to AKG increased the GltAB counts to 16 % (right panel).
(h-l) Non-hyperbolic progress curves displayed by GudB-GltAB complex in the presence of all the substrates except AKG. While at high glutamate concentrations GudB’s activity dominates (K), at low glutamate concentration the synthase activity takes over (G). At intermediate concentration of glutamate (H- J) the progress curve oscillates between GudB- and GltAB-predominant phases.
(m) Progress curves obtained using different amount of the GudB-GltAB complex at 37.5 mM glutamate (where oscillations are most pronounced; panel i). The reaction mixture contained all substrates for GudB and GltAB except AKG.
(n) Plotted are the slopes derived from phase 2 of the progress curves (GudB’s activity as shown in the inset) as a function of the complex concentration. The non-linear relationship suggest that association-dissociation of the complex plays a role in turning off-on GudB’s activity. Note also the elapsed time for phase 2 (the 2nd GudB activity phase) that becomes longer as the complex concentration decreases. The inset shows the phase 3 in each of the individual progress curves.
(o) Unlike phase 2, phase 3 that corresponds to GltAB’s activity shows linear dependence with enzyme concentration. The inset shows phase 3 in each of the individual progress curves. Data in panels g-o are from single experiment and are representative of at least 2 independent experiments.
Extended Data Fig. 4. Cyro-EM image processing for GudB6-GltA6B6.
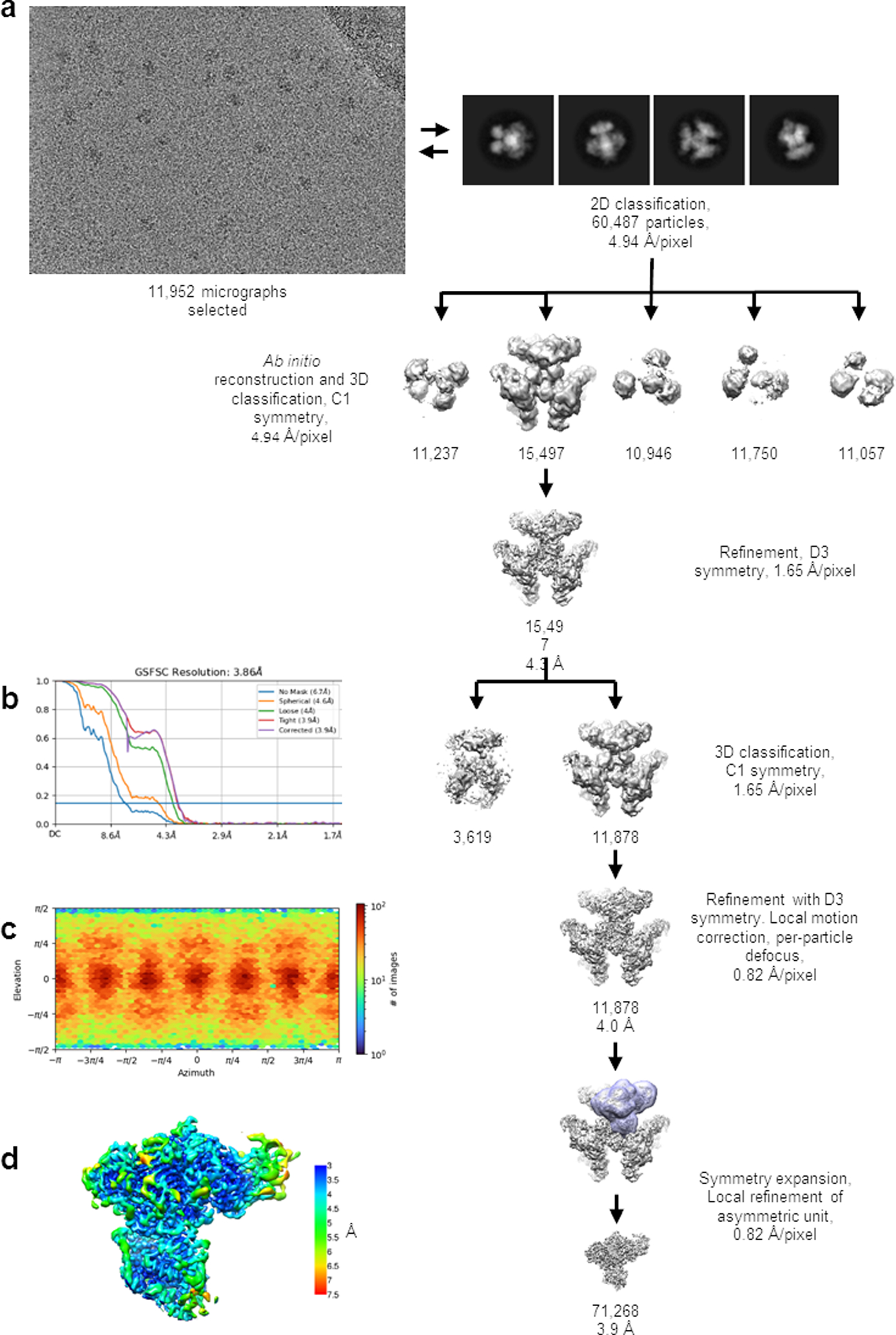
(a) Scheme for single particle cryo-EM analysis of the GudB6-GltA6B6. Details of the process are described in the Methods section. Briefly, particles were iteratively picked from selected micrographs using well resolved 2D class averages, followed by Ab initio 3D reconstruction and classification into five classes. The best resolved 3D class was refined with D3 symmetry imposed. In order to account for deviations from D3 symmetry, further refinement focused on single GudB-GltAB asymmetric units. The number of particles that are included in the maps are indicated, along with the estimated resolution where relevant.
(b) FSC curves for the refined GudB-GltAB asymmetric map.
(c) Angular distribution plot.
(d) 3D map colored according to local resolution estimate.
Extended Data Fig. 5. Native-MS and the corresponding particle types observed in cryo-EM of the GudB enriched preparation of the GudB-GltAB complex.
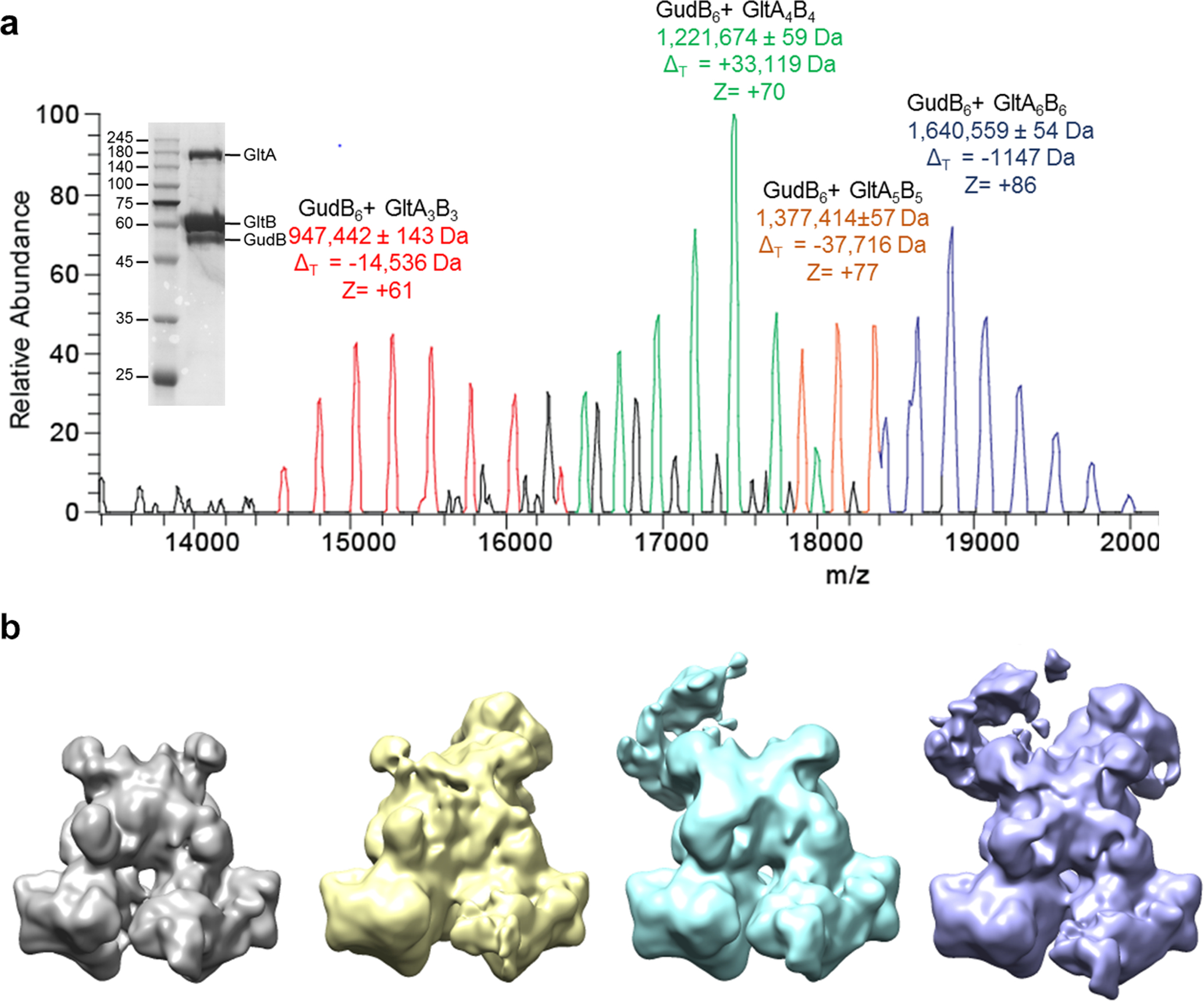
(a) Native-MS spectra showing different species of GudB-GltAB complex. These species primarily differ in the number of GltAB heterodimers attached to the GudB hexamer (from 3–6). Charge states (z) and the difference from theoretical mass (ΔT) is indicated for each species. While the mass of the fully assembled complex agreed well with the expected mass, high ΔT values of the other species could be because of degradation of some of the component proteins during the extended incubation step with E. coli lysate (Methods section) during the purification process. The inset shows the SDS-PAGE of GudB-GltAB complex used for the native-MS. The sample was prepared after enriching the B. subtilis lysate with recombinant GudB expressed and purified from E. coli (see Protein expression and purification, Methods). The image is from a single experiment and is a representative of at least two independent experiments. This sample contained a higher fraction of GudB and was also used for cryo-EM to obtain the high-resolution structure of GudB6-GltA2B2 complex (Figure 5).
(b) Preliminary cryo-EM maps corresponding to different particle types (GudB6-GltA2–4B2–4) observed. The key difference between particles is the number of GltAB subunits - the least being two (grey) and the maximum four (violet).
Extended Data Fig. 6. Single particle cryo-EM analysis of the GudB6-GltA2B2.
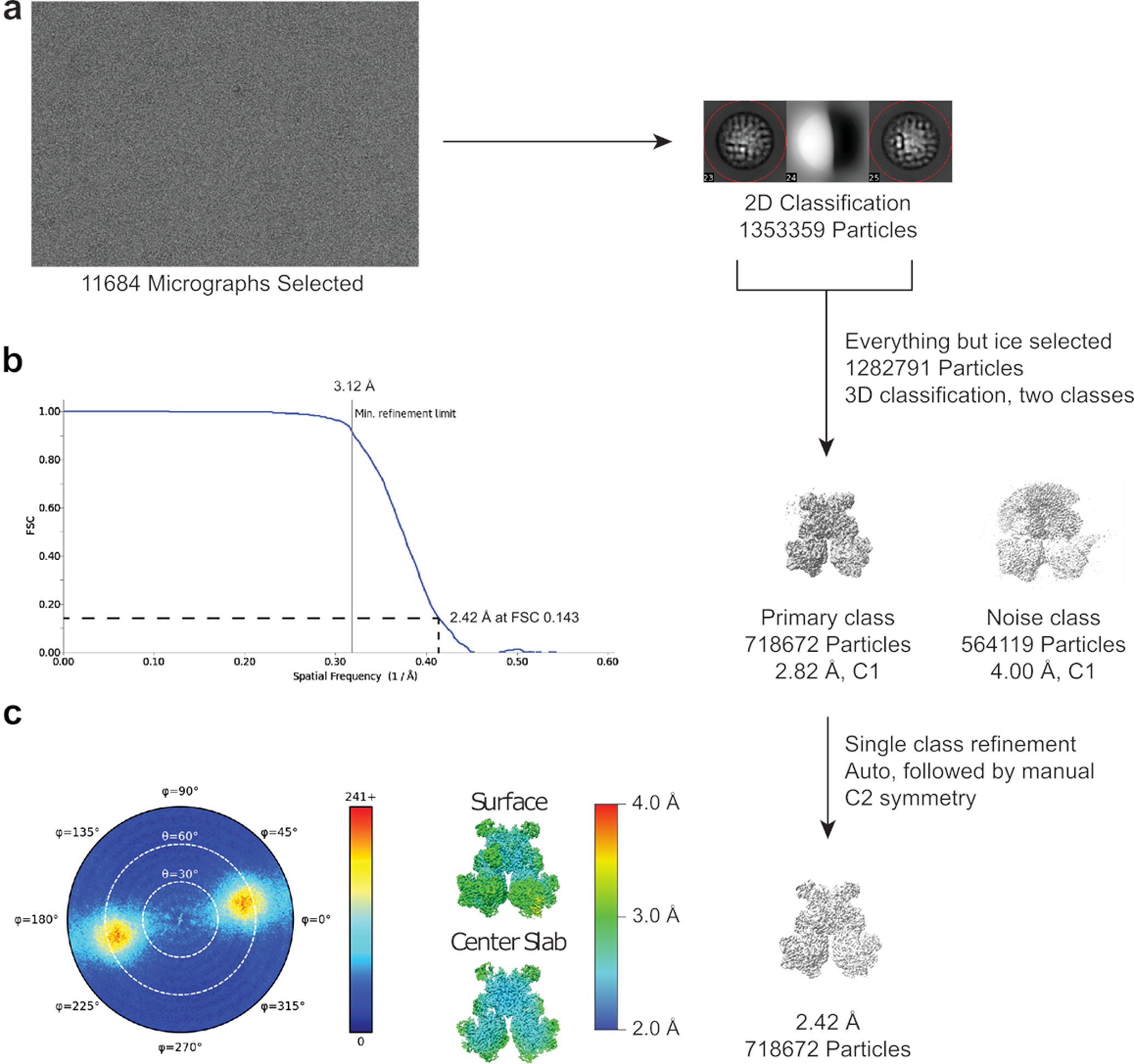
(a) The particles were iteratively picked from selected micrographs and classified. All non-ice particles were carried forward and subjected to a two-class 3D auto refinement in cisTEM using the preliminary GudB6-GltA2B2 reference. This yielded one noise class carrying unaligned particles and high frequency noise, and one class representing clear density for the GudB6-GltA2B2 species. This class was refined using auto and manual methods in cisTEM, with C2 symmetry applied. The number of particles and the estimated resolution are indicated.
(b) FSC curves for the refined GudB6-GltA2B2 map.
(c) Angular distribution plot and local resolution estimation (Relion3.1.2).
Extended Data Fig. 7. Effect of GltA binding on GudB.
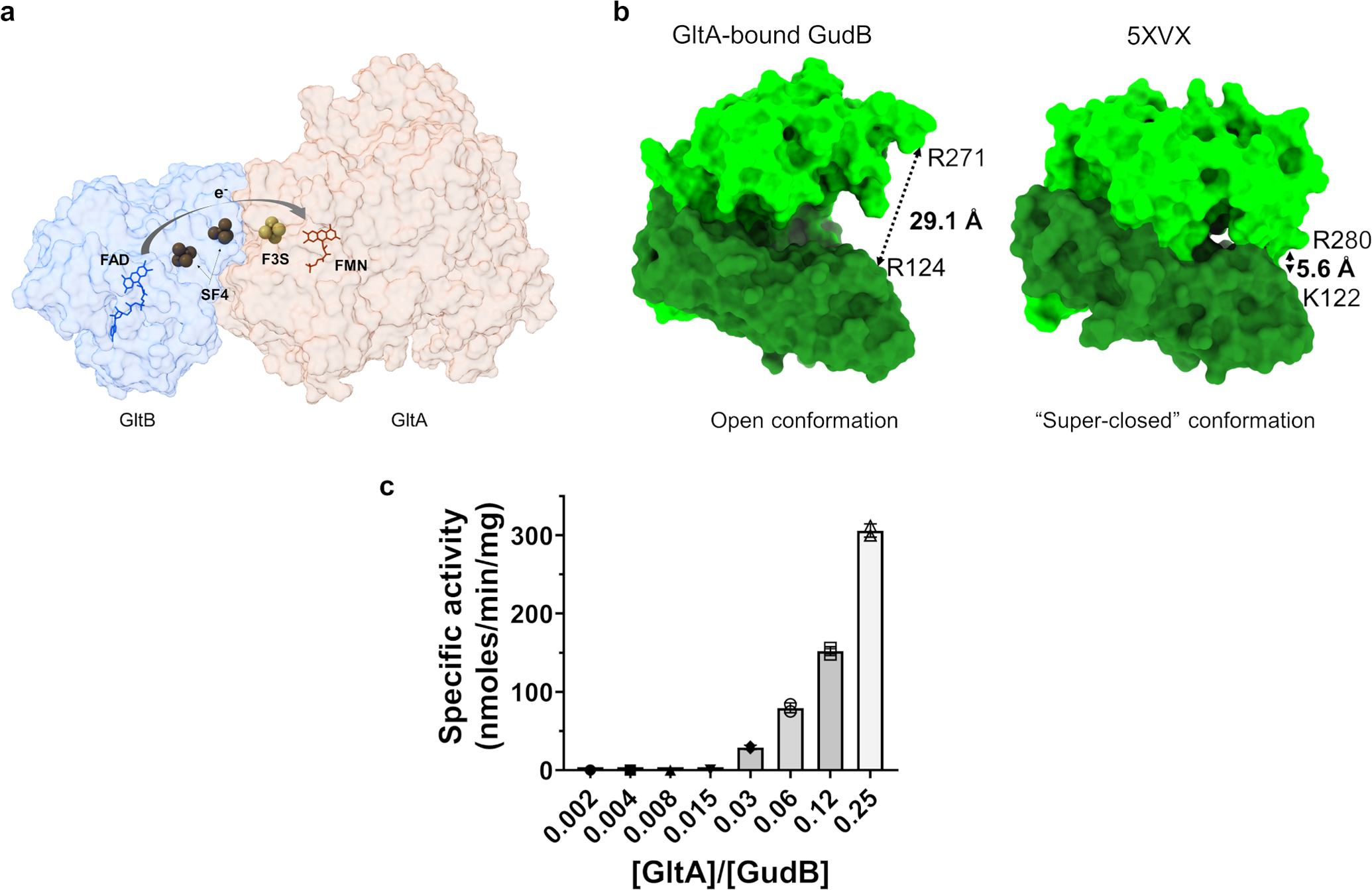
(a) The co-factors in GltAB: FAD, two 4Fe-4S clusters (SF4), 3Fe-4S cluster (F3S) and FMN. These co-factors are involved in shuttling of electrons from NADPH to 2-iminoglutarate along shown arrow. GltA and GltB are shown in a transparent surface representation.
(b) GltAB binding captures GudB in an “open” state with the distance of 29.1 Å between residues R280 (in the co-factor binding domain, shown in light green) and K122 (in the substrate binding domain, shown in green). These residues are equivalent to R271 and R124 in the model of a “super-closed” glutamate dehydrogenase (PDB 5XVX)30 (right panel).
(c) GltAB in sub-stoichiometric amounts promotes hexamerization of GudB by interacting with multiple GudB protomers (from different dimers, as shown in Figure 5c) and hence prevents loss of activity. The assay buffer contained 400 mM glutamate and 4 mM NAD+ and the reaction was initiated by the addition of an enzyme mix consisting of GudB pre-incubated with different amounts of GltAB (in all the reactions, GudB was present at a final concentration is 0.05 μM). GltAB prevents GudB inactivation in a concentration dependent manner. Individual data points are shown from two independent measurements and error bars indicate standard deviation of the mean.
Extended Data Fig. 8. Details of the GudB-GltAB interaction.
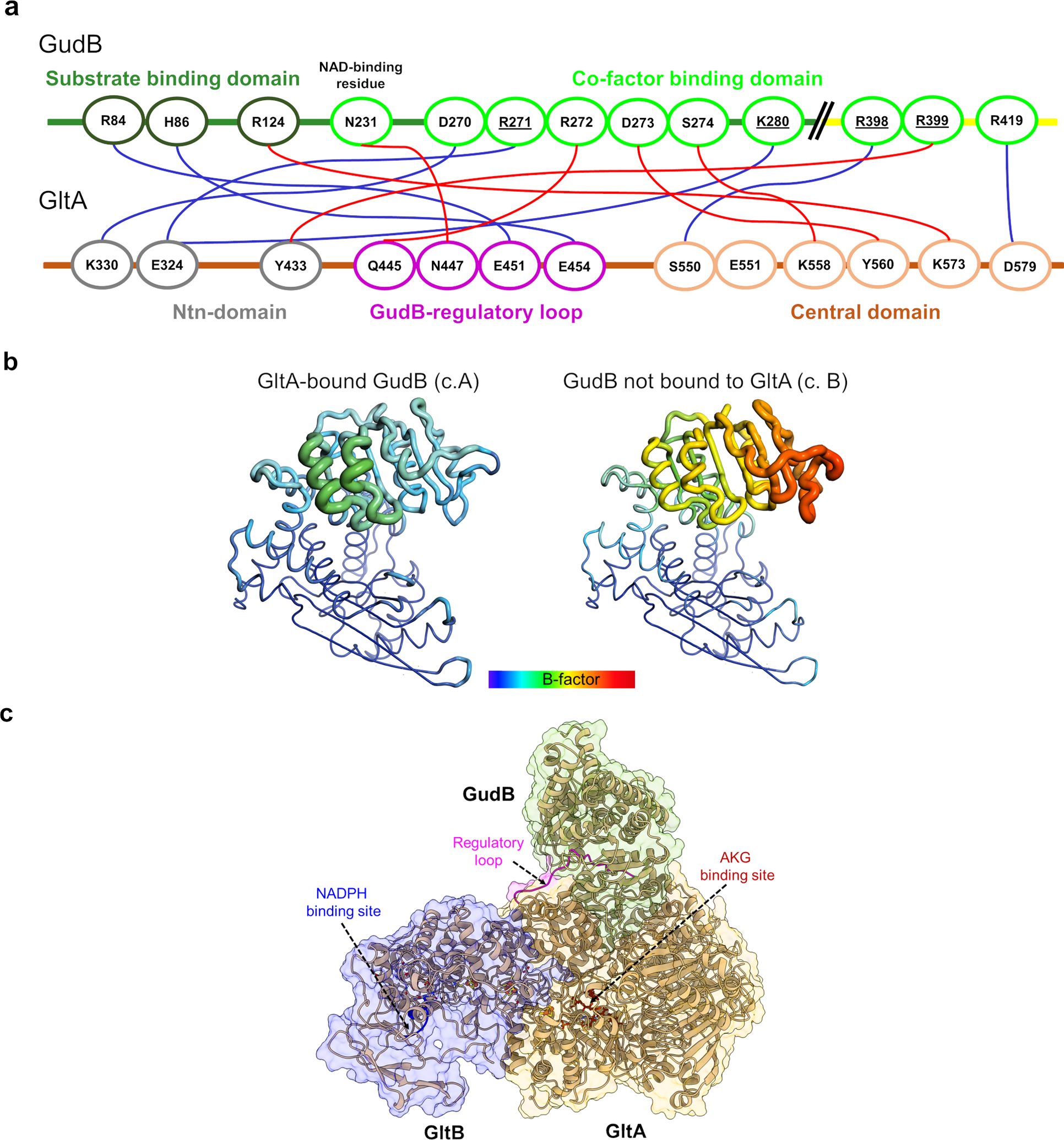
(a) Key interactions between GltA (ovals on orange line) and two GudB protomers (ovals on green/major interaction and yellow line/minor interaction); salt bridges are shown in red and hydrogen bonds in blue. The border of ovals are coloured based on domain/structural feature to which the residue belongs
(b) Binding to GltA stabilizes many loops in the cofactor binding domain of GudB. Shown are a GltA-bound GudB protomer (left, chain A in 7MFM) and a free GudB protomer in the same structure (right, chain B) in the “putty” representation as implemented in PyMol. The radius of the ribbon increases from low to high B-factor and the Cα B-factors are shown in dark blue (lowest B-factor, 54) to red (highest B-factor, 163).
(c) Location of NADPH binding site (the GXGXXG motif of the Rossmann fold is shown in blue) in GltB and AKG binding site (T1041, K948, S870, R968 shown in maroon) in GltA, with respect to the regulatory loop (shown in magenta) in GltA. AKG and NADPH binding site is located about 45 Å and 75 Å from the regulatory loop of GltA.
Extended Data Fig. 9. Biofilm growth and disruption phenotypes.
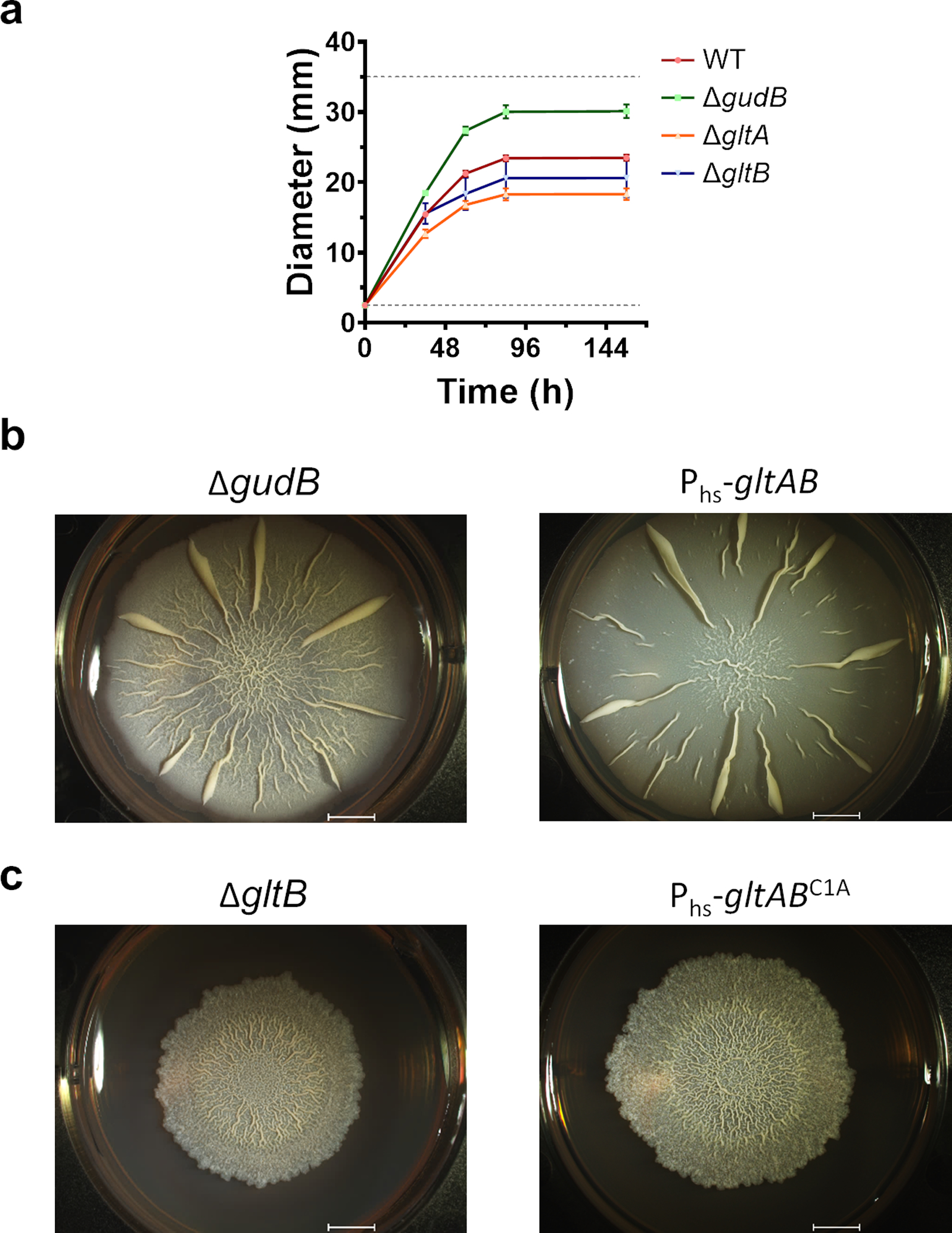
(a) Biofilm diameters measured at different time points. While ΔgudB biofilms were bigger in size and grew faster than wild-type biofilms, ΔgltA and ΔgltB biofilms were smaller. The dashed lines at 2.5 mm and 35 mm indicates the starting size of the biofilm, and the diameter of the well, used to grow the biofilm, respectively. The measurements were from four independent experiments (n=4). Data is shown as mean and error bars represent standard deviation.
(b) Similarity in biofilm morphology between ΔgudB and the Phs-gltAB strains (grown with 100 μM IPTG). Overexpression of GltAB in the latter increases in synthase activity and also silences GudB, thereby resembling the ΔgudB biofilm morphology. Both biofilms grew rapidly and had large wrinkles spreading from the interior to the periphery of the biofilm. All the images in this panel are reproduced from Figure 6 for better representation of the biofilm morphology.
(c) Similarity in biofilm morphology between ΔgltB and the Phs-gltABC1A strains (grown with 100 μM IPTG). In both the biofilms the wrinkles are restricted to the interior of the biofilm.
Supplementary Material
Acknowledgments:
We thank Kesava Phaneendra Cherukuri for help in synthesis of DSG, Alexander Leytens for assistance in cloning, and Yuval Kushmaro for screening different crosslinkers. We are grateful to Meital Kupervaser and Yishai Levin from The De Botton Protein Profiling institute of the Nancy and Stephen Grand Israel National Center for Personalized Medicine, Weizmann Institute of Science, for the proteomics analysis. We thank Dr. Adar Sonn Segev and Dr. Moshe Goldsmith for their assistance with the Mass photometry experiments. We thank Dr Sergey Malitsky and Dr. Maxim Itkin for the metabolomics analysis. We thank Prof. Uwe Sauer, Prof. Uri Alon, Dr. Nobuhiko Tokuriki and Dr. Sunil Laxman for their critical feedback on the manuscript. We thank Dr. Felix Jonas for valuable comments on the manuscript and Dr. Brian Ross for proofreading the manuscript. Funding by the Israel Science Foundation Grant No. 2575/20 is gratefully acknowledged. M.S. is the incumbent of the Aharon and Ephraim Katzir Memorial Professorial Chair. The research of S.V is supported by the Clore Israel Foundation. J.S.F is supported by NIH GM123159 and D.J.L is supported by NIH F32 AI148120. D.S.T is the incumbent of the Nella and Leon Benoziyo Professorial Chair.
Footnotes
Declaration of Interests:
The authors declare no conflict of interests
Data availability:
The proteomics data is provided as Excel sheets (Supplementary Datasets 1 and 2). The structural data is deposited in the PDB (PDB code: 7MFM and 7MFT).
References:
- 1.Chubukov V et al. Transcriptional regulation is insufficient to explain substrate‐induced flux changes in Bacillus subtilis. Mol. Syst. Biol 9, 709 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Metallo CM & Vander Heiden MG Understanding Metabolic Regulation and Its Influence on Cell Physiology. Molecular Cell vol. 49 388–398 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curi R et al. Regulatory principles in metabolism -Then and now. Biochemical Journal vol. 473 1845–1857 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Srere PA The metabolon. Trends in Biochemical Sciences vol. 10 109–110 (1985). [Google Scholar]
- 5.Zhang Y & Fernie AR Metabolons, Enzyme–Enzyme Assemblies that Mediate Substrate Channeling, and Their Roles in Plant Metabolism. Plant Commun 100081 (2020) doi: 10.1016/j.xplc.2020.100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sweetlove LJ & Fernie AR The role of dynamic enzyme assemblies and substrate channelling in metabolic regulation. Nature Communications vol. 9 1–12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin ECC, Lynch AS & Magasanik B Regulation of Nitrogen Assimilation. in Regulation of Gene Expression in Escherichia coli 281–290 (Springer; US, 1996). doi: 10.1007/978-1-4684-8601-8_13. [DOI] [Google Scholar]
- 8.Young VR & Ajami AM Glutamate: An amino acid of particular distinction. in Journal of Nutrition vol. 130 892S–900S (American Institute of Nutrition, 2000). [DOI] [PubMed] [Google Scholar]
- 9.Yan D, Ikeda TP, Shauger AE & Kustu S Glutamate is required to maintain the steady-state potassium pool in Salmonella typhimurium. Proc. Natl. Acad. Sci. U. S. A 93, 6527–6531 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bennett BD et al. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol 5, 593–599 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Eunen K et al. Measuring enzyme activities under standardized in vivo-like conditions for systems biology. FEBS J 277, 749–760 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Tempest DW, Meers JL & Brown CM Influence of environment on the content and composition of microbial free amino acid pools. J. Gen. Microbiol 64, 171–185 (1970). [DOI] [PubMed] [Google Scholar]
- 13.Huergo LF & Dixon R The Emergence of 2-Oxoglutarate as a Master Regulator Metabolite. Microbiol. Mol. Biol. Rev 79, 419–435 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noda‐Garcia L, Romero Romero ML, Longo LM, Kolodkin‐Gal I & Tawfik DS Bacilli glutamate dehydrogenases diverged via coevolution of transcription and enzyme regulation. EMBO Rep 18, 1139–1149 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Commichau FM, Gunka K, Landmann JJ & Stülke J Glutamate metabolism in bacillus subtilis: Gene expression and enzyme activities evolved to avoid futile cycles and to allow rapid responses to perturbations of the system. J. Bacteriol 190, 3557–3564 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belitsky BR & Sonenshein AL Role and regulation of Bacillus subtilis glutamate dehydrogenase genes. J. Bacteriol 180, 6298–6305 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Engel PC A marriage full of surprises; Forty-five years living with glutamate dehydrogenase. Neurochem. Int 59, 489–494 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Li M, Li C, Allen A, Stanley CA & Smith TJ The structure and allosteric regulation of mammalian glutamate dehydrogenase. Archives of Biochemistry and Biophysics vol. 519 69–80 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tomita T, Kuzuyama T & Nishiyama M Structural basis for leucine-induced allosteric activation of glutamate dehydrogenase. J. Biol. Chem 286, 37406–37413 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu J et al. Metabolic co-dependence gives rise to collective oscillations within biofilms. Nature 523, 550–554 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gunka K et al. Functional dissection of a trigger enzyme: Mutations of the bacillus subtilis glutamate dehydrogenase RocG that affect differentially its catalytic activity and regulatory properties. J. Mol. Biol 400, 815–827 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Picossi S, Belitsky BR & Sonenshein AL Molecular Mechanism of the Regulation of Bacillus subtilis gltAB Expression by GltC. J. Mol. Biol 365, 1298–1313 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smaldone GT et al. A global investigation of the Bacillus subtilis iron-sparing response identifies major changes in metabolism. J. Bacteriol 194, 2594–2605 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hart Y & Alon U The Utility of Paradoxical Components in Biological Circuits. Molecular Cell vol. 49 213–221 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Cottevieille M et al. The subnanometer resolution structure of the glutamate synthase 1.2-MDa hexamer by cryoelectron microscopy and its oligomerization behavior in solution: Functional implications. J. Biol. Chem 283, 8237–8249 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Swuec P, Chaves-Sanjuan A, Camilloni C, Vanoni MA & Bolognesi M Cryo-EM Structures of Azospirillum brasilense Glutamate Synthase in Its Oligomeric Assemblies. J. Mol. Biol 431, 4523–4526 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Vanoni MA & Curti B Glutamate synthase: A complex iron-sulfur flavoprotein. Cellular and Molecular Life Sciences vol. 55 617–638 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kameya M et al. A novel ferredoxin-dependent glutamate synthase from the hydrogen-oxidizing chemoautotrophic bacterium Hydrogenobacter thermophilus TK-6. J. Bacteriol 189, 2805–2812 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stillman TJ, Baker PJ, Britton KL & Rice DW Conformational Flexibility in Glutamate Dehydrogenase. J. Mol. Biol 234, 1131–1139 (1993). [DOI] [PubMed] [Google Scholar]
- 30.Prakash P, Punekar NS & Bhaumik P Structural basis for the catalytic mechanism and -ketoglutarate cooperativity of glutamate dehydrogenase. J. Biol. Chem 293, 6241–6258 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hassanov T, Karunker I, Steinberg N, Erez A & Kolodkin-Gal I Novel antibiofilm chemotherapies target nitrogen from glutamate and glutamine. Sci. Rep 8, 1–12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pisithkul T et al. Metabolic remodeling during biofilm development of bacillus subtilis. MBio 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang N et al. Whole transcriptomic analysis of the plant-beneficial rhizobacterium Bacillus amyloliquefaciens SQR9 during enhanced biofilm formation regulated by maize root exudates. BMC Genomics 16, 685 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu J et al. Coupling between distant biofilms and emergence of nutrient time-sharing. Science (80-. ) 356, 638–642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noyes RM & Field RJ Oscillatory Chemical Reactions. Annu. Rev. Phys. Chem 25, 95–119 (1974). [Google Scholar]
- 36.Goldbeter A Mechanism for oscillatory synthesis of cyclic AMP in Dictyostelium discoideum. Nature 253, 540–542 (1975). [DOI] [PubMed] [Google Scholar]
- 37.Rossomando EF & Sussman MA 5’-Adenosine Monophosphate-Dependent Adenylate Cyclase and an Adenosine 3’:5’-Cyclic Monophosphate-Dependent Adenosine Triphosphate Pyrophosphohydrolase in Dictyostelium discoideum. Proc. Natl. Acad. Sci 70, 1254–1257 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pálsson E A cAMP signaling model explains the benefit of maintaining two forms of phosphodiesterase in Dictyostelium. Biophys. J 97, 2388–2398 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Masaki N, Fujimoto K, Honda-Kitahara M, Hada E & Sawai S Robustness of self-organizing chemoattractant field arising from precise pulse induction of its breakdown enzyme: A single-cell level analysis of pde expression in dictyostelium. Biophys. J 104, 1191–1202 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakajima M et al. Reconstitution of circadian oscillation of cyanobacterial KaiC phosphorylation in vitro. Science (80-. ) 308, 414–415 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Wagner A Circuit topology and the evolution of robustness in two-gene circadian oscillators. Proc. Natl. Acad. Sci. U. S. A 102, 11775–11780 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Radde N The impact of time delays on the robustness of biological oscillators and the effect of bifurcations on the inverse problem. Eurasip J. Bioinforma. Syst. Biol 2009, 327503 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blanchini F, Cuba Samaniego C, Franco E & Giordano G Homogeneous Time Constants Promote Oscillations in Negative Feedback Loops. ACS Synth. Biol 7, 1481–1487 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Commichau FM, Herzberg C, Tripal P, Valerius O & Stülke J A regulatory protein–protein interaction governs glutamate biosynthesis in Bacillus subtilis: the glutamate dehydrogenase RocG moonlights in controlling the transcription factor GltC. Mol. Microbiol 65, 642–654 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Messenguy F & Wiame JM The control of ornithinetranscarbamylase activity by arginase in Saccharomyces cerevisiae. FEBS Lett 3, 47–49 (1969). [DOI] [PubMed] [Google Scholar]
- 46.Messenguy F, Penninckx M & Wiame J ‐M. Interaction between Arginase and Ornithine Carbamoyltransferase in Saccharomyces cerevisiae The Regulatory Site for Ornithine. Eur. J. Biochem 22, 277–286 (1971). [DOI] [PubMed] [Google Scholar]
- 47.Straube R Sensitivity and robustness in covalent modification cycles with a bifunctional converter enzyme. Biophys. J 105, 1925–1933 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilson GA & Bott KF Nutritional factors influencing the development of competence in the Bacillus subtilis transformation system. J. Bacteriol 95, 1439–1449 (1968). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Branda SS, González-Pastor JE, Ben-Yehuda S, Losick R & Kolter R Fruiting body formation by Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A 98, 11621–11626 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kavran JM & Leahy DJ Coupling antibody to cyanogen bromide-activated sepharose. in Methods in Enzymology vol. 541 27–34 (Academic Press Inc., 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kavran JM & Leahy DJ Silver staining of SDS-polyacrylamide gel. in Methods in Enzymology vol. 541 169–176 (Academic Press Inc., 2014). [DOI] [PubMed] [Google Scholar]
- 52.Elinger D, Gabashvili A & Levin Y Suspension Trapping (S-Trap) Is Compatible with Typical Protein Extraction Buffers and Detergents for Bottom-Up Proteomics. J. Proteome Res 18, 1441–1445 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Rueden CT et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, 529 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Konings S et al. Advances in Single Particle Analysis Data Acquisition. Microsc. Microanal (2019) doi: 10.1017/s1431927619005798. [DOI] [Google Scholar]
- 55.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol (2005) doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 56.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA CryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods (2017) doi: 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 57.Sonn-Segev A et al. Quantifying the heterogeneity of macromolecular machines by mass photometry. Nat. Commun. 2020 111 11, 1–10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The proteomics data is provided as Excel sheets (Supplementary Datasets 1 and 2). The structural data is deposited in the PDB (PDB code: 7MFM and 7MFT).