

Abstract
Driver mutations promote initiation and progression of cancer. Pharmacological treatment can inhibit the action of the mutant protein; however, drug resistance almost invariably emerges. Multiple studies revealed that cancer drug resistance is based upon a plethora of distinct mechanisms. Drug resistance mutations can occur in the same protein or in different proteins; as well as in the same pathway or in parallel pathways, bypassing the intercepted signaling. The dilemma that the clinical oncologist is facing is that not all the genomic alterations as well as alterations in the tumor microenvironment that facilitate cancer cell proliferation are known, and neither are the alterations that are likely to promote metastasis. For example, the common KRasG12C driver mutation emerges in different cancers. Most occur in NSCLC, but some occur, albeit to a lower extent, in colorectal cancer and pancreatic ductal carcinoma. The responses to KRasG12C inhibitors are variable and fall into three categories, (i) new point mutations in KRas, or multiple copies of KRAS G12C which lead to higher expression level of the mutant protein; (ii) mutations in genes other than KRAS; (iii) original cancer transitioning to other cancer(s). Resistance to adagrasib, an experimental antitumor agent exerting its cytotoxic effect as a covalent inhibitor of the G12C KRas, indicated that half of the cases present multiple KRas mutations as well as allele amplification. Redundant or parallel pathways included MET amplification; emerging driver mutations in NRAS, BRAF, MAP2K1, and RET; gene fusion events in ALK, RET, BRAF, RAF1, and FGFR3; and loss-of-function mutations in NF1 and PTEN tumor suppressors.
In the current review we discuss the molecular mechanisms underlying drug resistance while focusing on those emerging to common targeted cancer drivers. We also address questions of why cancers with a common driver mutation are unlikely to evolve a common drug resistance mechanism, and whether one can predict the likely mechanisms that the tumor cell may develop. These vastly important and tantalizing questions in drug discovery, and broadly in precision medicine, are the focus of our present review. We end with our perspective, which calls for target combinations to be selected and prioritized with the help of the emerging massive compute power which enables artificial intelligence, and the increased gathering of data to overcome its insatiable needs.
Keywords: Cancer, Chemotherapy, Drug resistance, Drug discovery, Precision medicine, Epigenetics, Transcriptomics, Interactome, Single cell, MAPK, Chromatin accessibility
1. Introduction
Cancer can evolve from somatic mutations (cancer.sanger.ac.uk) (Akdemir et al., 2020b; Martincorena and Campbell, 2015; Tate et al., 2019). These mutations frequently accumulate in oncogenes and tumor suppressor genes (Morjaria, 2021). Mutations accumulate over a relatively long period. Some mutations emerge randomly, some are inherited, whereas others develop due to mutagens. Estimates of the number of mutations that are required for a normal human cell to progress to advanced cancer, including by mathematical models based on the relation between age and incidence, vary (Tomasetti et al., 2015).
A driver mutation confers upon cancer cells a growth advantage for its neoplastic transformation (Pon and Marra, 2015). It differs from passenger mutations which do not contribute to the development and progression of the cancer. Only a small fraction of mutations identified in a cancer patient occur in driver genes. Drivers are commonly identified by their high frequencies of occurrence (Brown et al., 2019; Chen et al., 2020). However, they can also be rare (Nussinov et al., 2020b, 2019c). Drivers commonly lead to an observable conformational change. At the same time, ‘latent drivers’ may also contribute to a gain of function, even though the associated conformational change may be unobservable, and on their own, their contribution is minor (Nussinov et al., 2019a; Nussinov and Tsai, 2015). Rare latent drivers can also be preexisting drug resistance mutations. Driver mutations can propel cancer initiation and progression. They characterize molecular profiles of tumors and help in predicting clinical outcomes for the patients. They are sought after as druggable targets and are used in making therapeutic decisions (Nussinov et al., 2019b; Saleem et al., 2019; Zsakai et al., 2019). Their frequency points to their common occurrence. Not all driver mutations are drug resistance mutations and not all drug resistance mutations occur in driver genes (Beckman and Loeb, 2020; Loeb et al., 2019; Reiter et al., 2019). Driver mutations activate the protein even in the absence of an incoming activation signal, making it an escape regulatory control. On the other hand, drug resistance mutations can make a protein escape inhibition by certain drugs, commonly via changes in the active site shape or surface. In addition, alternative mechanisms of chemoresistance occur that are not associated with qualitative and/or quantitative changes in the driver genes; these include: impaired drug influx, enhanced drug efflux via multidrug resistance pumps of the ATP-Binding Cassette (ABC) superfamily, drug compartmentalization away from its target protein, metabolic drug inactivation, as well as induction of anti-apoptotic mechanisms (Assaraf et al., 2019; Gonen and Assaraf, 2012; Li et al., 2016; Shahar and Larisch, 2020; Wang et al., 2021; Wijdeven et al., 2016; Zhitomirsky and Assaraf, 2016). Resistance to imatinib (STI571), an orthosteric ATP-competitive drug that inhibits oncogenic mutants of Bcr-Abl fusion tyrosine kinase, is a classic example (Burchert, 2007; Gao et al., 2021) (see below). Mutations in Bcr-Abl’s ATP-binding site or the activation loop can result in steric clashes blocking imatinib binding, as observed upon T315I substitution (Nicolini et al., 2013; Zhang et al., 2010). The T315I mutation also blocks second-generation tyrosine kinase inhibitors (TKIs) such as nilotinib (AMN107) and dasatinib (BMS-354825) (O’Hare et al., 2005; Tamai et al., 2018). Another example is the case of epidermal growth factor receptor (EGFR) mutations T790M and C797S. In a second resistance scenario, gene amplification can spur an alternative signaling pathway (Genovese et al., 2017). Mesenchymal-epithelial transition factor (MET or c-MET, a.k.a. HGFR, hepatocyte growth factor receptor) encoded by the MET gene (Aissa et al., 2021), or other receptor tyrosine kinase (RTK) signaling pathways that can bypass the targeted RTK (Harbinski et al., 2012; Wilson et al., 2012), such as IGF-1R (insulin-like growth factor 1 receptor) (Crudden et al., 2015), FGFR1 (fibroblast growth factor receptor 1), HER2 (human epidermal growth factor receptor 2), and AXL (tyrosine-protein kinase receptor UFO), which share downstream pathways, PI3K/AKT/mTOR (PI3K, phosphatidylinositol 3-kinase; AKT, protein kinase B; mTOR, mechanistic target of rapamycin) and MAPK (mitogen-activated protein kinase), provide examples. Activation of EMT (epithelial-to-mesenchymal transition), NF-κB (nuclear factor kappa B), or AXL either by chemotherapy or a single small molecule inhibitor (Kim et al., 2019; Taniguchi et al., 2019; Zhu et al., 2019), dysregulation of growth factor cell signaling, a major driver of most human cancers (Gillis and McLeod, 2016), and loss of function of tumor suppressor genes (Gao et al., 2021) have all suggested that drug resistant cancer cells can exploit diverse and distinct mechanisms (Aissa et al., 2021).
In the current review we ask: Are drug resistance mechanisms that emerge following targeting of common driver mutations also common? That is, would targeting a certain Ras driver, e.g., KRasG12C, in different patients lead to the same drug resistance modality? This question portends critical drug treatment decisions (Nussinov et al., 2014). Below, we first outline our premise. We then briefly overview some drug resistance mechanisms. We follow with those exploited by human tumors harboring a common KRas mutation as examples. We further discuss why the mechanisms of resistance of common driver mutations are expected to differ. We next comment on genomic heterogeneity and the pre-existence of both driver and resistance mutations, and finally take up the key question of whether one can forecast the likely emerging resistance mechanisms. Drug resistance mechanisms are chiefly determined by genomics, pre-existing and evolving mutations (Aissa et al., 2021; Bar-Zeev et al., 2017; Friedman, 2016; Levin et al., 2021; Lim and Ma, 2019; Prieto-Vila et al., 2019; Taylor et al., 2015), and cell type and state which determine the gene expression status. Single cell transcriptomics can capture rare drug resistant cells in a tumor clone (Bhang et al., 2015; Hata et al., 2016; La Monica et al., 2019). Loss of H3K4me3 (histone 3 lysine 4 trimethylation) in EGFR mutant in erlotinib (Tarceva)-treated cells (Lantermann et al., 2015) along with changes in chromatin-modifying proteins HDAC9 (histone deacetylase 9), NCOR1 (nuclear receptor co-repressor 1), MLL1 (a histone methyltransferase), and EED (embryonic ectoderm development protein) demonstrate large transcriptomic alterations in drug resistant clones, emphasizing the cardinal role of epigenetics and chromatin organization (Nussinov et al., 2021a). Transcriptional changes associated with drug resistance, including those emerging from gene copy number mutations can seal cell fate, by allowing, or restricting, cancer re-growth, and as such can improve the predictive power of genomic sequencing for rare drug resistant mutations (Aissa et al., 2021; Sun et al., 2019). We end with a discussion of the almost limitless number of possible target combinations to address the many potential drug resistance mechanisms. In our view, the way to select and prioritize the combinations is by artificial intelligence (AI) strategies powered by the fast-increasing massive computational power and clinical and inhibitor databases (Kim, 2021; Nguyen et al., 2021). With the growing size of model parameters deep learning requires data-intensive computations. The rapid increase in the volume of data coupled with advanced hardware and software methods could break the performance wall of traditional approaches and provide more reliable target combination to be submitted for testing in the expensive clinical trials.
2. Our premise: cells with common driver mutations are unlikely to develop common drug resistance mechanisms
The reasons for this premise include: (i) the mutant protein may be in different cell (tissue) types and (ii) the cell state may differ. The cell state can be described by, e.g., its gene expression profile, abundance of other proteins in the corresponding pathway, its post-translational modifications status, and morphology. These are impacted by environmental cues and the cell developmental status. Even for a specific tissue, or cell type where the mutation emerged, the time window is a key consideration (Mitchell et al., 2018). (iii) Often overlooked, yet, with a profound influence on the mechanism of the emerging drug resistance are the identity and distinct patterns of background mutations. Background mutations (Beckman and Loeb, 2020; Nussinov et al., 2021b; Saito et al., 2020; Vasan et al., 2019b; Zhang et al., 2021), either pre-existing the emergence of cancer or emerging early in its evolution (Haupt et al., 2021; Johnson et al., 2014) are likely to vary among patients. Considering that tumors may initially respond to treatment but, as not all the neoplastic cells are decimated, the minor population, or rare remaining drug resistant cells housing these mutations can seed cancer regrowth (Figure 1). The mutations are likely to include driver mutations, or combinations of strong drivers, latent drivers, or weak drivers (Zhang et al., 2021). Rare cells are also likely to accommodate a preexisting drug resistance mutation, which may, or may not be a driver. In one example, it reactivates cellular proliferation through hypermutated Rb (retinoblastoma), which plays a key role in negatively regulating cell cycle progression from the G1 to S-phase through two positive regulators of cell cycle entry, E2F transcription factors and cyclin dependent kinases, and the AKT/mTOR, which is also required for cell cycle progression from G1 to S phase (Kumar et al., 2016). Since the expression levels of the corresponding genes need to be high, coupled mutations in chromatin remodelers is commonly observed (Akdemir et al., 2020a). Additional contributors not discussed here include parameters such as cell metabolism (Gremke et al., 2020; Knoechel and Aster, 2015; Zaal and Berkers, 2018). Resistance may not necessarily involve genetic changes. Some cancer cells may survive chemotherapy by exploiting nutrients in their tumor microenvironment (TME) (van Gastel et al., 2020). Thus, parameters related to the TME also play cardinal role [e.g., Refs. (Getzenberg and Coffey, 2011; Pontiggia et al., 2012; Wojtkowiak et al., 2011)].
Figure 1.
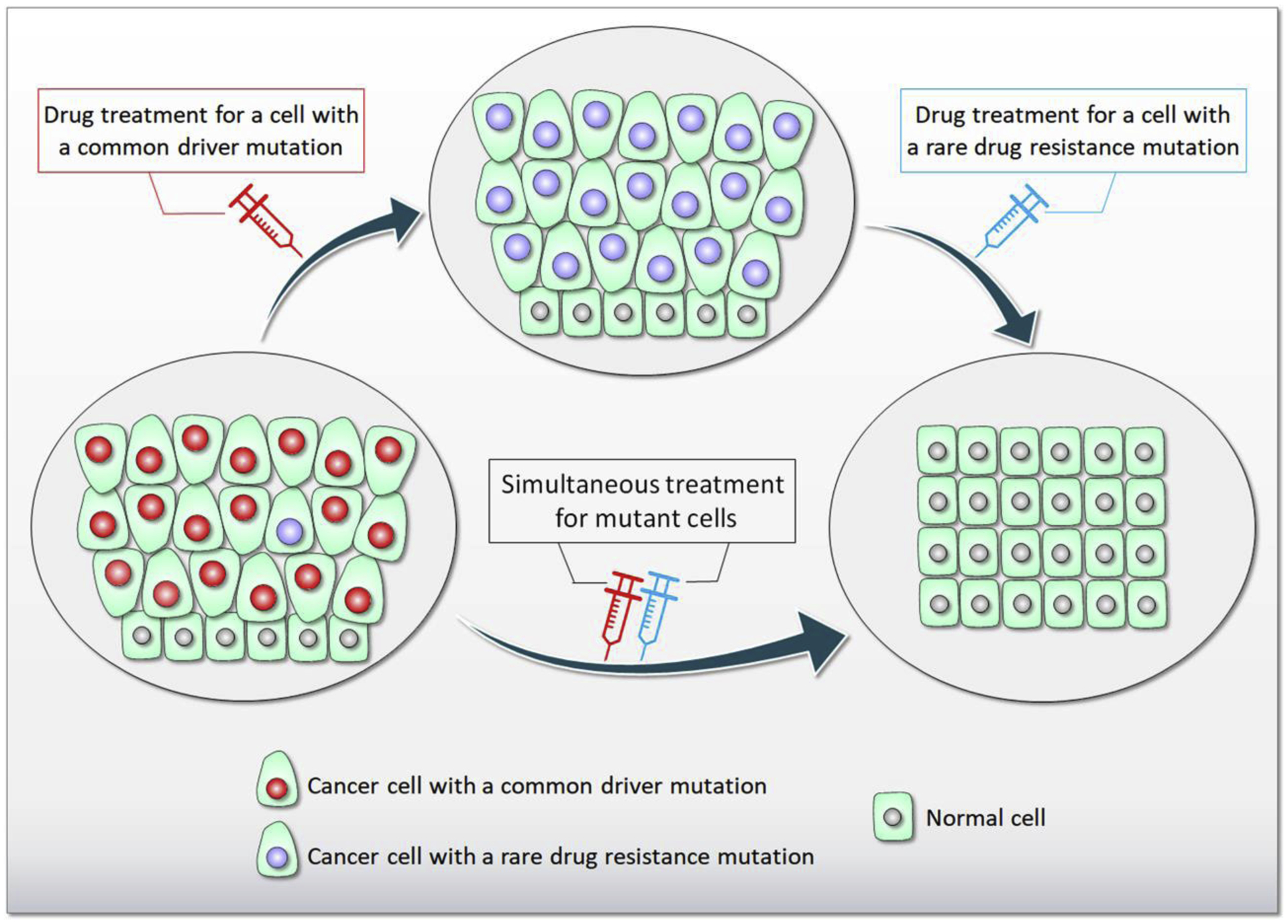
The role of heterogeneity in drug resistance. An example of cells in the tissue containing a vast number of common driver mutant (e.g., KRas4B G12C) and a small number of rare drug resistance mutant cells (lower left). When a drug is taken, all cells are decimated except those with rare resistance mutation for the particular drug, resulting that those cells with the rare mutation proliferate (middle). A subsequent drug will remove the rare drug resistance mutant cells in later stage of chemotherapy (lower right). However, simultaneously taking both drugs have a better chance of success.
As to background mutations, at the time of diagnosis, a tumor may already accumulate more than 1 billion cancer cells (Offord, 2020). The effective mutation rate was calculated to be 6 × 10−7 per gene per generation leading to an accumulation of mutational diversity beyond a critical tumor size (Beckman and Loeb, 2020). This is in line with the expectation that all drug resistant mutations pre-exist in the tumor in at least one cell at diagnosis, even though not all driver mutations are resistance mutations, and not all resistance mutations occur in drivers (Beckman and Loeb, 2020; Loeb et al., 2019; Reiter et al., 2019). Even rare cells harboring such mutations may proliferate, thereby seeding a tumor subclone resistant to a single therapy. However, the probability that they harbor co-existing mutations that simultaneously resist polypharmacology, that is, a combination of drugs, is low. These are expected to emerge during treatment. This argues that genetic alterations – pre-existing and emerging during tumor evolution – are a critical factor in drug resistance (Beckman and Loeb, 2020; Loeb et al., 2019; Reiter et al., 2019), including mutations in chromatin remodelers which play critical roles in protein expression. Rare mutations are vastly important contributors to cancer initiation and progression (Nussinov et al., 2019a, b; Nussinov et al., 2019c), and to resistance (Beckman and Loeb, 2020), making their identification a vital aim (Nussinov et al., 2020b).
3. Drug resistance mechanisms emerging to common targeted drivers reflect cell-type specific cancer evolution
Several excellent reviews on drug resistance mechanisms were published in the last few years that describe the multifactorial nature of cancer drug resistance [e.g., Refs. (Aleksakhina et al., 2019; Andrei et al., 2020; Assaraf et al., 2019; Das et al., 2021; Du et al., 2020; Dunnett-Kane et al., 2021; Friedman, 2016; Garraway and Janne, 2012; Gottesman, 2002; Haider et al., 2020; Housman et al., 2014; Hussein et al., 2021; Jiang et al., 2020; Lee et al., 2020; Li et al., 2020; Lim and Ma, 2019; Long et al., 2020; Narayanan et al., 2020; Rolfo et al., 2014; Ruan et al., 2020; Sabnis and Bivona, 2019; Sarmento-Ribeiro et al., 2019; Sciarrillo et al., 2020; Shahar and Larisch, 2020; Sun et al., 2019; Wang et al., 2019; Yan et al., 2020; Zhong and Virshup, 2020)]. Thus, we herein only touch on several key determinants, and proceed to our focus on drug resistance mechanisms that were observed to emerge following the drugging of common driver mutations in common target proteins in patients with the same or different tumor types, e.g., lung, colorectal, and breast cancer. Key determinants of drug resistance include (Vasan et al., 2019a) (i) Tumor burden, or tumor load, defined as the total amount of tumor (cells/mass) distributed in the patients’ body, or the sum of the longest diameters of all measurable lesions (Bousquet et al., 2013; Czarnecka et al., 2019), (ii) Tumor growth kinetics, defined as the ratio of the slope of tumor growth before treatment and the slope of tumor growth on treatment between the nadir and disease progression, calculated for each patient (Le Tourneau et al., 2012), (iii) Tumor heterogeneity (Lawrence et al., 2013), which captures the distinct phenotypic profiles of tumor cells, e.g., morphology, gene expression, metabolism, motility, proliferation, and (iv) Metastatic potential, the immune system, the TME, druggability, and more (Vasan et al., 2019a). Cancer drug resistance is a frequently a multifactorial phenomenon and hence can occur via multiple mechanisms, including multidrug resistance (Buck et al., 2021; Gottesman, 2002; Gottesman et al., 2002; Li et al., 2016; Robey et al., 2018; Su et al., 2021; Wang et al., 2021) and detoxification (Lee et al., 2012), cell death inhibiting (apoptosis suppression) (Eberle et al., 2007; Jiang et al., 2021; Shahar and Larisch, 2020), altered drug metabolism, epigenetic (Jiang et al., 2021), enhancing DNA repair (Chiappa et al., 2021; Pecoraro et al., 2021), gene amplification (Assaraf et al., 1989; Bram et al., 2007; Mansoori et al., 2017) as well as drug sequestration in lysosomes or vesicles away from the cellular drug target (Goler-Baron et al., 2012; Ifergan et al., 2005; Zhitomirsky and Assaraf, 2015, 2016).
At the same time, recent observations on the consequences of drugging KRasG12C (Figure 2) indicate that resistance mechanisms emerging in different cells vary, questioning the effectiveness and wisdom of adopting a common drug regime for a common driver mutation (Awad et al., 2021; Dana-Farber Cancer Institute, 2021). Considering the high frequency of common drivers, this dilemma of effective treatment for cancers harboring common drivers has far reaching consequences. The quandary facing the physician stems from the realization that the alterations in the genomic and cellular environment that permitted the cancer to proliferate are unknown, and neither are those that are likely to evolve. The common KRasG12C mutation emerged in different cancer types: even though most occurred in NSCLC (non-small-cell lung cancer), some were detected in colorectal cancer and pancreatic ductal adenocarcinoma (Seton-Rogers, 2020). The results from an analysis of patients with the KRasG12C mutation are revealing (Awad et al., 2021). They indicated that the response of patients to KRasG12C inhibitors led to cancer evolution that fell into the following categories (Figure 2), (i) Cancers with new mutations in KRas, at KRasG13, KRasR68, KRasH95, and KRasY96, or multiple copies of KRasG12C which lead to a higher expression level of the mutant protein; (ii) Mutations in genes other than KRAS, e.g., BRAF, MET, ALK, RET, MAP2K1; (iii) Transitioning to other cancers, e.g., transitioning from lung adenocarcinomas to squamous cell carcinomas. Overall, resistance to adagrasib (MRTX849), which earned an FDA ‘Breakthrough Therapy’ designation for KRasG12C NSCLC emerged in 17 out of the 38 patients, with half of these presenting multiple coincident mechanisms (Awad et al., 2021). Mutations included G12D/R/V/W, G13D, Q61H, R68S, H95D/Q/R, Y96C, and allele amplification. Redundant or parallel pathways included MET amplification; emerging driver mutations in NRAS, BRAF, MAP2K1, and RET; gene fusions events in ALK, RET, BRAF, RAF1, and FGFR3; and loss-of-function mutations in NF1 and PTEN tumor suppressors.
Figure 2.
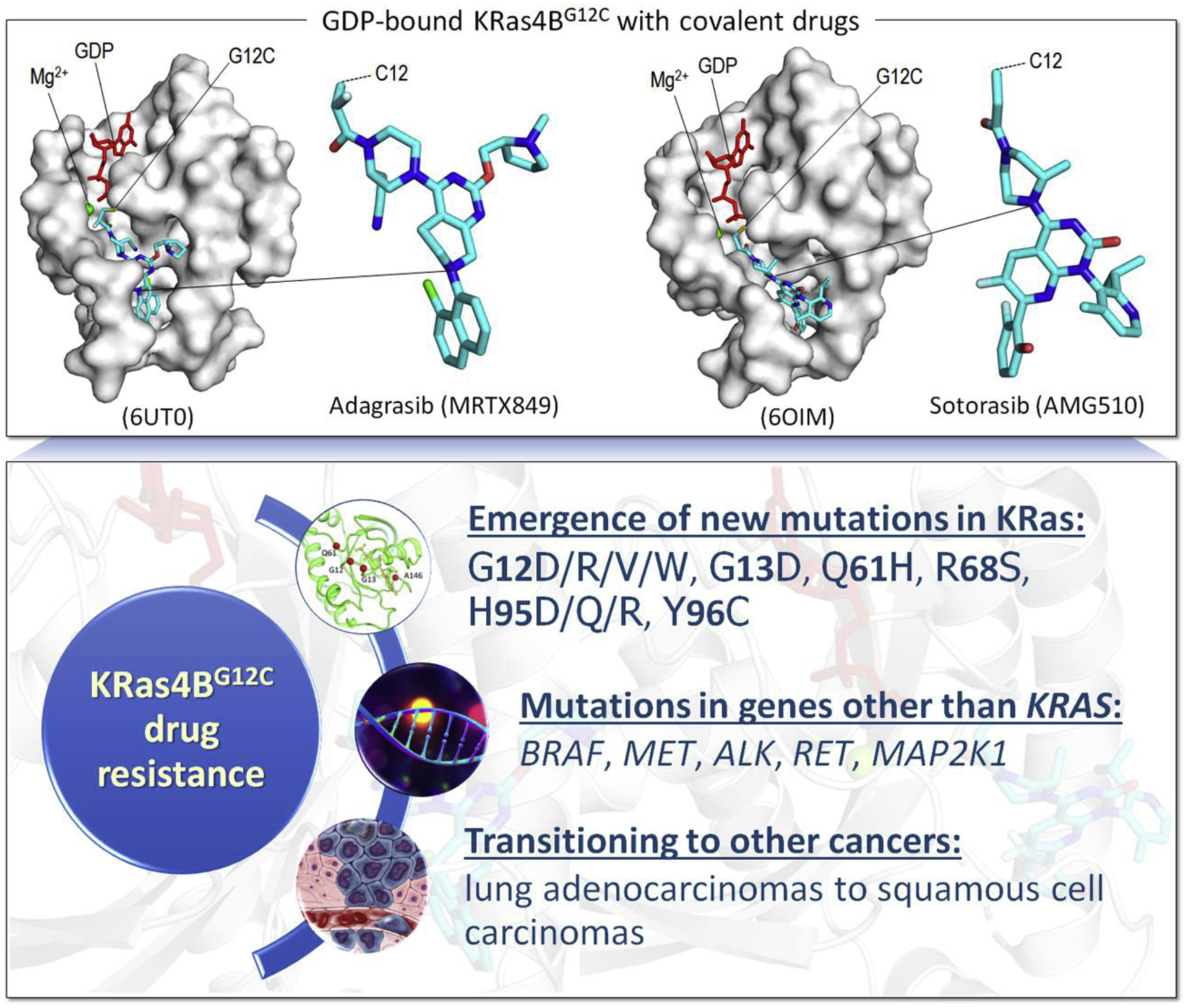
The mechanism of KRas4BG12C drug resistance. Examples of covalent drugs, adagrasib (MRTX849) and sotorasib (AMG510), bound to Cys12 of GDP-bound KRas4B (upper panel). The outcomes of drug resistance for a common driver mutation G12C in KRas4B (lower panel).
Cell type and background mutational load with the subsequent emergence and selection of new drivers critically influence the resistance mechanism. The pre-existing mutational burden already has more than a single driver, even though only the KRasG12C mutation was common in all patients. These mutations could be in the same gene (i.e., in cis), which is the case most of the time, or in different ones (in trans). In a pan-cancer analysis of 60,954 cancer samples Saito et al. (Saito et al., 2020) observed that in cis double/multiple driver mutations are more common than those in trans, in line with earlier observations (Kohsaka et al., 2017; Madsen et al., 2019). Especially, the background mutations need not be strong drivers. Functionally weak, or rare mutations, can confer enhanced oncogenicity in combination. Why the preference for the same gene versus different gene driver combinations? We believe that a second (or multiple) mutation emerging in the same gene has higher chances to elicit more potent outcome as compared to different genes, since many possible routes and key pathway nodes can engage in the same or parallel (or redundant) pathways (Nussinov et al., 2021a; Nussinov et al., 2020a). These relatively common mutational scenarios can clarify the clonal selection of suboptimal mutations that when coupled, can promote cancer emergence, tumor cell proliferation, progression and drug resistance.
The residual resistant population seeds regrowth of tumor cells that no longer respond to the drugs (Cree and Charlton, 2017). Even for a single cell, the resistance mechanism can be expected to be complex, encompassing both gene regulation at the chromatin level (Akdemir et al., 2020b) and resistance mutations. Drug resistance may be due to specific mutations, which in some cases it is, but in many others rapid resistance originates from multiple mechanisms (Di Nicolantonio et al., 2005; Glaysher et al., 2010; Glaysher et al., 2009), and especially as already detailed recently (Su et al., 2021), including also pre-existing resistance mutations. A tumor can compensate for EGFR (HER1 in humans) blockade through the activation of alternative signaling pathways such as amplification of MET as well as through changes in the TME (van der Wekken et al., 2016), or resistance to EGFR inhibition (van der Wekken et al., 2016), or neuroendocrine differentiation (Oser et al., 2015). Below we discuss regulation, the role of chromatin status and alternative signaling pathways which should be considered in combinatorial drug regimens as well as the heterogeneity of tumor clones and pre-existence of rare resistance mutations which might be captured by single-cell transcriptional changes associated with drug tolerance (Aissa et al., 2021). We also consider how can personalized strategies be predicted from pre-existing genomic and proteomic profiles.
4. The frequencies of common driver mutations vary with cell type and age
As examples, we focus on Ras drivers. Ras mutations have emerged in 27% of all human cancers (Hobbs et al., 2016). They occur in all Ras isoforms albeit at different frequencies (Altmuller et al., 2017; Bera et al., 2019; Blons et al., 2014; Bournet et al., 2016; Burd et al., 2014; Der, 2014; Lampson et al., 2013; Lu et al., 2016; Munoz-Maldonado et al., 2019; Newlaczyl, 2016; Nussinov et al., 2018; Nussinov et al., 2021c; Pellicer, 2011; Poulin et al., 2019; Prior et al., 2020; Prior et al., 2012; Pylayeva-Gupta et al., 2011; Rajasekharan and Raman, 2013; Raso, 2020; Rezaei Adariani et al., 2021; Russo et al., 2014; Simanshu et al., 2017; Stark et al., 2012; Stephens et al., 2017; Tsai et al., 2015; Xu et al., 2013; Yang and Kim, 2018). HRas is the least (4%), KRas is the most highly mutated (85%) whereas NRas is at 11% (Table 1). 98% of the mutations are at G12, G13, and Q61 positions in KRas. However, the mutation frequencies at these locations vary among the isoforms. G12 mutations predominate in KRas, whereas Q61 mutations are rare. On the other hand, Q61 is the dominant alteration in NRas. The relative frequencies of the other mutations and their ranked order vary as well among the isoforms. Even for the same isoform, the frequencies differ across cancer types. For example, NRas Q61 and G12 mutations are frequent in melanoma and acute myeloid leukemia (AML), respectively. On the other hand, among the KRas G12 mutations, G12D is the most common in pancreatic ductal adenocarcinoma (PDAC) followed by G12V, but not G13 and Q61. G12D is the predominant G12 mutation in KRas (41%) and NRas (52%), and G12V in HRas (57%) (Table 1). As to G13, G13D is the most frequent in KRas (89%) and NRas (50%) but it is rare in HRas (3%), where G13R (85%) dominates (Hobbs et al., 2016). KRas A146T mutation occurs in certain cancers but not in PDAC, and A146T is associated with better overall survival than G12 mutations of KRas. In general, mechanisms driving allelic selection in cancer reflect the activation mechanism of the mutation implicating the aggressiveness of the specific cancer (Poulin et al., 2019). G12V and G12D interfere with GTPase-activating protein (GAP)-assisted hydrolysis. Q61L mutation also reduces KRas intrinsic hydrolysis and GAP’s action and increases intrinsic nucleotide exchange (Hobbs et al., 2016; Smith et al., 2013). Likewise, G13D also interferes with GAP-mediated hydrolysis (Smith et al., 2013). Recent comprehensive characterization of Ras mutations in colon and rectal cancers in old and young patients (Serebriiskii et al., 2019) showed remarkable differences as well. Epidemiological stratification indicated that cancers expressing different mutant forms of KRas exhibit distinct clinical behaviors (Gibbs et al., 1984; Haigis, 2017; Hobbs et al., 2016; Ogura et al., 2013; Poulin et al., 2019; Witkiewicz et al., 2015).
Table 1.
Ras mutations in human cancer. The parentheses indicate the predominant mutations.
| Ras isoform | Frequency of mutations | G12 mutation | G13 mutation | Q61 mutation | Others |
|---|---|---|---|---|---|
| KRas | 85% | 83% (G12D 41%) | 14% (G13D 89%) | 2% (Q61H 58%) | 2% |
| NRas | 11% | 23% (G12D 52%) | 12% (G13D 50%) | 63% (Q61R 47%) | 3% |
| HRas | 4% | 33% (G12V 57%) | 27% (G13R 85%) | 37% (Q61R 43%) | 3% |
Tumors harbor tens to thousands of mutations (Mateo et al., 2020). The vast majority are passenger mutations, not observed to encode functional advantage. Analysis of over 9000 tumors from 33 tissues identified 258 genes with driver mutations (Bailey et al., 2018), 142 of which were from a single tumor type, and 87 from several types. The number of driver mutations in a single tumor type varies between 2 (kidney) to 55 (uterine). Especially relevant to our discussion, every patient has a unique combination of mutations and copy number variants (Rubio-Perez et al., 2015). These emphasize that tumor type and state, e.g., age as observed in Ras driver mutations (Serebriiskii et al., 2019). The differential background mutation load in the tumor cell are pivotal in tumor evolution (Gerstung et al., 2020), and we likewise expect in the emerging drug resistance mechanism.
5. Cell type, background mutation load and the complexity of forecasting emerging resistance mechanism
Vigorous divisions of a cell harboring a mutation that confers a selective advantage can make the cell an initiator of a fast-growing mutant clone. By the time the cancer is diagnosed, the tumor may already consist of ≥109 cells the replicating genomes of which accumulated a large number of mutations (Alberts et al., 2002; Beckman and Loeb, 2020; Offord, 2020). The emerging heterogeneous clonal populations can be viewed as substrates for cancer cell evolution, including those related to drug resistance. Thus, the rapid emergence of resistance to drugs, driven by the background mutational load, is not surprising. High drug doses may block the pathway. However, mutations that circumvent drug blockage and permit signaling – through the same pathway or via a by-pass pathway – can rapidly produce drug resistance, posing a massive therapeutic challenge. This underscores the critical question of forecasting drug resistance mechanism. Common driver mutations in a common protein, do not necessarily imply common resistance mechanisms (Figure 1).
To tackle drug resistance (National Cancer Institute, 2020; Volm and Efferth, 2015), efforts have been focusing on identifying mutations that impair drug binding in the active site cavity of the target protein, including the often so-called ‘gatekeeper’ residues, and on designing second generation drugs. Chemotherapy regimens mostly considered a combination of an orthosteric or allosteric drug targeting the mutant protein combined with drugs aiming at a downstream protein in the same pathway, or in an alternative signaling pathway that can take over in drug resistance (Nussinov et al., 2021b; Zhang et al., 2021, 2020). These additional proteins and pathways are largely selected based on experience, and drug availability (Cheng et al., 2019). EGFR mutants in NSCLC (~11%, (Neel and Bivona, 2017)) can provide an example (Lim and Ma, 2019). Secondary resistance mutations on the EGFR kinase domain can reduce the binding affinity of first- and second-generation drugs. A bypass pathway can take over. Sequentially prescribed drugs can temper resistance, only to cede to a small but fast growing subclonal population of resistant cells that becomes the dominant species. Additional examples include mutations in B-Raf (~7%), and gene rearrangements involving ALK and ROS1 (1–2%). These are protein kinases. The mutations hyperactivate downstream signaling promoting cell growth, proliferation, and survival (Neel and Bivona, 2017). Individualized genetic network approaches have been shown to reveal new therapeutic vulnerabilities in 6,700 cancer genomes promising new venue in such endeavors (Liu et al., 2020). Further, mutations that perturb protein-protein interaction networks were shown to be highly correlated with patient survival and drug resistance (and sensitivity) (Cheng et al., 2021).
The emerging mechanism of drug resistance depends on the cell type, cell state, and the set of background and evolving mutational load. A key factor that varies across cell types and dynamically fluctuates with cell state is epigenetics and chromatin accessibility of the regulatory regions of the respective genes (Akdemir et al., 2020b). Relatively lower density of chromatin can flag genes that can become available for transcription. In drug resistance, the expression level, and the potency of the mutation(s), which depends on the activation mechanism, are critical. Extremely high expression level – even on its own – can already drive cancer. However, it is not only the expression level of the mutant protein which is vital. The expression levels of all other protein nodes in the relevant signaling pathway should be at sufficiently high levels to allow the signal to propagate down to the cell cycle, to activate the corresponding transcription factors in gene expression.
6. Common driver mutation, genetic and epigenetic clone heterogeneity
Genomic instability and intratumoral heterogeneity are the driving force in drug resistance (Lim and Ma, 2019). Mutational burden, including in chromatin remodeler genes, and gene copy number alterations facilitate tumor evolution (Gerstung et al., 2020) and drug resistance. Even for a common driver mutation, such as the KRasG12C discussed above, the different tumor cell types (tissues) and states (in different patients) imply heterogeneity in epigenetics (Baylin and Jones, 2011; Bennett and Licht, 2018; Dawson and Kouzarides, 2012; Easwaran et al., 2014; Fardi et al., 2018; Feinberg et al., 2016; Flavahan et al., 2017; Lee et al., 2020; Nebbioso et al., 2018; Suva et al., 2013; You and Jones, 2012), chromatin organization and gene accessibility (Makova and Hardison, 2015). Heterogeneity implies that resistance mutations are likely to exist prior to treatment, including allosteric ones like the T315I (Abl1 isoform IA; T334I for Abl1 isoform IB) substitution that alters the ATP-binding site in the Bcr-Abl fusion tyrosine kinase discussed above which blocks the binding of imatinib, as well as second-generation TKIs such as nilotinib and dasatinib (Nicolini et al., 2013; O’Hare et al., 2005; Tamai et al., 2018; Zhang et al., 2010) (Figure 3). Screening drug libraries discovered the allosteric drug GNF-2 (Zhang et al., 2010) and second-generation GNF-5, as well as asciminib (ABL001) that bind to the Bcr-Abl C-terminal myristate pocket and re-sensitize it to imatinib and nilotinib. The allosteric agent GNF-5, together with imatinib, an ATP-competitive drug, overcame the drug resistance induced by T315I (Adrian et al., 2006; Wylie et al., 2017; Zhang et al., 2010). Thus, even though mutations at the myristate site confer resistance to asciminib, they retain sensitivity to nilotinib (or imatinib, or dasatinib), making their combined treatment successful (Schoepfer et al., 2018). Allosteric asciminib together with orthosteric ponatinib (AP24534) were found to strongly counter drug resistance mutations Y253H (Y272H for IB) and E255V (E274V for IB) (Eide et al., 2019). Thus, rare cells with rare pre-existing drug resistant mutations from the repertoire of background mutations can take over, and the mechanism of drug resistance that is again based on the T315I substitution, may or may not display common features.
Figure 3.
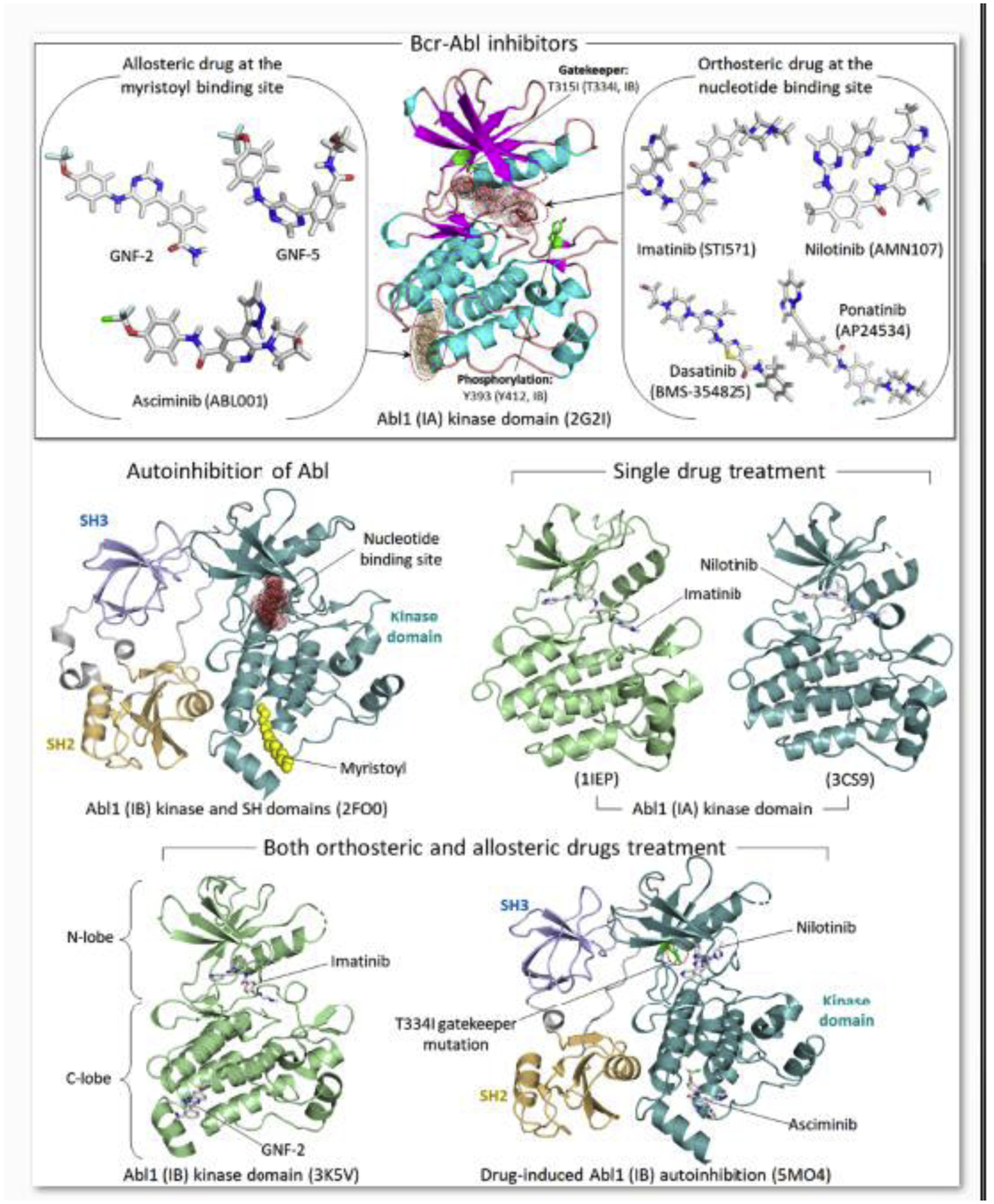
Inhibitors for Bcr-Abl fusion tyrosine kinase. Molecular structures of the drugs bound to the kinase domain of Abl (upper panel). Orthosteric ATP-competitive drugs, such as imatinib (STI571), nilotinib (AMN107), dasatinib (BMS-354825), and ponatinib (AP24534), can inhibit oncogenic Bcr-Abl kinase activity. However, drug resistance mutations in ATP-binding site, such as gatekeeper mutation T315I (Abl1 isoform IA; T334I for Abl1 isoform IB), or the activation loop can change conformation of the active site, preventing the orthosteric drugs from the oncogenic inhibition. Allosteric drugs bound to the myristate pocket, such as GNF-2, GNF-5 and asciminib (ABL001), can re-sensitize the orthosteric drugs, overcoming the drug resistant. Drug structures were obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov), a public chemical database at the National Library of Medicine (NLM) (Kim et al., 2021). Examples shown for the crystal structures of Abl1 autoinhibition, drug-bound kinase domains, and drug-induced Abl1 autoinhibition (lower panels).
7. Conclusions and perspectives
Resistance mechanisms include drug inactivation, qualitative and quantitative drug target alterations, drug efflux, enhanced DNA damage repair, cell death inhibition (Housman et al., 2014) as well as drug sequestration in lysosomes or vesicles away from the drug target. Recently, crosstalk with the TME has also been suggested to guide resistance evolution (Bhattacharya et al., 2021). However, the complexity of TME is daunting, and is only now beginning to be understood (Jin and Jin, 2020) and its pharmacology explored (Kaemmerer et al., 2021).
Resistance to kinase inhibitors is especially potent in the deadliest common cancers, such as pancreatic, lung, breast, colorectal, and prostate cancers (Geyer et al., 2006; Jonker et al., 2007; Knight et al., 2010; Miller et al., 2007; Moore et al., 2007; Sandler et al., 2006; Shepherd et al., 2005). Treatments which were given to those patients deemed with higher chances of favorable therapeutic response based on certain markers, fared better; however, treatments with only a single kinase inhibitor failed over time (Knight et al., 2010). This was not surprising since already early on, imatinib therapeutic regimen for Bcr-Abl fusion protein in chronic myeloid leukemia (CML) (Gorre et al., 2001), was observed to eventually lead to resistance mutations, such as the allosteric T315I mutation that interferes with imatinib binding as discussed above. ERBB2 (a.k.a. HER2) in breast cancer (Hudis, 2007), EGFR in lung cancer (Ciardiello and Tortora, 2008), and KRas in lung and colorectal cancer (Karapetis et al., 2008; Pao et al., 2005b) provide additional examples, as well as others [e.g., Refs. (Chen et al., 2004; Cools et al., 2003; Pao et al., 2005a; Tamborini et al., 2004)]. Emerging drug resistance can also involve combinations of mutations in the targeted kinase as well as with mutations in other proteins. These can involve constitutive mutations in a substitute kinase that can replace the drugged kinase, or alterations that lead to its overexpression. One such example involves the MET receptor tyrosine kinase in lung cancer cells becoming resistant to inhibitors following treatment of oncogenic mutant EGFR (Engelman et al., 2007). Alternatively, a mutation in the targeted kinase can couple with a mutation in a phosphatase whose inactivation can substitute the blocked kinase, as in the case of EGFR inhibitors in breast cancer cells (Sergina et al., 2007). Here, drug resistant cells respond by down regulating the phosphatase, thereby decreasing the potency of the kinase inhibitor. In another example, drug resistance to PI3K inhibitors can involve inactivation of phosphatase and tensin homolog (PTEN). The outcome of PTEN inactivation parallels that of PI3K activation (Carrera and Anderson, 2019; Jang et al., 2021; Lien et al., 2017; Papa and Pandolfi, 2019). Both result in an increase of the population of PIP3 (phosphatidylinositol 3,4,5-trisphosphate) signaling lipid molecules. Tumor cells also often overexpress redundant oncogenic signaling via multiple kinases, as in the case of multiple RTKs (Stommel et al., 2007). Activation of a downstream kinase can also decrease the action of an inhibitor, enabling the bypassing of the targeted kinase (Karapetis et al., 2008; Mellinghoff et al., 2005; Pao et al., 2005b). Thus, resistance can take place via multiple mechanisms including mutations in the respective targeted driver oncogene (Neel and Bivona, 2017), mutations that activate a protein downstream or a parallel pathway (Nussinov and Tsai, 2014; Nussinov et al., 2017), and pro-survival mutations via a different pathway. An independent class of drug resistance encompasses histological transformation from one tumor cell lineage to another, as discussed above for lung cancer.
Malignant cells within a tumor are not genetically identical, and this heterogeneity poses major problems in cancer therapy (Gatenby and Brown, 2018). The mutations may not emerge following drug treatment. Instead, at least some, are likely to preexist it. However, being rare in the population, they are undetected. Decimation of the sensitive population by first generation drugs leads to their proliferation, and drugs that could otherwise have prevented the cancers from recurring are largely ineffectual. As tumors evolve, they become more heterogeneous, with cancer cells acquiring new mutations that make them resistant to certain treatments. A comprehensive single-molecule sequencing study revealed patterns of preexisting drug resistance mutations that suggested treatment strategies in Philadelphia-positive (Ph+) leukemias (Schmitt et al., 2018). The study pointed to Abl1 resistance mutations unlikely present at the time of diagnosis, explaining the success of targeted therapy; at the same time, patients with Ph+ acute lymphoblastic leukemia (ALL) were observed to often harbor multiple preexisting resistant cells with single mutants which proliferated, following treatment with Abl1 inhibitor of cells harboring sensitive mutations.
Mutations in specific signaling nodes, such as kinases or receptors, often couple with alterations in chromatin structure and the transcription machinery. Mutations in the pioneer transcription factor, forkhead box protein A1 (FOXA1, a.k.a. HNF-3α, hepatocyte nuclear factor 3α), provide an example (Arruabarrena-Aristorena et al., 2020). The study uncovered mutations in two different regions that are localized at the C-terminal forkhead domain of FOXA1 in estrogen receptor-positive (ER+) breast cancers. The mutations in the Wing2 subdomain (residues 247–269) and SY242CS mutation in the third β strand, away from the Wing2 region, can lead to resistance to aromatase inhibitors in distinct ways. Wing2 or SY242CS mutations do not require estrogen receptor activation. Modeling suggested that the SY242CS mutation changes FOXA1 protein shape, apparently enabling chromatin exposure at altered sites, leading to altered gene expression. Wing2 mutations promote the ability of the cells to respond to the estrogen when available. To date, mechanistic details are unavailable. Importantly, FOXA1 mutations co-occur with other mutations in breast cancer-related genes (e.g., PIK3CA, AKT1, or ARID1A). Also notable is the involvement of alternative transcription factor motifs observed for other FOXA1 mutations (i.e., R219S and R219C) in prostate cancer as well as for other transcription factors (Arruabarrena-Aristorena et al., 2020). These lead to distinct patterns in chromatin accessibility.
Going forward (Knight et al., 2010), our perspective to alleviate resistance calls for targeting combinations of key nodes in tumor cells, adopting a multi-drug strategy that concomitantly targets multiple proteins (Pao et al., 2005b). The combination should consider targeting one kinase with multiple drugs, i.e., competitive and allosteric, targeting nodes in the signaling network in addition to the mutant protein, selecting drug targets from the same or from bypass (so-called redundant) pathways, and chromatin modifiers and RNA polymerases. The challenge is to predict the best combinations of targets and ranking them with some score. With the vast number of possible combinations, this is a formidable task. However, the evolution of artificial intelligence techniques (Nussinov et al., 2021a), and the increased gathering of data which their deep learning requires for reliable prediction, have been made possible by massive advances in compute intensive capabilities. That data is clinical, structural (of proteins and chromatin) and includes tested inhibitors in other settings, which can be used for drug repurposing. Altogether, such a challenging future perspective can become a reality. We believe that progress will come through massive computational involvement.
To conclude, treatment decisions in precision medicine are not made based on the location of the tumor, but on genomic sequencing and identification of the mutations. Here we reasoned that even though knowledge of the mutation is critical, the cell, or tumor type, is important to consider as well. Genomic sequences are essential to obtain all relevant data, chiefly of the driver mutations in question as well as the identity of the background mutational load. Data indicate that co-existing double/multiple mutations are more common in the same protein as compared to different proteins and pathways, and they demonstrate higher sensitivity to drugs. However, more complete information – if, or when, available – including epigenetic and chromatin accessibility, and clinical manifestation, can be powerful in forecasting the mechanisms underlying the expected emergence of drug resistance. The technology is not yet available for broad use, but future cell-specific developments portend more accurate predictions. Tumor evolution is a vastly complex process that is still not entirely understood. Here we highlighted the role of mutational load in tumor initiation, progression, and chemotherapy resistance, and argued for enlisting and levying the increasingly available massive computational power.
Acknowledgements
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261201500003I. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This Research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
The authors report no declarations of interest.
References
- Adrian FJ, Ding Q, Sim T, Velentza A, Sloan C, Liu Y, Zhang G, Hur W, Ding S, Manley P, Mestan J, Fabbro D, Gray NS, 2006. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat Chem Biol 2, 95–102. [DOI] [PubMed] [Google Scholar]
- Aissa AF, Islam A, Ariss MM, Go CC, Rader AE, Conrardy RD, Gajda AM, Rubio-Perez C, Valyi-Nagy K, Pasquinelli M, Feldman LE, Green SJ, Lopez-Bigas N, Frolov MV, Benevolenskaya EV, 2021. Single-cell transcriptional changes associated with drug tolerance and response to combination therapies in cancer. Nat Commun 12, 1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akdemir KC, Le VT, Chandran S, Li Y, Verhaak RG, Beroukhim R, Campbell PJ, Chin L, Dixon JR, Futreal PA, Group PSVW, Consortium P, 2020a. Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer. Nat Genet 52, 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akdemir KC, Le VT, Kim JM, Killcoyne S, King DA, Lin YP, Tian Y, Inoue A, Amin SB, Robinson FS, Nimmakayalu M, Herrera RE, Lynn EJ, Chan K, Seth S, Klimczak LJ, Gerstung M, Gordenin DA, O’Brien J, Li L, Deribe YL, Verhaak RG, Campbell PJ, Fitzgerald R, Morrison AJ, Dixon JR, Andrew Futreal P, 2020b. Somatic mutation distributions in cancer genomes vary with three-dimensional chromatin structure. Nat Genet 52, 1178–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P, Cancer as a Microevolutionary Process. Molecular Biology, Garland Science, New York: (2002). [Google Scholar]
- Aleksakhina SN, Kashyap A, Imyanitov EN, 2019. Mechanisms of acquired tumor drug resistance. Biochim Biophys Acta Rev Cancer 1872, 188310. [DOI] [PubMed] [Google Scholar]
- Altmuller F, Lissewski C, Bertola D, Flex E, Stark Z, Spranger S, Baynam G, Buscarilli M, Dyack S, Gillis J, Yntema HG, Pantaleoni F, van Loon RL, MacKay S, Mina K, Schanze I, Tan TY, Walsh M, White SM, Niewisch MR, Garcia-Minaur S, Plaza D, Ahmadian MR, Cave H, Tartaglia M, Zenker M, 2017. Genotype and phenotype spectrum of NRAS germline variants. Eur J Hum Genet 25, 823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrei L, Kasas S, Ochoa Garrido I, Stankovic T, Suarez Korsnes M, Vaclavikova R, Assaraf YG, Pesic M, 2020. Advanced technological tools to study multidrug resistance in cancer. Drug Resist Updat 48, 100658. [DOI] [PubMed] [Google Scholar]
- Arruabarrena-Aristorena A, Maag JLV, Kittane S, Cai Y, Karthaus WR, Ladewig E, Park J, Kannan S, Ferrando L, Cocco E, Ho SY, Tan DS, Sallaku M, Wu F, Acevedo B, Selenica P, Ross DS, Witkin M, Sawyers CL, Reis-Filho JS, Verma CS, Jauch R, Koche R, Baselga J, Razavi P, Toska E, Scaltriti M, 2020. FOXA1 Mutations Reveal Distinct Chromatin Profiles and Influence Therapeutic Response in Breast Cancer. Cancer Cell 38, 534–550 e539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assaraf YG, Brozovic A, Goncalves AC, Jurkovicova D, Line A, Machuqueiro M, Saponara S, Sarmento-Ribeiro AB, Xavier CPR, Vasconcelos MH, 2019. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist Updat 46, 100645. [DOI] [PubMed] [Google Scholar]
- Assaraf YG, Molina A, Schimke RT, 1989. Sequential amplification of dihydrofolate reductase and multidrug resistance genes in Chinese hamster ovary cells selected for stepwise resistance to the lipid-soluble antifolate trimetrexate. J Biol Chem 264, 18326–18334. [PubMed] [Google Scholar]
- Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, Johnson ML, Heist RS, Patil T, Riely GJ, Jacobson JO, Yang X, Persky NS, Root DE, Lowder KE, Feng H, Zhang SS, Haigis KM, Hung YP, Sholl LM, Wolpin BM, Wiese J, Christiansen J, Lee J, Schrock AB, Lim LP, Garg K, Li M, Engstrom LD, Waters L, Lawson JD, Olson P, Lito P, Ou SI, Christensen JG, Janne PA, Aguirre AJ, 2021. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N Engl J Med 384, 2382–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, Ng PK, Jeong KJ, Cao S, Wang Z, Gao J, Gao Q, Wang F, Liu EM, Mularoni L, Rubio-Perez C, Nagarajan N, Cortes-Ciriano I, Zhou DC, Liang WW, Hess JM, Yellapantula VD, Tamborero D, Gonzalez-Perez A, Suphavilai C, Ko JY, Khurana E, Park PJ, Van Allen EM, Liang H, Group, M.C.W., Cancer Genome Atlas Research, N., Lawrence MS, Godzik A, Lopez-Bigas N, Stuart J, Wheeler D, Getz G, Chen K, Lazar AJ, Mills GB, Karchin R, Ding L, 2018. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–385 e318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Zeev M, Livney YD, Assaraf YG, 2017. Targeted nanomedicine for cancer therapeutics: Towards precision medicine overcoming drug resistance. Drug Resist Updat 31, 15–30. [DOI] [PubMed] [Google Scholar]
- Baylin SB, Jones PA, 2011. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer 11, 726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman RA, Loeb LA, 2020. Rare Mutations in Cancer Drug Resistance and Implications for Therapy. Clin Pharmacol Ther 108, 437–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett RL, Licht JD, 2018. Targeting Epigenetics in Cancer. Annu Rev Pharmacol Toxicol 58, 187–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bera AK, Lu J, Wales TE, Gondi S, Gurbani D, Nelson A, Engen JR, Westover KD, 2019. Structural basis of the atypical activation mechanism of KRAS(V14I). J Biol Chem 294, 13964–13972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhang HE, Ruddy DA, Krishnamurthy Radhakrishna V, Caushi JX, Zhao R, Hims MM, Singh AP, Kao I, Rakiec D, Shaw P, Balak M, Raza A, Ackley E, Keen N, Schlabach MR, Palmer M, Leary RJ, Chiang DY, Sellers WR, Michor F, Cooke VG, Korn JM, Stegmeier F, 2015. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat Med 21, 440–448. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Mohanty A, Achuthan S, Kotnala S, Jolly MK, Kulkarni P, Salgia R, 2021. Group Behavior and Emergence of Cancer Drug Resistance. Trends Cancer 7, 323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blons H, Emile JF, Le Malicot K, Julie C, Zaanan A, Tabernero J, Mini E, Folprecht G, Van Laethem JL, Thaler J, Bridgewater J, Norgard-Petersen L, Van Cutsem E, Lepage C, Zawadi MA, Salazar R, Laurent-Puig P, Taieb J, 2014. Prognostic value of KRAS mutations in stage III colon cancer: post hoc analysis of the PETACC8 phase III trial dataset. Ann Oncol 25, 2378–2385. [DOI] [PubMed] [Google Scholar]
- Bournet B, Muscari F, Buscail C, Assenat E, Barthet M, Hammel P, Selves J, Guimbaud R, Cordelier P, Buscail L, 2016. KRAS G12D Mutation Subtype Is A Prognostic Factor for Advanced Pancreatic Adenocarcinoma. Clin Transl Gastroenterol 7, e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousquet G, Varna M, Ferreira I, Wang L, Mongiat-Artus P, Leboeuf C, de Bazelaire C, Faivre S, Bertheau P, Raymond E, Germain S, Janin A, 2013. Differential regulation of sunitinib targets predicts its tumor-type-specific effect on endothelial and/or tumor cell apoptosis. Cancer Chemother Pharmacol 72, 1183–1193. [DOI] [PubMed] [Google Scholar]
- Bram EE, Ifergan I, Grimberg M, Lemke K, Skladanowski A, Assaraf YG, 2007. C421 allele-specific ABCG2 gene amplification confers resistance to the antitumor triazoloacridone C-1305 in human lung cancer cells. Biochem Pharmacol 74, 41–53. [DOI] [PubMed] [Google Scholar]
- Brown AL, Li M, Goncearenco A, Panchenko AR, 2019. Finding driver mutations in cancer: Elucidating the role of background mutational processes. PLoS Comput Biol 15, e1006981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck SAJ, Koolen SLW, Mathijssen RHJ, de Wit R, van Soest RJ, 2021. Cross-resistance and drug sequence in prostate cancer. Drug Resist Updat 56, 100761. [DOI] [PubMed] [Google Scholar]
- Burchert A, 2007. Roots of imatinib resistance: a question of self-renewal? Drug Resist Updat 10, 152–161. [DOI] [PubMed] [Google Scholar]
- Burd CE, Liu W, Huynh MV, Waqas MA, Gillahan JE, Clark KS, Fu K, Martin BL, Jeck WR, Souroullas GP, Darr DB, Zedek DC, Miley MJ, Baguley BC, Campbell SL, Sharpless NE, 2014. Mutation-specific RAS oncogenicity explains NRAS codon 61 selection in melanoma. Cancer Discov 4, 1418–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrera AC, Anderson R, 2019. The cell biology behind the oncogenic PIP3 lipids. J Cell Sci 132, [DOI] [PubMed] [Google Scholar]
- Chen H, Li J, Wang Y, Ng PK, Tsang YH, Shaw KR, Mills GB, Liang H, 2020. Comprehensive assessment of computational algorithms in predicting cancer driver mutations. Genome Biol 21, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LL, Trent JC, Wu EF, Fuller GN, Ramdas L, Zhang W, Raymond AK, Prieto VG, Oyedeji CO, Hunt KK, Pollock RE, Feig BW, Hayes KJ, Choi H, Macapinlac HA, Hittelman W, Velasco MA, Patel S, Burgess MA, Benjamin RS, Frazier ML, 2004. A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer Res 64, 5913–5919. [DOI] [PubMed] [Google Scholar]
- Cheng F, Liang H, Butte AJ, Eng C, Nussinov R, 2019. Personal Mutanomes Meet Modern Oncology Drug Discovery and Precision Health. Pharmacol Rev 71, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F, Zhao J, Wang Y, Lu W, Liu Z, Zhou Y, Martin WR, Wang R, Huang J, Hao T, Yue H, Ma J, Hou Y, Castrillon JA, Fang J, Lathia JD, Keri RA, Lightstone FC, Antman EM, Rabadan R, Hill DE, Eng C, Vidal M, Loscalzo J, 2021. Comprehensive characterization of protein-protein interactions perturbed by disease mutations. Nat Genet 53, 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappa M, Guffanti F, Bertoni F, Colombo I, Damia G, 2021. Overcoming PARPi resistance: Preclinical and clinical evidence in ovarian cancer. Drug Resist Updat 55, 100744. [DOI] [PubMed] [Google Scholar]
- Ciardiello F, Tortora G, 2008. EGFR antagonists in cancer treatment. N Engl J Med 358, 1160–1174. [DOI] [PubMed] [Google Scholar]
- Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, Kutok J, Clark J, Galinsky I, Griffin JD, Cross NC, Tefferi A, Malone J, Alam R, Schrier SL, Schmid J, Rose M, Vandenberghe P, Verhoef G, Boogaerts M, Wlodarska I, Kantarjian H, Marynen P, Coutre SE, Stone R, Gilliland DG, 2003. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med 348, 1201–1214. [DOI] [PubMed] [Google Scholar]
- Cree IA, Charlton P, 2017. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 17, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crudden C, Ilic M, Suleymanova N, Worrall C, Girnita A, Girnita L, 2015. The dichotomy of the Insulin-like growth factor 1 receptor: RTK and GPCR: friend or foe for cancer treatment? Growth Horm IGF Res 25, 2–12. [DOI] [PubMed] [Google Scholar]
- Czarnecka AM, Brodziak A, Sobczuk P, Dendek C, Labochka D, Korniluk J, Bartnik E, Szczylik C, 2019. Metastatic Tumor Burden and Loci as Predictors of First Line Sunitinib Treatment Efficacy in Patients with Renal Cell Carcinoma. Sci Rep 9, 7754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana-Farber Cancer Institute, New research uncovers how cancers with common mutation develop resistance to targeted drugs. Medical Research News, Medical Xpress, Science X Network (2021), p. Science X Network. [Google Scholar]
- Das T, Anand U, Pandey SK, Ashby CR Jr., Assaraf YG, Chen ZS, Dey A, 2021. Therapeutic strategies to overcome taxane resistance in cancer. Drug Resist Updat 55, 100754. [DOI] [PubMed] [Google Scholar]
- Dawson MA, Kouzarides T, 2012. Cancer epigenetics: from mechanism to therapy. Cell 150, 12–27. [DOI] [PubMed] [Google Scholar]
- Der CJ, Are All RAS Proteins Created Equal in Cancer?, National Cancer Institute; (2014). [Google Scholar]
- Di Nicolantonio F, Mercer SJ, Knight LA, Gabriel FG, Whitehouse PA, Sharma S, Fernando A, Glaysher S, Di Palma S, Johnson P, Somers SS, Toh S, Higgins B, Lamont A, Gulliford T, Hurren J, Yiangou C, Cree IA, 2005. Cancer cell adaptation to chemotherapy. BMC Cancer 5, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Gu J, Li J, 2020. Mechanisms of drug resistance of pancreatic ductal adenocarcinoma at different levels. Biosci Rep 40, BSR20200401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunnett-Kane V, Nicola P, Blackhall F, Lindsay C, 2021. Mechanisms of Resistance to KRAS(G12C) Inhibitors. Cancers (Basel) 13, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easwaran H, Tsai HC, Baylin SB, 2014. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell 54, 716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle J, Kurbanov BM, Hossini AM, Trefzer U, Fecker LF, 2007. Overcoming apoptosis deficiency of melanoma-hope for new therapeutic approaches. Drug Resist Updat 10, 218–234. [DOI] [PubMed] [Google Scholar]
- Eide CA, Zabriskie MS, Savage Stevens SL, Antelope O, Vellore NA, Than H, Schultz AR, Clair P, Bowler AD, Pomicter AD, Yan D, Senina AV, Qiang W, Kelley TW, Szankasi P, Heinrich MC, Tyner JW, Rea D, Cayuela JM, Kim DW, Tognon CE, O’Hare T, Druker BJ, Deininger MW, 2019. Combining the Allosteric Inhibitor Asciminib with Ponatinib Suppresses Emergence of and Restores Efficacy against Highly Resistant BCR-ABL1 Mutants. Cancer Cell 36, 431–443 e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA, 2007. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Fardi M, Solali S, Farshdousti Hagh M, 2018. Epigenetic mechanisms as a new approach in cancer treatment: An updated review. Genes Dis 5, 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP, Koldobskiy MA, Gondor A, 2016. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet 17, 284–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan WA, Gaskell E, Bernstein BE, 2017. Epigenetic plasticity and the hallmarks of cancer. Science 357, eaal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman R, 2016. Drug resistance in cancer: molecular evolution and compensatory proliferation. Oncotarget 7, 11746–11755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Wu ZX, Assaraf YG, Chen ZS, Wang L, 2021. Overcoming anti-cancer drug resistance via restoration of tumor suppressor gene function. Drug Resist Updat 57, 100770. [DOI] [PubMed] [Google Scholar]
- Garraway LA, Janne PA, 2012. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov 2, 214–226. [DOI] [PubMed] [Google Scholar]
- Gatenby R, Brown J, 2018. The Evolution and Ecology of Resistance in Cancer Therapy. Cold Spring Harb Perspect Med 8, [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Genovese I, Ilari A, Assaraf YG, Fazi F, Colotti G, 2017. Not only P-glycoprotein: Amplification of the ABCB1-containing chromosome region 7q21 confers multidrug resistance upon cancer cells by coordinated overexpression of an assortment of resistance-related proteins. Drug Resist Updat 32, 23–46. [DOI] [PubMed] [Google Scholar]
- Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Rosebrock D, Mitchell TJ, Rubanova Y, Anur P, Yu K, Tarabichi M, Deshwar A, Wintersinger J, Kleinheinz K, Vazquez-Garcia I, Haase K, Jerman L, Sengupta S, Macintyre G, Malikic S, Donmez N, Livitz DG, Cmero M, Demeulemeester J, Schumacher S, Fan Y, Yao X, Lee J, Schlesner M, Boutros PC, Bowtell DD, Zhu H, Getz G, Imielinski M, Beroukhim R, Sahinalp SC, Ji Y, Peifer M, Markowetz F, Mustonen V, Yuan K, Wang W, Morris QD, Evolution P, Heterogeneity Working G, Spellman PT, Wedge DC, Van Loo P, Consortium P, 2020. The evolutionary history of 2,658 cancers. Nature 578, 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getzenberg RH, Coffey DS, 2011. Changing the energy habitat of the cancer cell in order to impact therapeutic resistance. Mol Pharm 8, 2089–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, Jagiello-Gruszfeld A, Crown J, Chan A, Kaufman B, Skarlos D, Campone M, Davidson N, Berger M, Oliva C, Rubin SD, Stein S, Cameron D, 2006. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med 355, 2733–2743. [DOI] [PubMed] [Google Scholar]
- Gibbs JB, Sigal IS, Poe M, Scolnick EM, 1984. Intrinsic GTPase activity distinguishes normal and oncogenic ras p21 molecules. Proc Natl Acad Sci U S A 81, 5704–5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis NK, McLeod HL, 2016. The pharmacogenomics of drug resistance to protein kinase inhibitors. Drug Resist Updat 28, 28–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaysher S, Gabriel FG, Johnson P, Polak M, Knight LA, Parker K, Poole M, Narayanan A, Cree IA, Oncology, N.H.S.C.R.P.f.P., 2010. Molecular basis of chemosensitivity of platinum pre-treated ovarian cancer to chemotherapy. Br J Cancer 103, 656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaysher S, Yiannakis D, Gabriel FG, Johnson P, Polak ME, Knight LA, Goldthorpe Z, Peregrin K, Gyi M, Modi P, Rahamim J, Smith ME, Amer K, Addis B, Poole M, Narayanan A, Gulliford TJ, Andreotti PE, Cree IA, 2009. Resistance gene expression determines the in vitro chemosensitivity of non-small cell lung cancer (NSCLC). BMC Cancer 9, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goler-Baron V, Sladkevich I, Assaraf YG, 2012. Inhibition of the PI3K-Akt signaling pathway disrupts ABCG2-rich extracellular vesicles and overcomes multidrug resistance in breast cancer cells. Biochem Pharmacol 83, 1340–1348. [DOI] [PubMed] [Google Scholar]
- Gonen N, Assaraf YG, 2012. Antifolates in cancer therapy: structure, activity and mechanisms of drug resistance. Drug Resist Updat 15, 183–210. [DOI] [PubMed] [Google Scholar]
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL, 2001. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 293, 876–880. [DOI] [PubMed] [Google Scholar]
- Gottesman MM, 2002. Mechanisms of cancer drug resistance. Annu Rev Med 53, 615–627. [DOI] [PubMed] [Google Scholar]
- Gottesman MM, Fojo T, Bates SE, 2002. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2, 48–58. [DOI] [PubMed] [Google Scholar]
- Gremke N, Polo P, Dort A, Schneikert J, Elmshauser S, Brehm C, Klingmuller U, Schmitt A, Reinhardt HC, Timofeev O, Wanzel M, Stiewe T, 2020. mTOR-mediated cancer drug resistance suppresses autophagy and generates a druggable metabolic vulnerability. Nat Commun 11, 4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider T, Pandey V, Banjare N, Gupta PN, Soni V, 2020. Drug resistance in cancer: mechanisms and tackling strategies. Pharmacol Rep 72, 1125–1151. [DOI] [PubMed] [Google Scholar]
- Haigis KM, 2017. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 3, 686–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbinski F, Craig VJ, Sanghavi S, Jeffery D, Liu L, Sheppard KA, Wagner S, Stamm C, Buness A, Chatenay-Rivauday C, Yao Y, He F, Lu CX, Guagnano V, Metz T, Finan PM, Hofmann F, Sellers WR, Porter JA, Myer VE, Graus-Porta D, Wilson CJ, Buckler A, Tiedt R, 2012. Rescue screens with secreted proteins reveal compensatory potential of receptor tyrosine kinases in driving cancer growth. Cancer Discov 2, 948–959. [DOI] [PubMed] [Google Scholar]
- Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, Maruvka YE, Ji F, Bhang HE, Krishnamurthy Radhakrishna V, Siravegna G, Hu H, Raoof S, Lockerman E, Kalsy A, Lee D, Keating CL, Ruddy DA, Damon LJ, Crystal AS, Costa C, Piotrowska Z, Bardelli A, Iafrate AJ, Sadreyev RI, Stegmeier F, Getz G, Sequist LV, Faber AC, Engelman JA, 2016. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med 22, 262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt S, Zeilmann A, Ahadova A, Blaker H, von Knebel Doeberitz M, Kloor M, Heuveline V, 2021. Mathematical modeling of multiple pathways in colorectal carcinogenesis using dynamical systems with Kronecker structure. PLoS Comput Biol 17, e1008970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs GA, Der CJ, Rossman KL, 2016. RAS isoforms and mutations in cancer at a glance. J Cell Sci 129, 1287–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S, 2014. Drug resistance in cancer: an overview. Cancers (Basel) 6, 1769–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudis CA, 2007. Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med 357, 39–51. [DOI] [PubMed] [Google Scholar]
- Hussein NA, Malla S, Pasternak MA, Terrero D, Brown NG, Ashby CR Jr., Assaraf YG, Chen ZS, Tiwari AK, 2021. The role of endolysosomal trafficking in anticancer drug resistance. Drug Resist Updat 57, 100769. [DOI] [PubMed] [Google Scholar]
- Ifergan I, Scheffer GL, Assaraf YG, 2005. Novel extracellular vesicles mediate an ABCG2-dependent anticancer drug sequestration and resistance. Cancer Res 65, 10952–10958. [DOI] [PubMed] [Google Scholar]
- Jang H, Smith IN, Eng C, Nussinov R, 2021. The mechanism of full activation of tumor suppressor PTEN at the phosphoinositide-enriched membrane. iScience 24, 102438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Chen X, Ren X, Yang JM, Cheng Y, 2021. Emerging role of autophagy in anti-tumor immunity: Implications for the modulation of immunotherapy resistance. Drug Resist Updat 56, 100752. [DOI] [PubMed] [Google Scholar]
- Jiang W, Xia J, Xie S, Zou R, Pan S, Wang ZW, Assaraf YG, Zhu X, 2020. Long non-coding RNAs as a determinant of cancer drug resistance: Towards the overcoming of chemoresistance via modulation of lncRNAs. Drug Resist Updat 50, 100683. [DOI] [PubMed] [Google Scholar]
- Jin MZ, Jin WL, 2020. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther 5, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, Asthana S, Jalbert LE, Nelson SJ, Bollen AW, Gustafson WC, Charron E, Weiss WA, Smirnov IV, Song JS, Olshen AB, Cha S, Zhao Y, Moore RA, Mungall AJ, Jones SJM, Hirst M, Marra MA, Saito N, Aburatani H, Mukasa A, Berger MS, Chang SM, Taylor BS, Costello JF, 2014. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343, 189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes RJ, Tebbutt NC, van Hazel G, Wierzbicki R, Langer C, Moore MJ, 2007. Cetuximab for the treatment of colorectal cancer. N Engl J Med 357, 2040–2048. [DOI] [PubMed] [Google Scholar]
- Kaemmerer E, Loessner D, Avery VM, 2021. Addressing the tumour microenvironment in early drug discovery: a strategy to overcome drug resistance and identify novel targets for cancer therapy. Drug Discov Today 26, 663–676. [DOI] [PubMed] [Google Scholar]
- Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, Langer C, Moore MJ, Zalcberg JR, 2008. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 359, 1757–1765. [DOI] [PubMed] [Google Scholar]
- Kim D, Bach DH, Fan YH, Luu TT, Hong JY, Park HJ, Lee SK, 2019. AXL degradation in combination with EGFR-TKI can delay and overcome acquired resistance in human non-small cell lung cancer cells. Cell Death Dis 10, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J-Y, 2021. Chapter Five - FPGA based neural network accelerators. Advances in Computers 122, 135–165. [Google Scholar]
- Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, Li Q, Shoemaker BA, Thiessen PA, Yu B, Zaslavsky L, Zhang J, Bolton EE, 2021. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res 49, D1388–D1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight ZA, Lin H, Shokat KM, 2010. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer 10, 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoechel B, Aster JC, 2015. Metabolic Mechanisms of Drug Resistance in Leukemia. Cell Metab 22, 759–760. [DOI] [PubMed] [Google Scholar]
- Kohsaka S, Nagano M, Ueno T, Suehara Y, Hayashi T, Shimada N, Takahashi K, Suzuki K, Takamochi K, Takahashi F, Mano H, 2017. A method of high-throughput functional evaluation of EGFR gene variants of unknown significance in cancer. Sci Transl Med 9, eaan6566. [DOI] [PubMed] [Google Scholar]
- Kumar R, Li DQ, Muller S, Knapp S, 2016. Epigenomic regulation of oncogenesis by chromatin remodeling. Oncogene 35, 4423–4436. [DOI] [PubMed] [Google Scholar]
- La Monica S, Minari R, Cretella D, Flammini L, Fumarola C, Bonelli M, Cavazzoni A, Digiacomo G, Galetti M, Madeddu D, Falco A, Lagrasta CA, Squadrilli A, Barocelli E, Romanel A, Quaini F, Petronini PG, Tiseo M, Alfieri R, 2019. Third generation EGFR inhibitor osimertinib combined with pemetrexed or cisplatin exerts long-lasting anti-tumor effect in EGFR-mutated pre-clinical models of NSCLC. J Exp Clin Cancer Res 38, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampson BL, Pershing NL, Prinz JA, Lacsina JR, Marzluff WF, Nicchitta CV, MacAlpine DM, Counter CM, 2013. Rare codons regulate KRas oncogenesis. Curr Biol 23, 70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantermann AB, Chen D, McCutcheon K, Hoffman G, Frias E, Ruddy D, Rakiec D, Korn J, McAllister G, Stegmeier F, Meyer MJ, Sharma SV, 2015. Inhibition of Casein Kinase 1 Alpha Prevents Acquired Drug Resistance to Erlotinib in EGFR-Mutant Non-Small Cell Lung Cancer. Cancer Res 75, 4937–4948. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, Drier Y, Zou L, Ramos AH, Pugh TJ, Stransky N, Helman E, Kim J, Sougnez C, Ambrogio L, Nickerson E, Shefler E, Cortes ML, Auclair D, Saksena G, Voet D, Noble M, DiCara D, Lin P, Lichtenstein L, Heiman DI, Fennell T, Imielinski M, Hernandez B, Hodis E, Baca S, Dulak AM, Lohr J, Landau DA, Wu CJ, Melendez-Zajgla J, Hidalgo-Miranda A, Koren A, McCarroll SA, Mora J, Crompton B, Onofrio R, Parkin M, Winckler W, Ardlie K, Gabriel SB, Roberts CWM, Biegel JA, Stegmaier K, Bass AJ, Garraway LA, Meyerson M, Golub TR, Gordenin DA, Sunyaev S, Lander ES, Getz G, 2013. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Tourneau C, Servois V, Dieras V, Ollivier L, Tresca P, Paoletti X, 2012. Tumour growth kinetics assessment: added value to RECIST in cancer patients treated with molecularly targeted agents. Br J Cancer 106, 854–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Raffaghello L, Longo VD, 2012. Starvation, detoxification, and multidrug resistance in cancer therapy. Drug Resist Updat 15, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Rauch J, Kolch W, 2020. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int J Mol Sci 21, 1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin M, Stark M, Ofran Y, Assaraf YG, 2021. Deciphering molecular mechanisms underlying chemoresistance in relapsed AML patients: towards precision medicine overcoming drug resistance. Cancer Cell Int 21, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Jiang J, Assaraf YG, Xiao H, Chen ZS, Huang C, 2020. Surmounting cancer drug resistance: New insights from the perspective of N(6)-methyladenosine RNA modification. Drug Resist Updat 53, 100720. [DOI] [PubMed] [Google Scholar]
- Li W, Zhang H, Assaraf YG, Zhao K, Xu X, Xie J, Yang DH, Chen ZS, 2016. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist Updat 27, 14–29. [DOI] [PubMed] [Google Scholar]
- Lien EC, Dibble CC, Toker A, 2017. PI3K signaling in cancer: beyond AKT. Curr Opin Cell Biol 45, 62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ZF, Ma PC, 2019. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J Hematol Oncol 12, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Zhao J, Lu W, Dai Y, Hockings J, Zhou Y, Nussinov R, Eng C, Cheng F, 2020. Individualized genetic network analysis reveals new therapeutic vulnerabilities in 6,700 cancer genomes. PLoS Comput Biol 16, e1007701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb LA, Kohrn BF, Loubet-Senear KJ, Dunn YJ, Ahn EH, O’Sullivan JN, Salk JJ, Bronner MP, Beckman RA, 2019. Extensive subclonal mutational diversity in human colorectal cancer and its significance. Proc Natl Acad Sci U S A 116, 26863–26872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long L, Assaraf YG, Lei ZN, Peng H, Yang L, Chen ZS, Ren S, 2020. Genetic biomarkers of drug resistance: A compass of prognosis and targeted therapy in acute myeloid leukemia. Drug Resist Updat 52, 100703. [DOI] [PubMed] [Google Scholar]
- Lu S, Jang H, Muratcioglu S, Gursoy A, Keskin O, Nussinov R, Zhang J, 2016. Ras Conformational Ensembles, Allostery, and Signaling. Chem Rev 116, 6607–6665. [DOI] [PubMed] [Google Scholar]
- Madsen RR, Knox RG, Pearce W, Lopez S, Mahler-Araujo B, McGranahan N, Vanhaesebroeck B, Semple RK, 2019. Oncogenic PIK3CA promotes cellular stemness in an allele dose-dependent manner. Proc Natl Acad Sci U S A 116, 8380–8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makova KD, Hardison RC, 2015. The effects of chromatin organization on variation in mutation rates in the genome. Nat Rev Genet 16, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B, 2017. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv Pharm Bull 7, 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Campbell PJ, 2015. Somatic mutation in cancer and normal cells. Science 349, 1483–1489. [DOI] [PubMed] [Google Scholar]
- Mateo L, Duran-Frigola M, Gris-Oliver A, Palafox M, Scaltriti M, Razavi P, Chandarlapaty S, Arribas J, Bellet M, Serra V, Aloy P, 2020. Personalized cancer therapy prioritization based on driver alteration co-occurrence patterns. Genome Med 12, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH, Chute DJ, Riggs BL, Horvath S, Liau LM, Cavenee WK, Rao PN, Beroukhim R, Peck TC, Lee JC, Sellers WR, Stokoe D, Prados M, Cloughesy TF, Sawyers CL, Mischel PS, 2005. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 353, 2012–2024. [DOI] [PubMed] [Google Scholar]
- Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, Shenkier T, Cella D, Davidson NE, 2007. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med 357, 2666–2676. [DOI] [PubMed] [Google Scholar]
- Mitchell TJ, Turajlic S, Rowan A, Nicol D, Farmery JHR, O’Brien T, Martincorena I, Tarpey P, Angelopoulos N, Yates LR, Butler AP, Raine K, Stewart GD, Challacombe B, Fernando A, Lopez JI, Hazell S, Chandra A, Chowdhury S, Rudman S, Soultati A, Stamp G, Fotiadis N, Pickering L, Au L, Spain L, Lynch J, Stares M, Teague J, Maura F, Wedge DC, Horswell S, Chambers T, Litchfield K, Xu H, Stewart A, Elaidi R, Oudard S, McGranahan N, Csabai I, Gore M, Futreal PA, Larkin J, Lynch AG, Szallasi Z, Swanton C, Campbell PJ, Consortium TRR, 2018. Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell 173, 611–623 e617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W, National Cancer Institute of Canada Clinical Trials, G., 2007. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 25, 1960–1966. [DOI] [PubMed] [Google Scholar]
- Morjaria S, 2021. Driver mutations in oncogenesis. International Journal of Molecular & Immuno Oncology 6, 100–102. [Google Scholar]
- Munoz-Maldonado C, Zimmer Y, Medova M, 2019. A Comparative Analysis of Individual RAS Mutations in Cancer Biology. Front Oncol 9, 1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan S, Cai CY, Assaraf YG, Guo HQ, Cui Q, Wei L, Huang JJ, Ashby CR Jr., Chen ZS, 2020. Targeting the ubiquitin-proteasome pathway to overcome anti-cancer drug resistance. Drug Resist Updat 48, 100663. [DOI] [PubMed] [Google Scholar]
- National Cancer Institute, Cancer Drug Resistance: Unraveling Its Complexity. National Cancer Institute; (2020). [Google Scholar]
- Nebbioso A, Tambaro FP, Dell’Aversana C, Altucci L, 2018. Cancer epigenetics: Moving forward. PLoS Genet 14, e1007362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neel DS, Bivona TG, 2017. Resistance is futile: overcoming resistance to targeted therapies in lung adenocarcinoma. NPJ Precis Oncol 1, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newlaczyl AU, Isoform-specific Ras expression and signalling. University of Liverpool, Liverpool L69 3BX, United Kingdom: (2016). [Google Scholar]
- Nguyen G, Šipková V, Dlugolinsky S, Nguyen BM, Tran V, Hluchý L, 2021. A comparative study of operational engineering for environmental and compute-intensive applications. Array 12, 100096. [Google Scholar]
- Nicolini FE, Ibrahim AR, Soverini S, Martinelli G, Muller MC, Hochhaus A, Dufva IH, Kim DW, Cortes J, Mauro MJ, Chuah C, Labussiere H, Morisset S, Roche-Lestienne C, Lippert E, Hayette S, Peter S, Zhou W, Maguer-Satta V, Michallet M, Goldman J, Apperley JF, Mahon FX, Marin D, Etienne G, 2013. The BCR-ABLT315I mutation compromises survival in chronic phase chronic myelogenous leukemia patients resistant to tyrosine kinase inhibitors, in a matched pair analysis. Haematologica 98, 1510–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Jang H, Nir G, Tsai CJ, Cheng F, 2021a. A new precision medicine initiative at the dawn of exascale computing. Signal Transduct Target Ther 6, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Jang H, Tsai CJ, 2014. The structural basis for cancer treatment decisions. Oncotarget 5, 7285–7302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Jang H, Tsai CJ, Cheng F, 2019a. Precision medicine review: rare driver mutations and their biophysical classification. Biophys Rev 11, 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Jang H, Tsai CJ, Cheng F, 2019b. Review: Precision medicine and driver mutations: Computational methods, functional assays and conformational principles for interpreting cancer drivers. PLoS Comput Biol 15, e1006658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, 2015. ‘Latent drivers’ expand the cancer mutational landscape. Curr Opin Struct Biol 32, 25–32. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, 2014. Unraveling structural mechanisms of allosteric drug action. Trends Pharmacol Sci 35, 256–264. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, Jang H, 2020a. Are Parallel Proliferation Pathways Redundant? Trends Biochem Sci 45, 554–563. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, Jang H, 2020b. Autoinhibition can identify rare driver mutations and advise pharmacology. FASEB J 34, 16–29. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, Jang H, 2017. A New View of Pathway-Driven Drug Resistance in Tumor Proliferation. Trends Pharmacol Sci 38, 427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, Jang H, 2018. Oncogenic Ras Isoforms Signaling Specificity at the Membrane. Cancer Res 78, 593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, Jang H, 2019c. Why Are Some Driver Mutations Rare? Trends Pharmacol Sci 40, 919–929. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Zhang M, Maloney R, Jang H, 2021b. Drugging multiple same-allele driver mutations in cancer. Expert Opin Drug Discov 16, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Zhang M, Maloney R, Jang H, 2021c. Ras isoform-specific expression, chromatin accessibility, and signaling. Biophys Rev 13, 489–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hare T, Walters DK, Stoffregen EP, Jia T, Manley PW, Mestan J, Cowan-Jacob SW, Lee FY, Heinrich MC, Deininger MW, Druker BJ, 2005. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res 65, 4500–4505. [DOI] [PubMed] [Google Scholar]
- Offord C, Cancer Researchers Use Evolution to Target Drug Resistance. The Scientist (2020). [Google Scholar]
- Ogura T, Yamao K, Hara K, Mizuno N, Hijioka S, Imaoka H, Sawaki A, Niwa Y, Tajika M, Kondo S, Tanaka T, Shimizu Y, Bhatia V, Higuchi K, Hosoda W, Yatabe Y, 2013. Prognostic value of K-ras mutation status and subtypes in endoscopic ultrasound-guided fine-needle aspiration specimens from patients with unresectable pancreatic cancer. J Gastroenterol 48, 640–646. [DOI] [PubMed] [Google Scholar]
- Oser MG, Niederst MJ, Sequist LV, Engelman JA, 2015. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol 16, e165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H, 2005a. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2, e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG, Varmus HE, 2005b. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa A, Pandolfi PP, 2019. The PTEN(−)PI3K Axis in Cancer. Biomolecules 9, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecoraro C, Faggion B, Balboni B, Carbone D, Peters GJ, Diana P, Assaraf YG, Giovannetti E, 2021. GSK3beta as a novel promising target to overcome chemoresistance in pancreatic cancer. Drug Resist Updat 58, 100779. [DOI] [PubMed] [Google Scholar]
- Pellicer A, RAS Genes, Springer, Berlin, Heidelberg: (2011). [Google Scholar]
- Pon JR, Marra MA, 2015. Driver and passenger mutations in cancer. Annu Rev Pathol 10, 25–50. [DOI] [PubMed] [Google Scholar]
- Pontiggia O, Sampayo R, Raffo D, Motter A, Xu R, Bissell MJ, Joffe EB, Simian M, 2012. The tumor microenvironment modulates tamoxifen resistance in breast cancer: a role for soluble stromal factors and fibronectin through beta1 integrin. Breast Cancer Res Treat 133, 459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin EJ, Bera AK, Lu J, Lin YJ, Strasser SD, Paulo JA, Huang TQ, Morales C, Yan W, Cook J, Nowak JA, Brubaker DK, Joughin BA, Johnson CW, DeStefanis RA, Ghazi PC, Gondi S, Wales TE, Iacob RE, Bogdanova L, Gierut JJ, Li Y, Engen JR, Perez-Mancera PA, Braun BS, Gygi SP, Lauffenburger DA, Westover KD, Haigis KM, 2019. Tissue-Specific Oncogenic Activity of KRAS(A146T). Cancer Discov 9, 738–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto-Vila M, Usuba W, Takahashi RU, Shimomura I, Sasaki H, Ochiya T, Yamamoto Y, 2019. Single-Cell Analysis Reveals a Preexisting Drug-Resistant Subpopulation in the Luminal Breast Cancer Subtype. Cancer Res 79, 4412–4425. [DOI] [PubMed] [Google Scholar]
- Prior IA, Hood FE, Hartley JL, 2020. The Frequency of Ras Mutations in Cancer. Cancer Res 80, 2969–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior IA, Lewis PD, Mattos C, 2012. A comprehensive survey of Ras mutations in cancer. Cancer Res 72, 2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D, 2011. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 11, 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekharan SK, Raman T, 2013. Ras and Ras mutations in cancer. Cent Eur J Biol 8, 609–624. [Google Scholar]
- Raso E, 2020. Splice variants of RAS-translational significance. Cancer Metastasis Rev 39, 1039–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JG, Baretti M, Gerold JM, Makohon-Moore AP, Daud A, Iacobuzio-Donahue CA, Azad NS, Kinzler KW, Nowak MA, Vogelstein B, 2019. An analysis of genetic heterogeneity in untreated cancers. Nat Rev Cancer 19, 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezaei Adariani S, Kazemein Jasemi NS, Bazgir F, Wittich C, Amin E, Seidel CAM, Dvorsky R, Ahmadian MR, 2021. A comprehensive analysis of RAS-effector interactions reveals interaction hotspots and new binding partners. J Biol Chem 296, 100626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM, 2018. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer 18, 452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolfo C, Fanale D, Hong DS, Tsimberidou AM, Piha-Paul SA, Pauwels P, Van Meerbeeck JP, Caruso S, Bazan V, Cicero G, Russo A, Giovannetti E, 2014. Impact of microRNAs in resistance to chemotherapy and novel targeted agents in non-small cell lung cancer. Curr Pharm Biotechnol 15, 475–485. [DOI] [PubMed] [Google Scholar]
- Ruan T, Liu W, Tao K, Wu C, 2020. A Review of Research Progress in Multidrug-Resistance Mechanisms in Gastric Cancer. Onco Targets Ther 13, 1797–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Perez C, Tamborero D, Schroeder MP, Antolin AA, Deu-Pons J, Perez-Llamas C, Mestres J, Gonzalez-Perez A, Lopez-Bigas N, 2015. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell 27, 382–396. [DOI] [PubMed] [Google Scholar]
- Russo AL, Borger DR, Szymonifka J, Ryan DP, Wo JY, Blaszkowsky LS, Kwak EL, Allen JN, Wadlow RC, Zhu AX, Murphy JE, Faris JE, Dias-Santagata D, Haigis KM, Ellisen LW, Iafrate AJ, Hong TS, 2014. Mutational analysis and clinical correlation of metastatic colorectal cancer. Cancer 120, 1482–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabnis AJ, Bivona TG, 2019. Principles of Resistance to Targeted Cancer Therapy: Lessons from Basic and Translational Cancer Biology. Trends Mol Med 25, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Koya J, Araki M, Kogure Y, Shingaki S, Tabata M, McClure MB, Yoshifuji K, Matsumoto S, Isaka Y, Tanaka H, Kanai T, Miyano S, Shiraishi Y, Okuno Y, Kataoka K, 2020. Landscape and function of multiple mutations within individual oncogenes. Nature 582, 95–99. [DOI] [PubMed] [Google Scholar]
- Saleem H, Kulsoom Abdul U, Kucukosmanoglu A, Houweling M, Cornelissen FMG, Heiland DH, Hegi ME, Kouwenhoven MCM, Bailey D, Wurdinger T, Westerman BA, 2019. The TICking clock of EGFR therapy resistance in glioblastoma: Target Independence or target Compensation. Drug Resist Updat 43, 29–37. [DOI] [PubMed] [Google Scholar]
- Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, Lilenbaum R, Johnson DH, 2006. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 355, 2542–2550. [DOI] [PubMed] [Google Scholar]
- Sarmento-Ribeiro AB, Scorilas A, Goncalves AC, Efferth T, Trougakos IP, 2019. The emergence of drug resistance to targeted cancer therapies: Clinical evidence. Drug Resist Updat 47, 100646. [DOI] [PubMed] [Google Scholar]
- Schmitt MW, Pritchard JR, Leighow SM, Aminov BI, Beppu L, Kim DS, Hodgson JG, Rivera VM, Loeb LA, Radich JP, 2018. Single-Molecule Sequencing Reveals Patterns of Preexisting Drug Resistance That Suggest Treatment Strategies in Philadelphia-Positive Leukemias. Clin Cancer Res 24, 5321–5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepfer J, Jahnke W, Berellini G, Buonamici S, Cotesta S, Cowan-Jacob SW, Dodd S, Drueckes P, Fabbro D, Gabriel T, Groell JM, Grotzfeld RM, Hassan AQ, Henry C, Iyer V, Jones D, Lombardo F, Loo A, Manley PW, Pelle X, Rummel G, Salem B, Warmuth M, Wylie AA, Zoller T, Marzinzik AL, Furet P, 2018. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J Med Chem 61, 8120–8135. [DOI] [PubMed] [Google Scholar]
- Sciarrillo R, Wojtuszkiewicz A, Assaraf YG, Jansen G, Kaspers GJL, Giovannetti E, Cloos J, 2020. The role of alternative splicing in cancer: From oncogenesis to drug resistance. Drug Resist Updat 53, 100728. [DOI] [PubMed] [Google Scholar]
- Serebriiskii IG, Connelly C, Frampton G, Newberg J, Cooke M, Miller V, Ali S, Ross JS, Handorf E, Arora S, Lieu C, Golemis EA, Meyer JE, 2019. Comprehensive characterization of RAS mutations in colon and rectal cancers in old and young patients. Nat Commun 10, 3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, Moasser MM, 2007. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 445, 437–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seton-Rogers S, 2020. KRAS-G12C in the crosshairs. Nat Rev Cancer 20, 3. [DOI] [PubMed] [Google Scholar]
- Shahar N, Larisch S, 2020. Inhibiting the inhibitors: Targeting anti-apoptotic proteins in cancer and therapy resistance. Drug Resist Updat 52, 100712. [DOI] [PubMed] [Google Scholar]
- Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, Tu D, Johnston D, Bezjak A, Clark G, Santabarbara P, Seymour L, National Cancer Institute of Canada Clinical Trials, G., 2005. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 353, 123–132. [DOI] [PubMed] [Google Scholar]
- Simanshu DK, Nissley DV, McCormick F, 2017. RAS Proteins and Their Regulators in Human Disease. Cell 170, 17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MJ, Neel BG, Ikura M, 2013. NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc Natl Acad Sci U S A 110, 4574–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark Z, Gillessen-Kaesbach G, Ryan MM, Cirstea IC, Gremer L, Ahmadian MR, Savarirayan R, Zenker M, 2012. Two novel germline KRAS mutations: expanding the molecular and clinical phenotype. Clin Genet 81, 590–594. [DOI] [PubMed] [Google Scholar]
- Stephens RM, Yi M, Kessing B, Nissley DV, McCormick F, 2017. Tumor RAS Gene Expression Levels Are Influenced by the Mutational Status of RAS Genes and Both Upstream and Downstream RAS Pathway Genes. Cancer Inform 16, 1176935117711944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, Stegh AH, Bradner JE, Ligon KL, Brennan C, Chin L, DePinho RA, 2007. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 318, 287–290. [DOI] [PubMed] [Google Scholar]
- Su Z, Dong S, Zhao SC, Liu K, Tan Y, Jiang X, Assaraf YG, Qin B, Chen ZS, Zou C, 2021. Novel nanomedicines to overcome cancer multidrug resistance. Drug Resist Updat 58, 100777. [DOI] [PubMed] [Google Scholar]
- Sun T, Zhao Q, Zhang C, Cao L, Song M, Maimela NR, Liu S, Wang J, Gao Q, Qin G, Wang L, Zhang Y, 2019. Screening common signaling pathways associated with drug resistance in non-small cell lung cancer via gene expression profile analysis. Cancer Med 8, 3059–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suva ML, Riggi N, Bernstein BE, 2013. Epigenetic reprogramming in cancer. Science 339, 1567–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamai M, Inukai T, Kojika S, Abe M, Kagami K, Harama D, Shinohara T, Watanabe A, Oshiro H, Akahane K, Goi K, Sugihara E, Nakada S, Sugita K, 2018. T315I mutation of BCR-ABL1 into human Philadelphia chromosome-positive leukemia cell lines by homologous recombination using the CRISPR/Cas9 system. Sci Rep 8, 9966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamborini E, Bonadiman L, Greco A, Albertini V, Negri T, Gronchi A, Bertulli R, Colecchia M, Casali PG, Pierotti MA, Pilotti S, 2004. A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient. Gastroenterology 127, 294–299. [DOI] [PubMed] [Google Scholar]
- Taniguchi H, Yamada T, Wang R, Tanimura K, Adachi Y, Nishiyama A, Tanimoto A, Takeuchi S, Araujo LH, Boroni M, Yoshimura A, Shiotsu S, Matsumoto I, Watanabe S, Kikuchi T, Miura S, Tanaka H, Kitazaki T, Yamaguchi H, Mukae H, Uchino J, Uehara H, Takayama K, Yano S, 2019. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat Commun 10, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, Fish P, Harsha B, Hathaway C, Jupe SC, Kok CY, Noble K, Ponting L, Ramshaw CC, Rye CE, Speedy HE, Stefancsik R, Thompson SL, Wang S, Ward S, Campbell PJ, Forbes SA, 2019. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 47, D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S, Spugnini EP, Assaraf YG, Azzarito T, Rauch C, Fais S, 2015. Microenvironment acidity as a major determinant of tumor chemoresistance: Proton pump inhibitors (PPIs) as a novel therapeutic approach. Drug Resist Updat 23, 69–78. [DOI] [PubMed] [Google Scholar]
- Tomasetti C, Marchionni L, Nowak MA, Parmigiani G, Vogelstein B, 2015. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc Natl Acad Sci U S A 112, 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai FD, Lopes MS, Zhou M, Court H, Ponce O, Fiordalisi JJ, Gierut JJ, Cox AD, Haigis KM, Philips MR, 2015. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc Natl Acad Sci U S A 112, 779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wekken AJ, Saber A, Hiltermann TJ, Kok K, van den Berg A, Groen HJ, 2016. Resistance mechanisms after tyrosine kinase inhibitors afatinib and crizotinib in non-small cell lung cancer, a review of the literature. Crit Rev Oncol Hematol 100, 107–116. [DOI] [PubMed] [Google Scholar]
- van Gastel N, Spinelli JB, Sharda A, Schajnovitz A, Baryawno N, Rhee C, Oki T, Grace E, Soled HJ, Milosevic J, Sykes DB, Hsu PP, Vander Heiden MG, Vidoudez C, Trauger SA, Haigis MC, Scadden DT, 2020. Induction of a Timed Metabolic Collapse to Overcome Cancer Chemoresistance. Cell Metab 32, 391–403 e396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasan N, Baselga J, Hyman DM, 2019a. A view on drug resistance in cancer. Nature 575, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasan N, Razavi P, Johnson JL, Shao H, Shah H, Antoine A, Ladewig E, Gorelick A, Lin TY, Toska E, Xu G, Kazmi A, Chang MT, Taylor BS, Dickler MN, Jhaveri K, Chandarlapaty S, Rabadan R, Reznik E, Smith ML, Sebra R, Schimmoller F, Wilson TR, Friedman LS, Cantley LC, Scaltriti M, Baselga J, 2019b. Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kalpha inhibitors. Science 366, 714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volm M, Efferth T, 2015. Prediction of Cancer Drug Resistance and Implications for Personalized Medicine. Front Oncol 5, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JQ, Yang Y, Cai CY, Teng QX, Cui Q, Lin J, Assaraf YG, Chen ZS, 2021. Multidrug resistance proteins (MRPs): Structure, function and the overcoming of cancer multidrug resistance. Drug Resist Updat 54, 100743. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhang H, Chen X, 2019. Drug resistance and combating drug resistance in cancer. Cancer Drug Resistance 2, 141–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijdeven RH, Pang B, Assaraf YG, Neefjes J, 2016. Old drugs, novel ways out: Drug resistance toward cytotoxic chemotherapeutics. Drug Resist Updat 28, 65–81. [DOI] [PubMed] [Google Scholar]
- Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, Ribas A, Li J, Moffat J, Sutherlin DP, Koeppen H, Merchant M, Neve R, Settleman J, 2012. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 487, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, Choti MA, Yeo CJ, McCue P, White MA, Knudsen ES, 2015. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 6, 6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtkowiak JW, Verduzco D, Schramm KJ, Gillies RJ, 2011. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol Pharm 8, 2032–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie AA, Schoepfer J, Jahnke W, Cowan-Jacob SW, Loo A, Furet P, Marzinzik AL, Pelle X, Donovan J, Zhu W, Buonamici S, Hassan AQ, Lombardo F, Iyer V, Palmer M, Berellini G, Dodd S, Thohan S, Bitter H, Branford S, Ross DM, Hughes TP, Petruzzelli L, Vanasse KG, Warmuth M, Hofmann F, Keen NJ, Sellers WR, 2017. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 543, 733–737. [DOI] [PubMed] [Google Scholar]
- Xu J, Haigis KM, Firestone AJ, McNerney ME, Li Q, Davis E, Chen SC, Nakitandwe J, Downing J, Jacks T, Le Beau MM, Shannon K, 2013. Dominant role of oncogene dosage and absence of tumor suppressor activity in Nras-driven hematopoietic transformation. Cancer Discov 3, 993–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Lin M, Pan S, Assaraf YG, Wang ZW, Zhu X, 2020. Emerging roles of F-box proteins in cancer drug resistance. Drug Resist Updat 49, 100673. [DOI] [PubMed] [Google Scholar]
- Yang IS, Kim S, 2018. Isoform specific gene expression analysis of KRAS in the prognosis of lung adenocarcinoma patients. BMC Bioinformatics 19, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You JS, Jones PA, 2012. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell 22, 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaal EA, Berkers CR, 2018. The Influence of Metabolism on Drug Response in Cancer. Front Oncol 8, 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, Sim T, Powers J, Dierks C, Sun F, Guo GR, Ding Q, Okram B, Choi Y, Wojciechowski A, Deng X, Liu G, Fendrich G, Strauss A, Vajpai N, Grzesiek S, Tuntland T, Liu Y, Bursulaya B, Azam M, Manley PW, Engen JR, Daley GQ, Warmuth M, Gray NS, 2010. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature 463, 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Jang H, Nussinov R, 2021. PI3K Driver Mutations: A Biophysical Membrane-Centric Perspective. Cancer Res 81, 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Jang H, Nussinov R, 2020. PI3K inhibitors: review and new strategies. Chem Sci 11, 5855–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhitomirsky B, Assaraf YG, 2015. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget 6, 1143–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhitomirsky B, Assaraf YG, 2016. Lysosomes as mediators of drug resistance in cancer. Drug Resist Updat 24, 23–33. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Virshup DM, 2020. Wnt Signaling and Drug Resistance in Cancer. Mol Pharmacol 97, 72–89. [DOI] [PubMed] [Google Scholar]
- Zhu C, Wei Y, Wei X, 2019. AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer 18, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zsakai L, Sipos A, Dobos J, Eros D, Szantai-Kis C, Banhegyi P, Pato J, Orfi L, Matula Z, Mikala G, Keri G, Petak I, Valyi-Nagy I, 2019. Targeted drug combination therapy design based on driver genes. Oncotarget 10, 5255–5266. [DOI] [PMC free article] [PubMed] [Google Scholar]