

Abstract
Nucleic acid delivery has applications ranging from tissue engineering to vaccine development to infectious disease. Cationic polymer condensed nucleic acids have been used with surface coated porous scaffolds and are able to promote long term gene expression. However, due to surface loading of the scaffold, there is a limit to the amount of nucleic acid that can be loaded, resulting in decreasing expression rate over time. In addition, surface coated scaffolds are generally non-injectable. Here, we demonstrate that cationic polymer condensed nucleic acids can be effectively loaded into injectable granular hydrogel scaffolds by stabilizing the condensed nucleic acid into a lyophilized powder, loading the powder into a bulk hydrogel, and then fragmenting the loaded hydrogel. The resulting hydrogel microparticles contain non-aggregated nucleic acid particles, can be annealed post-injection to result in an injectable microporous hydrogel, and can effectively deliver nucleic acids to embedded cells with a constant expression rate. Due to the nature of granular hydrogels, we demonstrate that mixtures of loaded and unloaded particles and spatially resolved gene expression can be easily achieved. The ability to express genes long term from an injectable porous hydrogel will further open the applications of nucleic acid delivery.
Keywords: gene delivery, non-viral, FLIP hydrogel, porous, DNA/PEI particle
Graphical Abstract
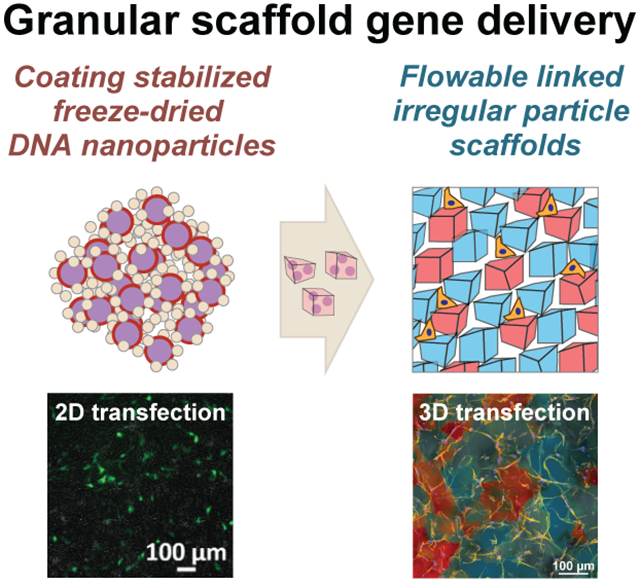
Freeze-dried hyaluronic acid coated DNA/PEI particles result in the same or improved levels of transgene expression in cultured cells and can be incorporated into hydrogel scaffolds. DNA loaded hydrogel scaffolds can then be fragmented and reassembled into a tunable, microporous Flowable Linked Irregular Particle scaffold that achieves high levels of transfection over time.
Nucleic acid delivery has applications ranging from tissue engineering to vaccine development and infectious disease. Viruses or plasmids can deliver transgenes encoding recombinant proteins or native bioactive signals[1,2], as well as gene regulatory systems such as RNA interference or gene editing technologies[3,4]. Most approaches to deliver nucleic material involve the systemic intravenous (IV) delivery of condensed nucleic acids (e.g. within a virus or a synthetic particle). However, this approach suffers from immune recognition, accumulation in first pass organs, and rapid clearance[5]. This includes the FDA approved gene therapies onasemnogene abeparvovec (intravenous AAV) and patisiran (intravenous siRNA lipid nanoparticle), in addition to the recently authorized SARS-CoV-2 vaccines (intramuscular injection of mRNA lipid nanoparticles or adenovirus). In the cases for nanoparticles, the nucleic acids are intended to enter circulation, either to accumulate in the liver for therapeutic delivery[6,7] or to interact with the immune system and elicit a response[8,9]. While viruses can be engineered for tissue-specific tropism, nonviral methods have largely been limited to short-term delivery applications due to their rapid clearance[5]. To circumvent these limitations and make nonviral delivery applicable for long-term, therapeutic gene expression, local delivery approaches that inject and retain the nucleic acid cargo at the desired tissue are beneficial. Hydrogel-mediated nucleic acid delivery has been investigated for decades, with different levels of success in pre-clinical models. The main reasons for lack of success are immune recognition of the nucleic acid or delivery vector[10–12], aggregation of condensed particles within the scaffold[13–16], lack of cellular infiltration into the scaffold, and insufficient nucleic acid release[17,18]. Thus, approaches to improve local delivery require addressing both the incorporation and release of active nucleic acids from scaffolds and facilitating cellular infiltration.
Here, we report on efficient nucleic acid delivery using a combined stable lyophilized nanoparticle formulation and an injectable microporous hydrogel scaffold, assembled from nucleic acid-loaded building blocks. Although direct incorporation of naked nucleic acid into hydrogels works in vivo[19] for transfection, immune system recognition prevented clinical success. The use of a condensation agent or vehicle to generate nanosized particles may improve transfection efficiency and prevent immune recognition[20–23], but challenges exist when combining with hydrogels for delivery. Most vehicles result in positively charged particles, which tend to aggregate under physiological conditions[24,25], at high concentrations[26,27], during incorporation into hydrogel scaffolds[14,16,27,28], and are actively recognized and silenced by the immune system[5,12,29]. There has been success with vehicles that contain poly(ethylene glycol)[15,24,25] or zwitterionic coronas[30–32] to promote stabilization and prevent immune cell recognition. However, these modifications generally result in lower gene transfer efficiency than the parent delivery vehicle, and thus lower transgene expression when incorporated into hydrogels. Approaches to surface coat porous scaffolds rather than encapsulate nucleic acid particles have resulted in superior transgene expression than the same particles delivered as a bolus[13,33–35]. Cationic polymer nucleic acid delivery from surface coated porous scaffolds also results in longer-term gene expression[36]. However, surface-loading of the scaffold limits the amount that can be adsorbed, resulting in decreased expression rates over time. In addition, cationic polymer nucleic acid-loaded scaffolds are generally non-injectable[15], limiting their applications.
Thus, our first goal was to generate a coating technology for positively charged, condensed nucleic acids that maintains the transfection efficiency of the parent vector, while allowing for concentration into a hydrogel precursor solution to yield non-aggregated, active particles. We used the common linear poly(ethylene imine) (PEI) polymer to condense plasmid DNA, as these particles are known to be highly charged, unstable at physiological conditions and at concentrations higher than 0.1 μg/μL, yet are highly effective at delivering nucleic acids[37,38]. For in vivo transfection, doses of DNA are generally in the 0.1-1.0 mg/kg for IV administration and 0.5-5.0 mg/kg for local delivery, such as intramuscular injection. Thus, concentrations above 0.5 μg/μL within hydrogels are desirable, which are difficult to achieve with surface-loading. Past developments in hydrogel-loading with nucleic acids were limited to 0.02-0.1 mg/mL due to aggregation at higher concentrations[15,27,39]. Previously we developed a caged nanoparticle encapsulation approach that protected condensed nucleic acids from aggregation during lyophilization and allowed effective incorporation of the nucleic acid inside hydrogel scaffolds at concentrations up to 5 μg/μL[27,40]. While this was effective in scaffolds comprised of collagen, fibrin, or PEG, the use of inert, insoluble agarose as a stabilizing agent increased the viscosity of resuspended DNA/PEI particles, which impacted both the scaffold mechanical properties and cellular infiltration. Eliminating agarose while optimizing sucrose concentration improved particle stability and distribution in bulk gels but failed to transfect cells efficiently. Thus, alternative formulations were needed to both reduce particle aggregation and improve transfection ability.
We reasoned that using hyaluronic acid (HA), a native extracellular matrix component and common material used in hydrogels, rather than agarose could effectively coat DNA/PEI particles, improve solubility, reverse or neutralize the charge of the particles, and maintain transfection efficiency of the parent PEI vector. HA has been previously used to stabilize a variety of condensed nucleic acid particles, including chitosan[26,41,42], PAMAM dendrimers[43,44], and PEI[45,46]. In each case, HA reduced toxicity of the parent vector and improve particle stability. Here we explored acrylamide and norbornene modified HA, which are commonly used to generate hydrogels through click-chemistry via Michael addition or radical-induced crosslinking, respectively[47–49]. While non-modified HA can interact with cationic nanoparticles electrostatically, functional groups such as norbornene may introduce a hydrophobicity effect, as has been found with other transfection reagents, compared to the less polar acrylamide modification. There are also benefits of using norbornene as it can participate in biorthogonal chemistries that do not react with biologically occurring functional groups, through the cycloaddition reaction with tetrazine. We first assessed whether HA-coated DNA/PEI particles (HA-DNA/PEI) retained their physical properties. We used dynamic light scattering (DLS) and electrophoretic light scattering (ELS) to determine particle size and zeta potential. DNA/PEI particles were coated with three types of HA ranging in degree of COOH modification and molecular weight. The percent-free COOH groups changed from 100% for native non-modified HA to 49% for norbornene modified HA (HA-NB), and 17% for acrylamide modified (HA-AC), respectively. Note that HA-AC requires adipic dihydrazine (ADH) to be conjugated to HA prior to the addition of AC, so the actual modification was first 83% with ADH, and then 28% for AC, which resulted in the remaining 17% free COOH. The functionalization altered the molecular weight from native 79 kDa to 88 kDa and 112 kD for HA-NB and HA-AC, respectively [Figure 1a, Figure S1a–b]. From ELS on the three HA materials (1 mg/mL, 15 mM NaCl), it was determined that their zeta potentials were −32.0±2.1 mV, −15.9±0.9 mV, and −20.0±1.2 mV, respectively [Figure S1c–d].
Figure 1.
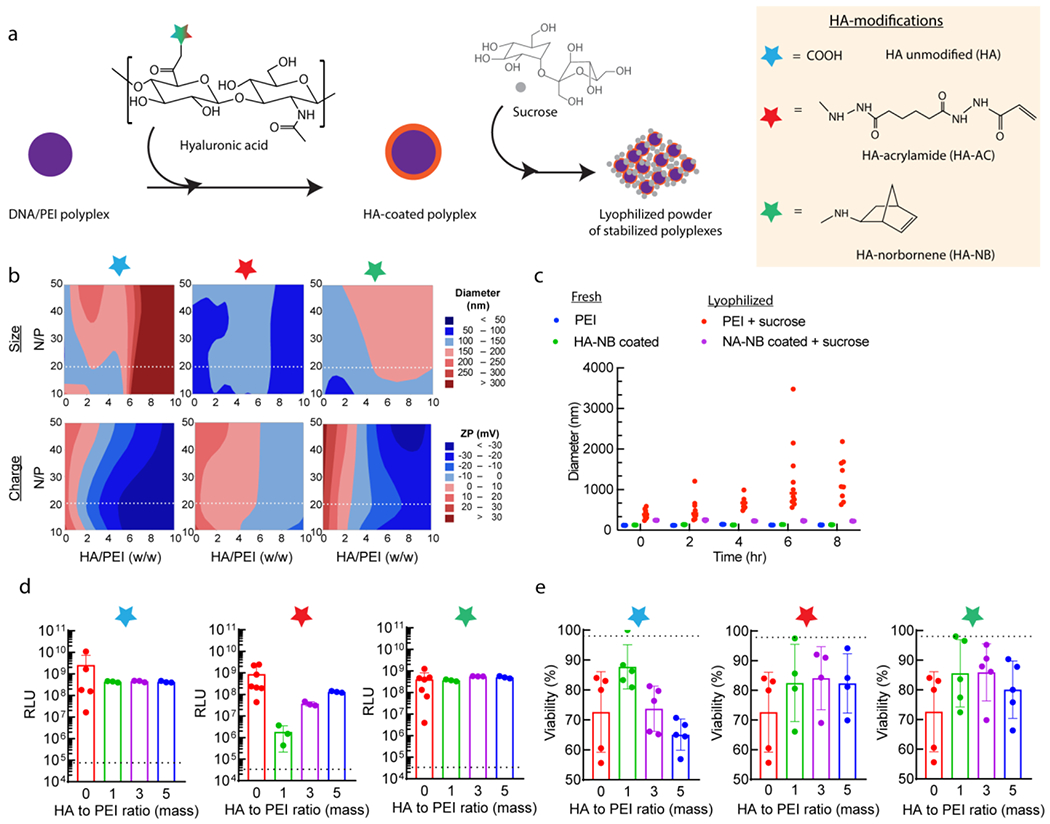
DNA/PEI particle coating strategy overview. (a) DNA/PEI particles were formed through mixing DNA/PEI and subsequently coated using hyaluronic acid, either as its non-modified state, or modified with acrylamide (Ac) and norbornene (NB). These nanoparticles are then cryoprotected with sucrose before lyophilizing for concentration and storage. (b) Design of experiment approach to optimizing the coating formulation, from the N/P ratio of PEI to DNA, and the mass ratio of HA to PEI. Parameters were optimized for the particle size and charge to ensure successful transfection, with the dashed line in each plot corresponding to the best N/P ratio, at 20, which was used for all subsequent analyses. (c) Particle stability following reconstitution in buffer, as assessed for 5:1 w/w ratio HA-NB to L-PEI, 87.5 μg sucrose / μg pDNA, at 2.5 μg DNA / μL gel. Freshly prepared DNA/PEI particles were compared to those reconstituted in a 10 μg/mL solution. (d) Transfection of lyophilized coated particles in mouse mesenchymal stem cells. Note that the non-coated (zero mass ratio) condition is a positive control, consisting of freshly prepared DNA/PEI particles. All other mass ratio conditions (1, 3, and 5) are for the lyophilized and reconstituted particles. (e) Viability measured as metabolic activity from PrestoBlue assay for the 2D transfection in (e). For both (d) and (e), the dashed line represents the cell-only background level. X-axis corresponds to HA/PEI coating mass ratios for samples that have been lyophilized and resuspended in 150mM NaCl, except for the “0” control, which are non-coated, freshly prepared DNA/PEI particles as a positive control. N=3, with one-way ANOVA and Tukey’s HSD (p<0.01).
We determined that none of the HA coatings significantly affected particle size, but they significantly reduced the charge of the DNA/PEI particles from cationic to anionic [Figure 1b]. We used design of experiments (DOE) to explore how the DNA-to-PEI ratio and HA coating amount affected particles size, charge, and stability. PEI nitrogen to DNA phosphate (N/P) molar ratios from 10 to 50, as well as PEI to HA mass ratios from 0 to 10, were explored for each coating type. The N/P ratio was chosen based on past studies with L-PEI transfection, where the highest N/P ratio before adverse toxicity ranges from 20-25 for plasmid transfection[50,51]. To see if coating with HA can reduce toxicity at higher N/P ratios, we expanded this to be the median point of the DOE space. Likewise, studies that explored HA coatings varied greatly on the mass and molar ratios used, largely based on the cationic vehicle. The most common ratio for PEI was equivalent to a mass ratio of 5 (for HA to PEI)[52,53], which we again used as a median point to get a wider study range and optimize the formulation. We observed that particles less than 300 nm could be obtained with most HA/PEI coating ratios at an N/P ratio of 15-20. Only non-modified HA induced aggregation of >300 nm at higher PEI to HA ratios, likely due to being the most negatively charged. In general, HA coatings resulted in more negatively charged particles as a function of degree of HA modification, excluding any differences between functional groups for the modifications themselves. Non-modified HA shifted the particles towards neutral charge with the lowest mass ratio, while the most modified coating, HA-AC, required the highest PEI/HA mass ratio to achieve neutrality.
We next aimed to determine if HA coating prevented aggregation during lyophilization. We used sucrose as a cryoprotectant during lyophilization (previously optimized at 350 μg sucrose / μg pDNA[15]). Our reported method for caged nanoparticle encapsulation resulted in stable particles following lyophilization only when combined with low-melting point agarose. However, these particles failed to reconstitute easily and formed a viscous solution that was prone to aggregation when encapsulated within our hydrogel precursor for microgel applications [Figure S2c], thus not being compatible with granular scaffold formation. Freshly prepared DNA/PEI particles (both coated and non-coated) were stable and did not aggregate during prolonged buffer incubation [Figure 1c, Figure S1e]. Without the HA coating, lyophilized DNA/PEI particles rapidly aggregated upon reconstitution in buffer [Figure 1c, Figure S1f]. However, HA-NB coated DNA/PEI particles remained stable after lyophilization and reconstitution for most ratios between 2-5 and an N/P of at most 20 [Figure 1c]. Therefore, HA-NB coating at 5:1 HA/PEI ratio and N/P of 20 was best for small (<300 nm) particles with slightly cationic or anionic charge and suitable for transfection. It is worth noting that while our focus is solely on HA-based hydrogels, other platforms could theoretically benefit from the stabilized particle formulation, such as PEG-based gels, but the formulation would likely need tailoring to the particular material. Alternative coatings and modifications could also be implemented, using a similar rationale design approach from our study to optimize particle stability, opening opportunities to introduce unique properties beyond those explored here.
To assess the biological properties of lyophilized DNA/PEI particles, we transfected a mouse mesenchymal stem cell (MSC) line (D1 cells) to assess transfection efficiency and viability. We found that freshly prepared DNA/PEI particles with HA coating resulted in enhanced gene transfer efficiency compared to non-coated DNA/PEI particles [Figure 1d] and reduced the toxicity of DNA/PEI particles [Figure 1e], consistent with similar reports for HA-coated cationic vehicles[26,43]. We found that lyophilized DNA/PEI particles without HA coating resulted in poor or no transfection. With HA coating, reconstituted lyophilized particles were able to transfect cells with similar efficiencies as freshly complexed particles, as compared to a fresh non-coated bolus control (zero mass ratio condition). In addition, coating and lyophilization did not significantly decrease viability compared to the freshly made non-coated particles, and in some cases trended towards improved viability, which may allow for higher amounts of nucleic acid to be delivered to cells without adverse effects. Balancing transfection efficiency with viability, HA-NB at an HA/PEI ratio of 3-5 gave the best result in mouse MSCs and was consistent across other cell lines. This was also demonstrated with minicircle DNA (MC), which is preferred clinically for its low immunogenicity and higher transfection ability in vitro and in vivo[54–56]. Several coating formulations were selected for size, charge, and transfection ability, again with DOE-optimization of N/P ratio and HA to PEI mass ratio [Figure S3a–e]. Higher transfection rates were observed by using lyophilized MC, compared to their plasmid equivalent, when coated with HA-NB. However, viability was relatively consistent for coated and non-coated samples, both freshly prepared and lyophilized, unlike the trends seen with plasmid DNA. In general, optimal transfection conditions were likely due to reducing the nanoparticle charge and resulting electrostatic interactions, aside from specific uptake and viability concerns.
Lyophilized DNA/PEI particles coated with HA and cryoprotected with sucrose resulted in a powder that could be incorporated inside hydrogels without aggregation while retaining activity [Figure 2a, Figure S1g]. Briefly, a nonporous hydrogel was generated through Michael Addition of HA-AC and a di-cysteine containing MMP-labile peptide as previously described[57], using a 3.5 wt% HA-AC solution and crosslinker molar ratio of 19.00 (free thiol to HA monomer). The stabilized powder was mixed with the hydrogel precursor and then crosslinked, resulting in a nonporous gel containing distributed DNA/PEI particles [Figure 2b]. Particle aggregation and distribution were dependent on coating formulation, with only HA-AC at w/w 3 and HA-NB at w/w 5 giving homogenous distribution, while all non-modified HA conditions resulted in aggregation. We further improved DNA/PEI particle concentration within hydrogel precursor solution by balancing the sucrose amount [Figure 2c]. We previously determined from using different combinations of sucrose and agarose that the agarose component could be eliminated without detrimental effects on particle size and stability. However, having a high sucrose concentration still poses toxicity concerns for a translatable formulation, as seen with our previous sucrose-agarose formulations[27]. As such, we looked to optimize the sucrose concentration in the top-performing HA-NB coating. At an N/P of 20 and HA/PEI ratio of 5, up to 5 μg particle/μL gel was loaded homogenously by using a reduced 87.5 μg sucrose/ μg particle, significant lower than the previous 350 μg/μg sucrose/DNA without any HA coating[27]. This reduction could allow for improved loading and translation in vivo. It is possible that the other HA coatings could be formulated using less sucrose, which in turn could improve viability for cell culture. However, as only HA-NB particles gave favorable transfection, we pursued this system for subsequent studies.
Figure 2.
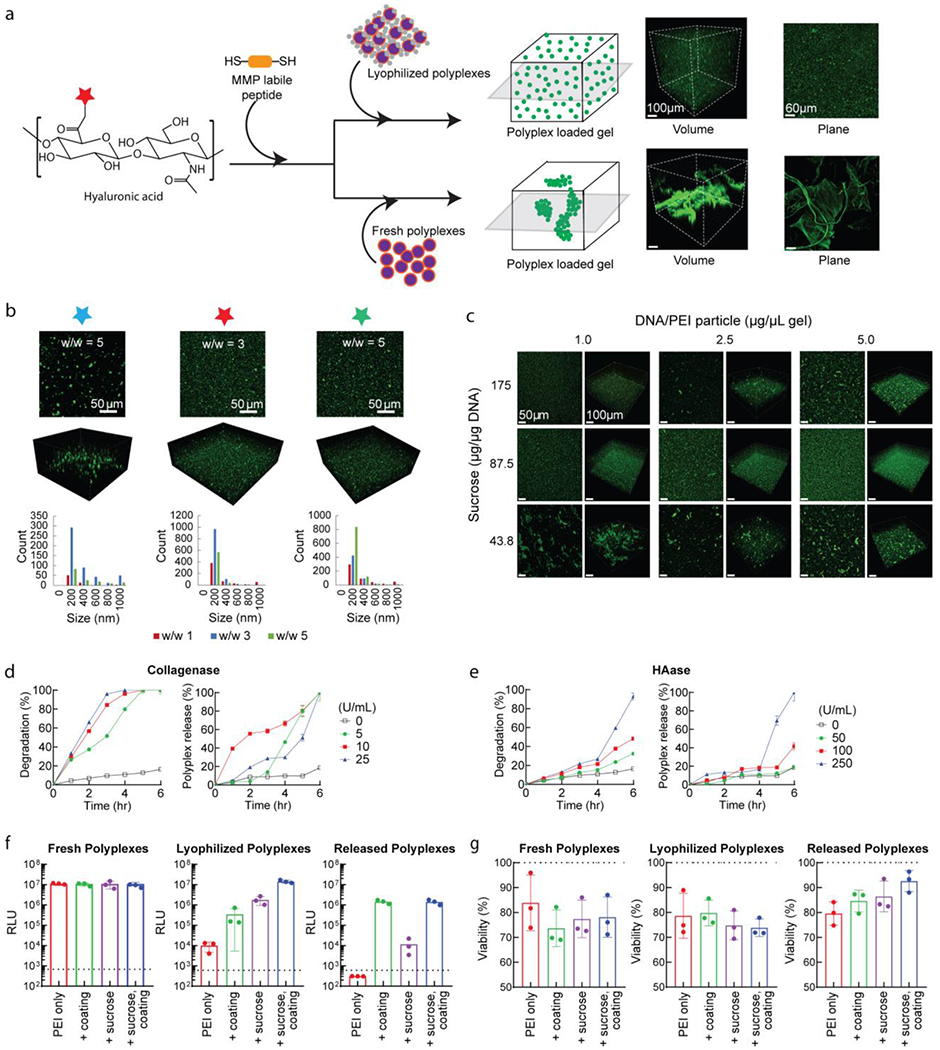
Loading and transfection of lyophilized particles from bulk HA-AC hydrogels. (a) Overview of hydrogel formation and DNA/PEI particle loading, with acrylated HA crosslinked through Michael addition chemistry via an MMP-cleavable dithiol peptide. The precursor solution is mixed directly with the lyophilized or fresh DNA/PEI particles to encapsulate prior to crosslinking. DNA was stained with YOYO-1 dye and imaged on confocal microscopy. (b) Comparison of HA coatings, with non-modified HA, HA-AC, and HA-NB for their top performing conditions from DOE-optimization and 2D transfection studies. Bar plots correspond to nanoparticle size distributions, based on particle analysis in ImageJ (n=3). (c) Further optimization of HA-NB coated particles by altering the sucrose concentration to balance precursor solution viscosity and aggregation within the HA-AC material with increased particle loading. (d) Minimal release of loaded DNA/PEI particles (HA-NB coating, sucrose lyophilization) in PBS solution, with and without collagenase degradation. Plasmids were labelled with P32 for scintillation, while gels were labelled with a fluorescent dye for microplate measurements. (e) Similar to (d), but demonstrating controlled released from hyaluronidase degradation of gels containing DNA/PEI particles. (f) Transfection of cells in 2D cell culture using loaded gels degraded by collagenase and hyaluronidase to release the DNA/PEI particles. (g) Viability data for the transfection in (f). For both (f) and (g), samples were compared for effect of sucrose cryoprotection and HA-NB coating at their optimized conditions (HA/PEI w/w 5, 87.5 μg sucrose / μg DNA, and loading at 2.5 μg/μL gel).
Because the mesh size of these hydrogels (1-100 nm) is smaller than the DNA/PEI particles (100-400 nm), particle release requires gel degradation rather than passive diffusion to allow for transfection. We exposed our hydrogels to collagenase and hyaluronidase to promote degradation of the MMP-liable crosslinker and HA backbone. Characterization of loaded scaffolds was performed with radiolabeling and scintillation [Figure 2d,e], demonstrating that without enzymatic degradation, HA-NB coated particles were well-retained over time, with minimal background release from possible hydrolytic degradation, given the assumed non-degradative nature of the scaffold in the absence of enzymes. It was observed that the nanoparticle release profiles did not trend with the degradation profiles, based on conjugated fluorescence dye release, and nearly total degradation was required to release the majority of particles. While scintillation gave the more accurate measurement for particle release over time, there are many parameters that could influence gel degradation, from the buffer composition, temperature, degree of protease-mediated degradation, and crosslinking mesh size and diffusion. During degradation, the hydrogel swells as more crosslinks are broken, likely increasing release from passive diffusion, although the effect is still primarily due to the enzyme treatment. The degree and type of coating modification may also play a role in regulating the particle release rate. This could result from possible coating interference on enzyme performance, as with hyaluronidase; however, this requires further investigation.
Under enzymatic conditions with collagenase to cleave the MMP crosslinker, released particles were able to effectively transfect cells with similar efficiency as fresh and lyophilized coated DNA/PEI particles [Figure 2f,g], while improving viability compared to freshly prepared particles. Compared to non-coated particles, both with and without sucrose and loaded into bulk gel, the HA coating significantly improved the released particle transfection-ability, likely from the enhanced stability with optimized size and charge properties. It was observed that the released HA-coated particles resulted in a 10-fold lower luciferase activity than direct transfection, which may be in part due to residual enzyme from the degradation process interfering with particle uptake. Here, the digest enzyme was only collagenase to avoid possible effects from hyaluronidase on coatings and particle stability. However, hyaluronidase could interfere in the protective ability and should be considered when translating in vivo, even though the physiologically-relevant conditions explored for release did not result in quick degradation. This also suggests the potential to control the release rate by partially degrading the scaffold prior to cell culture or implantation, although further studies are required to determine if such treatments affect cell viability and long-term transfection.
We took advantage of the success with bulk hydrogel loading and directly converted those gels into microgels through fragmentation to generate injectable microporous scaffolds and enhance transfection of infiltrating cells. The hydrogel precursor solution itself is technically injectable prior to crosslinking, but direct injection of the nanoparticle-mixed precursor would not afford the microporous architecture needed to promote rapid cell infiltration and transfection. While we and other labs have previously reported on emulsion and microfluidic methods to generate granular scaffolds composed of micron-sized spherical hydrogel particles[18,58,59], the challenge has been loading charged nanoparticles within the precursor while avoiding aggregation and inactivation. Here, even though our enhanced HA-coated DNA/PEI particle formulation was optimized for bulk gel loading, it did improve particle distribution within microfluidic generated particles as well [Figure S2c]. However, this approach resulted in heterogeneous microgel size and less homogenous distribution of the nanoparticles than in bulk. To overcome this challenge, an alternative method for granular scaffold fabrication is hydrogel fragmentation, which is a widely used technique for injectable biomaterials[59] and can be found in FDA-approved products such as soft tissue fillers. Fragmentation is achieved by breaking up a hydrogel using blades, passing the bulk gel through a filter or sieve[60,61], or extruding through nozzles of different sizes[62,63]. These methods generally do not control the resulting microgel size, thereby forming irregular particles of wide size distributions. However, they allow for a microporous scaffold from imperfect packing of the microgels, which can then be annealed via different secondary crosslinking schemes to fix the scaffold structure and be suitable for cell culture applications[64].
We sought to use a controlled fragmentation method on our DNA/PEI particle loaded HA gels to form shredded microgels. Briefly, the nonporous bulk hydrogel was generated as before [Figure 2a], but for cell culture applications the HA-AC backbone was also modified with cysteine-containing peptides to introduce integrin binding sites (RGD) and enzymatic crosslinking sites (Q and K peptides for Factor XIII/thrombin transamination). The crosslinked nonporous gels were then passed through a filter to generate “shredded hydrogel microparticles” (sHMP) [Figure 3a]. By using a fixed gel thickness and varying the sieve size, we determined the effect on microgel size and the resulting physical properties on cell response. We compared sieving filters with mean sieve size of 40, 70, or 100 μm, which yielded irregular microgels between 5,000 to 25,000 μm2 (cross-section area) based on confocal imaging. We observed that increased sieve size resulted in increased microgel size, although distributions spread widely, as expected from fragmentation.
Figure 3.
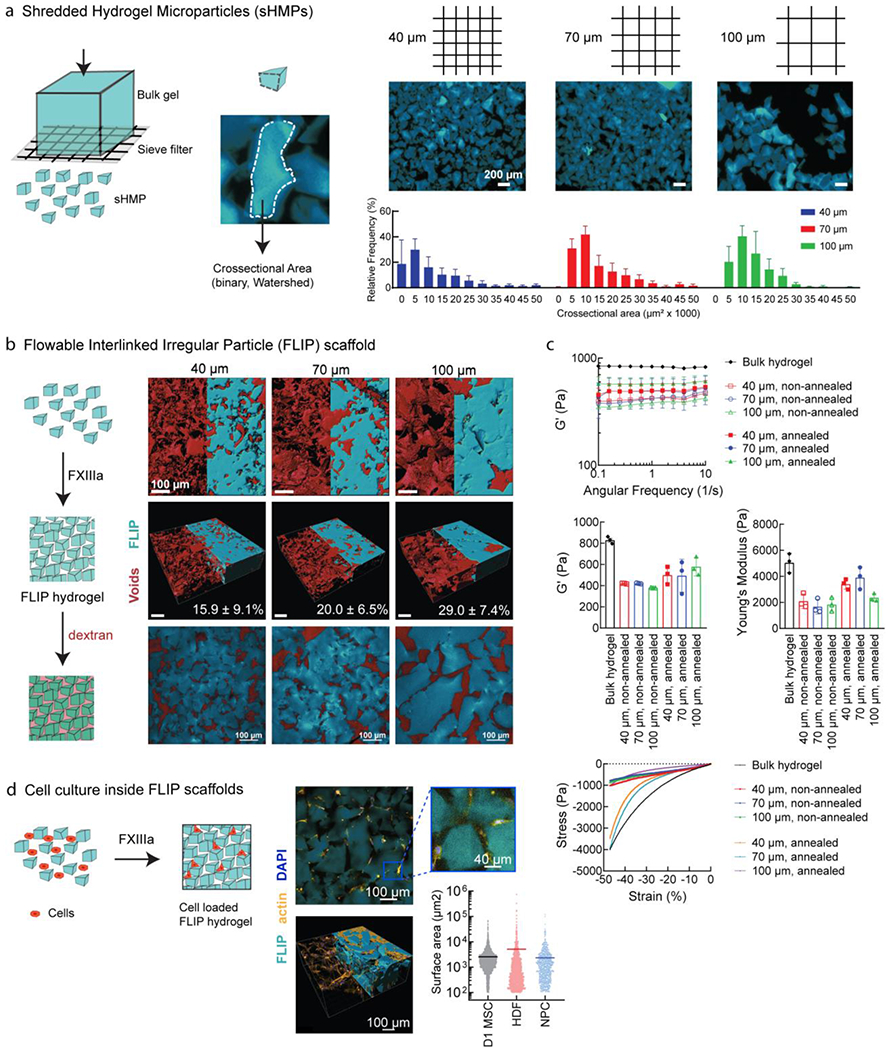
FLIP scaffold formation, mechanical properties, and 3D cell culture. (a) Formation of sHMP (shredded hydrogel microparticles) by centrifuging bulk gel disks on a cell sieve at a range of sizes (40 μm, 70 μm, and 100 μm sieve size). The resulting irregular sHMP distribution was quantified based on surface area from confocal microscopy to generate distributions. (b) Annealed FLIP scaffolds from different sieve sizes (blue), with dextran imaging of the interstitial void space (red) to quantify porosity, n = 3. Renderings performed from confocal microscopy images in IMARIS, with half corresponding to maximum intensity projection, and half from model volume filling. The third figure corresponds to the median z-slice. (c) FLIP scaffold mechanical properties. Rheology of FLIP scaffolds from 40, 70, and 100 μm sieving, with and without annealing chemistry (lack of Q/K peptides for FXIII-mediated annealing), as compared to standard bulk nanoporous HA-AC hydrogels. Frequency sweeps were performed for the stiffness (G’) while compression testing was performed for the Young’s modulus and stress-strain profiles. Sample size n=3, with error bars corresponding to S.D. (d) Cells culture is supported without loss of viability. Top-down view of actin stained cells for spreading within the FLIP scaffold, imaged half-way into the gel following seeding on top following annealing. IMARIS rendering of confocal images across a z-stack array to observe cell infiltration and spreading within the annealed scaffold. Images shown correspond to HDF culture, but data is presented as well for D1 MSCs and primary neural progenitor cells (NPC), with the horizontal bar corresponding to the mean surface area spreading and data points for each cell measured, as determined by IMARIS.
Given that cells infiltrate granular scaffolds based on the connecting voids between microgels[65], we sought to understand the porosity of the annealed scaffold to facilitate cell culture. sHMP can be annealed together with secondary crosslinking peptides in the presence of Factor XIII, thrombin, and calcium, which generates a stable, porous scaffold. In the absence of excess buffer, sHMP can be loaded into a syringe and ejected out as a fluid-like granular material, giving it a “flowable” property [Supplemental Video 1, Figure S2a]. Following ejection into a mold [Supplemental Video 2, Figure S2b] and covalent annealing upon incubation under physiological conditions, the resulting packed structure is fixed. Thus, we call the overall microporous hydrogel a “flowable linked, irregular particle” or FLIP scaffold, for short. There has been increased interest in the specific void geometry and its effect on cell interactions in the 3D microenvironment[66,67], and in granular scaffolds, porosity changes with size of the microgel building blocks, from their irregular packing and displaced void space. For our FLIP scaffolds, this was shown to range from 10-35% void volume, based on dextran diffusion and confocal imaging [Figure 3b]. From volume-rendering and measuring the void space, we observed that increased sieve size, and thus microgel size, resulted in increased porosity, with 100 μm sieves giving the largest void fraction and microgel sizes, while 40 μm gave the smallest voids and microgel sizes. The range of void fractions was similar to that observed with our previous 100 μm spherical microporous annealed particle (MAP) scaffolds (29.2±5.4%)[18]. However, no significant trend was observed between the different sieve sizes due to the irregular microgel packing fluctuating with individual samples of same sieve size.
Void space may be reduced with soft, compressible scaffolds. For gel stiffness, our therapeutic target has been ischemic stroke[68,69], which can benefit from an injectable, porous hydrogel to fill the infarct region and engage surrounding tissue, encouraging cellular infiltration and scar reduction. Injectable materials for the brain range in stiffness from 300-500 Pa[69,70]. FLIP scaffolds demonstrated appropriate stiffness and Young’s modulus relative to non-annealed scaffolds and the bulk hydrogel precursor [Figure 3c]. However, we did not observe significant differences in mechanical properties across different sieve sizes, despite the scaffolds having different porosities. As such, to have consistent gel size and void space, we selected the 70 μm sieve as our standard platform for generating sHMP, as its size distribution was similar to what we observed from 100 μm MAP gels[18] for surface-coated transfection, which exhibited adequate cell spreading and biocompatibility. However, porosity could also be tuned by mixing different ratios of microgels at different sieve sizes and/or stiffness, although that was not explored in this study.
To demonstrate that FLIP scaffolds support 3D cell culture, we utilized several cell types, both primary and cell lines, seeded within and on-top of the scaffold. Prior to secondary annealing, cells can be mixed with the flowable microgels. Upon annealing, cells are then entrapped within the porous gel structure and able to spread outwards, which may be suitable for cell therapy applications. More relevant to the application of gel injection for tissue repair and local cell infiltration into the scaffold, we explored cells seeded on-top of the annealed scaffold and their resulting distribution after several days of culture [Figure 3d]. Seeded cells were able to infiltrate, spread, proliferate, and remain viable, consistent with the notion that the material was non-toxic. This was observed across MSCs, dermal fibroblasts, and neural progenitor cells (NPCs), among other cell types. Additionally, the culture of NPCs and astrocytes was improved by inclusion of additional laminin-derived peptides, IKVAV and YIGSR[70], although concentrations were not optimized beyond previous work.
To generate sHMP for transfection, we generated DNA/PEI particle-loaded nonporous hydrogels as before, and subjected them to hydrogel fragmentation [Figure 4a]. The resulting microgels preserved the particle distribution present in bulk gels [Figure 4b], demonstrating that the particles are not physically altered by the microgel formation process. The mechanical properties and microstructure of particle-loaded gels was similar to that of non-loaded gels [Figure 4c–e] based on annealing, rheology, and compression testing.
Figure 4.
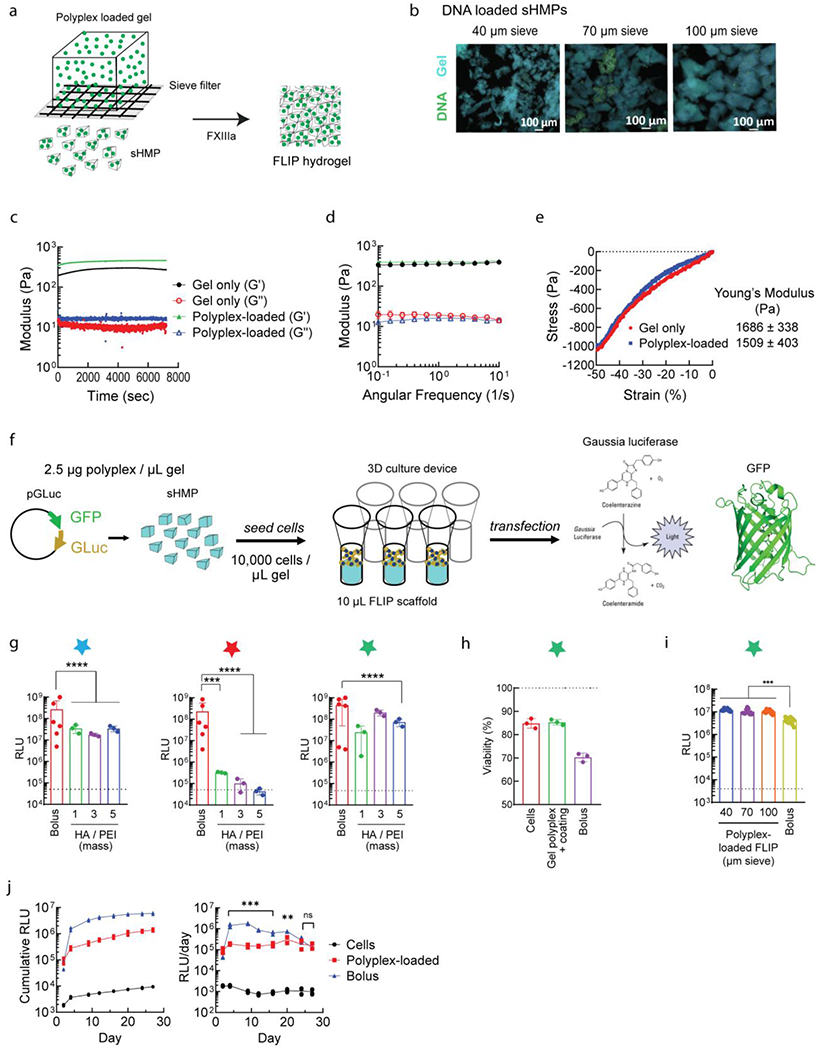
Nucleic acid incorporation into FLIP scaffolds. (a) Process of converting DNA/PEI particle-loaded bulk HA-AC gels to sHMP, again using the sieving process, and then annealing the microgels together into the microporous FLIP scaffold. (b) Retention of lyophilized DNA/PEI particles within FLIP scaffolds for different sieve sizes and the HA-NB coated particles. (c-e) Annealing and mechanical properties of FLIP scaffolds loaded with DNA/PEI particles. (c) Jamming study to obverse annealing time from Q/K peptides FXIII crosslinking. (d) Shear rheology on annealed gels following 1 hour incubation at 37°C to derive the storage (G’) and loss (G”) moduli. (e) Comparison of loaded and non-loaded annealed gels for Young’s modulus from microstrain compression testing. Values and error bars correspond to S.D, n=3. (f) Overview of 3D transfection process, with loading coated, lyophilzied DNA/PEI particles into bulk gels, converting to sHMP, and annealing into FLIP within a custom 3D cell culture chamber. Cells are then seeded on top or within the scaffold for exposure to the microgels and degradation to release the DNA/PEI particles. Transfection is measured based on GLuc and GFP reporter genes. (g) Lyophilized coated DNA/PEI particles and FLIP transfection in mouse mesenchymal stem cells. The dashed line represents the background levels from the cell-only negative control, while “bolus” is positive control transfected cells from fresh DNA/PEI particles when seeded into the gels (the equivalent of cells being exposed to all of the particles at once). The w/w HA/PEI represents different coating ratios by weight and are all samples that have been lyophilized and resuspended in the hydrogel HA-AC precursor solution. n=3-5, with one-way ANOVA and Tukey’s HSD (p<0.01). (h) Viability is also improved by the HA-NB coating, when compared to bolus transfection from fresh DNA/PEI particles as a control. (i) Comparison of FLIP from sHMP of different sieve size, loaded with HA-NB coated particles. (j) Time study of loaded FLIP scaffolds (HA-NB coated DNA/PEI particles, 2.5 μg/μL), of D1 MSCs, with GLuc expression monitored every 2-3 days post seeding, compared to a bolus transfection. Loaded FLIP gave sustained transfection levels over the month long study, while bolus dissipated over time, despite higher overall RLU levels. n=5, with one-way ANOVA and Tukey’s HSD (p<0.01). For all figures, p <0.05 (*), <0.01 (**), <0.005 (***), and <0.001 (****).
Turning to 3D cell culture, we explored how the different HA coatings influenced transfection and viability [Figure 4f]. The coating formulation for DNA/PEI particles was optimized by balancing the mass ratio of the HA coating to PEI (1, 3, or 5), for a fixed N/P ratio between PEI and DNA at 20. As before, cells were seeded on top of the scaffold, following sHMP annealing within a 3D cell culture device [Supplemental Video 2, Figure S2b]. Freshly prepared, non-coated DNA/PEI nanoparticles were administered in bolus to the media as a postive control. After a 48 hour incubation to allow for spreading and transfection, we generally observed that cells infiltrated the nucleic acid-loaded FLIP scaffolds akin to the non-loaded scaffold, and that they were transfected at levels similar to that from fresh bolus transfection [Figure 4g]. Of the three coatings tested, HA-NB coating gave the best improvement in transfection in 3D culture, specifically at an HA/PEI ratio of 3-5, followed by non-modified HA, while HA-AC demonstrated poor transfection. This differed from 2D culture results, where there was no significant difference between the optimized formulation for each coating, and also differed from the general particle distribution in nonporous gels, in which non-modified HA showed the most aggregation. Such an outcome is not atypical when translating cell studies from 2D to 3D environments, and further mechanistic studies could help to better understand the coating effect on transfection pathways between 2D and 3D. We demonstrated this previously when characterizing pathway differences from 2D and surface-coated transfection in our spherical MAP scaffolds[71], in addition to others who have investigated various forms of granular scaffold transfection[72]. However, it is likely in the case of the HA-AC coating that decreased transfection with increased coating resulted from the coating itself being crosslinked or conjugated to the scaffold material due to having the same functional group, reducing particle uptake. Between the physical properties and transfection results in 2D and 3D culture, HA-NB was selected as the final optimized formulation, at 87.5 μg sucrose per μg DNA, 5 w/w HA/PEI, and N/P of 20, with gels loaded at 2.5 μg DNA / μL of gel. This formulation demonstrated high cell viability compared to non-transfected and bolus transfected cells [Figure 4h], supporting that entrapped particles were largely non-toxic. However, as with mechanical properties, there was no difference observed when comparing transfection from FLIP with microgels of different sieve size [Figure 4i], suggesting that the void space may not play as significant of a role in fragmented hydrogels scaffolds as it did in our previous spherical MAP scaffolds[18].
A key feature of FLIP scaffolds is the MMP-cleavable crosslinkers used to promote cell infiltration and hydrogel degradation or remodeling. This allows the loaded DNA/PEI particles to be released and locally transfect cells over time. Comparing the HA-NB coated formulation to simple bolus transfection in 3D culture, we observed that only the particle-loaded scaffolds were capable of sustained transfection over a one-month period, from prolonged GLuc expression [Figure 4j]. The particles were capable of a constant rate of transfection, while bolus transfection dissipated after 48-72 hours post administration, despite having an initially higher rate. This is likely due to “re-transfection” events from the loaded scaffold acting as a reservoir for active DNA/PEI particles, as demonstrated from the 2D culture transfection and supported by previous work on MAP scaffolds[36]. The advantage of our fragmented scaffolds is the significantly higher loading capacity, giving higher overall transfection levels and a duration upwards of one month in D1 MSCs and human dermal fibroblasts (HDFs). The optimized coating formulation was also shown to be compatible for 3D transfection of minicircle DNA [Figure S3f–h]. Recall that in 2D cell culture, HA coating was required for improved lyophilized MC particle transfection, aside from sucrose cryoprotection. For 3D culture, we again observed that scaffolds loaded with coated MC particles gave higher levels of transfection and improved viability, relative to bolus transfected particles. However, it was noted that bolus administration of DNA/PEI particles gave a higher percentage of transfected cells, based on GFP expression [Figure S3g]. This is likely due to cells being exposed to all of the DNA at once, whereas the scaffold-loaded particles required gel degradation to be released. This was confirmed when assessing transfection over time [Figure S3i–l], and that as with plasmid DNA, the loaded MC particles gave better, prolonged rates of transfection and cell viability, outperforming bolus transfection after one month of 3D culture. As such, HA coating and lyophilization enhances MC transfection, in addition to giving higher transgene expression than the respective plasmid-loaded gels. Considering the eventual translation of our new scaffold platform for in vivo applications and serve as a local gene therapy, MC particle scaffolds could help to reduce associated immunogenicity during long-term transfection[54,73], based on the improved vector response. However, future biocompatibility studies will be required to see if an immune response to the scaffold is altered upon DNA/PEI particle incorporation and understand how it differs between plasmid and MC particles as compared to the non-loaded scaffold, and determine if further optimization is needed for efficient, sustained transfection.
While we demonstrated here the use of our optimized HA coating and microporous scaffolds for DNA vectors (plasmid and minicircle), the system could be optimized for many other therapeutic vectors and vehicles, from DNA and RNA, and alternative cationic polymers and lipids. While not explored here, this would require separate optimization of the HA/polymer and polymer/nucleic acid ratios, based on the vector size and polymer properties. mRNA carries the advantage of direct translation in the nucleus and transfection of non-mitotic cells, which is relevant for our application in ischemic stroke repair. There has been some progress for hydrogel-mediated delivery of RNA[14,74,75], particularly as a vaccine from pre-cast porous gels, although direct incorporation of cationic nanoparticles into the hydrogel precursor remains a challenge. While we have preliminarily explored the use of mRNA within our system, further work is needed to select the best vehicle and coating parameters to enhance mRNA delivery from FLIP scaffolds. However, with the success of minicircles in the clinic, our system is still able to accommodate therapeutic DNA delivery and could be suitable for injection in ischemic wounds to target actively dividing and infiltrating cells within the localized scaffold environment and promote tissue repair.
In line with the translational application for ischemic tissue repair, particularly with injectable therapies for ischemic stroke, we demonstrated that the annealed FLIP scaffolds were compatible for transfection of neural cell types, including human astrocytes and primary mouse NPCs [Figure 5a]. Both cell types showed transgene expression that was significantly higher than background, although we observed that relative luminescence was approximately 100-1000 fold lower than that from bolus administration. This was in contrast to transfections with MSCs and fibroblasts, which gave as-good or only 10-fold lower expression than bolus, depending on the formulation. The particle-loaded scaffolds likely require further modification and optimization for improved transfection of neural cells. This is especially needed when translating to in vivo stroke mouse models, as the scaffold injection volume is generally smaller than the already low-volume (10 μL) 3D cultures demonstrated here.
Figure 5.
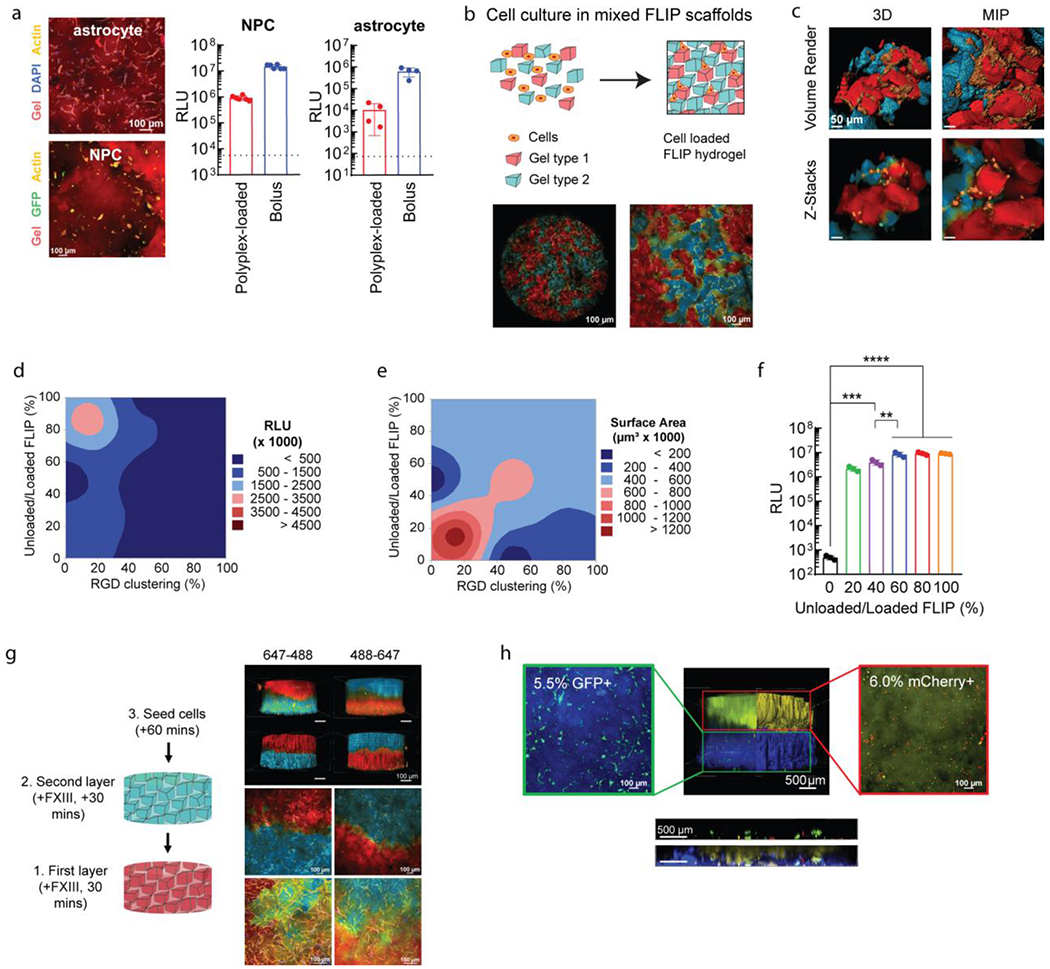
Further applications of FLIP scaffolds for plasmid transfection. (a) Transfection of relevant CNS cell types in 3D culture. Confocal images of stained cell cultures for human astrocytes and mouse-derived primary neural progenitor cells (NPCs). Cells were transfected using HA-NB coated DNA/PEI particles in FLIP (n=4), compared to bolus transfection and the cell control (dotted line), after 48 hours. (b) Altering FLIP properties from sHMP mixing, with confocal imaging for MSCs seeded within mixed colored FLIP scaffolds. Cell spreading within FLIP scaffolds comprised of two different precursor solutions, stained for 488 and 647, with cell membrane staining of live cells. Note that the 488 channel is pseudocolored blue, and the 647 channel red. (c) High-resolution confocal imaging of the mixed scaffold, showing the interface of cells spreading between the two types of sHMP. Both z-stacks and volume renderings are shown, from a 3D orthogonal view and from maximum-intensity projection (MIP). (d-e) DOE optimization of cell transfection (d) and spreading or surface area (e) based on RGD peptide clustering on sHMP and ratio of nanoparticle-loaded to non-loaded sHMP within FLIP scaffolds. Transfection was assessed with GLuc. RGD clustering corresponds to the ratio of gel exposed to reactive RGD peptide at the time of precursor formulation, with a fixed 1 mM concentration. 100% modified RGD-modified HA meant that the scaffold was homogenous (i.e. the entire HA-AC precursor was reacted with 1 mM RGD), while 10% modified and 90% non-modified meant that there was 10% clustering on the microgels, and 0% referred to a gel without any RGD (0 mM). Surface area was assessed based on confocal imaging of the 3D cultures and rending the z-stacks in IMARIS for volume filling and surface area measurements from actin labeling. (f) An example of the transgene output is shown in for a fixed 20% RGD clustering and was determined that 60-80% nanoparticle content and 20% RGD clustering was the optimal balance between transfection and spreading/viability of cells. A one-way ANVOA with Tukey HSD was performed on the samples (n=3), with significance reported at p <0.05 (*), <0.01 (**), <0.005 (***), and <0.001 (****). (g) FLIP gel layering based on sequencing syringe loading and injection into the 3D culture mold, with the ability to alternate layers of 488 or 647 stained gels without merging or loss of gels due to the annealing chemistry. Again, the 488 channel is pseudocolored blue, and the 647 channel red. 4X images were processed for volume rendering to show the entire scaffold distribution. 10X and 20X images were taken at the interface of the scaffold layers to demonstrate cell infiltration across the layers, shown as MIPs. (h) Application of FLIP scaffolds for “domain transfection,” in which different gel layers contained DNA/PEI particles with transgenes for either GFP (AF305-labelled layer, blue) or mCherry (AF647-labelled layer, yellow). Transgene expression was assessed with flow cytometry on degraded gels for cell extraction (separated by layer), showing highly specific gene expression for only the given reporter in each layer (denoted in the microscopy image of reporter-expressing cells by the percentage). The FLIP scaffolds were also imaged for z-stacks at 4X across the layer interface to assess transfection of GFP and mCherry in the region, shown with the horizontal ortho-projection, both with and without the gel channels present.
Apart from the inclusion of laminin peptides to aid in neural cell culture, the granular nature of fragmented scaffolds can be used to direct cellular behavior and improve transfection. Aside from the explored physical properties (microgel size, stiffness, degradability), we can alter the composition of the scaffold and microgels to regulate the presentation of certain peptides or the amount of DNA/PEI particle-loaded gels [Figure 5b–c]. To modulate cell response, we previously explored how the concentration and distribution of RGD adhesion peptide through “clustering” affected viability and gene transfer in bulk hydrogels[76] and polyplex surface-coated MAP scaffolds[18]. For our FLIP scaffolds, it was not known if cells required homogenous RGD presentation on the microgel surface, or if a specific degree of clustering was ideal for receptor interaction and improve transfection and viability. Thus, we used a DOE approach to balance the amount of DNA/PEI particle-loaded microgels with non-loaded microgels (the mixed scaffold approach), under the assumption that the particle-loaded gels were the main influence on viability, and also varied the degree of RGD clustering for a fixed total concentration of 1 mM RGD in the precursor solutions [Figure 5e–f]. The clustering method was achieved as previously described[76], in which the HA precursor solution is first partitioned off for RGD-modification before pooling back into the total solution and crosslinking, and then compare the effects from different ratios of RGD-modification. Following sieving, the DNA/PEI particle-loaded and non-loaded sHMP were mixed at different gel ratios (percent total scaffold, volume-basis) to form a FLIP scaffold, and cells were seeded following annealing as before. Based on transgene expression and cell spreading, we determined that optimal RGD clustering was at 15-20%, which gave the best cell spreading and viability [Figure 5e]. This was similar to our past MAP scaffolds, where 20% RGD clustering worked best for surface-coated transfection[18]. However, the main improvement here was the ratio of DNA/PEI particle-loaded and non-loaded microgels. We determined that scaffolds only required 70-80% particle-loaded microgels (at 2.5 μg/μL loading) to achieve maximum transfection levels [Figure 5d]. It is likely that having 100% of the scaffold comprising DNA-loaded gels, as used in the coating optimization studies, can still transfect cells with adequate viability, but that the scaffold can be improved by reducing presentation of loaded particles to the infiltrating cells. This was further demonstrated for loaded and non-loaded ratios at a fixed 20% RGD clustering [Figure 5g], where we observed that having a scaffold of 60-80% particle-loaded microgels was optimal for peak transgene expression and cell viability. While not explored here, varying RGD clustering and/or loading concentration of different microgels, rather than uniformly, within the scaffold could further improve transfection and viability, or confer unique properties such as selective transfection for different cell types. Future studies could also utilize a similar DOE approach to optimize laminin peptide concentration and clustering necessary for neural cells, similar to past work with spherical MAP scaffolds[70].
Alternatively, rather than mix a ratio of gels homogenously to create a scaffold, fixed gel layers can be made of uniform fragmented gels to spatially guide cell culture and function. Again, with the granular nature and annealing chemistry of FLIP scaffolds, individual microgel solutions could be loaded into a syringe and injected to create a layered 3D culture environment, as we previously explored with spherical MAP scaffolds in cell culture and stroke mouse models[69]. This can also be accomplished by sequential addition of layers in a 3D culture device, which we used here to allow for well-defined scaffold layers [Figure 5g, Figure S2d–e]. Applying this concept to transfection, we explored the possibility of “domain transfection,” in which each gel layer contains DNA/PEI particles for only one specific reporter transgene. Here, we generated a two-layered FLIP scaffold, with the lower layer containing particles for a GFP plasmid, while the upper layer contained particles for an mCherry plasmid. We demonstrated that the layered scaffold gave specific transfection of cells seeded within only a particular layer prior to annealing, as supported from flow cytometry [Figure 5h]. It was observed that some cells along the interface region were transfected by one or both plasmids, although the vast majority of cells within each layer only were positive for the respective reporter. This is likely due to the local release and uptake of DNA/PEI particles as cells remodel the scaffold, as opposed to any diffusion of released nanoparticles into different layers, although future studies could help to clarify if diffusion and cell migration play a minor role in transfection, from alternating layers of loaded and non-loaded gels. This controlled delivery can be beneficial for spatially regulating gene expression both in vitro and in vivo, overcoming the current limitations from simple bolus administration. Further work is needed prior to translation to determine how domain transfection can direct cell culture through patterning the expression of therapeutic proteins, such as VEGF or BDNF, although we did not explore therapeutic gene delivery here.
Conclusion
In this study, we performed in vitro characterization to develop and demonstrate the use of flowable linked irregular particle, or FLIP scaffolds, as a novel granular hydrogel platform for highly efficient gene delivery. The primary advantages of microporous FLIP scaffolds are high DNA/PEI particle loading capacity, improved cell viability, and duration of transfection, in addition to the benefits of being a tunable, granular scaffold. Compared to past work from our lab using MAP scaffolds for surface-coated or bolus transfection[36], we demonstrated that a gel fragmentation approach could easily produce microgels loaded with non-aggregated DNA/PEI particles, in part from the reduced sucrose formulation and HA polymer coating, and with desirable mechanical properties. Our previous work accomplished DNA particle loading in bulk hydrogels via agarose fixation, but this prevented adequate reconstitution within the precursor and could not yield highly-loaded microgels from emulsion or microfluidics. Further, the previous method failed to transfect cells in 2D culture when directly reconstituted in buffer, which negated the ability to use the formulation for stable storage of DNA/PEI particles. While native, non-modified HA coatings on cationic particles have been explored by our lab and others, the comparison here of different functional group modifications and DOE on the HA/PEI ratio and N/P ratio likely resulted in our success to identify an optimal formulation for stable lyophilization and reconstitution, in addition to giving increased transgene delivery in both 2D and 3D cell culture. Our optimized nanoparticle formulation can allow for not only high concentration loading within the hydrogel precursor, but more broadly allows for a stable, lyophilized product for nucleic acid delivery in cell culture. FLIP provides a prolonged delivery of transgenes, from gradual but constant re-transfection events as cells infiltrate and degrade the scaffold. As FLIP scaffolds are formed by simply shredding bulk gel, the process is significantly easier and faster to produce than traditional microporous scaffolds, allowing for a lower-cost injectable therapy and easier scale-up for commercial manufacturing. This could provide a method to promote tissue regeneration, such as in the ischemic stroke infarct, by injecting the DNA/PEI particle-loaded scaffold and activating therapeutic genes, resolving current clinical need for neuroprotective therapies. Overall, our system will advance the state of local, nonviral gene therapies.
Experimental Section/Methods
Preparation of hyaluronic acid-acrylamide (HA-AC)
To modify hyaluronic acid (HA) to contain acrylamide functional groups, 1 g of 70 kDa sodium hyaluronan (Contipro, 50-90 kDa) was dissolved in 200 mL DI water (1 g/200 mL). Adipic dihydrazide (ADH, Fisher Scientific) was added for a 1:40 molar ratio with HA, at 18.35 g, to add amines to the carboxylic acid side chains, and pH adjusted with 1 M HCl to 4.75 while stirring to dissolve. 2.02 g N-(3-Dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDAC HCl, VWR) was added for a molar ratio of 1:4 for HA to EDC to activate the carboxylic acids. pH was maintained for 4 hours before allowing the reaction to proceed overnight at 25°C with constant stirring. The reaction solution was then transferred to dialysis tubing (Fisherbrand, 6000-8000 Da), and dialyzed over 3 days in NaCl solutions of decreasing concentration, starting at 100 mM NaCl and ending with 24 hours of DI water. The final product was then filtered, flash-frozen, and lyophilized. The extent of ADH modification was confirmed via 1H-NMR spectrometry. The integrations of the peaks were normalized to the peak corresponding to the methyl group on the HA monomer at δ = 2.0 ppm to determine percent of HA monomers modified to contain ADH groups. After this, the HA-ADH was modified with 2.23 g N-Succinimidyl Acrylate (NHS-AC, TCI Chemicals) for a molar ratio of 1:5 for HA to NHS-AC. The HA-ADH was resuspended in 200 mL 10 mM HEPES (1 g/200 mL) with 150 mM NaCl, 10 mM EDTA, at pH 7.4. NHS-AC was dissolved in DMSO (100 mg/mL) and added to the HA solution, lowering the pH to 6.0 and maintain for 4 hours while stirring before reacting overnight at 25°C. As before, the reaction solution was transferred to dialysis tubing and dialyzed over 3 days before the product was filtered, flash-frozen, and lyophilized. The extent of Ac modification was also confirmed via 1H-NMR spectrometry, normalizing to the HA peak.
Preparation of hyaluronic acid-norbornene (HA-NB)
To modify hyaluronic acid (HA) to contain norbornene functional groups, 1 g of 70 kDa sodium hyaluronan (Contipro, 50-90 kDa) and 3.111 g 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride (DMTMM) (Thermo Fisher Scientific, Waltham, MA) were each dissolved in 40 mL 200 mM MES buffer pH 5.5 (molar ratio of ~1:633 for HA to DMTMM). The two solutions were combined and stirred for 10 minutes to allow for activation of the carboxylic acid. 0.677 mL 5-norbornene-2-methylamine (TCI America, Portland, OR) was added dropwise to the reaction mixture (molar ratio of ~1:343 for activated HA to NMA), which was then allowed to react overnight at 25°C with constant stirring. The reaction product was then precipitated in ethanol, filtered to collect the solid, dissolved in 2 M NaCl in water, and dialyzed under running deionized water for 24 hours. The final product was then filtered, flash-frozen, and lyophilized. The extent of modification was confirmed via 1H-NMR spectrometry. 1H-NMR shifts of attached norbornene groups in the product in D2O are δ = 6.33 and 6.02 (vinyl protons, endo), and 6.26 and 6.23 ppm (vinyl protons, exo). The integrations of these peaks were normalized to the peak corresponding to the methyl group on the HA monomer at δ = 2.0 ppm to determine percent of HA monomers modified to contain norbornene groups.
Vector design and generation (plasmid, minicircle)
All plasmid vectors were based on the CMV promoter with polycistronic GFP-P2A-GLuc for GFP and Gaussia luciferase (GLuc) expression for in vitro studies. SV40 poly(A) tail and the Woodchuck Hepatitis Virus (WHP) Posttranscriptional Regulatory Element (WPRE) were used downstream the gene to enhance expression. Plasmids used for in vivo used Firefly luciferase (FLuc) in place of GLuc, while plasmids with non-GFP fluorescent reporters used either DsRed, tdTomato, or mCherry. In addition, pcDNA3.1(+)/Luc2=tdT was a gift from Christopher Contag (Addgene plasmid #32904), encoding for enhanced luciferase (Luc2) with a C-terminal fusion of tdTomato under the CMV promoter.
For studies with minicircles (MC), the CMV vectors with GLuc or FLuc were used, with the promoter and genes flanked by attP/attB recombinase sites for excising the bacterial elements via ZYCY10P3S2T E. coli (System Biosciences). MC production was induced with arabinose and cultured for 3 hours prior to plasmid prep. Purity was determined with agarose gel electrophoresis and AFM. For cases in while completely pure MC product was desired, prepped products were treated with restriction enzymes that targeted the bacterial backbone, followed by exonuclease treatment and column purification.
DNA/PEI particle formation and HA coating assessment
DNA/PEI particles were prepared by complexing plasmid DNA encoding for Gaussia luciferase (GLuc) and eGFP with linear polyethylenimine (L-PEI, 25 kDa, Polyscience) at N/P ratios of 5, 7, 10, and 20. Briefly, 1 μg DNA was diluted in 10 μL of 150 mM NaCl and the corresponding amount of L-PEI was diluted in a separate tube in 10 μL of 150 mM NaCl. The L-PEI solution was then added to the DNA solution, immediately vortexed, and allowed to incubate for 15 min at 25°C to allow for complexation. In the case of HA coating, non-modified HA (70 kDa), HA-AC, or HA-NB were added to the DNA/PEI solution following incubation at w/w ratios (HA to PEI) of 2, 5, or 10, and then incubated another 15 min. Size and charge of the particles was assessed with dynamic light scattering (DLS) and electrophoretic light scattering (ELS) on a Malvern ZetaSizer ZS or Anton Paar Litesizer 500 instrument, to determine if the cationic particles are prone to aggregation. Measurements were assessed in triplicate using the default run parameters, at up to 150 scans for ELS and 20 for DLS, for stabilized measurements to derive the zeta potential, hydrodynamic diameter, and polydispersity index (PDI). Trends were compared across all N/P and w/w ratios for each HA coating condition using contour modelling in Minitab
Lyophilized particle formulation and bulk HA gel distribution
To load DNA/PEI particles into HA scaffolds, a method for “caged nanoparticle encapsulation” was developed from our lab’s previous work[15,27]. Instead of having both low-melting point (LMP) agarose and sucrose in solution, only sucrose was used at concentrations from 45 to 350 μg sucrose/μg DNA. Particles were prepared similar to before for up to 250 μg DNA, pre-mixed with sucrose and diluted 1:40 in nuclease-free water. No salt solution was used. Following L-PEI complexing and HA coating, DNA/PEI particle solutions were flash-frozen and lyophilized.
To prepare bulk gels with and without lyophilized particles, 3.5 wt% HA-AC gels were made with a matrix-metalloproteinase (MMP) dithiol crosslinker (Ac-GCRDGPQGIWGQDRCG-NH2, Genscript). HA-AC was dissolved in 0.3 M triethanolamine (TeOA), pH 8.8. Crosslinker was prepared for a SH/HA monomer ratio of 19.00, dissolving in DI water. In the case of particle-loaded bulk hydrogels, the lyophilized DNA/PEI particles were resuspended in the volume of nuclease-free water for the crosslinker solution and subsequently used directly to resuspend the crosslinker peptide. Both solutions were then combined and used to make 35 μL bulk gels by sandwiching with Sigmacoted glass slides using a 1 mm Teflon spacer, and then incubating at 37°C for 60 mins. Following incubation, gels were transferred to 1X PBS solution containing AlexaFlour 647-NHS (Thermo Fisher Scientific, 1:1000 dilution) to stain the HA using the residual ADH groups and YOYO-1 (Thermo Fisher Scientific, 1:10,000 dilution) to stain the DNA, swelling at 4°C overnight.
Following swelling, gels were imaged using a confocal microscope (Nikon C2 scanning confocal) at 20X and 40X magnification across z-stacks (300-500 μm, at 25-50 slices) to visualize the DNA/PEI particle distribution. Z-stacks were then assessed in IMARIS (Bitplane) to generate 3D renders of the distribution and assess aggregation. Maximum intensity projections (MIPs) were generated to view the general particle size, and imported to ImageJ (FIJI) for particle size analysis. Briefly, images were converted to binary and assessed using the built-in Watershed analysis tool to identify particles, and then measured with the built-in Particle analyzer tool to quantify the cross-section area. Assuming spherical particles, the diameter was calculated for the particle size distribution.
Particle loading and release from bulk scaffolds
To assess the degree of lyophilized DNA/PEI particle loading in the scaffolds, and subsequent release rates upon degradation, we used scintillation on P32-labelled plasmid DNA. Briefly, P32-dCTP was added into plasmid DNA using a nick-translation kit (Roche), with purification using a DNA concentrator kit (Zymo). The labeled DNA was diluted to a 0.5% P32-DNA solution (by mass) in the stock plasmid. Lyophilized particle prep was prepared as before with HA-NB coating. The particles were then reconstituted in HA-AC solution with the MMP-dithiol crosslinker also as before. The HA-AC was labelled using AF350-NHS for fluorescence tracking upon degradation. The nucleic acids were also labelled using YOYO-1 dye as a secondary fluorescence measurement to confirm particle concentration. Gels were diluted 1:10 by volume in digest buffer, degraded from incubation at 37°C using a range of concentrations of either Type IV Collagenase (Worthington Biochemical) or Hyaluronidase (Worthington Biochemical) in PBS. At each time point, samples were centrifuged and the entire supernatant collected, before replacing with fresh digest buffer. The supernatant was split in half and diluted 1:20 in scintillation fluid, and then read on a Beckman-Coulter LS 6500 scintillation counter. Raw CPM readings were converted to nucleic acid concentration based on a standard curve. This was compared to the total DNA loaded within the hydrogel at time 0 by diluting the hydrogel in scintillation fluid, to determine the cumulative release rate. Readings were normalized to background levels from the individual digest buffers to account for low levels of radioactivity from the enzymes. For gel degradation tracking, the remaining supernatant was used to measure fluorescence, read directly on a plate reader (Tecan Spark), normalized to background from treatment buffers, and cumulative release tracked as before.
sHMP-DNA generation
From the bulk gels prepared with and without DNA/PEI particles, shredded hydrogel microparticles (sHMP) with prepared by sieving the swelled gels stacked on either a 40, 70, or 100 μm cell sieve (pluriSelect-USA, Mini-Strainer, PET sieve) and washing with 1X PBS. The resulting flow-through was centrifuged at maximum speed (~18,000g) to collect the particles and imaged on a fluorescent microscope (Zeiss Observer Z1) to determine if the nanoparticle distribution was preserved. Gel size distributions were quantified using binary converted images and cross-section area measurements in ImageJ, similar to what was done for the particles themselves for their size analysis.
For gels to be used in vitro with 3D culture, HA-AC nanoporous gels were prepared similar to before, but the HA solution was modified with an RGD ligand (RGDSP, 1 mM) to improve cell adhesion, in addition to modification with Q-peptide (Ac-NQEQVSPLGGERCG-NH2, 0.75 mM) and K-peptide (Ac-FKGGERCG-NH2, 0.75 mM) for Factor XIII (FXIII)/Thrombin transamination to anneal the microgels into FLIP scaffolds. Peptide modification was clustered as previously described[76,77] by initially reacting with 20% of the HA precursor for 15 mins at 25°C before pooling back with the original solution.
FLIP scaffold annealing and void space analysis
Non-loaded sHMP were combined with Factor XIII (Fibrogammin 1250, CSL Behring) and Thrombin (with calcium, from bovine plasma, Sigma Life Science) at 0.01 U/uL gel and 0.002 U/uL gel respectively, mixed thoroughly by pipetting, and residual buffer aspirated following centrifugation. The scaffolds were allowed to anneal for 1 hour at 37°C under humidified conditions to avoid drying out. The resulting FLIP scaffolds, comprised of sHMP at various sizes, were incubated with PBS containing 1 μg/mL 500 kDa tetramethylrhodamine isothiocyanate-dextran (TRITC-dextran) (Sigma-Aldrich, St. Louis, MO) to fill the void space in between microgels, as it is too large to penetrate the microgel polymer network. The labelled void space was imaged using Nikon Ti Eclipse equipped with C2 laser LED excitation to obtain 200-μm z-stacks. The z-stacks were imported into IMARIS to generate surface renders, and void space volumes were quantified as a fraction of the total volume represented by the z-stack. A minimum of four measurements were made for each scaffold.
Oscillation rheometry and compression testing
Stiffness of both nonporous HA-AC hydrogels and annealed FLIP scaffolds was measured as the storage modulus (G’) using a plate-to-plate rheometer (Physica MCR, Anton Paar, Ashland, VA). A frequency sweep was performed on the hydrogels using a strain of 0.2% with an angular frequency range of 0.1 to 10 rad/s. To measure the storage modulus of an annealed FLIP scaffold, 50 μL microgels with FXIII/Thrombin were pipetted directly onto the rheometer stage. The measuring position was set to 1 mm and the gel was allowed to incubate with humidity at 37°C for 1 hour to allow for annealing. Once the gel was annealed, a frequency sweep was performed on the hydrogels using a strain of 1% with an angular frequency range of 0.1 to 10 rad/s.
Compression testing was performed using a microstrain analyzer (TA Instruments RSA III). As with the shear rheometer, 50 μL gels (bulk or annealed) were loaded on an 8 mm stage, and compressed at a rate of 0.5 mm/min for half of the total gel height to observe deformation and strain. The Young’s modulus was derived from the stress-strain curves.
Preparation of cell culturing devices
A custom negative mold was printed using a 3D, Form 2 stereolithography printer (Formlabs, Inc.). Cell culture devices were cast using soft lithography to produce a PDMS reservoir for cell culture. The culture wells were composed of a cylindrical culture section (3 mm in diameter and 5 mm tall), enabling a maximum of 35 μL of volume. Additionally, a conical media reservoir above the cylindrical culturing section was able to contain up to 150 μL of media. Specific dimensions of the mold, and subsequently the PDMS wells. To fabricate PDMS culturing devices, 70 g of Sylgard 184 PDMS (Dow Corning) was preparing according to the manufacturer’s instructions and poured into a 10 cm x 10 cm square dish. The mold was placed in the PDMS, and the PDMS was degassed by applying a vacuum for 1 hour. Subsequently, the PDMS was allowed to cure at 60°C for 4 hours in a convection oven. The PDMS slab was then cut into three-well pieces and plasma-bonded to cover glass slides using a corona plasma gun. PDMS triplicate well-slides were then autoclaved prior to use for cell culture and experimental evaluation.
Cell culture and seeding in FLIP scaffolds
Human dermal fibroblasts (HDF) or D1 mouse mesenchymal stem cells (Cell Applications, Inc., San Diego, CA) were maintained in culture in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific) containing 10% fetal bovine serum (Thermo Fisher Scientific) at 37°C and 5% CO2. Human astrocytes were cultured using astrocyte growth media (Lonza), with all provided bullet kit supplements. Primary mouse neural precursor cells (NPCs) were cultured in DMEM containing 5% FBS (Hyclone), pituitary extract (13.6 μg/μL), EGF (0.1 μg/μL), bFGF (0.1 μg/μL) and N-2 formulation (100x, Gibco). Human umbilical vein cells (HUVECs) were cultured in endothelial cell media (Lonza) with bullet kit supplements except for VEGF. All media for cells contained 1% Penn/Step. Media was changed every 2-3 days. To seed cells in FLIP scaffolds, 20 μL microgels were first equilibrated in supplemented media for 30 minutes before pelleting and removing supernatant. In some cases, cells were pre-stained with CellTracker Orange (Thermo Fisher Scientific), according to the manufacturer protocol, to allow for live monitoring of the cells as they spread in the annealed gels. Briefly, cells were stained with CellTracker at 10 μM and incubated 37°C for 30 mins before replacing the media. Cells were trypsinized and 1.0 x 105 cells/10 μL gel were pelleted by centrifugation at 250 x g for 5 minutes. Media supernatant was aspirated and equilibrated microgels in FXIII and Thrombin (at 0.01 U/uL gel and 0.002 U/uL gel respectively) were then added to the cell pellet and mixed thoroughly by pipetting. Importantly, prior to gel/cell seeding, 6 μL of sterile 1% agarose in PBS was added to the wells to coat the glass surface and allowed to cool to 25°C to prevent cell attachment to glass. 10 μL of gel plus cells was then pipetted into each well in the PDMS culturing device. The sHMP gel was allowed to anneal for 1 hour at 37°C. After annealing, the wells were filled with 150 μL supplemented media and incubated for 48 hours.
3D culture imaging and spreading assessment
For improved imaging resolution and visualizing cell spreading, in cases without live image assessment as described above, annealed gels with cells cultured for 2 days were fixed in 1% paraformaldehyde for 15 minutes at 25°C. The cultures were permeabilized in 0.1% Triton X-100 in PBS and stained using DAPI (Sigma-Aldrich) for cell nuclei and rhodamine-phalloidin (Thermo Fisher) for cell actin per manufacturer’s guidelines for 1 hour. Gels were washed with PBS before z-stack imaging with a Nikon confocal. To quantify cell spreading, the z-stacks were imported into IMARIS to generate surface renders of cell actin for surface area quantification and to count nuclei.
Transfection of FLIP gel culture and assay for transgene expression
Transfection from loaded FLIP scaffolds was performed as described in the main text and above methods. Scaffolds were annealed as before, and cells seeded into the scaffolds. Bolus transfection was performed two days after seeding cells to allow for adequate infiltration and spreading. For bolus controls, DNA/PEI particles were prepared as previously described for L-PEI conditions with and without HA coating prior to 3D culture preparation. Cells were seeded into non-loaded scaffold, similar to the cell-only control, before adding in the fresh particle solution. Amounts were scaled up depending on DNA dose and number of wells, but the particle volume administered to each well remained constant (20 μL of particles were added to each well as a bolus administration). After 4 hours of DNA/PEI particle exposure, the particle-containing media was removed and replenished with fresh media, as in 2D culture.
Transfection was quantified by measuring expression of GLuc 48 hours after 3D culture seeding, using the FLASH Gaussia Luciferase assay kit (NanoLight) per manufacturer’s protocol. Conditioned media was collected from each well at each time point. Briefly, 20 μL of each sample was mixed with 50 μL of diluted substrate solution, pipetted for 2 to 3 seconds to mix, and read for luminescence with a 5 second integration time using a plate reader (Tecan Spark).
Cells were also processed for flow cytometry (BD Accuri™ C6 Plus) quantification of transgene expression (GFP). Briefly, cells were extracted from gels using a digestion protocol (200 U/mL Type IV Collagenase, Hyaluronidase, 125 U/mL DNase I) in RPMI media (no serum). Following 30 minutes incubation and wash steps in PBS, cells were for flow. Gating was based on 2D controls from trypsinized samples for cells/debris, singlets, and GFP expressing cells, and controls gated against gels without any cells. Samples were run for 20,000 cell singlets, n = 3-5.
Cell viability
Cell viability was quantified using the PrestoBlue assay (Thermo Fisher Scientific) per manufacturer’s instructions. 2D culture in 48 and 24 well plates was assessed with a 1:5 dilution of PrestoBlue in serum-containing media, followed by 2 hours incubation at 37°C. For 3D culture, the media in each well was replaced with a solution of 10 μL of the PrestoBlue reagent mixed with 90 μL of media and incubated for 3 hours. For both models, 90 μL from each well was transferred into a 96-well plate and absorbance was read at 570 nm, normalized to 600 nm, using the plate reader (Tecan Spark). Viability (metabolic activity) was calculated using a blank control and cell-only control, normalized across the remaining sample conditions.
For 3D culture, cell viability was also assessed using LIVE/DEAD Viability/ Cytotoxicity Kit (Thermo). Briefly, cells were stained at 0.5 μM Calcein AM (Live stain, 488 nm) and 2 μM Ethidium homodimer-1 (Dead stain, 555 nm) in sterile 1X PBS for 30 minutes at room temperature. Cells were washed in 1X PBS prior to imaging on a Nikon Ti Eclipse equipped with C2 laser LED excitation. Z-stacks were performed within 60 minutes of staining to reduce cell death and sample bias. Live and dead channels were assessed with IMARIS for relative quantification of cell population based on volume-filling.
Cells were also processed for flow cytometry (BD Accuri™ C6 Plus) quantification of viability. Briefly, cells were extracted from gels using a digestion protocol (200 U/mL Type IV Collagenase, Hyaluronidase, 125 U/mL DNase I) in RPMI media (no serum). Following 30 minutes incubation and wash steps, cells were stained with Propidium Iodide (PI) at 3 μM and incubating at room temperature for 15 minutes. Gating was based on 2D controls from trypsinized samples for cells/debris, singlets, and GFP expressing cells, and controls gated against gels without any cells. Samples were run for 20,000 cell singlets, n = 3-5.
Statistical analysis
Statistical analysis and plotting were performed using Graphpad Prism 8 and Minitab 17. Unless stated otherwise, experiments were repeated at least three times in duplicate (3D culture) or triplicate (2D culture) and/or with three independent gel samples in each experiment (mechanical studies). It was assumed that samples, which were prepared independently, were statistically independent from each other. Simple study designs were assessed using a 95% confidence interval using a one-way ANOVA where appropriate, and followed by Tukey post-hoc test. All error is reported as the standard deviation (SD). Design of experiment (DOE) studies were based on either a factorial design (3x3 or 4x4) or a surface-response design (central composite, two-block orthogonal), based on suggestions from Minitab software for the parameter ranges of interest. For these experiments, design-optimization within Minitab was used to determine the optimal conditions, with α of 0.05 and target RMSE of 2-3 SD for predicted parameters. Last, for time-series studies, a repeated-measured (RM) ANOVA was performed for luciferase data, since from the same sample over time, while flow analysis (GFP, viability) was performed with a standard one-way ANOVA, since endpoints were independent samples (separate devices, with cells extracted at each time point). In all cases, significance (p<) is represented as **** < 0.001, *** < 0.005, ** < 0.01, and * < 0.05.
Supplementary Material
Funding/Acknowledgements
This invention was made with Government support under Federal Grant No. R01NS094599 awarded by the National Institutes of Health, as well as the T32 training grant from the National Institute of General Medical Sciences (NIGMS) NIH National Service Research Awards (NSRA) Program under Duke University’s Cell and Biomolecular Tissue Engineering training grant (T32GM008555). The Federal Government has certain rights to this invention. Special thanks to the staff at Duke’s Shared Materials instrumentation Facility (SMIF), with their support on the microstrain analyzer and the atomic force and electron microscopes.
Footnotes
The supplemental results give more detail on the design of experiment approaches used to optimize several parameters of the FLIP system. The data also describes the optimization used for minicircle DNA to improve transfection efficiency, compared to plasmid methods.
Conflicts of Interest
The authors have no conflicts of interest related to the described work.
References
- [1].Keeler AM, Flotte TR, Annu. Rev. Virol 2019, 6, 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Anselmo AC, Mitragotri S, Bioeng. Transl. Med 2019, DOI 10.1002/btm2.10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chin JS, Chooi WH, Wang H, Ong W, Leong KW, Chew SY, Acta Biomater. 2019, 90, 60. [DOI] [PubMed] [Google Scholar]
- [4].Wang F, Qi LS, Trends Cell Biol. 2016, 26, 875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Li SD, Huang L, Gene Ther. 2006, 13, 1313. [DOI] [PubMed] [Google Scholar]
- [6].Li L, Hu S, Chen X, Biomaterials 2018, 171, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kozielski KL, Tzeng SY, Green JJ, Wiley Interdiscip. Rev. Nanomedicine Nanobiotechnology 2013, 5, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hassett KJ, Benenato KE, Jacquinet E, Lee A, Woods A, Yuzhakov O, Himansu S, Deterling J, Geilich BM, Ketova T, Mihai C, Lynn A, McFadyen I, Moore MJ, Senn JJ, Stanton MG, Almarsson Ö, Ciaramella G, Brito LA, Mol. Ther. - Nucleic Acids 2019, 15, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R, Nat. Rev. Drug Discov 2020, DOI 10.1038/s41573-020-0090-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Spivack K, Muzzelo C, Neely C, Vanzelli J, Kurt E, Elmer J, bioRxiv 2018, 338707. [Google Scholar]
- [11].Dna B, 2014, 4, 2010. [Google Scholar]
- [12].Williams JA, Carnes AE, Hodgson CP, Biotechnol. Adv 2009, 27, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kowalczewski CJ, Saul JM, Acta Biomater. 2015, 25, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen R, Zhang H, Yan J, Bryers JD, Gene Ther. 2018, 25, 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Siegman S, Truong NF, Segura T, Acta Biomater. 2015, 28, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Villate-Beitia I, Truong NF, Gallego I, Zárate J, Puras G, Pedraz JL, Segura T, RSC Adv. 2018, 8, 31934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ehsanipour A, Nguyen T, Aboufadel T, Sathialingam M, Cox P, Xiao W, Walthers CM, Seidlits SK, Cell. Mol. Bioeng 2019, 12, DOI 10.1007/s12195-019-00593-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Truong NFNF, Kurt E, Tahmizyan N, Cai Lesher-Pérez S, Chen M, Darling NJNJ, Xi W, Segura T, Lesher-Pérez SC, Chen M, Darling NJNJ, Xi W, Segura T, Acta Biomater. 2019, 94, DOI 10.1016/J.ACTBIO.2019.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bonadio J, Smiley E, Patil P, Goldstein S, Nat. Med 1999, 5, 753. [DOI] [PubMed] [Google Scholar]
- [20].Debus H, Beck-Broichsitter M, Kissel T, Lab Chip 2012, 12, 2498. [DOI] [PubMed] [Google Scholar]
- [21].Pezzoli D, Giupponi E, Mantovani D, Candiani G, Sci. Rep 2017, 7, 44134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Grigsby CL, Ho Y-P, Lin C, Engbersen JFJ, Leong KW, Sci. Rep 2013, 3, 3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lu M, Ho Y-P, Grigsby CL, Ahsan Nawaz A, Leong KW, Huang TJ, 2013, DOI 10.1021/nn404193e. [DOI] [Google Scholar]
- [24].Werfel TA, Jackson MA, Kavanaugh TE, Kirkbride KC, Miteva M, Giorgio TD, Duvall C, J. Control. Release 2017, 255, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Adolph EJ, Nelson CE, Werfel TA, Guo R, Davidson JM, Guelcher SA, Duvall CL, J. Mater. Chem. B 2014, 2, 8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Veilleux D, Gopalakrishna Panicker RK, Chevrier A, Biniecki K, Lavertu M, Buschmann MD, J. Colloid Interface Sci 2018, 512, 335. [DOI] [PubMed] [Google Scholar]
- [27].Lei Y, Rahim M, Ng Q, Segura T, J. Control. Release 2011, 153, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wang LL, Burdick JA, Adv. Healthc. Mater 2017, 6, DOI 10.1002/adhm.201601041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hajj KA, Whitehead KA, Nat. Rev. Mater 2017, 2, 17056. [Google Scholar]
- [30].Cheng Q, Wei T, Farbiak L, Johnson LT, Dilliard SA, Siegwart DJ, Nat. Nanotechnol 2020, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jackson MA, Werfel TA, Curvino EJ, Yu F, Kavanaugh TE, Sarett SM, Dockery MD, V Kilchrist K, Jackson AN, Giorgio TD, Duvall CL, ACS Nano 2017, 11, 5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].García KP, Zarschler K, Barbaro L, Barreto JA, O’Malley W, Spiccia L, Stephan H, Graham B, Small 2014, 10, 2516. [DOI] [PubMed] [Google Scholar]
- [33].Youngblood RL, Truong NF, Segura T, Shea LD, Mol. Ther 2018, 26, 2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Schwabe K, Ewe A, Kohn C, Loth T, Aigner A, Hacker MC, Schulz-Siegmund M, Int. J. Pharm 2017, 526, 178. [DOI] [PubMed] [Google Scholar]
- [35].Saul JM, Linnes MP, Ratner BD, Giachelli CM, Pun SH, Biomaterials 2007, 28, 4705. [DOI] [PubMed] [Google Scholar]
- [36].Truong NF, Segura T, ACS Biomater. Sci. Eng 2018, 4, 981. [DOI] [PubMed] [Google Scholar]
- [37].Mukalel AJ, Evans BC, Kilchrist KV, Dailing EA, Burdette B, Cheung-Flynn J, Brophy CM, Duvall CL, Control J. Release 2018, 282, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jung S, Lodge TP, Reineke TM, J. Phys. Chem. B 2018, 122, 2449. [DOI] [PubMed] [Google Scholar]
- [39].Jeon O, Krebs M, Alsberg E, J. Biomed. Mater. Res. Part A 2011, 98A, 72. [DOI] [PubMed] [Google Scholar]
- [40].Lei Y, Segura T, Biomaterials 2009, 30, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Oliveira AV, Bitoque DB, Silva GA, J. Nanomater 2014, 2014, DOI 10.1155/2014/246347. [DOI] [Google Scholar]
- [42].Zaki NM, Nasti A, Tirelli N, Macromol. Biosci 2011, 11, 1747. [DOI] [PubMed] [Google Scholar]
- [43].Martens TF, Remaut K, Deschout H, Engbersen JFJ, Hennink WE, Van Steenbergen MJ, Demeester J, De Smedt SC, Braeckmans K, J. Control. Release 2015, 202, 83. [DOI] [PubMed] [Google Scholar]
- [44].Urbiola K, Sanmartín C, Blanco-Fernández L, Tros de Ilarduya C, Nanomedicine 2014, 9, 2787. [DOI] [PubMed] [Google Scholar]
- [45].He Y, Cheng G, Xie L, Nie Y, He B, Gu Z, Biomaterials 2013, 34, 1235. [DOI] [PubMed] [Google Scholar]
- [46].Wang Y, Xu Z, Zhang R, Li W, Yang L, Hu Q, Colloids Surfaces B Biointerfaces 2011, 84, 259. [DOI] [PubMed] [Google Scholar]
- [47].Kwon MY, Wang C, Galarraga JH, Puré E, Han L, Burdick JA, Biomaterials 2019, 222, 119451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kharkar PM, Rehmann MS, Skeens KM, Maverakis E, Kloxin AM, ACS Biomater. Sci. Eng 2016, 2, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gramlich WM, Kim IL, Burdick JA, Biomaterials 2013, 34, 9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Choosakoonkriang S, Lobo BA, Koe GS, Koe JG, Middaugh CR, J. Pharm. Sci 2003, 92, 1710. [DOI] [PubMed] [Google Scholar]
- [51].Wightman L, Kircheis R, Rössler V, Garotta S, Ruzicka R, Kursa M, Wagner E, J. Gene Med 2001, 3, 362. [DOI] [PubMed] [Google Scholar]
- [52].Feng C-L, Han Y-X, Guo H-H, Ma X-L, Wang Z-Q, Wang L-L, Zheng W-S, Jiang J-D, 2017, DOI 10.1080/10717544.2017.1386732. [DOI] [Google Scholar]
- [53].Jiang G, Park K, Kim J, Kim KS, Oh EJ, Kang H, Han S-E, Oh Y-K, Park TG, Kwang Hahn S, Biopolymers 2008, 89, 635. [DOI] [PubMed] [Google Scholar]
- [54].Hardee CL, Arévalo-Soliz LM, Hornstein BD, Zechiedrich L, Genes (Basel). 2017, 8, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Cheng C, Tang N, Li J, Cao S, Zhang T, Wei X, Wang H, J. Med. Genet 2019, 0, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Osborn MJ, McElmurry RT, Lees CJ, DeFeo AP, Chen Z-Y, Kay MA, Naldini L, Freeman G, Tolar J, Blazar BR, Mol. Ther 2011, 19, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Griffin DR, Weaver WM, Scumpia P, Di Carlo D, Segura T, Nat. Mater 2015, 14, 737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Pruett L, Koehn H, Martz T, Churnin I, Ferrante S, Salopek L, Cottler P, Griffin DR, Daniero JJ, Laryngoscope 2020, 130, 2432. [DOI] [PubMed] [Google Scholar]
- [59].Daly AC, Riley L, Segura T, Burdick JA, Nat. Rev. Mater 2020, 5, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gehlen DB, Jürgens N, Omidinia-Anarkoli A, Haraszti T, George J, Walther A, Ye H, De Laporte L, Macromol. Rapid Commun 2020, 41, 2000191. [DOI] [PubMed] [Google Scholar]
- [61].Sinclair A, O’Kelly MB, Bai T, Hung H-C, Jain P, Jiang S, Adv. Mater 2018, 30, 1803087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Cho IS, Ooya T, J. Biomater. Sci. Polym. Ed 2018, 29, 145. [DOI] [PubMed] [Google Scholar]
- [63].Muir VG, Qazi TH, Shan J, Groll J, Burdick JA, ACS Biomater. Sci. Eng 2021, acsbiomaterials.0c01612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Hinton TJ, Jallerat Q, Palchesko RN, Park JH, Grodzicki MS, Shue HJ, Ramadan MH, Hudson AR, Feinberg AW, Sci. Adv 2015, 1, e1500758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Cam C, Segura T, Acta Biomater. 2014, 10, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Seymour AJ, Shin S, Heilshorn SC, Adv. Healthc. Mater 2021, 2100644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ehsanipour A, Nguyen T, Aboufadel T, Sathialingam M, Cox P, Xiao W, Walthers CM, Seidlits SK, Cell. Mol. Bioeng 2019, 12, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Nih LR, Sideris E, Carmichael ST, Segura T, Adv. Mater 2017, 29, 1606471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Darling NJ, Sideris E, Hamada N, Carmichael ST, Segura T, Adv. Sci 2018, 5, 1801046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Nih LR, Moshayedi P, Llorente IL, Berg AR, Cinkornpumin J, Lowry WE, Segura T, Carmichael ST, Data Br. 2017, 10, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Truong NFNF, Lesher-Pérez SCSC, Kurt E, Segura T, Bioconjug. Chem 2018, 30, acs. bioconjchem.8b00696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Mantz A, Pannier AK, Biomaterial Substrate Modifications That Influence Cell-Material Interactions to Prime Cellular Responses to Nonviral Gene Delivery, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Johnston CD, Cotton SL, Rittling SR, Starr JR, Borisy GG, Dewhirst FE, Lemon KP, Proc. Natl. Acad. Sci 2019, 116, 11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Steinle H, Ionescu T-M, Schenk S, Golombek S, Kunnakattu S-J, Özbek MT, Schlensak C, Wendel HP, Avci-Adali M, Int. J. Mol. Sci 2018, 19, 1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Yan J, Chen R, Zhang H, Bryers JD, Macromol. Biosci 2019, 19, 1800242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Lei Y, Gojgini S, Lam J, Segura T, Biomaterials 2011, 32, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Lam J, Segura T, Biomaterials 2013, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.