

Abstract
Intraoperative bioprinting (IOB), which refers to the bioprinting process performed on a live subject in a surgical setting, has made it feasible to directly deliver gene-activated matrices into craniomaxillofacial (CMF) defect sites. In this study, we demonstrated a novel approach to overcome the current limitations of traditionally fabricated non-viral gene delivery systems through direct IOB of bone constructs into defect sites. We used a controlled co-delivery release of growth factors from a gene-activated matrix (an osteogenic bioink loaded with plasmid-DNAs (pDNA)) to promote bone repair. The controlled co-delivery approach was achieved from the combination of platelet-derived growth factor-B encoded plasmid-DNA (pPDGF-B) and chitosan-nanoparticle encapsulating pDNA encoded with bone morphogenetic protein-2 (CS-NPs(pBMP2)), which facilitated a burst release of pPDGF-B in 10 days, and a sustained release of pBMP-2 for 5 weeks in vitro. The controlled co-delivery approach was tested for its potential to repair critical-sized rat calvarial defects. The controlled-released pDNAs from the intraoperatively bioprinted bone constructs resulted in ~40% bone tissue formation and ~90% bone coverage area at 6 weeks compared to ~10% new bone tissue and ~25% total bone coverage area in empty defects. The delivery of growth factors incorporated within the intraoperatively bioprinted constructs could pose as an effective way to enhance bone regeneration in patients with cranial injuries in the future.
Keywords: Intraoperative bioprinting, in-situ delivery, controlled co-delivery, plasmid-DNAs
INTRODUCTION
Craniomaxillofacial (CMF) abnormalities, including genetic mutations or chromosome abnormalities [1] and traumatic injuries like combat injuries [2], affect thousands of people every year around the world [3]. Patients with craniofacial disorders often require several surgical procedures for the reconstruction of the crania and are often left with poor aesthetic and functional results [4]. Intraoperative bioprinting (IOB) technology can accurately capture and acquire defect information by processing digital data in real-time to accurately deliver regenerative constructs to defect site(s). Compared to implantation of prefabricated tissue constructs, IOB technology is greatly beneficial as it can facilitate the regeneration of bone defects occuring naturally with irregular topographies with minimum risk of contamination and manual interventions such as in vitro culturing bioprinted scaffolds, transportation during surgery, or modifying bioprinted scaffolds conforming the defect shape [5]. Thus, IOB can be considered a promising method for the immediate delivery of anatomically-correct bone constructs into the defect site for CMF reconstruction.
The necessity for accelerated bone repair has led to the investigation of growth factor/gene-based therapies with recombinant proteins and growth factors including bone morphogenetic protein-2 (BMP-2) and platelet-derived growth factor (PDGF) within bone constructs to promote osteogenesis and mitogenesis in vivo, respectively [6–8]. BMP-2 is a clinically available protein and improved bone regeneration has been demonstrated when BMP-2 is employed in vitro and in vivo [9–11]. PDGF, on the other hand, shows an angiogenic effect through the upregulation of vascular endothelial growth factor (VEGF) expression, promotes osteoblast cell proliferation, and enhances cell migration (chemotaxis) and proliferation (mitogenesis) [6,12–14]. However, the high cost of these growth-factor based therapies, their short half-lives, and the need for supra-physiological dosages for clinical effectiveness limit their clinical use [15,16]. Additionally, high doses of recombinant human protein are linked to several side effects such as soft tissue swelling and ectopic bone formation [17] generating demand for the development of alternative growth factor delivery methods.
To date, both in-vivo and ex-vivo gene therapy has been used in calvarial bone tissue repair [18] as evident in a convincing proof-of-principle study that gene therapy successfully heals the damaged bone tissue [19]. Gene-based growth factors using non-viral gene therapy have been used due to their feasibility, safety, and potential for clinical translational for bone regeneration applications [13,20], yet relatively low transfection efficiency has restricted the usage of non-viral vehicles as an ideal gene transfection vector [7,21]. New release mechanisms and strategies have been thus investigated for gene-activated matrices, such as nano/microparticle encapsulated plasmid-DNAs (pDNAs) or various hydrogels loaded with genes [19]. The delivered gene-based growth factors can result commitment of differentiating autologous cells into multiple lineages [22]. Recent literature illustrates the enhancement of bone regeneration using multiple growth factors through synergistic mechanisms [8]. The combinatorial treatment of PDGF and VEGF was shown to enhance bone regeneration compared to either alone [23]; however, drug combinations may exhibit antagonistic effects and necessitate the controlled delivery, which has been demonstrated to enhance tissue formation previously [24]. The controlled release of growth factors has been employed to guide the formation of biomimetic tissues and improve tissue engineering outcomes by manipulating overlaps or timing of the release. [25–28]. For example, the combinatorial treatment of PDGF with fibroblast growth factor (FGF) did not significantly improve vascularization, but the controlled delivery of FGF followed by PDGF improved the vessel formation as compared to their simultaneous delivery [29]. The sequence of PDGF followed by BMP-2 also demonstrated more vascularized bone tissue formation compared to, simultaneous delivery of these two growth factors [24]. Although the majority of research endeavors has focused on signaling pathways and delivery systems, this study utilized the strategy of delivering PDGF first followed by BMP-2 through the design of our gene-activated matrix (bioink) to investigate its potential in bone regeneration in vivo. Considering the extensive evidence in the literature, a platform for the controlled plasmid delivery encoding PDGF and BMP-2 as a non-viral gene delivery reservoir was investigated by introducing a novel extrudable bioink under the IOB technology.
In this study, we aimed to investigate in-situ controlled co-delivery of pDNAs via IOB technology for the first time, to repair critical-sized rat calvarial defects. We utilized a gene activated matrix as an in-situ delivery reservoir for pDNAs encoding BMP-2 and PDGF-B. pBMP-2, encapsulated in chitosan nanoparticles (CS-NPs), and pPDGF-B were loaded in an osteogenic bioink and directly bioprinted into critical-sized calvarial defects on a rat model as shown in Fig.1. We expect that pPDGF releasing from the bioprinted bone constructs transfecting endogenous cells, which will in turn encode PDGF resulting in improved recruitment of progenitors to the defect site. With the sustained and delayed release of pBMP-2, host cells are expected to be transfected an encoded BMP-2 which can differentiate recruited endogenous progenitors into osteogenic lineage and improve bone regeneration in vivo. The outcomes of the controlled co-delivery of pDNAs from bioprinted bone constructs were assessed both in vitro and in vivo, and intraoperatively bioprinted rat calvarial defects were investigated using functional characterization for potential therapeutic efficacy of cranial injuries.
Fig 1.
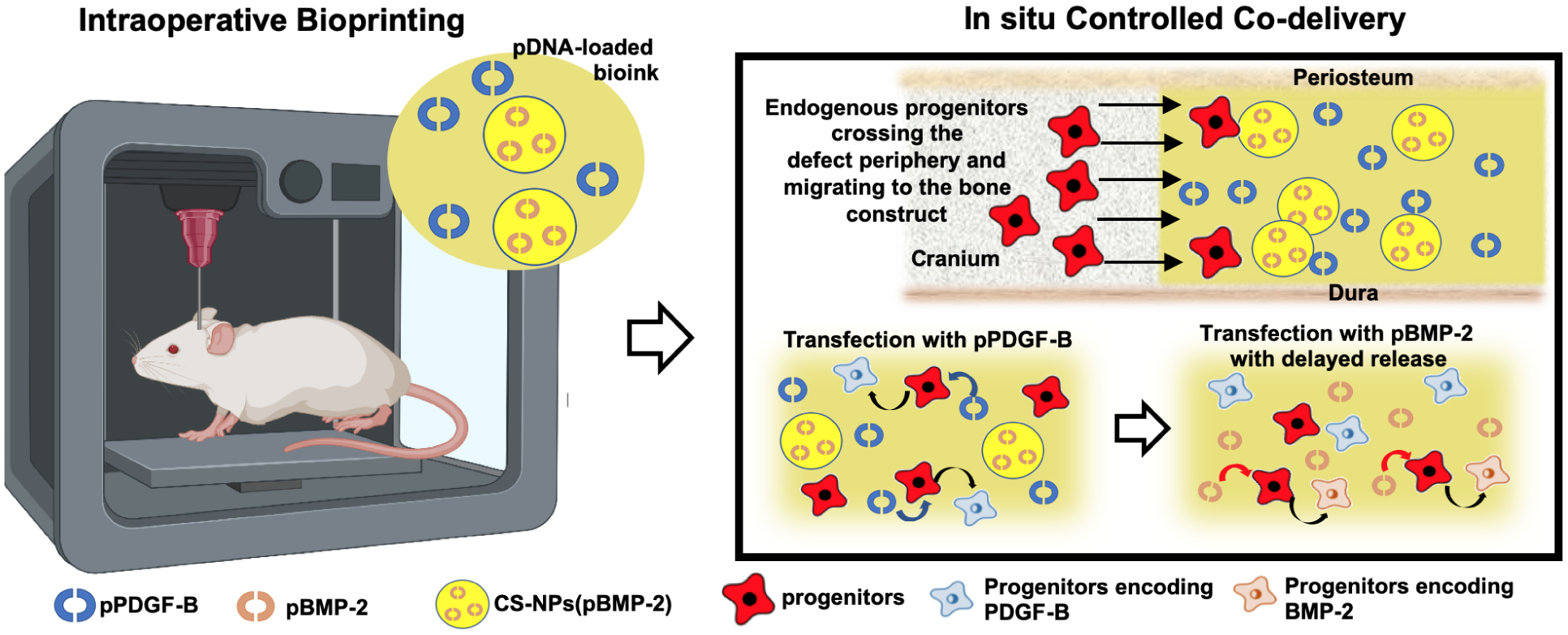
Schematic overview of intraoperative bioprinting of bone constructs with an osteogenic bioink utilized as a controlled co-delivery release platform for the repair of critical-sized rat calvarial defects (intraoperative bioprinting figure was created using BioRender).
MATERIALS AND METHODS
2.1. Preparation of chitosan nanoparticles with and without pDNA-BMP2 encapsulation
Chitosan nanoparticles (CS-NPs) were formed based on an ionic gelation method using sodium tripolyphosphate (TPP, Sigma-Aldrich, cat. no. 238503) with deacetylated chitosan (CS, DD: 75% and MW:190–375 kDa, Sigma-Aldrich, cat. no. 417963) as described previously [30]. CS and TPP solutions were prepared in three different concentrations of 0.50, 0.75, and 1.00 (mg/mL) by dissolving CS in 0.1M HCL acid and TPP in distilled water. All solutions were filtered through a 0.22 μm filter (Millipore) for sterilization. Prepared CS solutions were then mixed with TPP solutions at a volumetric ratio of 2:1 (v/v) (CS:TPP) under magnetic stirring at 500 rpm at room temperature for 15–20 min according to our previous study [30]. Next, the nanoparticles were purified by centrifuging at 10,000 rpm for 15 min at 4 °C. All groups for nanoparticle formulation were presented in Table 1.
Table 1.
DLS results of CS-NPs* (data were presented as mean ± s.dev., n=3).
| CS (mg/mL) | TPP (mg/mL) | Average Dia. of CS-NPs (nm) | PDI | ||
|---|---|---|---|---|---|
| 0.50 | 0.50 | 230.8±7.6 | 0.331±0.032 | ||
| 0.50 | 0.75 | 2755.3±153.4 | 1.000±0.000 | ||
| 0.50 | 1.00 | 7721.0±205.1 | 0.964±0.052 | ||
| 0.75 | 0.50 | 134.2±2.8 | 0.384±0.018 | ||
| 0.75 | 0.75 | 577.6±9.1 | 0.698±0.032 | ||
| 0.75 | 1.00 | 6030.0±3388.5 | 1.000±0.000 | ||
| 7 | 1.00 | 0.50 | 113.3±0.6 | 0.472±0.052 | 30.3±1.2 |
| 1.00 | 0.75 | 725.4±20.6 | 0.416±0.112 | ||
| 1.00 | 1.00 | 5304.0±NA | 1.000±0.000 |
CS:TPP (2:1) (v/v)
pDNA was prepared based on our previously published study [31]. Briefly, the chemically competent DH5α™ bacterial strain (Escherichia Coli species) was transformed with either pPDGF-B (cat. no. BC064056, Origene Technologies, Rockville, MD) or pBMP-2 (cat. no. SC119392, Origene technologies). The further information about the inserted genes, the plasmid vectors and the maps can be can be reached from Origene technologies (Rockville, MD, USA). The transformed cultures were then expanded in Lennox L Broth (Research products incorporated, MT. Prospect, IL) overnight at 37 °C in an incubator shaker at 300 rpm. pDNA was extracted using a GenElute™ HP endotoxin-free plasmid maxiprep kit (cat. no. NA0410–1KT, Sigma Aldrich) and analyzed for purity using a Zetasizer Nano-ZS (Malvern Instruments, Westborough, MA) by measuring the ratio of absorbance (A260 nm/A280 nm).
Similarly, pBMP-2 encapsulated chitosan nanoparticles (CS-NPs(pBMP2)) were prepared. Briefly, 0.75 ml of pBMP-2 (500 μg/ml) was mixed manually with 0.25 ml of TPP solution (0.25 and 0.50 mg/mL) in an Eppendorf tube. Next, CS-NPs were formed by adding TPP+pBMP-2 solution into CS solution (1 mg/mL) at a ratio of 2:1 (v/v) (chitosan:(TPP+pBMP-2)) stirred at a rate of 500 rpm. The pH of the solution was adjusted to be 4.6–5.4 by adding 0.1N NaOH. Supernatants were discarded by centrifuging at 10,000 rpm for 15 min at 4 °C. The remaining precipitate was washed twice with PBS, lyophilized, and then stored at −20 °C for further use. The groups used for the formation of CS-NPs(pBMP-2) were enumerated in Table 2.
Table 2.
DLS results and the encapsulation efficiency of CS-NPs(pBMP-2) * (data were presented as mean ± s.dev., n=3).
| CS (mg/mL) | TPP (mg/mL) | Average Dia. Of CS-NPs(pBMP-2) (nm) | PDI | Zeta Potential (mV) | ||
|---|---|---|---|---|---|---|
| 1 | 0.50 | 2553±114.9 | 0.509±0.095 | 19.8±0.2 | ||
| 2 | 1 | 0.25 | 308.1±4.1 | 0.265±0.053 | 28.8±0.9 | 77.3±0.13 |
CS:TPP (2:1) (v/v)
2.2. Characterization of nanoparticles
The mean particle size and size distribution of CS-NPs with and without pBMP-2 were measured using Dynamic Light Scattering (DLS) (Zetasizer Nano ZS, Worcestershire, UK. Polydispersive index (PDI) of nanoparticles was recorded from DLS measurements.
The zeta potential of CS-NPs with and without pBMP-2 was analyzed using a Malvern Zetasizer Nano ZS instrument (Worcestershire, UK) at 25 °C. The encapsulation efficiency of prepared nanoparticles was determined by measuring the supernatants of chitosan:(TPP+pBMP-2) solutions spectrophotometrically using Nanodrop ND-1000 Spectrophotometer (Wilmington) with an absorbance ratio of absorbance A260 nm/A280 nm. Encapsulation efficiency (EE) for pBMP-2 encapsulated CS-NPs was calculated as follows:
| (Eq. 1) |
where W0 was the total amount of pDNA and W1 was the measured free pDNA, which was not encapsulated in CS-NPs. All measurements were performed in triplicates.
CS-NPs with and without pBMP-2 were visualized using scanning electron microscopy (SEM) (Nova NanoSEM, Milpitas, CA). Diluted CS-NPs solutions were manually pipetted onto a carbon tape. After air drying, samples were iridium coated with a sputter coater (EM ACE600 Leica Microsystems) and then imaged under high vacuum mode using a Through the Lens Detector (TLD) at an accelerating voltage of 5.0 keV with a working distance from 3.2 to 4.3 mm.
Characteristic functional groups of CS-NPs with and without pBMP-2 were determined using attenuated total reflectance fourier transform infrared spectrophotometry (ATR-FTIR) (Bruker Vertex V70 FTIR spectrometer, The Woodlands, TX, US) as described in a previous study [32].
2.3. Preparation of the osteogenic bioink
The osteogenic bioink was prepared according to the following procedure. Briefly, 56% (w/v) β-glycerophosphate (βGP) solution was combined with 2% (w/v) chitosan solution (CS) at a volumetric ratio of 1:9 (v/v). CS and βGP facilitates temperature-induced physical crosslinking and a rapid solution-to-gelation phase transition when the temperature goes over 34 °C. The 56% βGP has been exclusively used to crosslink with chitosan in the literature [33–37]. Type-I collagen (extracted from rat tail according to the protocol described in [38]) was dissolved in 0.02N acetic acid solution to prepare a 9 mg/ml collagen solution and then mixed with neutralized CS-βGP solution at a ratio of 1:4 (v/v). Freeze-dried collagen sponges were then added to CS-BGP+Coll solution to achieve a final concentration of 26.6 mg/ml collagen sponge, and was mixed for 30 min at room temperature using an in-house developed paste mixer until homogenity was achieved. The increasing collagen concentration did not affect pH as the collagen was added into solution in dried form. Furthermore, the final pH of the bioink was neutral and it did not cause a toxic effect on the cells as we demonstrated recently [39]. 160 mg of sterile nano-hydroxyapatite particles (nHAp) (sifted with 106-micron filter) (Fluidinova, Portugal), which was made upon evaporation of 15% (w/v) aqueous hydroxyapatite solution (nanoXIM-HAp202), were added to the 1.5 ml of the overall mixture and stirred for an additional 10 – 15 min. The final amount of 1.5 ml bioink was mixed with freeze-dried CS-NPs(pBMP-2) to form Bioink+CS-NPs(pBMP-2). The aqueous form of pPDGF-B (2000 μg /ml) was mixed at the same concentration of CS-NPs(pBMP2) with bioink to form Bioink+pPDGF-B+CS-NPs(pBMP-2). For controlled co-delivery, freeze-dried CS-NPs(pBMP-2) and aqueous form of pPDGF-B were simultaneously mixed with the bioink to form Bioink+ pPDGF-B+CS-NPs(pBMP-2).
CS powder and nHAp were sterilized using gamma-ray irradiation (25 kGy (± 10%) [40], Cobalt-60, Gamma Irradiation Facility, Radiation Science and Engineering Center, The Pennsylvania State University (PSU). β-GP solution was sterilized with 0.22 μm filters (Millipore, Burlington, MA, US). Collagen solution was sterilized according to our previously published study [41]. Collagen sponges were sterilized using an Anprolene gas sterilizer with ethylene oxide [42] and stored at −80 °C.
2.4. Fabrication of 3D bioprinted bone constructs
Bone constructs were 3D bioprinted using the in-house developed Multi-Arm BioPrinter (MABP) [43]. Bioink was loaded and then extruded pneumatically using an air dispenser (Nordson Corp., Westlake, OH, USA) on a glass cover slide, placed on a heated stage maintained at 37 °C. The constructs were designed with the same pore size and filament diameter of 0.5 mm and bioprinted in a lay-down pattern of 0°/90°. The extrusion pressure was set to be between 80 to 140 kPa. The inner nozzle diameter of needle was 410 μm and the bioprinting speed was 400 mm/min.
2.5. Characterization of 3D bioprinted constructs
Dual-layer bioprinted constructs were imaged using an EVOS FL auto cell imaging system (Thermo Fisher Scientific, Waltham, WA, US). The pore size and filament diameter were measured using the manufacturer’s software. Data were obtained for a total of five filament diameter values and five pores per construct, and each measurement was done in triplicates.
The morphological characteristics of 3D bioprinted constructs were visualized by SEM as described before. Bioink constructs with and without pDNA-loaded were imaged under the high vacuum mode at the accelerating voltage range of 3.0 to 5.0 keV with a working distance of 3.5 mm to 4.4 mm.
ATR-FTIR spectrum of the bioink and pDNA-loaded bioink constructs was measured using air-dried constructs to determine functional groups as described before. 100 scans per sample with a spectral range of 400–4000 cm−1 were taken to generate an FT-IR spectrum at a resolution of 0.48 cm−1.
2.7. In-vitro release of pDNAs and their stability
The release of pBMP-2 (with and without CS-NPs encapsulation) and pPDGF-B from the bioink were performed in PBS solution (pH:7.4) at 37 °C. Bioprinted constructs were prepared with predetermined initial concentrations of CS-NPs(pBMP-2) (25 and 50 μg per construct), pBMP-2 (25 μg per construct), and pPDGF-B (25 μg per construct), immersed in 1 mL buffered solution and incubated for 45 days. The release medium was replaced with fresh medium at predetermined time points and the amount of pDNA release was determined spectrophotometrically at 260 nm using a Nanodrop ND-1000 Spectrophotometer (Wilmington, DE, US). The percentage of cumulative release of pDNAs was calculated according to a previous study [44]. All measurements were performed in triplicates.
The protection efficiency of CS-NPs and the bioink for the pDNAs from nuclease degradation was evaluated using DNA digestion and gel electrophoresis as previously described [45]. Briefly, samples were digested with DNase I (0.02 U/μL) (New England Biolabs, Ipswich, MA) at 37 °C for 30 min to remove any unprotected DNA from samples. The DNase I was heat-inactivated with EDTA solution (Sigma Aldrich). The following reaction was set up on ice including resuspending ~10μg of RNA in DNase I Reaction Buffer (1X) to a final volume of 100μL Nuclease-free H2O. Next, 2 units of DNAse I (RNase-free) was mixed thoroughly and incubated at 37 °C for 10 min. Thereafter, 1 μL of 0.5 M EDTA (to a final concentration of 5 mM) was added and then followed with heat inactivation at 75 °C for 10 min. The remaining pDNA was released from the constructs with acetic acid (Fisher Scientific, Hampton, NH). The stability of pDNA was analyzed using 1% agarose gel (Research Products Incorporated) in gel electrophoresis (FB300, Fisher Scientific).
2.8. Cell Culture
Rat bone marrow mesenchymal stem cells (rBMSCs) were isolated from inbred 4 weeks-old male Fischer white rats (F344/DuCrl, Charles River Laboratories) according our previous work [41], cared for in the animal facility (Millennium Science Complex, PSU) according to American Association for Laboratory Animal Science (AALAS) and The Institutional Animal Care and Use Committee (IACUC protocol #46591). Briefly, the hindlimbs were removed after euthanizing the rats using the CO2 gas. Next, femur and tibias were separated from the adherent soft tissue and placed in 1X DPBS. The end of each bone pieces was removed and 22-gauge syringe needle (BD, Franklin Lakes, NJ) was inserted into the open shaft cavity of the bone and flush with Minimal Eagle’s Medium, Alpha modification (αMEM; Corning Cellgro, Manassas, VA) using a 10 mL syringe. After harvesting all of cells, the entire solution was filtered through 70-mm nylon mesh filter (BD Biosciences, Bedford, MA). Then, the filtered solution was centrifuged at 1,600 rpm for 5 min. After the centrifuge, the media was removed and replaced with growth media containing Minimal Eagle’s Medium, Alpha modification (α-MEM; Corning Cellgro, cat. no. 10–022-CV) including 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, cat. no. 35–010-cv), penicillin (100 IU/ml) –streptomycin (100 μg/ml) (Corning Life Sciences, cat. no. 30–002-CI) and 2.5 μg/ml fungizone (Thermo Fisher Scientific, cat. no. 15-290-026). The isolated rBMSCs were plated on 6-well plastic cell culture plates. At ~85% confluency, the culture was trypsinized (0.25% Trypsin/EDTA (Sigma-Aldrich)) for 5 min at 37 °C and the cell suspension was centrifuged at 1,600 rpm for 5 min at room temperature. The cells were sub-cultured until passage 4 for further studies. To determine the purity of isolated rBMSCs, Fluorescence-activated Cell Sorting (FACS) analysis was performed (Supplementary Information), where 96.5% of the cells had positive CD90 marker, with negative CD34 marker expression (Figure S1) demonstrating phenotypic characteristic of those specific surface protein markers expressed in mesenchymal stem cells [46].
2.8.1. Cell Proliferation Assay for 3D Bioprinted Constructs
Isolated rBMSCs were encapsulated within Bioink, Bioink+CS-NPs(pBMP2), Bioink+pPDGF-B and Bioink+pPDGF-B+CS-NPs(pBMP2) and bioprinted into 3D bone constructs (7 mm in diameter, 2 mm in thickness) with a density of 8×105 rBMSCs per construct. In this effort, we bioprinted cell-laden bone constructs although seeding cells around the rib of scaffolds would better mimic the calvarial defect conditions; however, migration of cells towards constructs from sides will be very slow and may not result in effective uptake of delivered genes by cells. Therefore, we embedded the cells inside the bioink.
Cell proliferation assays were performed using a cell counting kit-8 (CCK-8) (Dojindo Molecular Technologies, MD) on Days 1, 4, 7, 10, and 14, according to the manufacturer’s instructions. Constructs were washed thrice with DPBS and treated with 500 μL of growth media mixed with the CCK-8 solution at a ratio of 10:1 (v/v). After 2 h of incubation at 37 °C, 120 μL of the solution was pipetted into a new 96-well plate and read at 450 nm using a microplate scanning spectrophotometer (Bio-Tek PowerWave x340, Winooski, VT, US). The constructs were then washed thrice with DPBS and cultured with growth media for repeated measures until the next time point. All measurements were performed in triplicates and normalized with respect to Day 1.
2.8.2. Calcium content assay
Calcium ion concentration of the bioprinted constructs was determined using a Calcium Colorimetric Assay kit (Sigma-Aldrich, cat. no. MAK022–1KT) on Days 0, 7, and 21. The constructs were cultured in osteogenic medium. At Days 0, 7, and 21, the culture medium was removed and acetic acid (1% v/v) was added to each construct and sonicated. Constructs (n=3) were incubated overnight at 4°C to extract calcium and then centrifuged at 4°C at 12,000 rpm for 10 min. Supernatants were read spectrometrically at 575 nm (in three technical replicates) using the Calcium Colorimetric Assay kit according to the manufacturer’s instructions. To determine the calcium concentration, six serial dilutions of calcium standard solution were prepared ranging from 0 to 2.0 (μg/well). Concentrations were calculated according to the standard curve and normalized with respect to Day 0.
2.8.3. ELISA assay
The expression level of BMP-2 protein by rBMSCs in 3D bioprinted constructs was quantified by rat BMP-2 ELISA Kit (Abcam, cat. no. ab213900) according to the manufacturer’s protocol. Samples were cultured in osteogenic differentiation media (consisting of α-MEM, 10% FBS, 10 mM β-GP (Sigma-Aldrich), 50 μg/ml ascorbic acid (Sigma-Aldrich), 100 IU/ml penicillin - 100 μg/ml streptomycin (Sigma-Aldrich) and 100 nM dexamethasone (Sigma-Aldrich), 2.5 μg/ml fungizone). Supernatants of culture media were read in triplicates spectrometrically at 450 nm on Days 7, 10, and 14. A standard curve was developed using eight points of serial dilution of rat BMP-2, ranging from 0 to 2000 (pg/ml). Concentrations were calculated according to the standard curve.
2.8.4. Gene Expression using Quantitative Real-Time Polymerase Chain Reaction (RT-PCR)
Expression of osteogenic genes including runt-related transcription factor 2 (RUNX2), alkaline phosphatase (ALP), bone morphogenetic protein 2 (BMP-2), and osteocalcin (OCN) [sequence provided in Table 3], were quantified by RT-PCR on Days 7 and 21 according to our previous study [47]. 3D constructs were placed in TRIzol® reagent (Thermo Fisher Scientific) and RNA was isolated from constructs according to the procedure described before [47]. After measuring the collected RNA concentrations using a NanoDrop, complementary deoxyribonucleic acid (cDNA) was synthesized from 1 μg of RNA using a T100 Thermal Cycler (BioRad) according to the AccuPower® protocol. An RT-PCR assay was performed in a StepOnePlus Real-Time PCR System (Applied Biosystems) with four replicates per gene in a 96-well plate using the Power SYBRTM Green Master Mix (Thermo Fisher Science) via comparative cycle threshold method. 2−ΔΔCT method was used for normalization of genes, which is a common and convenient approach to analyze relative gene expression in RT-PCR experiments [48]. Briefly, ΔCT values were obtained for both target samples and reference samples (untreated rBMSCs at time 0) by the difference between the CT values of target and the reference (GAPDH) genes. The difference in ΔCT values between the target sample and the reference samples was used to obtain ΔΔCT values.
Table 3.
The targeted primers of the measured genes in this study.
| Gene | Forward primers | Reverse primers |
|---|---|---|
| GAPDH | 5’-GGC ATG GAC TGT GGT CAT GA-3’ | 5’-CAA CTC CCT CAA GAT TGT CAG CAA −3’ |
| RUNX2 | 5’-CCG ATG GGA CCG TGG TT-3’ | 5’-CAG CAG AGG CAT TTC GTA GCT-3’ |
| OCN | 5’-GAG CTG CCC TGC ACT GGG TG-3’ | 5’-TGG CCC CAG ACC TCT TCC CG-3’ |
| ALP | 5’-TCC GTG GGT CGG ATT CCT-3’ | 5’-GCC GGC CCA AGA GAG AA-3’ |
| BMP-2 | 5’- AAG GCA CCC TTT GTA TGT GG-3’ | 5’- CAT GCC TTA GGG ATT TTG GA-3’ |
2.8.6. Immunofluorescence Staining
To investigate the osteogenic differentiation, samples were visualized by confocal microscopy after staining with mouse anti-RUNX2 antibody (Abcam, cat. no. ab76956) as described in our previous work [47]. Briefly, samples were rinsed with PBS thrice on Day 21 and fixed in 4% paraformaldehyde (Sigma-Aldrich) overnight at 4 °C. Samples were permeabilized in 0.1% Triton X-100 (Sigma-Aldrich) for 30 min the following day and blocked with 2.5% normal goat serum (NGS) (Sigma-Aldrich) for 60 min at room temperature. Then, samples were incubated with mouse anti-RUNX2 antibody (1:100 (v/v) in 2.5% NGS) at 4 °C for overnight at dark, washed three times with DPBS, and incubated with goat anti-mouse Alexa Fluor 488 secondary antibody (1:250 in 2.5% NGS) for RUNX2 (green fluorophore), Phalloidin (Molecular Probes, cat. no. A12380) (1:100 in 2.5% NGS) and Hoechst nucleic acid staining (Life Technologies) (1:500 in 2.5% NGS) for 2 h at room temperature in dark. Cellular morphologies and cytoskeletal organization were imaged using a laser excitation wavelength of Alexa Fluor 488 (green fluorophore), 568 (red fluorophore) and 405 (blue fluorophore) wavelengths, for RUNX2, F-actin and DAPI staining, respectively. Stained samples were mounted using Neo-Mount® anhydrous mounting medium (Millipore, Burlington, MA, US) and then imaged using a Zeiss LSM880 confocal microscopy (Oberkochen, Germany) with a Plan-Apochromat 20x/0.80 M27 objective via ZEN 2.3 SP1 software.
2.9. Surgical Procedure
A total of nine inbred 12 weeks-old male Fischer 344 white rats (Charles River Laboratories, Wilmington, MA, US) were cared for in our animal facility (Millennium Science Complex, PSU, PA) according to AALAS and IACUC (protocol #46591). Rats were anesthetized with an intraperitoneal injection of ketamine (Midwest Veterinary, Lakeville, MN, US) mixed with xylazine (LLOYD Inc., Shenandoah, IA, US) at a dose of 100 mg/kg for ketamine and 10 mg/kg for xylazine after inhalation of isoflurane (2–5%) over 1–2 mins. When animals were fully anesthetized, artificial tears (Rugby Laboratories, Livonia, MI, US) were placed on both eyes of the rats, and their heads were shaved and treated with betadine surgical scrub followed by ethanol. Bupivacaine (0.015 mg/kg) (Centralized Biological Laboratory, PSU) at a concentration of 2.5 mg/mL was injected at the site of incision under the skin (~0.15 – 0.2 ml) prior to the surgery.
Next, a sagittal incision was made through the periosteum, which was retracted to expose the calvarium. Two critical-sized calvarial defects (5 mm in diameter) were drilled into the parietal bone on each side of the rat skull using a trephine bit, taking care to keep the Dura mater intact. Power analysis was performed to minimize the use of animals while ensuring adequate power for statistical significance. Sample size calculation was performed using LaMorte’s power calculations [49] with independent study groups, an α error of 0.05 (p=0.05) and a power of 95 or 90%, resulting in a minimum of four or three samples, respectively. The Bioink (n=4), Bioink+CS-NPs(pBMP-2) (n=4), Bioinkink+pPDGF-B (n=4), Bioink+pPDGF-B+CS-NPs(pBMP-2) (n=3) were extruded directly into critical-size calvarial defects using the MABP following a spiral deposition pattern at a bioprinting speed of 60 mm/min. An empty defect group (n=3) was also used as a negative control. During the bioprinting process, established sterilization practices were followed in the operation room according to our institution standards and IACUC protocol. After bioprinting, periosteum and skin were closed with interrupted 5–0 monocryl (Ethicon Inc., Somerville, NJ, US) and 4–0 vicryl (Ethicon Inc.) sutures, respectively.
Postsurgery, animals were placed on a warming pad allowing for recovery. The animals were observed closely until they awoke from sternal recumbency. Following post-surgery recovery, buprenorphine (0.015 mg/kg) was administered twice up to 24 h for all animals. Depending on the pain management, some animals required an additional dose of painkiller post 24h. The animals were observed and weighed daily for at least 10 days post-surgery and weighed once a week after removing the sutures on Day 10. Animals were euthanized 6 weeks after the surgery by CO2 inhalation of 2 L per min until breathing cessation and subsequently confirmed with decapitation.
2.9.1. Micro-computed tomography (μCT)
μCT scanning of calvarial defects was performed with a vivaCT 40 scanner (Scanco Medical, Switzerland) (17.5 μm isometric voxels, 70 kV energy, 114 μA intensity, and 200 ms integration time) [47]. Image analysis was conducted using a cylindrical 3D oriented volume of interest with a radius of 2.5 mm and a height of 1 mm in Avizo (FEI Company, Hillsboro, OR), MATLAB (Natick, MA) after applying a gaussian smoothing filter (sigma 0.9) and a threshold cutoff of 300 mgHA/cm3. Thereafter, the bone volume divided by total volume (BV/TV %), normalize bone mineral density (BMD %) and bone coverage area (%) were determined from the filtered and processed data.
2.9.2. Histological and Immunohistochemistry (IHC) Staining
Calvarial explants, extracted from rat skulls 6 weeks post surgery, were rinsed with DPBS and fixed with 4% paraformaldehyde for 2 days at 4 °C. Explants were decalcified with 0.5 M ethylenediaminetetraacetic acid (EDTA) disodium salt (Research Products International) for 6 weeks at 4°C. After the decalcification process, samples were embedded in O.C.T. cyromatrix (ThermoFisher Scientific) embedding resin, sectioned using Leica CM1950 Cryostat (Leica Biosystems, Wetzlar, Germany) at −32 °C with 18 μm thickness, and stained in Hematoxylin and Eosin (H&E) automated staining platform (Leica Auto Stainer XL, Leica Biosystems) without applying any heat during the staining process. H&E stained sections were mounted using Neo-Mount® anhydrous mounting medium (Millipore, Burlington, MA, US) and imaged using the Keyence BZ-9000 fluorescence microscope (Keyence Corporation of America, Elmwood Park, NJ, US) under bright field using a neutral density optical filter. Then, H&E stained images were reprocessed via applying glowing edges filter for visualization of mislabeling of tissues with the overlapped color spectra.
To visualize the mature and immature bone formation histomorphometrically, sectioned samples were stained using Masson’s Trichrome staining (MTS) kit (Sigma-Aldrich, cat. no. HT15–1KT) based on the manufacturer’s instructions. Samples were dehydrated gradually with ethanol, mounted using Neo-Mount® anhydrous mounting medium (Millipore), and imaged using the Keyence BZ-9000 microscope under the bright field using a neutral density optical filter.
Immunohistochemical (IHC) staining of cryosectioned tissue samples was performed similarly to the immunofluorescence staining protocol as described before. Section samples were stained for nuclei, F-actin, and RUNX2 to visualize osteogenically-differentiating progenitors in the defect side at Week 6. Stained samples were imaged using the Zeiss LSM880 confocal microscopy using a Plan-Apochromat 20x/0.80 M27 objective via ZEN 2.3 SP1 software.
RUNX2 Fluorescence intensity was determined from IHC images using Image J software (National Institutes of Health (NIH)). Images were converted to RGB color and the green channel mean gray value was determined from histogram plots. The percentage of cell nuclei density was calculated from DAPI stained nuclei images using Image J. RGB Images were converted into a binary image and threshold of ‘2’ pixels was applied to eliminate the black noise. Then, the “watershed” tool was used to separate seemingly touching cell nuclei from each other. Next, “analyze particles” tool was used to determine the total cell nuclei area by setting the particle size range from 5 to 50 μm2 and the circularity range from 0 to 1. Cell nuclei density (%) was determined by dividing the cell nuclei area by the total area. A total of five images were used per group for the measurements.
2.10. Statistical Analysis
The data were presented as mean ± s.e.m unless stated otherwise. To compare two groups, data were analyzed using a two-tailed Student’s t-test with a 95% confidence interval assuming equal variances. Two-way ANOVA with repeated measurements was used to determine cell proliferation studies at a 95% confidence level. Data were analyzed by two-sided one-way ANOVA with the posthoc Fisher’s individual test for differences of means at a 95% confidence level for comparing among three or more groups, assuming the data were to be normally distributed. Differences were considered significant at p***<0.001, p**<0.01, p*<0.05. All statistical analysis was performed using Minitab 17.3 (Minitab Inc.. State College, PA, US).
RESULTS AND DISCUSSION
CS-NPs Delivery for Gene-Based Growth Factors
CS-NPs loaded with pDNA were synthesized using negatively charged polyanions (TPP) and positively charged CS [30] via the ionotropic gelation technique that relies on immediate ionic complexation (Fig. 2A). It is well known that TPP has an excellent ability to form nanoparticles with CS for sustained release [50,51], making it an ideal candidate for gene delivery systems. Furthermore, the addition of TPP was shown to improve oligonucleotides encapsulation efficiency [52] and form round, spherical nanoparticles on incorporation with CS [53]. Nanoscale particles are crucial for stable delivery of gene-based growth factors [54]. To achieve optimal nanoparticle formation, appropriate concentrations of CS and TPP need to be determined depending on their volumetric mixing ratios [30,53]. Therefore, we investigated different combination of CS and TPP at concentrations of 0.5, 0.75, and 1 (mg/mL) for each solution to design an optimal CS-NP delivery vehicle for gene-based growth factors. Table 1 shows the average diameter and particle size distribution of CS-NPs, which were largely dependent on the concentration of CS and TPP solutions. The nanoscale particle size was achieved when the TPP concentration did not exceed the concentration of CS. Additionally, the mean particle size of CS-NPs was increased with increasing TPP concentrations. CS-NPs, prepared at a concentration of 1.0:0.5 (mg/mL) (CS:TPP), resulted in the smallest average particle diameter of 113.3±0.6 nm. However, pBMP-2 encapsulated CS-NPs at this concentration resulted in an average particle diameter of 2553±115 nm with a PDI value of 0.509±0.095. The particles are known to have a wider particle size distribution as the PDI value approaches 1 and a PDI value of lesser than 0.3 denotes a homogeneously distributed particle size [55]. When a TPP concentration of 0.25 mg/mL was used, PDI and zeta potential of CS-NPs(pBMP-2) were determined to be 0.27±0.05 and 28.8±0.9 mV, respectively (Table 2). Also, the average diameter of CS-NPs was 308.1±4.1 nm (Figure 2B), which is desirable for uptake by cells [56]. Therefore, we selected the combination of 1:0.25 (mg/mL) (CS:TPP) for the rest of the work. CS-NPs with and without pBMP-2 encapsulation were also visualized under SEM, where the particles exhibited aggregation (Fig. 2C).
Fig 2.

(Ai.) An illustration scheme of chitosan nanoparticles (CS-NPs) combining chitosan (CS) with tripolyphosphate (TPP) via ionic gelation method. (Aii.) Formation of pBMP-2 encapsulated CS-NPs by ionic gelation. (B) Particle size distribution profiles and peak zeta potential of CS-NPs and CS-NPs(pBMP-2) (PDI: poly dispersive index) (n=3). (C) SEM images showing the morphology of formed CS-NPs and CS-NPs(pBMP-2). (D) ATR-FTIR spectra of CS-NPs and CS-NPs(pBMP-2).
Encapsulation efficiency (EE) of CS-NPs(pBMP2) was determined to be ~77%, which is similar to those reported in previous studies by Li [57] and Gaspar [53] (80 and 75%, respectively). EE of pDNA is one of the important parameters to design tailored characteristics to deliver growth factors successfully in controlled dosages [57]. The functional groups were investigated using ATR-FTIR to verify the pBMP-2 encapsulation in CS-NPs. The characteristic IR bands of pDNA, at 1210 cm−1 and 1051 cm−1 referring PO stretching [58], indicated the presence of pBMP2 in CS-NPs (Fig. 2D).
3D Bioprinted Bone Constructs and controlled co-delivery of pDNAs
Using the osteogenic bioink, bone constructs were 3D bioprinted via pneumatic extrusion generating a well-defined porous architecture (Fig. 3A). The constructs exhibited a filament thickness of 497 ± 53 μm and a pore size of 421 ± 98 μm with a raster gap of 1 mm. Furthermore, the morphological characterization of 3D bioprinted constructs demonstrated a straight filament morphology and an interconnected porous architecture with a pore size of >300 μm. Moreover, CS-NPs(pBMP2) and pPDGF-B were visible in SEM images of 3D bioprinted constructs (Fig. 3B).
Fig 3.
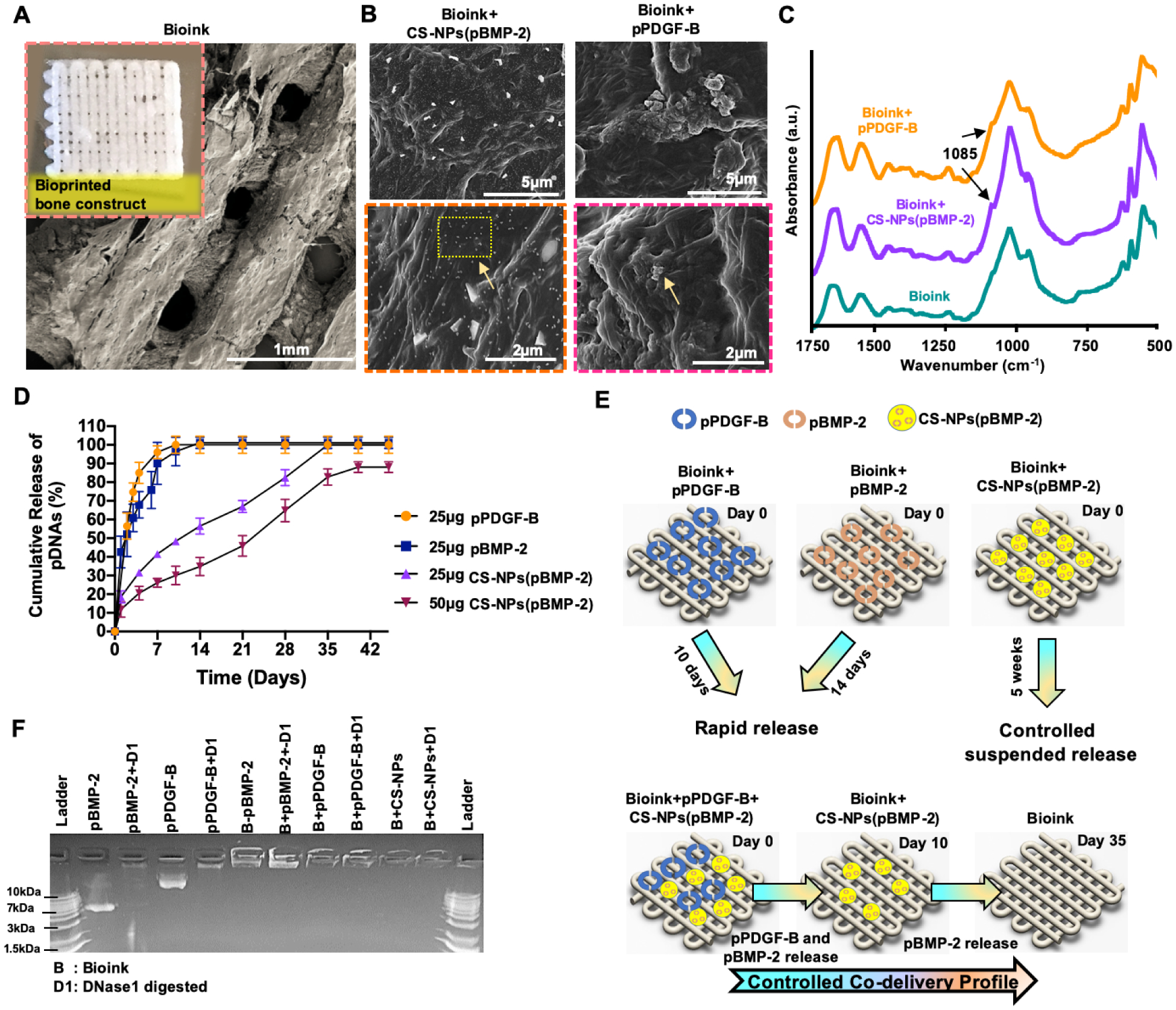
(A) 3D Bioprinted bone construct with well-defined porous architecture and its morphological display under SEM. (B) SEM images of encapsulated CS-NPs(pBMP-2) and (pPDGF-B). (C) ATR-FTIR spectra of the bioink, bioink+CS-NPs(pBMP-2) and bioink+pPDGFB. (D) In-vitro cumulative pDNA release profiles from 3D bioprinted constructs. (E) The release strategy of pDNAs from 3D bioprinted construct. (F) The agarose gel retardation assay demonstrating the stability of NPs and pDNAs in 3D bioprinted constructs. Data were presented as mean ± s.dev (n=3).
Successful loading of CS-NPs(pBMP-2) and pPDGF-B into 3D bioprinted constructs was also confirmed by ATR-FTIR spectra (Fig. 3C). The bioink, based on collagen and chitosan, showed peaks at 1649 cm−1 and 1548 cm−1, which were characteristic to the amide I and amide II for collagen [59]. The peaks at 1025 cm−1 and 960 cm−1 were the characteristic of phosphate stretching vibration, whereas, the peaks at 598 cm−1 and 557 cm−1 were attributed to the phosphate bending vibration bands indicating the presence of nHAp in the bioink [60]. The single peak observed at 1085 cm−1 referred to PO stretching IR bands of pDNAs and confirmed their presence pDNA within the bioprinted constructs.
The release profiles of pDNAs from 3D bioprinted constructs were evaluated for pPDGF-B and CS-NPs(pBMP-2) (Fig. 3D). pPDGF-B was completely released from the constructs within the first 10 days of incubation demonstrating a noticeable burst effect. Similarly, pDNA-BMP-2 without nanoparticle encapsulation, was released within 14 days of incubation. Two different loading dosages of 25 and 50 μg pDNA-BMP2 were tested for the nanoparticle encapsulation according to the previous studies, where dosages 10 to 50 μg pBMP-2 demonstrated a significant enhancement in bone regeneration in vivo [8,61]. When 25 μg of pBMP-2 per construct were encapsulated in CS-NPs, a prolonged release of pDNAs was achieved with a complete release over five weeks. The constructs loaded with 50 μg of CS-NPs(pBMP-2) exhibited a sustained release profile with 91% cumulative release over six weeks. The literature suggests that a controlled delivery of growth factors to the wound site can improve tissue regeneration outcomes [8,57,61–63]. The constructs containing both pPDGF-B and CS-NPs(pBMP-2) demonstrated a rapid release of pPDGF-B and a sustained release of pBMP-2 confirming controlled co-delivery (Fig. 3E).
The functionality and transfection efficiency of pDNA were described in our previous work [7, 8] and the plasmid activity was consistent with our previous transfection results using pDNA encoding BMP-2 and PDGF-B. Briefly, pDNA encoding BMP-2 generated 111 pg/ml of BMP-2 when transfected in BMSCs and pDNA PDGF-B generated 138 pg/ml of PDGF-B when transfected in BMSCs. The success of gene-based growth factors relies on pDNA being protected from DNase digestion in vivo. In this regard, integrity of pDNAs was evaluated by DNase I protection assay resulting in a stable microenvironment with the possibility of increasing its short half-life time for constructs loaded with naked pDNAs or constructs loaded with CS-NPs encapsulating pDNAs (Fig. 3F). Additionally, the bioink preparation did not damage pDNAs confirming the suitability of the bioink for successful gene delivery.
In vitro Growth and Differentiation Capability of rBMSCs in 3D Bioprinted pDNA-loaded Constructs
In our previous work [39], we confirmed the cellular viability of extruded rBMSCs within bioink by performing LIVE/DEAD staining on Day 1, where both the bioprinting process or the bioink did not harm the cells. In this work, the proliferation capability of rBMSCs within 3D bioprinted constructs was depicted in Fig. 4A. All of the groups had a positive proliferation trend and cellular growth over time. The Bioink and Bioink+CS-NPs(pBMP-2) showed a similar proliferation trend up to Day 7. From Day 7 to 14, the slower cell proliferation rate observed for Bioink+CS-NPs(pBMP-2) group, whereas Bioink group continued to increase its cellular proliferation. The decrease in proliferation rate for the Bioink+CS-NPs(pBMP-2) group might be associated with the increase in osteogenic activities. As a result of incorporating pPDGF-B, the cell proliferation rate for Bioink+pPDGF-B and Bioink+pPDGF-B+CS-NPs(pBMP-2) groups were significantly enhanced starting as early as from Day 4 (p<0.001) compared to the other groups. It is clear that PDGF containing groups resulted in increase in cell proliferation rate, whereas BMP-2 group demonstrated the slowest rate of proliferation due to increase in differentiation over time. This observation could be due to PDGF’s role in inducing mitogenesis, which would result in an increased proliferation rate [7,12].
Fig 4.
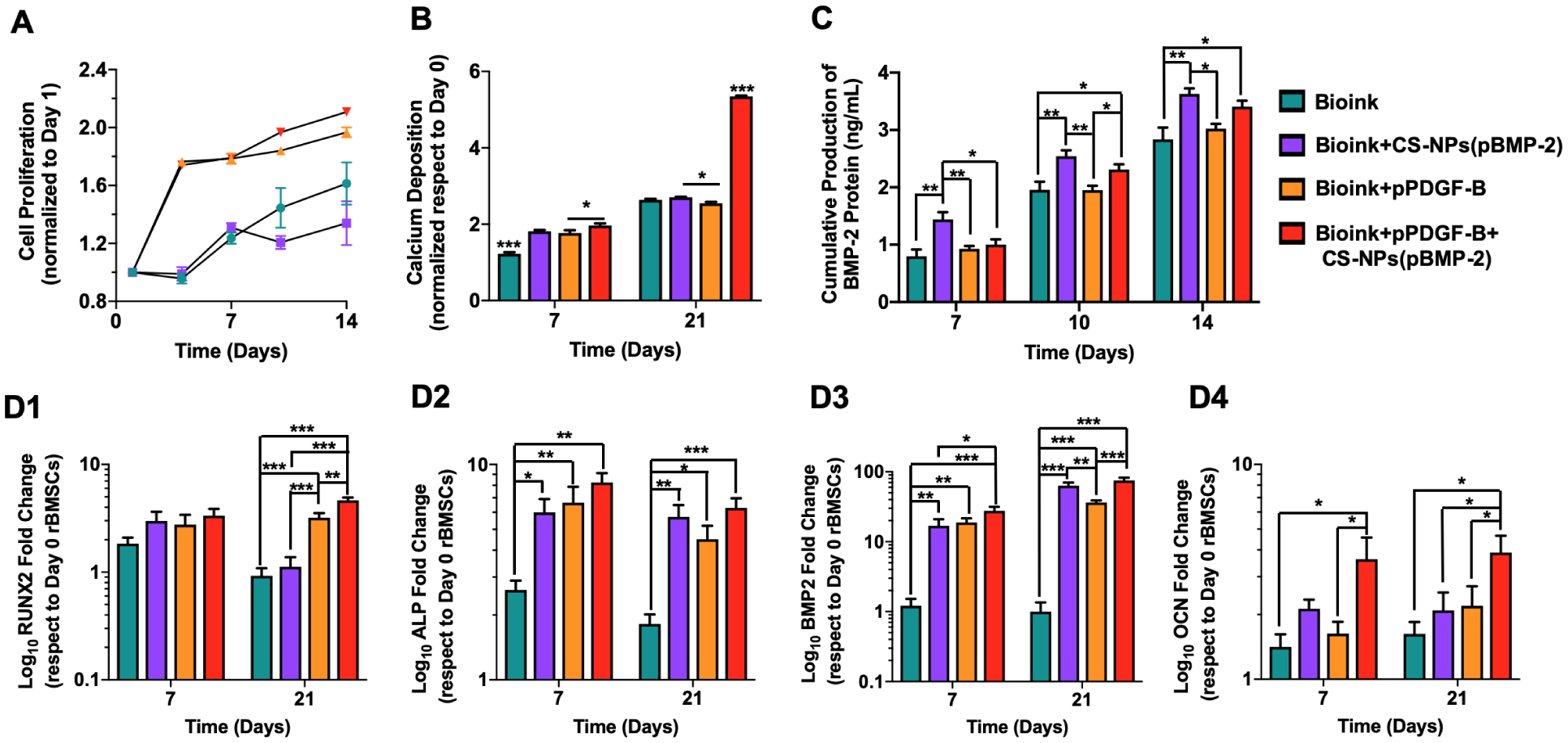
(A) CCK-8 assay results showing the proliferation of rBMSCs within 3D bioprinted constructs on Days 1, 4, 7, 10, and 14 (n=3). (B) Deposited calcium contents by rBMSCs on 3D bioprinted constructs determined on Days 7 and 21 (n=3). (C) ELISA assay was performed to demonstrate BMP-2 protein expression levels of rBMSCs on Days 7, 10, and 14 (n=3). RT-PCR assay demonstrating gene expression levels of (D1) RUNX2, (D2) ALP, (D3) BMP-2, and (D4) OCN on Days 7 and 21 for rBMSCs within 3D bioprinted constructs cultured in osteogenic differentiation media. Data were presented as mean ± s.e.m. p*<0.05, p**<0.01, p***<0.001.
Calcium ion deposition was determined on Days 7 and 21. The Bioink group produced a significantly lower amount of calcium deposition on Day 7 compared to the other groups (p<0.001) (Fig. 4B). The Bioink+CS-NPs(pBMP-2) and Bioink+pPDGF-B groups showed similar calcium deposition amount, which could be attributed to the BMP-2 release and an increase in cell proliferation, respectively. However, the controlled co-delivery group, Bioink+CS-NPs(pBM-2)+pPDGF-B, had a significant increase (p=0.028) in calcium ion deposition compared to the Bioink+pPDGF-B group. When the constructs were cultured in osteogenic media for three weeks, the Bioink group resulted in a similar calcium content as the Bioink+CS-NPs(pBMP-2) and Bioink+pPDGF-B groups. The calcium amount significantly (p=0.01) increased in the Bioink+CS-NPs(pBMP-2) group compared to the Bioink+pPDGF-B group at Week 3 due to the release of pBMP-2, which could facilitate improved differentiation of rBMSCs. Despite the increase in proliferation rate and having a similar trend to the controlled co-delivery group, the measured calcium amount in the Bioink+pPDGF-B group was not higher than the controlled co-delivery group. The potential reason for the increase in calcium content for the controlled co-delivery group despite having a similar proliferation trend to the Bioink+pPDGF-B group could be the slower release of pBMP-2 over time increased differentiation of cells; and supported deposition of mineralized bone matrix. The controlled co-delivery group resulted in the most significant increase (~2-fold) in calcium ion deposition in Week 3 compared to other groups (p<0.001), providing on optimal extracellular matrix (ECM) environment for the initiation of mineralization [63].
The production of BMP-2 protein by rBMSCs in bioprinted constructs was assessed by ELISA assay over the 2-week incubation period (Fig. 4C). Overall, an increase in BMP-2 protein production was observed over two weeks for all groups. On Day 7, the Bioink+CS-NPs(pBMP-2) group had the highest BMP-2 release resulting in ~1.8-, 1.6-, and 1.4-folds increment in cumulative BMP-2 production compared to the Bioink (p=0.002), Bioink+pPDGF-B (p=0.007) and Bioink+pPDGF-B+CS-NPs(pBMP-2) (p=0.015) groups, respectively. The Bioink+CS-NPs(pBMP-2) had a higher BMP-2 expression compared to the other groups on Days 10 and 14 with no significant difference compared to the controlled co-delivery group. CS-NPs encapsulation of pBMP-2 did not significantly reduce BMP-2 production by Day 10.
ECM plays an important role in the osteogenic differentiation process, with structural ECM proteins, cytokines secreted in ECM, and ECM-degrading proteases and their inhibitors potentially contributing towards the regulation of osteoblastic cells [8]. It is well known that BMP-2 plays a significant role in stimulating the differentiation of progenitors into osteoblasts through stimulating Smad signal pathway and regulating the transcription of osteogenesis-related genes, such as ALP, Type-I collagen, osteocalcin, and bone sialoprotein genes [64]. In addition, the role of PDGF and mechanism in bone regeneration is accomplished by increasing the population of progenitors and then activating them to turn into osteoblasts. Then more importantly, PDGF induce mitogenesis, angiogenesis and macrophage activation for continued repair and bone regeneration [65]. Osteogenic differentiation of rBMSCs due to the pDNA incorporation within the constructs was evaluated by determining the gene expression levels of an early-stage osteogenic differentiation marker, RUNX2, (Fig. 4D1), mid-stage markers ALP and BMP-2 (Fig. 4D2–3) and a late-stage marker of OCN (Fig. 4D4) on Days 7 and 21. We observed no statistical difference in the expression of RUNX2 on Day 7 among the groups that could be due to the lower calcium ion deposition, lesser BMP-2 production, and slower release of pBMP2. However, by Day 21, groups loaded with pPDGF-B had significantly higher expression of RUNX2 compared to the Bioink and Bioink+CS-NPs(pBMP-2) groups as consistent with a previously reported study [66]. After 21 days, the Bioink+PDGF-B+CS-NPs(BMP-2) resulted in the highest expression level of RUNX2, increased by ~5-, 4-, and 1.5-folds compared to the Bioink (p<0.001), Bioink+CS-NPs(pBMP-2) (p<0.001), and Bioink+pPDGF-B (p=0.002) groups, respectively. All the groups loaded with pBMP-2 and/or pPDGF-B had upregulated expression for ALP and BMP-2 on Days 7 and 21 as compared to the Bioink group. OCN is an important protein in osteogenic differentiation cascade, which can regulate mineralization process during the osteogenic differentiation of progenitors. The expression level of OCN was determined to be the highest for the Bioink+pPDGF-B+CS-NPs(pBMP-2) group on both Days 7 and 21. On Day 21, OCN expression from the Bioink+pPDGF-B+CS-NPs(pBMP-2) group resulted in a ~2.4-, 1.9-, and a 1.8-fold increase compared to the Bioink (p=0.010), Bioink+CS-NPs(pBMP-2) (p=0.032), and Bioink+pPDGF-B (p=0.041) groups, respectively. All osteogenic regulator genes indicated that the Bioink+pPDGF-B+CS-NPs(pBMP-2) promoted the most accelerated osteogenic differentiation compared to other groups, due to the controlled co-delivery of pDNA from bioprinted constructs.
Cytoskeleton organization and osteogenic differentiation of rBMSCs within the bioprinted constructs were also evaluated by immunofluorescent imaging (Fig. 5). All of the groups showed RUNX2 protein expression, where the controlled co-delivery group exhibited the strongest expression of RUNX2 among all groups at Day 21. F-actin staining of rBMSCs within the controlled co-delivery group displayed a remarkable cuboidal-shaped cellular morphology similar to that for osteoblasts [67].
Fig 5.
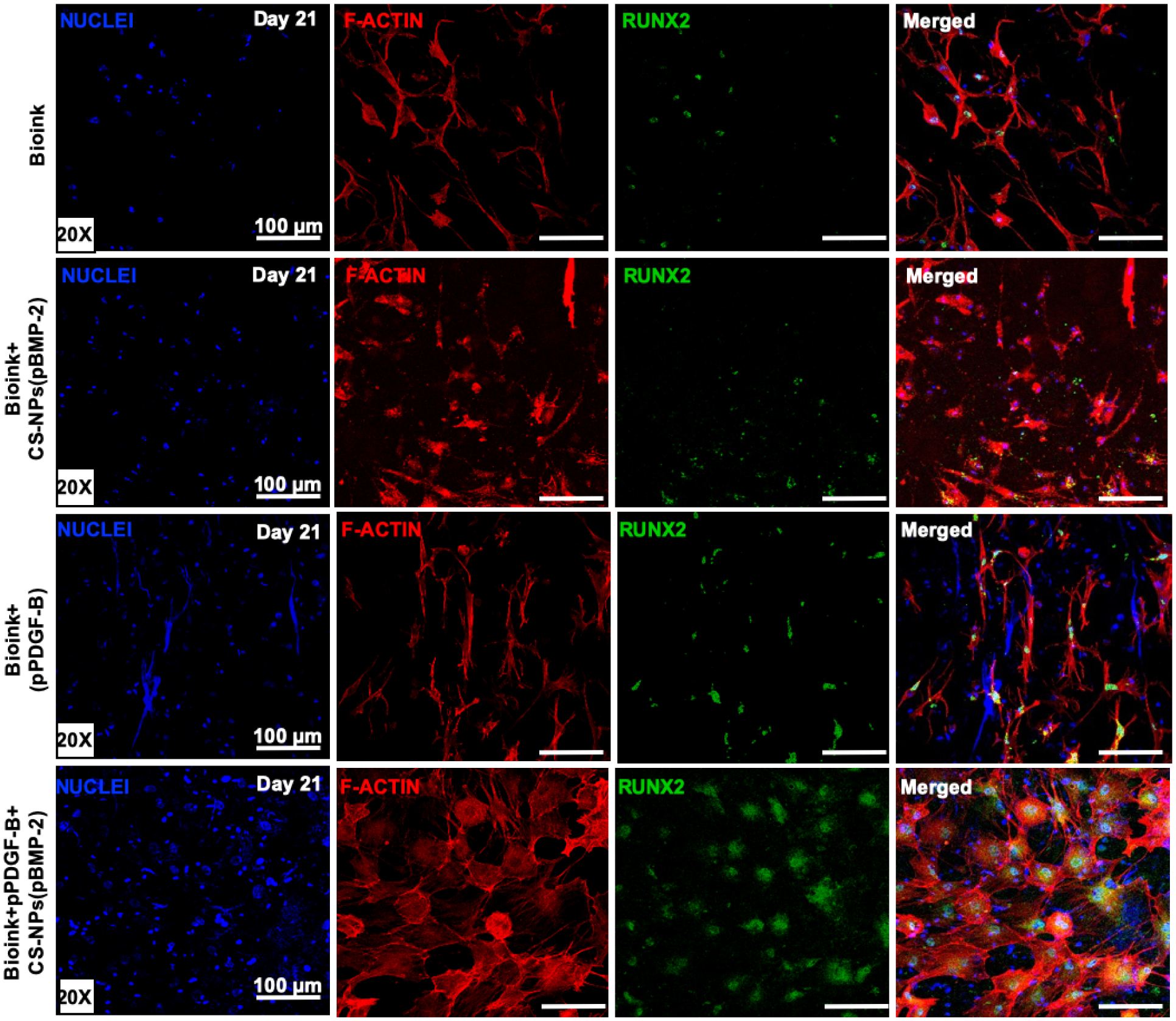
Immunofluorescent images of rBMSCs on Day 21 in 3D bioprinted constructs cultured in osteogenic differentiation media stained with nuclei (blue), F-actin (red), and RUNX2 (green).
In-situ Delivery of Controlled Released pDNA via Intraoperative Bioprinting
Non-viral gene delivery has gained great attention due to ease of production, storage stability, and low immunogenicity [68]. Multiple reports cautioned against utilizing BMP-2 to facilitate bone repair [69,70] due to carcinogenicity concerns; however, in the context of using gene therapy to enhance the production of BMP-2 has yet to be evaluated in terms of carcinogenicity. It appears that the carcinogenicity of BMP-2 depends on the dose, so with gene therapy significantly reducing the bolus dose of BMP-2 present in the defect, it is unlikely to cause any concerns [69]. Non-viral gene delivery has the potential to reprogram the host cells via in-vivo transfection by rapid or controlled delivery of gene-activated matrixes within the defect site [15]. Therefore, it is important to develop novel bone grafts to promote effective delivery and use of non-viral genes for stimulating osteogenesis and calvarial bone repair in an accelerated manner. In this study, a non-viral based gene therapy offered an exciting platform for delivery of gene-based growth factors, which was mediated by the in-situ delivery of controlled-released pDNAs from intraoperatively bioprinted constructs. To evaluate the role of controlled release in bone tissue repair, a well-established dual critical-sized calvarial defect model on rats was used [71]. In this study, in vitro experiments were used to assess the cellular activities of encapsulated rBMSCs. Then, the bioprinting parameters for the bioink loaded with rBMSCs were optimized in order to bioprint porous constructs. However, cells were not used for intraoperative bioprinting purposes and the non-flat defect in the cranium presented unique reconstructive challenges. To able to intraoperatively deliver the bone constructs, different design parameters were utilized. Bone constructs made of Bioink, Bioink+CS-NPs(pBMP-2), Bioink+pPDGF-B and Bioink+pPDGF-B+CS-NPs(pBMP-2) (also known as the controlled co-delivery group) were directly bioprinted into defect in a controlled manner by following a spiral deposition plan (Fig. 6A). Empty defects were used as a negative control.
Fig 6.

(A) Intraoperative bioprinting of bone constructs into critical-sized rat calvarial defects. (B) μCT images of calvarial defects at Week 6. (C) BV/TV (%), (D) Normalized BMD (%) and (E) bone coverage area (%) for empty (n=3), bioink (n=4), bioink+CS-NPs(pBMP-2) (n=4), bioink+pPDGF-B (n=4) and bioink+pPDGF-B+CS-NPs(pBMP-2) (n=3) groups obtained from the quantitative analysis of μCT scans. Data were presented as mean ± s.e.m. (*: compared to the empty group, p*<0.05, p**<0.01, p***<0.001). (#: compared to the bioink group, p#<0.05, p##<0.01, p###<0.001).
The presented IOB approach differs from manual deposition of bioinks such as handheld systems by simply filling the defects. Manual deposition of the bioink for the repair of calvarial defect may not be ideal due to the manual maneuvers preventing deposition of uniform filaments and precise amount of bioink and pDNA, which are critical for durable and safe reconstruction of cranial defects [62]. The bioprinting process to deliver bone construct for a single defect took around 1–2 min per defect, which can be considered highly short time compared to the entire duration of surgery. The stability and shape fidelity of the bioink post extrusion was achieved by temperature-induced physical crosslinking of CS-βGP where chitosan–chitosan interactions become dominant resulting in a phase transition from a liquid to a gel [63].
Six weeks post-surgery, bone regeneration was visualized and quantified by μCT scanning (Fig. 6B). Limited bone regeneration was observed for the Empty group and newly formed bone was mainly observed at the edges of the surrounding defect without forming any bridges across the defects. A significant amount of newly formed mineralized bone was detected in all other groups compared to the Empty group. As compared to the Empty group, the bone volume/total volume (BV/TV) (%) was increased by ~2.1-, 3.2-, 3.4- and 3.7-fold for the Bioink (p=0.03), Bioink+CS-NPs(pBMP-2) (p<0.001), Bioink+pPDGF-B (p<0.001), and the controlled co-delivery group (p<0.001), respectively (Fig. 6C). All groups containing pDNA had a significantly higher normalized bone mineral density (BMD) (%) compared to the Empty and Bioink groups. The controlled co-delivery group showed the highest mineralization density compared to the Empty (~2.4-fold) and Bioink (~1.4-fold) groups (Fig. 6D). There was a remarkable increase in the percentage of bone coverage area (~90%) for the controlled co-delivery group, which nearly filled the entire defect with significantly increment compared to the Empty (~ 3-fold) and Bioink (~1.3-fold) groups (Fig. 6E). Although the controlled co-delivery group resulted in the highest absolute value in Figs 6C, 6D, and 6E, no significant difference (p>0.05) was observed compared to delivered plasmid-only groups.
H&E staining of calvarial explants confirmed that fibrous soft tissue (ST) was formed in empty defects (Fig. 7A). The Bioink group primarily displayed a thin layer of newly regenerated bone (RG) but thicker RG tissue was formed in pDNA-involved groups. A dense layer of RG with distinguishable dark purple staining closer to host bone (HB) was observed in the pPDGF-B-loaded groups. The controlled co-delivery group exhibited the most apparent newly formed RB and this observation was consistent for all samples. In order to delineate RB from ST more clearly, H&E images were processed to represent higher-quality digital images (Fig 7A1). Fibrous ST represents green areas, which can be clearly distinguished from osteoblastic tissue (pink-purple areas). MTS images demonstrated mature and immature collagen deposition within the defect sides (Fig. 7B). PDNA-loaded groups displayed the most collagen deposition, suggesting the formation of immature bone tissue.
Fig 7.
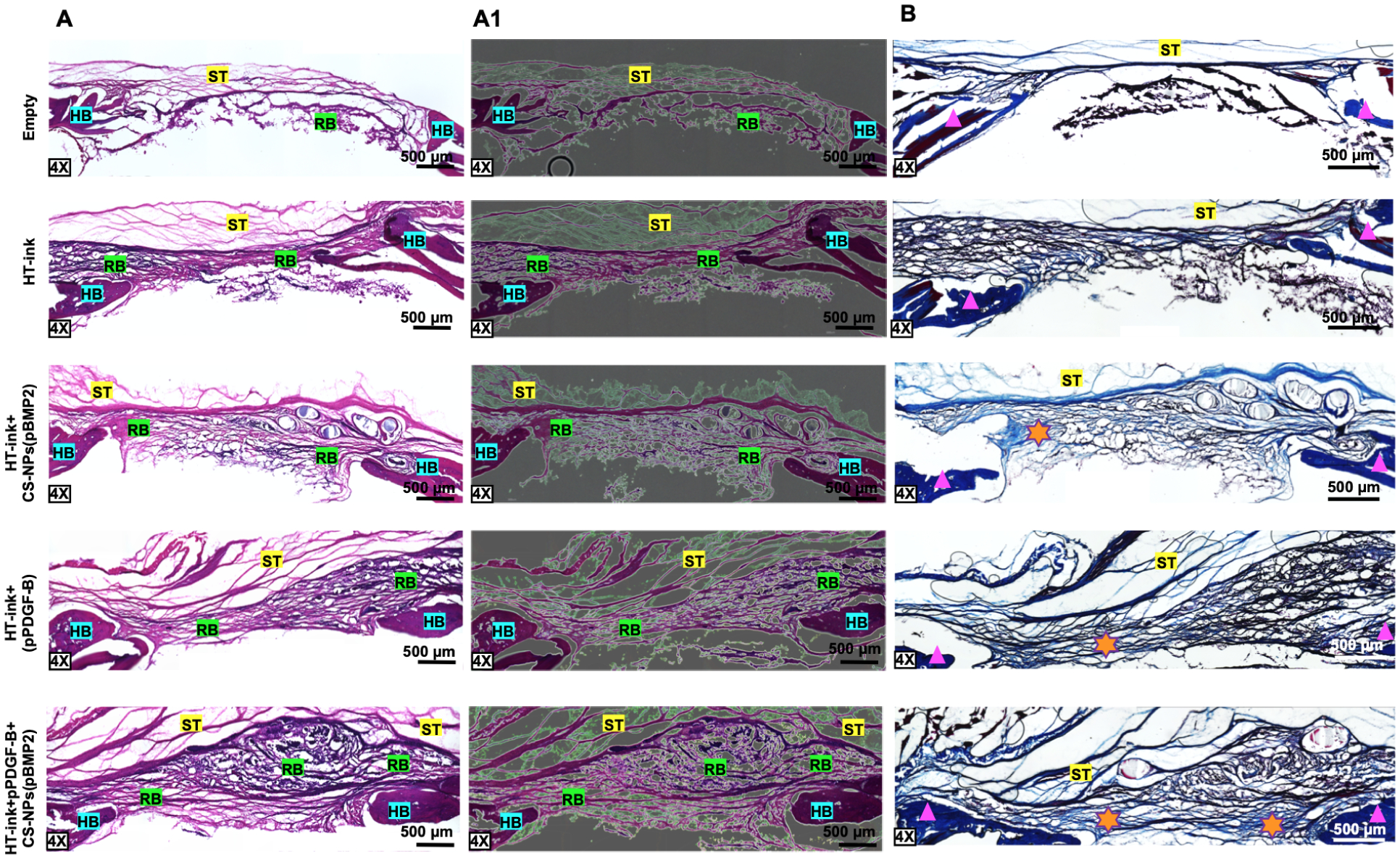
(A) H&E and (A1) image-corrected H&E (B) Masson’s Trichrome staining of sectioned calvarial defects at Week 6 post bioprinting (ST: soft tissue, RB: regenerated bone, HB: host bone, Triangle: mature bone, Star: immature bone). Image correction was performed via applying filters for high-quality digital images (green area: ST, pink-purple area: RB, purple: HB).
IHC stained images demonstrated the expression of RUNX2 in calvarial defects (Fig. 8A), where significantly higher RUNX2 fluorescence intensity was observed in the controlled co-delivery group compared to the Empty (~7-fold), Bioink (~2 fold), Bioink+CS-NPs(pBMP-2) (~1.4-fold), and Bioink+pPDGF-B (~1.7-fold) groups (Fig. 8B). RUNX2 is a nuclear transcriptional factor, which overlaps with that of nuclei. Due to the auto-fluorescence effect of collagen interfering with cellular signals, the overlap was not detected clearly. Therefore, merged images were provided at a higher magnification demonstrating that RUNX2 overlaps with nuclei clearly. The groups treated with pBMP-2 had a higher mean intensity compared to the other groups, which could be attributed to the differentiation capability of cells with the presence of pBMP-2 in vivo [8]. Furthermore, pPDGF-B loaded groups had the highest cell nuclei density (%) compared to the other groups (Fig. 8C), which could be due to mitogenesis as a result of the increased proliferation caused by the pPDGF-B release in the defect site.
Fig 8.
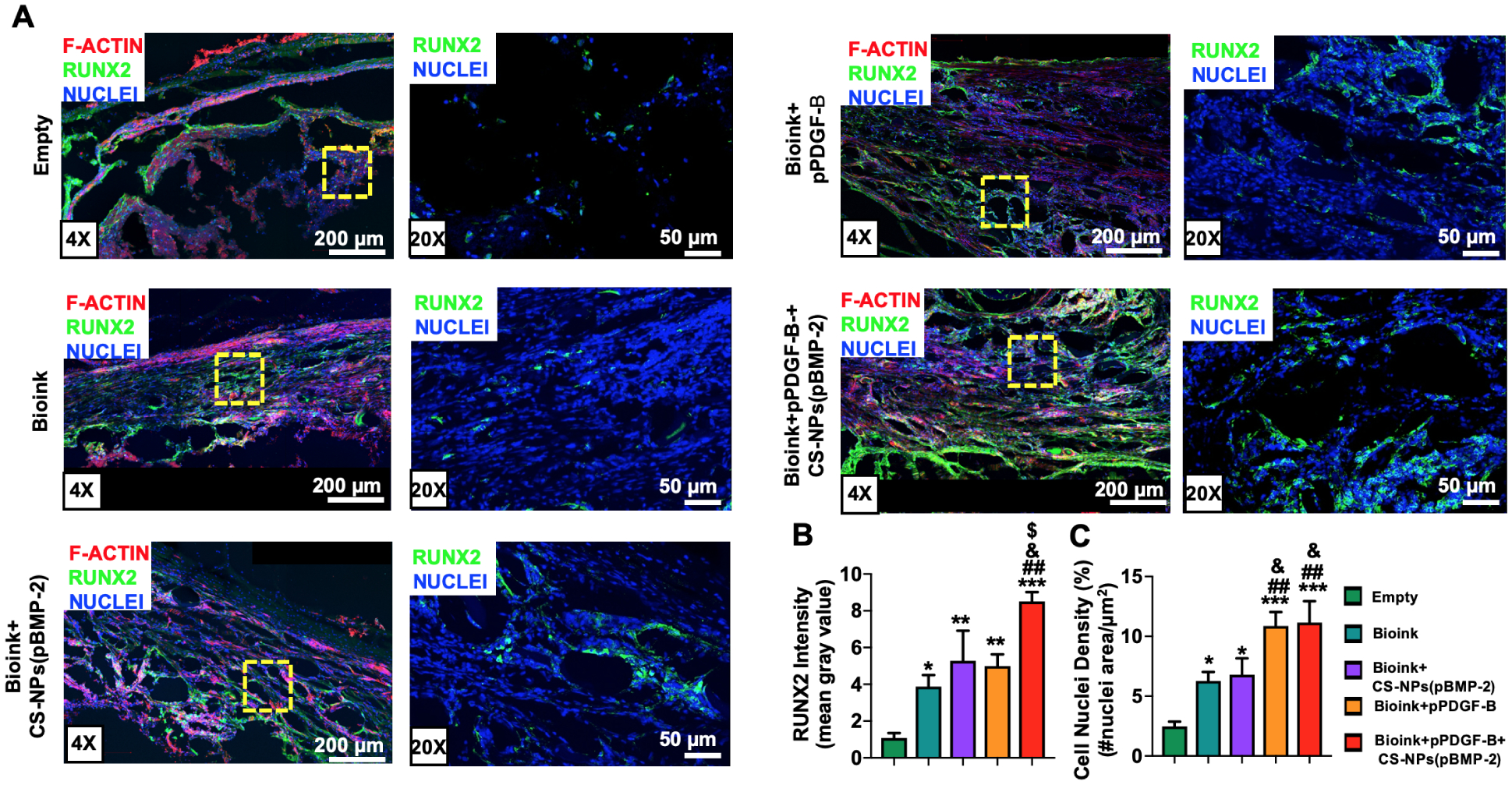
(A) IHC staining of sectioned calvarial defects at Week 6 stained with nuclei (blue), F-actin (red) and RUNX2 (green). (B) RUNX2 Fluorescence intensity determined based on mean gray value from RUNX2 stained images at Week 6. (C) Cell nuclei density (%) determined from nuclei-stained fluorescence images at Week 6. Data were presented as mean ± s.e.m (n=5). (*: compared to the empty group, p*<0.05, p**<0.01, p***<0.001). (#: compared to bioink group, p#<0.05, p##<0.01, p###<0.001). (&: compared to the bioink+CS-NPs(pBMP-2 group), p&<0.05, p&&<0.01, p&&&<0.001). ($: compared to the bioink+pPDGF-B group, p$<0.05, p$ $<0.01, p$ $ $<0.001).
Overall, the controlled co-delivery of pPDGF-B and pBMP-2 from intraoperatively bioprinted constructs facilitated improved repair of calvarial bone defects in 6 weeks inducing the highest amount of newly formed mineralized bone tissue and bone coverage area compared to other groups. Bone repair involves early steps of cell recruitment, due to PDGF, and then osteoblast proliferation and mineralization are upregulated by BMP-2 [14]. Thus, the importance of order in growth factor delivery is crucial, and importantly, an antagonistic effect may exist between PDGF and BMP-2 signaling, as the simultaneous delivery of these growth factors results in significantly less tubule formation when compared to the controlled growth factor delivery [8]. This underscores the importance of custom design and optimization of the delivery system based on plasmid therapy and for specific clinical applications. In particular, when considering bone repair, our results suggest that the controlled co-delivery of pPDGF-B and pBMP-2 are beneficial for cell recruitment, proliferation, and mineralization stages of healing.
CONCLUSION
In this study, we presented a non-viral delivery method for gene-based growth factors through combining controlled co-delivery release with an in-situ delivery approach with the use of IOB technology. The controlled co-delivery strategy presented in this study contained naked pPDGF-B, for faster release to recruit surrounding endogenous cells into the defect site, and pBMP-2 encapsulated CS-NPs for a controlled sustained release to maintain sustainable production of BMP-2 to facilitate the osteogenic differentiation of progenitors in vivo. These pDNAs were directly bioprinted into the defect site to demonstrate the in-situ delivery potential of gene-based growth factors, which significantly improved bone regeneration compared to the Bioink and Empty group. Therefore, the presented approach could be a promising means to promote quick treatment and accelerated bone tissue repair, and has great potential for translating bioprinting technologies from bench to bed side in the foreseeable future.
Supplementary Material
Acknowledgements
This work was supported by the International Team for Implantology Award #1275_2017 (I.T.O.), National Institute of Health Award #R01DE028614 (I.T.O.), National Science Foundation Award #1600118 (I.T.O.), and Osteology Foundation Award #15-042 (I.T.O.). The authors are also thankful to Dr. Dishary Banerjee and Dr. Miji Yeo for their technical support in FACS analysis, PSU institutions, including Materials Research Institute and The Huck Institute of The Life Sciences, for the generous facility support. Dr. R. Seda Tigli Aydın acknowledges the support from the International Postdoctoral Research Scholarship Program (BIDEP 2219) of the Scientific and Technological Research Council of Turkey (TUBITAK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Ethics declarations
The authors declare no competing interests.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data Availability
All data needed to evaluate the conclusions in the paper are present in the paper. Additional data related to this paper may be requested from the authors.
References
- [1].Johnson D, Wilkie AOM, Craniosynostosis, Eur. J. Hum. Genet 19 (2011) 369–376. 10.1038/ejhg.2010.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kumar AR, Tantawi D, Armonda R, Valerio I, Advanced Cranial Reconstruction Using Intracranial Free Flaps and Cranial Bone Grafts: An Algorithmic Approach Developed from the Modern Battlefield, Plast. Reconstr. Surg 130 (2012). https://journals.lww.com/plasreconsurg/Fulltext/2012/11000/Advanced_Cranial_Reconstruction_Using_Intracranial.22.aspx. [DOI] [PubMed] [Google Scholar]
- [3].Szpalski C, Barr J, Wetterau M, Saadeh PB, Warren SM, Cranial bone defects: current and future strategies, Neurosurg. Focus FOC 29 (2010) E8. 10.3171/2010.9.FOCUS10201. [DOI] [PubMed] [Google Scholar]
- [4].Brown Baer PR, Wenke JC, Thomas SJ, Hale CRG, Investigation of Severe Craniomaxillofacial Battle Injuries Sustained by U.S. Service Members: A Case Series, Cranial Maxillofac Trauma Reconstr. 05 (2012) 243–252. 10.1055/s-0032-1329542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wu Y, Ravnic DJ, Ozbolat IT, Intraoperative Bioprinting: Repairing Tissues and Organs in a Surgical Setting, Trends Biotechnol. (2020). 10.1016/j.tibtech.2020.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hollinger JO, Hart CE, Hirsch SN, Lynch S, Friedlaender GE, Recombinant Human Platelet-Derived Growth Factor: Biology and Clinical Applications, JBJS. 90 (2008). https://journals.lww.com/jbjsjournal/Fulltext/2008/02001/Recombinant_Human_Platelet_Derived_Growth_Factor_10.aspx. [DOI] [PubMed] [Google Scholar]
- [7].Elangovan S, D’Mello SR, Hong L, Ross RD, Allamargot C, V Dawson D, Stanford CM, Johnson GK, Sumner DR, Salem AK, The enhancement of bone regeneration by gene activated matrix encoding for platelet derived growth factor, Biomaterials. 35 (2014) 737–747. 10.1016/j.biomaterials.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Khorsand B, Nicholson N, Do A-V, Femino JE, Martin JA, Petersen E, Guetschow B, Fredericks DC, Salem AK, Regeneration of bone using nanoplex delivery of FGF-2 and BMP-2 genes in diaphyseal long bone radial defects in a diabetic rabbit model, J. Control. Release 248 (2017) 53–59. 10.1016/j.jconrel.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Devescovi V, Leonardi E, Ciapetti G, Cenni E, Growth factors in bone repair, Chir. Organi Mov 92 (2008) 161–168. 10.1007/s12306-008-0064-1. [DOI] [PubMed] [Google Scholar]
- [10].Stevens MM, Marini RP, Schaefer D, Aronson J, Langer R, Shastri VP, In vivo engineering of organs: The bone bioreactor, Proc. Natl. Acad. Sci. U. S. A 102 (2005) 11450 LP–11455. 10.1073/pnas.0504705102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hunt NC, Grover LM, Cell encapsulation using biopolymer gels for regenerative medicine, Biotechnol. Lett 32 (2010) 733–742. 10.1007/s10529-010-0221-0. [DOI] [PubMed] [Google Scholar]
- [12].Canalis E, Varghese S, McCarthy TL, Centrella M, Role of platelet derived growth factor in bone cell function, Growth Regul. 2 (1992) 151–155. http://europepmc.org/abstract/MED/1290951. [PubMed] [Google Scholar]
- [13].D’Mello SR, Elangovan S, Hong L, Ross RD, Sumner DR, Salem AK, A Pilot Study Evaluating Combinatorial and Simultaneous Delivery of Polyethylenimine-Plasmid DNA Complexes Encoding for VEGF and PDGF for Bone Regeneration in Calvarial Bone Defects, Curr. Pharm. Biotechnol 16 (2015) 655–660. 10.2174/138920101607150427112753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kanczler JM, Oreffo RO., Osteogenesis and angiogenesis: the potential for engineering bone, Eur Cell Mater. 15 (2008) 100–114. [DOI] [PubMed] [Google Scholar]
- [15].D’Mello S, Atluri K, Geary SM, Hong L, Elangovan S, Salem AK, Bone Regeneration Using Gene-Activated Matrices, AAPS J. 19 (2017) 43–53. 10.1208/s12248-016-9982-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Woo EJ, Adverse Events Reported After the Use of Recombinant Human Bone Morphogenetic Protein 2, J. Oral Maxillofac. Surg 70 (2012) 765–767. 10.1016/j.joms.2011.09.008. [DOI] [PubMed] [Google Scholar]
- [17].Shimer AL, Öner FC, Vaccaro AR, Spinal reconstruction and bone morphogenetic proteins: Open questions, Injury. 40 (2009) S32–S38. 10.1016/S0020-1383(09)70009-9. [DOI] [PubMed] [Google Scholar]
- [18].Kay MA, State-of-the-art gene-based therapies: the road ahead, Nat. Rev. Genet 12 (2011) 316–328. 10.1038/nrg2971. [DOI] [PubMed] [Google Scholar]
- [19].Evans CH, Gene therapy for bone healing, Expert Rev. Mol. Med 12 (2010) e18. 10.1017/S1462399410001493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Evans C, Gene therapy for the regeneration of bone, Injury. 42 (2011) 599–604. 10.1016/j.injury.2011.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Elangovan S, Karimbux N, Review Paper: DNA Delivery Strategies to Promote Periodontal Regeneration, J. Biomater. Appl 25 (2010) 3–18. 10.1177/0885328210366490. [DOI] [PubMed] [Google Scholar]
- [22].Evans CH, Gene delivery to bone, Adv. Drug Deliv. Rev 64 (2012) 1331–1340. 10.1016/j.addr.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].De la Riva B, Sánchez E, Hernández A, Reyes R, Tamimi F, López-Cabarcos E, Delgado A, Évora C, Local controlled release of VEGF and PDGF from a combined brushite–chitosan system enhances bone regeneration, J. Control. Release 143 (2010) 45–52. 10.1016/j.jconrel.2009.11.026. [DOI] [PubMed] [Google Scholar]
- [24].Bayer EA, V Fedorchak M, Little SR, The Influence of Platelet-Derived Growth Factor and Bone Morphogenetic Protein Presentation on Tubule Organization by Human Umbilical Vascular Endothelial Cells and Human Mesenchymal Stem Cells in Coculture, Tissue Eng. Part A 22 (2016) 1296–1304. 10.1089/ten.TEA.2016.0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tengood JE, Kovach KM, Vescovi PE, Russell AJ, Little SR, Sequential delivery of vascular endothelial growth factor and sphingosine 1-phosphate for angiogenesis, Biomaterials. 31 (2010) 7805–7812. 10.1016/j.biomaterials.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kim S, Kang Y, Krueger CA, Sen M, Holcomb JB, Chen D, Wenke JC, Yang Y, Sequential delivery of BMP-2 and IGF-1 using a chitosan gel with gelatin microspheres enhances early osteoblastic differentiation, Acta Biomater. 8 (2012) 1768–1777. 10.1016/j.actbio.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang S, Ju W, Shang P, Lei L, Nie H, Core–shell microspheres delivering FGF-2 and BMP-2 in different release patterns for bone regeneration, J. Mater. Chem. B 3 (2015) 1907–1920. 10.1039/C4TB01876A. [DOI] [PubMed] [Google Scholar]
- [28].Raiche AT, Puleo DA, In vitro effects of combined and sequential delivery of two bone growth factors, Biomaterials. 25 (2004) 677–685. 10.1016/S0142-9612(03)00564-7. [DOI] [PubMed] [Google Scholar]
- [29].Tengood JE, Ridenour R, Brodsky R, Russell AJ, Little SR, Sequential Delivery of Basic Fibroblast Growth Factor and Platelet-Derived Growth Factor for Angiogenesis, Tissue Eng. Part A 17 (2011) 1181–1189. 10.1089/ten.tea.2010.0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Aydin M, and Pulat R, 5-Fluorouracil encapsulated chitosan nanoparticles for pH-stimulated drug delivery: evaluation of controlled release kinetics., J. Nanomater 2012 (2012) 42. https://www.hindawi.com/journals/jnm/2012/313961/cta/. [Google Scholar]
- [31].Acri TM, Laird NZ, Geary SM, Salem AK, Shin K, Effects of calcium concentration on nonviral gene delivery to bone marrow-derived stem cells, J. Tissue Eng. Regen. Med 13 (2019) 2256–2265. 10.1002/term.2971. [DOI] [PubMed] [Google Scholar]
- [32].Moncal KK, Heo DN, Godzik KP, Sosnoski DM, Mrowczynski OD, Rizk E, Ozbolat V, Tucker SM, Gerhard EM, Dey M, Lewis GS, Yang J, Ozbolat IT, 3D printing of poly(ε-caprolactone)/poly(D,L-lactide-co-glycolide)/hydroxyapatite composite constructs for bone tissue engineering, J. Mater. Res 33 (2018) 1972–1986. 10.1557/jmr.2018.111. [DOI] [Google Scholar]
- [33].Liu W-F, Kang C-Z, Kong M, Li Y, Su J, Yi A, Cheng X-J, Chen X-G, Controlled release behaviors of chitosan/α, β-glycerophosphate thermo-sensitive hydrogels, Front. Mater. Sci 6 (2012) 250–258. 10.1007/s11706-012-0169-1. [DOI] [Google Scholar]
- [34].Zhen Y, Xu K, Chen X, Zhao C, Wang B, Zhang D, Wang H, Xu N, Yu J, Luo Q, [Embolization of aneurysm by chitosan-glycerophosphate-fibroblast tissue hydrogel, a tissue engineering material: experiment with rabbits]., Zhonghua Yi Xue Za Zhi. 89 (2009) 727–731. [PubMed] [Google Scholar]
- [35].Wu S, Zhou Y, Yu Y, Zhou X, Du W, Wan M, Fan Y, Zhou X, Xu X, Zheng L, Evaluation of Chitosan Hydrogel for Sustained Delivery of VEGF for Odontogenic Differentiation of Dental Pulp Stem Cells, Stem Cells Int. 2019 (2019) 1515040. 10.1155/2019/1515040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Qi B, Yu A, Zhu S, Chen B, Li Y, The preparation and cytocompatibility of injectable thermosensitive chitosan/poly(vinyl alcohol) hydrogel, J. Huazhong Univ. Sci. Technol. [Medical Sci 30 (2010) 89–93. 10.1007/s11596-010-0116-2. [DOI] [PubMed] [Google Scholar]
- [37].Moreira CDF, Carvalho SM, Mansur HS, Pereira MM, Thermogelling chitosan-collagen-bioactive glass nanoparticle hybrids as potential injectable systems for tissue engineering., Mater. Sci. Eng. C. Mater. Biol. Appl 58 (2016) 1207–1216. 10.1016/j.msec.2015.09.075. [DOI] [PubMed] [Google Scholar]
- [38].Rajan N, Habermehl J, Coté M-F, Doillon CJ, Mantovani D, Preparation of ready-to-use, storable and reconstituted type I collagen from rat tail tendon for tissue engineering applications, Nat. Protoc 1 (2007) 2753. 10.1038/nprot.2006.430. [DOI] [PubMed] [Google Scholar]
- [39].Moncal KK, Gudapati H, Godzik KP, Heo DN, Kang Y, Rizk E, Ravnic DJ, Wee H, Pepley DF, Ozbolat V, Lewis GS, Moore JZ, Driskell RR, Samson TD, Ozbolat IT, Intra-Operative Bioprinting of Hard, Soft, and Hard/Soft Composite Tissues for Craniomaxillofacial Reconstruction, Adv. Funct. Mater (2021). 10.1002/adfm.202010858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Nguyen H, Morgan DAF, Forwood MR, Sterilization of Allograft Bone: is 25 kGy the Gold Standard for Gamma Irradiation?, Cell Tissue Bank. 8 (2006) 81. 10.1007/s10561-006-9019-7. [DOI] [PubMed] [Google Scholar]
- [41].Moncal KK, Ozbolat V, Datta P, Heo DN, Ozbolat IT, Thermally-controlled extrusion-based bioprinting of collagen, J. Mater. Sci. Mater. Med 30 (2019) 55. 10.1007/s10856-019-6258-2. [DOI] [PubMed] [Google Scholar]
- [42].Noah EM, Chen J, Jiao X, Heschel I, Pallua N, Impact of sterilization on the porous design and cell behavior in collagen sponges prepared for tissue engineering, Biomaterials. 23 (2002) 2855–2861. 10.1016/S0142-9612(01)00412-4. [DOI] [PubMed] [Google Scholar]
- [43].Ozbolat IT, Chen H, Yu Y, Development of ‘Multi-arm Bioprinter’ for hybrid biofabrication of tissue engineering constructs, Robot. Comput. Integr. Manuf 30 (2014) 295–304. 10.1016/j.rcim.2013.10.005. [DOI] [Google Scholar]
- [44].S.M.& S.A.D. Chandrasekaran Arcot Ravindran, Jia Chan Yoke, Theng Choong Sheau, Muniandy Teeba, Invitro Studies and Evaluation of Metformin Marketed Tablets-Malaysia, Int. J. Pharm. Res. Technol 3 (2011) 5–8. https://pdfs.semanticscholar.org/ebbb/d1a12dbe21b93ecb0396098284d27296462a.pdf?_ga=2.60612973.1588088683.1575647787-83280966.1546442228. [Google Scholar]
- [45].K KE. and Subramanian A, Selective Modification of Chitosan to Enable the Formation of Chitosan-DNA Condensates by Electron Donator Stabilization, Int. J. Carbohydr. Chem 2011 (2011) 11. [Google Scholar]
- [46].Tan S-L, Ahmad TS, Selvaratnam L, Kamarul T, Isolation, characterization and the multi-lineage differentiation potential of rabbit bone marrow-derived mesenchymal stem cells., J. Anat 222 (2013) 437–450. 10.1111/joa.12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Moncal KK, Aydin RST, Abu-Laban M, Heo DN, Rizk E, Tucker SM, Lewis GS, Hayes D, Ozbolat IT, Collagen-infilled 3D printed scaffolds loaded with miR-148b-transfected bone marrow stem cells improve calvarial bone regeneration in rats, Mater. Sci. Eng. C 105 (2019). 10.1016/j.msec.2019.110128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Livak KJ, Schmittgen TD, Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method, Methods. 25 (2001) 402–408. 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- [49].Sample Size Calculations (IACUC), (n.d.). https://www.bu.edu/researchsupport/compliance/animal-care/working-with-animals/research/sample-size-calculations-iacuc/ (accessed December 5, 2021).
- [50].Jiaofeng S. Peng and Xinli Xing and Kemin Wang and Weihong Tan and Xiaoxiao He and Huang, Influence of Anions on the Formation and Properties of Chitosan-DNA Nanoparticles, J. Nanosci. Nanotechnol 5 (2005) 713–717. https://doi.org/doi: 10.1166/jnn.2005.091. [DOI] [PubMed] [Google Scholar]
- [51].Tran Huu H. Dung and Seung-Rok Lee and Suhk-Dong Han and Seon-Jeong Kim and Yeon-Mi Ju and Myong-Soo Kim and Yoo, Chitosan-TPP Nanoparticle as a Release System of Antisense Oligonucleotide in the Oral Environment, J. Nanosci. Nanotechnol 7 (2007) 3695–3699. https://doi.org/doi: 10.1166/jnn.2007.041. [DOI] [PubMed] [Google Scholar]
- [52].Janes KA, Calvo P, Alonso MJ, Polysaccharide colloidal particles as delivery systems for macromolecules, Adv. Drug Deliv. Rev 47 (2001) 83–97. 10.1016/S0169-409X(00)00123-X. [DOI] [PubMed] [Google Scholar]
- [53].Gaspar VM, Sousa F, Queiroz JA, Correia IJ, Formulation of chitosan{\textendash}{TPP}{\textendash}{pDNA} nanocapsules for gene therapy applications, Nanotechnology. 22 (2010) 15101. 10.1088/0957-4484/22/1/015101. [DOI] [PubMed] [Google Scholar]
- [54].Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG, Non-viral vectors for gene-based therapy, Nat. Rev. Genet 15 (2014) 541–555. 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- [55].Bayat A, Larijani B, Ahmadian S, Junginger HE, Rafiee-Tehrani M, Preparation and characterization of insulin nanoparticles using chitosan and its quaternized derivatives, Nanomedicine Nanotechnology, Biol. Med 4 (2008) 115–120. 10.1016/j.nano.2008.01.003. [DOI] [PubMed] [Google Scholar]
- [56].Kou L, Sun J, Zhai Y, He Z, The endocytosis and intracellular fate of nanomedicines: Implication for rational design, Asian J. Pharm. Sci 8 (2013) 1–10. 10.1016/j.ajps.2013.07.001. [DOI] [Google Scholar]
- [57].Li D-D, Pan J-F, Ji Q-X, Yu X-B, Liu L-S, Li H, Jiao X-J, Wang L, Characterization and cytocompatibility of thermosensitive hydrogel embedded with chitosan nanoparticles for delivery of bone morphogenetic protein-2 plasmid DNA, J. Mater. Sci. Mater. Med 27 (2016) 134. 10.1007/s10856-016-5743-0. [DOI] [PubMed] [Google Scholar]
- [58].Lucotti A, Tommasini M, Pezzoli D, Candiani G, Molecular interactions of DNA with transfectants: a study based on infrared spectroscopy and quantum chemistry as aids to fluorescence spectroscopy and dynamic light scattering analyses, RSC Adv. 4 (2014) 49620–49627. 10.1039/C4RA08845J. [DOI] [Google Scholar]
- [59].Fernandes LL, Resende CX, Tavares DS, Soares GA, Castro LO, Granjeiro JM, Cytocompatibility of chitosan and collagen-chitosan scaffolds for tissue engineering, PolÃ\-Meros. 21 (2011) 1–6. http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0104-14282011000100003&nrm=iso. [Google Scholar]
- [60].Takallu S, Mirzaei E, Azadi A, Karimizade A, Tavakol S, Plate-shape carbonated hydroxyapatite/collagen nanocomposite hydrogel via in situ mineralization of hydroxyapatite concurrent with gelation of collagen at pH = 7.4 and 37°C, J. Biomed. Mater. Res. Part B Appl. Biomater 107 (2019) 1920–1929. 10.1002/jbm.b.34284. [DOI] [PubMed] [Google Scholar]
- [61].Wegman F, van der Helm Y, Öner FC, Dhert WJA, Alblas J, Bone Morphogenetic Protein-2 Plasmid DNA as a Substitute for Bone Morphogenetic Protein-2 Protein in Bone Tissue Engineering, Tissue Eng. Part A 19 (2013) 2686–2692. 10.1089/ten.tea.2012.0569. [DOI] [PubMed] [Google Scholar]
- [62].Wegman J, F., Bijenhof A, Schuijff L, Oner FC, Dhert WJ and Alblas, Osteogenic differentiation as a result of BMP-2 plasmid DNA based gene therapy in vitro and in vivo, Eur Cell Mater. 21 (2011) 230–242. [DOI] [PubMed] [Google Scholar]
- [63].Kolambkar YM, Boerckel JD, Dupont KM, Bajin M, Huebsch N, Mooney DJ, Hutmacher DW, Guldberg RE, Spatiotemporal delivery of bone morphogenetic protein enhances functional repair of segmental bone defects, Bone. 49 (2011) 485–492. 10.1016/j.bone.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Yang J, Shi P, Tu M, Wang Y, Liu M, Fan F, Du M, Bone morphogenetic proteins: Relationship between molecular structure and their osteogenic activity, Food Sci. Hum. Wellness 3 (2014) 127–135. 10.1016/j.fshw.2014.12.002. [DOI] [Google Scholar]
- [65].Shah P, Keppler L, Rutkowski J, A Review of Platelet Derived Growth Factor Playing Pivotal Role in Bone Regeneration, J. Oral Implantol 40 (2014) 330–340. 10.1563/AAID-JOI-D-11-00173. [DOI] [PubMed] [Google Scholar]
- [66].Davies OG, Grover LM, Lewis MP, Liu Y, PDGF is a potent initiator of bone formation in a tissue engineered model of pathological ossification, J. Tissue Eng. Regen. Med 12 (2018) e355–e367. 10.1002/term.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Maynard RL, Downes N, Chapter 3 - Introduction to the Skeleton: Bone, Cartilage and Joints, in: Maynard RL, Downes N (Eds.), Anat. Histol. Lab. Rat Toxicol. Biomed. Res, Academic Press, 2019: pp. 11–22. 10.1016/B978-0-12-811837-5.00003-4. [DOI] [Google Scholar]
- [68].Ramamoorth M, Narvekar A, Non viral vectors in gene therapy- an overview, J. Clin. Diagn. Res 9 (2015) GE01–6. 10.7860/JCDR/2015/10443.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Cahill KS, McCormick PC, Levi AD, A comprehensive assessment of the risk of bone morphogenetic protein use in spinal fusion surgery and postoperative cancer diagnosis, J. Neurosurg. Spine SPI 23 (2015) 86–93. 10.3171/2014.10.SPINE14338. [DOI] [PubMed] [Google Scholar]
- [70].Devine JG, Dettori JR, France JC, Brodt E, McGuire RA, The use of rhBMP in spine surgery: is there a cancer risk?, Evid. Based. Spine. Care. J 3 (2012) 35–41. 10.1055/s-0031-1298616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Shang Q, Wang Z, Liu W, Shi Y, Cui L, Cao Y, Tissue-Engineered Bone Repair of Sheep Cranial Defects with Autologous Bone Marrow Stromal Cells, J. Craniofac. Surg 12 (2001). https://journals.lww.com/jcraniofacialsurgery/Fulltext/2001/11000/Tissue_Engineered_Bone_Repair_of_Sheep_Cranial.17.aspx. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper. Additional data related to this paper may be requested from the authors.